
Abstract
Oligodendrocyte progenitor cells (OPCs) play pivotal roles in myelin formation and phagocytosis, communicating with neighboring cells and contributing to the integrity of the blood–brain barrier (BBB). However, under the pathological circumstances of Alzheimer’s disease (AD), the brain’s microenvironment undergoes detrimental changes that significantly impact OPCs and their functions. Starting with OPC functions, we delve into the transformation of OPCs to myelin-producing oligodendrocytes, the intricate signaling interactions with other cells in the central nervous system (CNS), and the fascinating process of phagocytosis, which influences the function of OPCs and affects CNS homeostasis. Moreover, we discuss the essential role of OPCs in BBB formation and highlight the critical contribution of OPCs in forming CNS-protective barriers. In the context of AD, the deterioration of the local microenvironment in the brain is discussed, mainly focusing on neuroinflammation, oxidative stress, and the accumulation of toxic proteins. The detrimental changes disturb the delicate balance in the brain, impacting the regenerative capacity of OPCs and compromising myelin integrity. Under pathological conditions, OPCs experience significant alterations in migration and proliferation, leading to impaired differentiation and a reduced ability to produce mature oligodendrocytes. Moreover, myelin degeneration and formation become increasingly active in AD, contributing to progressive neurodegeneration. Finally, we summarize the current therapeutic approaches targeting OPCs in AD. Strategies to revitalize OPC senescence, modulate signaling pathways to enhance OPC differentiation, and explore other potential therapeutic avenues are promising in alleviating the impact of AD on OPCs and CNS function. In conclusion, this review highlights the indispensable role of OPCs in CNS function and their involvement in the pathogenesis of AD. The intricate interplay between OPCs and the AD brain microenvironment underscores the complexity of neurodegenerative diseases. Insights from studying OPCs under pathological conditions provide a foundation for innovative therapeutic strategies targeting OPCs and fostering neurodegeneration. Future research will advance our understanding and management of neurodegenerative diseases, ultimately offering hope for effective treatments and improved quality of life for those affected by AD and related disorders.
Similar content being viewed by others


Background
The cellular components of the mammalian central nervous system (CNS) include neurons and glial cells [1]. In the past, neurons were considered signaling cells, and glia were given an under-appreciated name suggesting that glial cells were merely the glue that held the cells together, keeping the nervous system’s architecture intact [2]. However, increasing evidence has shown the essential role of glial cells in the sophisticated structure and dynamics of neural networks [3, 4], including oligodendrocyte progenitor cells (OPCs), one of the precursor cells of glial cells [5]. OPCs, also known as oligodendrocyte precursor cells, NG2-glia, O2A cells, or polydendrocytes, are abundant in both the white matter and the gray matter of the adult CNS, and named for their essential role as a precursor to oligodendrocytes [6,7,8].
In the past, molecular mechanistic studies on Alzheimer’s disease (AD) were predominantly centered around amyloid plaques and neurofibrillary tangles (NFTs) [9,10,11]. The intricate interplay between OPCs and the pathogenesis of neurodegenerative disorders [12,13,14], particularly AD, has received significant attention in recent research [15,16,17,18]. OPCs, a type of glial cells that have long been considered merely precursors to oligodendrocytes, have emerged as a key player in various essential functions within the CNS [19]. In this review, we discuss the multifaceted roles of OPCs, including the crucial roles in myelin formation, cellular signaling within the CNS, phagocytic activity, and blood–brain barrier (BBB) formation and repair. Moreover, we delve into the deteriorating microenvironment in the AD brain, highlighting the alterations of OPC-related events and their potential implications in AD prevention and treatment.
Unveiling the intricate functions of OPCs may open new avenues for understanding the underlying mechanisms of neurodegeneration and identifying potential therapeutic targets. Several critical questions remain unanswered, warranting further investigation and exploration. (1) How does the dynamic interplay between OPCs and their surrounding environment influence OPC differentiation into myelin-forming oligodendrocytes? (2) How does the aging process affect OPC function? (3) Will approaches targeting OPC senescence-related changes provide potential therapeutic strategies for AD and related disorders?
By addressing these complex and unresolved questions, we can gain deeper insights into the intricate roles of OPCs in AD and pave the way for developing novel therapeutic approaches targeting these cells. This review aims to provide a comprehensive overview of the current knowledge regarding OPCs in AD and discuss the potential future directions for AD prevention and treatment from the perspective of OPC changes.
OPC functions
OPCs, known as CNS resident stem cells, originate from the ventricular zone of the brain and spinal cord, and proliferate and migrate to populate the CNS [20]. Strikingly, OPCs are distributed in the CNS and represent a group of migratory and proliferating adult progenitor cells that can differentiate into oligodendrocytes [19]. OPCs express A2B5, as well as oligodendroglial cell lineage markers Olig1, Olig2, Sox10, GPR17, and Nkx2.2, but they are typically characterized by PDGFR-α and NG2 [19, 21]. The most specific OPC marker is PDGFR-α, a receptor for platelet-derived growth factor (PDGF) A [22,23,24,25]. PDGF is the most potent OPC mitogen and survival factor produced by astrocytes, endothelial cells, and neurons [22,23,24,25,26]. The functions of OPCs include involvement in the processes of myelination [27], signal transmission [28] and synaptic pruning [29], and differentiation into other types of glial cells [30].
Cellular physiology of OPCs: proliferation and differentiation to myelinating oligodendrocytes
The primary function of oligodendrocytes is to produce myelin, the elongated cell membrane that tightly surrounds axons to provide support and insulation [19, 31]. Myelin sheaths provide electrical insulation to axons and allow faster nerve signal transmission [32]. In rodents, the optimal functionality of CNS myelin sheaths is observed when the thickness of the myelin sheath remains stable at a G-ratio (inner diameter/outer diameter) of 0.77 [33]. Deviations from this ratio may potentially lead to initiation and development of neurological disorders [34]. Mature oligodendrocytes can myelinate up to 50 axonal segments, although the actual number may vary depending on the specific regions in the CNS [35].
Initially, OPCs, which are multipotent cells, are present throughout the brain, including in the hippocampus and in all layers of the cortex [36]. During embryonic development, OPCs utilize blood vessels as a “pathway” to migrate in a single-cell fashion toward neurons, forming myelin sheaths [37]. During this process, the signaling of the CXC chemokine receptor CXCR2 plays a crucial role in regulating the number of OPCs and facilitating OPC migration [38, 39]. Myelination during development is an inherent process of CNS maturation guided by genetic programming [40]. After their rapid growth and spread during brain development, the cell division and movement rate of OPCs decreased substantially. However, the OPCs remain among the most actively dividing cell populations in the adult CNS [41].
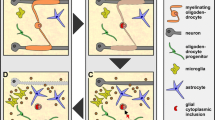
Under appropriate conditions, OPCs receive cues to proliferation and differentiate into oligodendrocytes. As shown in Fig. 1, the OPC differentiation to myelinating cells can be classified into four stages: proliferative OPC stage, pre-oligodendrocyte stage, differentiated oligodendrocyte stage, and myelinating cell stage. The transition between these stages is orchestrated through the intricate interplay of various molecular factors and signaling pathways [42, 43]. As a key step of myelination, OPC differentiation following OPC proliferation and migration are complicated and regulated by a large number of secreted signaling factors that are essential for myelination and remyelination [44,45,46]. Once OPCs receive differentiation signals, they undergo morphological and molecular changes [47]. The extended processes of OPCs are termed oligodendrocyte processes or lamellipodia, which develop and wrap around axons [19]. The processes gradually compact and form the myelin sheath, a fatty insulating layer surrounding the axon [48].

The process of oligodendrocyte progenitor cell (OPC) myelination varies during different stages. a During development, OPCs generated from neural stem cells rapidly form myelin sheaths, and a fraction of OPCs will be reserved in the stem cell pool. b In adulthood, upon demyelination, OPCs residing in the stem cell pool are swiftly recruited to the site of injury along the vasculature, where they proliferate and differentiate to initiate remyelination
During myelination, the OPCs undergo a maturation process characterized by expression of specific markers and activation of myelin-related genes [49, 50]. As mentioned previously, the myelin sheath wraps around axons of neurons, resembling the lamellipodia of a ‘liquid croissant’ enveloping the axon [51]. In this configuration, the inner tongue of the lamellipodia wraps around the axon faster than the outer tongue, thereby generating growth polarity and ultimately forming a multi-layered, closed-loop myelin sheath [52]. This process can be initiated by elevated concentrations of phosphatidylinositol-(3,4,5)-triphosphate [52]. The myelination process occurs in spatially and temporally determined sequences [53]. Remarkably, microglia can refine myelin sheaths during development by engaging in phagocytic activity, contributing to the functional modulation of neuronal activity [54].
OPCs are also essential in maintaining the population of oligodendrocytes in remyelination [55]. In response to injury or disease, OPCs differentiate into mature oligodendrocytes to replace damaged or lost myelin [5]. In adulthood, the capacity for migration along blood vessels can be reactivated following myelin damage, allowing OPCs to participate in myelin repair [56]. Remyelination is initiated in response to pathological conditions, including demyelination resulting from injuries, neuroinflammation, or diseases such as multiple sclerosis [57]. Fyn, which belongs to the Src family of kinases, plays a crucial role in facilitating the migration of OPCs by mediating the activation of cyclin-dependent kinase 5 through PDGF signaling. Fyn also contributes to reorganization of the actin cytoskeleton, an essential step in promoting efficient cell migration [58].
Unlike establishing stable and functional myelin during embryonic development, myelin regeneration aims to repair and replace damaged myelin to restore normal impulse conduction. However, the thickness of newly formed myelin sheaths during remyelination is usually thinner than those formed during development, which is one of the characteristics widely used to distinguish between developmental myelination and remyelination [59, 60]. Moreover, the OPCs that are involved in myelination and remyelination are different. The OPCs can be classified into neonatal OPCs (nOPCs) and OPCs that persist into adulthood, named adult OPCs (aOPCs). The expression profile of resting aOPCs is different from that of the nOPCs. However, aOPCs can be activated and revert to a nOPC-like transcriptome. However, the nOPCs generate new oligodendrocytes for myelination, and the aOPCs generate that for remyelination [59, 60].
OPCs possess strict intrinsic proliferation control mechanisms that promote proper cell proliferation and cease cell division while initiating differentiation at the appropriate time [61]. OPC proliferation necessitates the involvement of a diverse range of signaling molecules. Notably, the activation of extracellular signal-regulated protein kinase(s), phosphatidylinositol-3 kinase (PI3K), and p38 mitogen-activated protein kinase is known to trigger the swiftest cellular responses to growth and differentiation factors, as well as a myriad of external stimuli [62,63,64,65]. During myelination, the OPCs undergo an extensive and coordinated migration along blood vessels as their physical substrate [66, 67]. During this process, the intrinsic Wnt tone is an essential signal for attracting OPCs to the blood vessels, and the endothelial cells and their products inhibit OPC differentiation during migration [66, 67]. After that, the astrocytes and astrocyte-derived semaphorins 3a and 6a repel the OPCs from the blood vessels, allowing the OPCs to escape from the inhibitory endothelial niche [66, 67]. During the transformation from OPC proliferation to differentiation, several factors are involved in the OPC exit from cell cycle and initiation of differentiation, including the miR-297c-5p targets cyclin T2 (CCNT2), c-Myc, and E2F1 [44,45,46]. Overall, myelin generation from OPCs to mature oligodendrocytes is a tightly regulated and dynamic process critical for proper functioning of the CNS. Interestingly, it is essential to note that under certain circumstances, OPCs can also give rise to other types of glial cells, which suggests a robust stem cell-like nature of OPCs [6, 68].
Cellular physiology of OPCs: interaction with other cells in the CNS
OPCs play a role in signal transmission by interaction with other cell types, including neurons and glial cells [66, 69]. The established connections with neurons create an environment for optimal neuronal function [70, 71]. OPCs activate nearby neurons and modulate synaptic transmission by secreting glutamate [72]. The synaptic signaling between neurons and OPCs has been extensively documented in rodent brain regions and human white matter [6, 68, 70, 71]. The synaptic plasticity observed in the neuronal-OPC synapses is similar to that of neuronal-neuronal synapses. Rapid neuron-glial synaptic transmission has been identified between hippocampal neurons and NG2 cells, exhibiting activity-dependent modifications similar as long-term potentiation (LTP) observed in excitatory synapses, a hallmark of neuronal plasticity [73]. However, the distinction sets the neuronal-NG2 synapses apart from numerous other neuronal synapses, as the induction of LTP in these synapses is not accompanied by an increase in the permeability of AMPA receptors to Ca2+ [73].
Also, OPCs are critical in maintaining microglial homeostasis within the CNS. Depletion of OPCs leads to microglial activation and exacerbates neuroinflammation [74]. Interestingly, interactions between OPCs and microglia under neuroinflammatory conditions are enhanced [75]. OPCs are capable of producing chemokines that recruit and activate microglia. The interaction between OPCs and microglia is a crucial aspect of the immune response within the CNS [75].
Furthermore, there is an interconnection between OPCs and astrocytes, which has been implicated in brain diseases [76]. For instance, in mice and humans, a neuroglial signaling niche exists between reactive astrocytes and OPCs in white matter stroke [77]. Astrocytes regulate the expression of Sp1R3 on OPCs and promote OPC proliferation through Cx47 signaling in pharmacological analysis [78]. These findings imply that OPCs are “versatile relay stations” for various neuronal signaling pathways.
Cellular physiology of OPCs: immunomodulatory function
As previously mentioned, the reciprocal interactions regarding neuroinflammation between OPCs and microglial cells suggest that OPCs also play a role in the immune system of the CNS, and some studies have detected the immunomodulatory function of OPCs in CNS diseases [79, 80]. In the experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis (MS), interleukin (IL)-17 induces a cascade of crucial immune responses by interacting with OPCs within the CNS [81]. IL-17 induces a robust pathogenic inflammatory response in OPCs, leading to significantly elevated release of inflammatory cytokines and exacerbating disease progression [81]. Moreover, in a disease context, OPCs exhibit enhanced expression of genes involved in antigen processing and presentation via major histocompatibility complex (MHC)-II, suggesting the immunomodulatory function of OPCs [82]. In addition, the study also found that OPCs have phagocytosis capacity and MHC-II-expressing OPCs contribute to the activation of memory CD4+ T cells, suggesting that OPCs may work as an active immunomodulator in MS [82].
However, the CNS environment in demyelination is detrimental for OPCs. The upregulation of interferon-gamma (IFNγ) increases immunoproteasomes and MHC-I molecules, further reducing OPC numbers. These changes cause antigen cross-presentation by OPCs to cytotoxic CD8+ T cells, resulting in OPC death [83]. Interestingly, OPCs lacking low-density lipoprotein receptor-related protein 1 (LRP1) in the demyelinated CNS exhibit an anti-inflammatory solid phenotype [80]. The LRP1-deficient OPCs display impaired antigen presentation mechanisms, indicating the inability of the inflammatory response to propagate, thus facilitating faster remyelination and neuroprotection [80]. Furthermore, within the demyelinated environment, OPCs are activated through increased expression of chemokine (C–C motif) ligand 2 and IL-1β to enhance their responsiveness to injury [84]. These changes increase OPC motility and terminal differentiation, contributing to remyelination following demyelinated insults [84]. Together, these observations underscore the essential role of OPCs in immune regulation, neural protection, and the modulation of inflammatory environments.
Cellular physiology of OPCs: phagocytosis
OPCs are not traditionally considered phagocytic cells, but recent studies have suggested that OPCs are capable of engulfment and pruning under certain conditions [69]. Most of the structural pruning of neurons is believed to be primarily mediated by microglia [85, 86]. However, it has also been found that OPCs possess phagolysosomes and are even more abundant than microglia [69]. In a previous study, high-resolution transmission electron microscopy images showed that the developing mouse cortical OPCs actively participate in axonal pruning [69]. Furthermore, the findings from single-nucleus RNA sequencing demonstrated that OPCs at this stage express crucial phagocytic genes and neuronal transcripts, aligning with active axonal engulfment [69].
Further research revealed that the OPC-mediated synaptic pruning is involved in responses to sensory experience in the developing and adult mouse visual cortex by facilitating thalamocortical synaptic pruning [29]. Therefore, the phagocytic function of OPCs appears to be enhanced by sensory experiences, such as visual deprivation/stimulation paradigms [29]. Interestingly, while participating in shaping neural circuits in the brain, OPCs are also subject to phagocytosis by microglial cells, leading to the formation of intricately refined myelin sheaths and synapses [87]. Notably, NG2 cells cluster around amyloid plaques in APP/PS1 mice and clear β-amyloid peptides through endocytic and autophagic processes in response to the initiation of AD [88]. These findings have opened up new research avenues to better understand the role of OPCs in the brain.
Cellular physiology of OPCs: contribution to BBB formation and repair
Previous studies suggested that the BBB primarily comprises endothelial cells, glial cells, and pericytes [89]. However, OPCs have been considered as a novel component of the BBB in recent studies [90, 91]. Perivascular OPCs are connected to the brain vasculature endothelial cells through the basement membrane. Therefore, the OPCs are now recognized as an additional cellular constituent of the BBB [91]. OPCs maintain the integrity of BBB by upregulating the expression of tight junction proteins in endothelial cells through TGF-β signaling [92]. During embryonic development, OPCs arise between the perivascular cells and glial cells surrounding the blood vessels, directly participating in the formation of the BBB [91]. Once the BBB is formed, endothelial cells and perivascular cells regulate the proliferation, survival, and differentiation of OPCs by releasing nutrients and signaling molecules [93, 94]. The vascular endothelial growth factor A secreted by endothelial cells regulates the migration of OPCs, which is crucial for the relocation and functions of OPCs [95].
Conversely, OPCs release regulatory factors that modulate the proliferation of perivascular cells and the expression of functionally relevant proteins in endothelial cells [93]. Under normal conditions, most OPCs reside in the parenchymal regions of the adult brain. However, when the BBB is compromised, parenchymal OPCs transform towards perivascular OPCs, significantly increasing perivascular OPCs and the accompanying vascular regeneration [91]. These findings imply that maintaining normal OPC functions is essential during embryonic BBB development, and OPCs play a crucial role in interacting with other vascular cells and spatial migration for the preservation and protection of BBB, and repair of BBB damage.
AD pathology and harsh microenvironment
AD is the most common neurodegenerative disorder [96]. With the aging of the global population, AD has become a significant burden on the global healthcare system and the leading cause of death among adults older than 65 [97, 98]. AD is characterized by specific neuropathological and biomarker changes, including extracellular accumulation of amyloid-beta (Aβ) peptides and intraneuronal NFTs formed by aggregation of hyperphosphorylated or abnormally-phosphorylated tau proteins [99, 100]. The amyloid hypothesis posits that the Aβ accumulation initiates AD pathogenesis, leading to NFTs, neuronal dysfunction, and dementia [10]. The Tau hypothesis poses that tau hyperphosphorylation induces tau dissociation from the microtubules and aggregation into NFTs, which initiate AD pathology [10]. However, the amyloid and tau hypotheses support that mitochondrial dysfunction, excessive release of neuroinflammatory cytokines, gliosis, and oxidative stress exacerbate AD pathology and induce a harsh microenvironment around cells [10]. The harsh environment affects the cellular physiology of OPCs and neuronal myelination [101].
At the cellular level, the microenvironment can be understood as the extracellular matrix, neighboring cells, bioactive agents such as cytokines, and mechanical forces that collectively influence the functioning of the individual cell [102]. Neuroinflammation, oxidative stress, and mitochondrial damage are significantly elevated in the altered microenvironment of the AD brain [103]. In the presence of multiple pathological features, the microenvironment of the AD brain becomes intricately complex.
Neurotoxicity-related neuroinflammation, oxidative stress, and cytokine release are pathological factors in AD [104, 105]. The accumulation of Aβ and NFTs, along with neuroinflammation and oxidative damage, leads to progressive neurodegeneration [10, 106]. Neuroinflammation is a crucial complex biological process in age-related cerebrovascular and neurodegenerative diseases, such as brain ischemia, AD, and Parkinson’s disease [107]. Studies in AD mouse models have confirmed the significant involvement of neuroinflammatory responses, which induce elevated levels of peripheral inflammatory cytokines and chemokines [108]. AD-related inflammatory components that contribute to neuroinflammation include microglia and astrocytes in the brain, the complement system, and various cytokines and chemokines [109]. The secretion of pro-inflammatory cytokines such as IL-6, IL-1 and tumor necrosis factor-alpha accelerates the progression of inflammation [110]. Interestingly, microglial activation contributes to the clearance of Aβ via phagocytosis and degradation [11]. However, prolonged activation of brain immune cells leads to the release of pro-inflammatory cytokines, initiating an inflammatory cascade and ultimately exacerbating neurodegeneration and neuronal death [111].
Furthermore, oxidative stress serves as an early event in AD and is also regarded as a primary factor contributing to the formation of NFTs in AD [112, 113]. Studies have demonstrated that throughout the disease, the brains of AD patients are exposed to elevated oxidative stress, resulting in lipid oxidation, protein oxidation, a further decline in antioxidant capacity, and increased susceptibility of high-energy-demanding neurons to cell death [112, 114]. Mitochondrial dysfunction drives oxidative stress and inflammation in cells of the brain, shaping the challenging microenvironment in brain injuries and neurodegenerative diseases [115, 116]. Aging neurons exhibit severe mitochondrial fragmentation and cell death, leading to calcium overload and exacerbating microenvironmental deterioration [117].
Moreover, it is crucial to note that mitochondrial dysfunction and structural alterations are prominent features of AD [118]. In AD, there is an aberration in mitochondrial dynamics, shifting from fusion to fission, leading to excessive mitochondrial fragmentation [119]. Additionally, the activities of enzymes related to the oxidative phosphorylation, such as cytochrome c oxidase and pyruvate dehydrogenase, are attenuated, indicating decreased ATP production and mitochondrial dysfunction [120]. The structural and functional changes caused by mitochondrial fragmentation lead to release of contents such as mitochondrial DNA (mtDNA) and reactive oxygen species (ROS), which can act as damage-associated molecular patterns (DAMPs) recognized by pattern recognition receptors. The increased DAMPs trigger excessive activation of immune cells in the brain, including astrocytes and microglia, leading to the release of inflammatory cytokines [121].
Traditionally, astrocytes and microglia are the primary immune cells in the CNS [10]. Previous studies have divided the activated astrocytes and microglia into two phenotypes: the neurotoxic pro-inflammatory subtypes A1 (astrocytes) and M1 (microglia), as well as the neuroprotective anti-inflammatory subtypes A2 (astrocytes) and M2 (microglia) [122, 123]. However, single-cell sequencing technology did not detect the existence of classic M1 and M2 phenotypes in vivo [124, 125]. Instead, single-cell analysis has revealed multiple subtypes as well as cellular heterogeneity of microglia and astrocytes [126,127,128]. Although microglia and astrocytes exhibit changes in transcriptional profile, morphology, and function in diseases, these alterations are similar across various neurodegenerative diseases [128]. Therefore, the activated microglia and astrocytes are called “disease- or degeneration-associated microglia (DAM)” or “disease- or degeneration-associated astrocytes (DAA)” [128]. Like in other neurodegenerative diseases, DAM and DAA have also been identified through single-cell sequencing technology in AD model mice [129, 130]. The pro-inflammatory DAM contribute to the harsh microenvironment by releasing pro-inflammatory cytokines and appear at early AD disease stages, demonstrating better predictive value for pathology even before a cognitive decline occurs [131]. The pro-inflammatory DAM are characterized by expression of pro-inflammatory genes (e.g., Tlr2, Ptgs2, Il12b, and Il1b), while the anti-inflammatory DAM are characterized by phagocytic genes (Igf1, Apoe, and Myo1e) [131]. The DAAs exhibit similar transcriptomics as the DAMs and are observed also before AD-related cognitive decline [129].
A recent study revealed that pathogenic tau proteins can enter microglia, leading to mitochondrial dysfunction and subsequent leakage of mtDNA [132]. This detrimental process exacerbates the microenvironment, further impairing neuronal repair and regeneration [132]. Also, the impairment of the BBB contributes to the onset of the disease [133]. The accumulation of Aβ at sites of vascular leakage leads to inflammatory reactions and cytotoxicity, further exacerbating BBB permeability and accelerating the deterioration of the cellular microenvironment, thus advancing the pathological progression of AD [134]. Therefore, inflammation is not only a passive consequence of the AD process, but also a contributing factor to exacerbating AD pathology [135]. For example, excessive inflammatory cytokine release leads to cellular senescence in various cell types, including OPC senescence [136]. In the process of brain aging in AD, there is an accumulation of iron, which can be attributed to exposure to low levels of H2O2, leading to worsening of oxidative stress and an impairment of cellular function, further deteriorating the microenvironment of the AD brain [137, 138]. It is worth noting that mechanical force changes also contribute to the deterioration of the microenvironment surrounding OPCs. With age, the “niche” of the brain becomes stiffer, and these mechanical alterations are substantial enough to result in age-related functional decline in OPCs [139]. These findings suggest that the pathological changes in AD are exceedingly complex, wherein the mitochondria-associated changes and excessively activated glial cells form a microenvironment in the brain that fosters a decline in cellular vitality and facilitates aging.
Alterations in OPC-related events under pathological circumstances of AD
Both OPCs and oligodendrocytes are closely associated with inflammatory pathology and the toxicity of Aβ, highlighting their involvement in the disease process [140]. Under the harsh microenvironment, as previously mentioned, the biological activity of OPCs is reduced, and the possibility of OPC senescence is significantly increased [141]. The alterations of cellular activity of OPCs under AD pathology primarily result in impaired neuronal axonal health and compromised myelin sheath formation (Fig. 2).

AD pathology and alterations in OPC-related events under pathological circumstances of AD. Under AD pathological conditions, alterations in mitochondrial structure and function result in an overproduction of reactive oxygen species (ROS), leading to oxidative damage of subcellular structures and fragmentation of mitochondria. The oxidative damage and mitochondrial fragmentation further induce inflammatory responses, involving changes in microglial and astrocyte phenotypes. Pro-inflammatory DAM type microglia and DAA type astrocytes release pro-inflammatory cytokines, exacerbating mitochondrial dysfunction and oxidative stress, culminating in a vicious cycle termed “mitochondrial dysfunction-oxidative stress-inflammation response.” Consequently, these changes exacerbate the AD pathology. Within unfavorable cerebral microenvironment, OPC myelination-related events are adversely affected to varying degrees
Changes in OPC migration and proliferation
OPCs migrate to the injured area before undergoing proliferation [47]. Insufficient migration of OPCs to the injured area following demyelination is a determining factor that hinders the timely repair of myelin [142]. Currently, there is a dearth of research on the migratory capacity of OPCs in the AD brain. However, the migration ability of neural stem cells (NSCs), which also possess pluripotency and have the ability to differentiate into OPCs, is compromised due to the senescence of NSCs, suggesting the possibility of impaired OPCs in AD due to cellular senescence [143,144,145]. Intriguingly, a study on MS, a demyelinating disease that shares similar environments with AD, has found a decline in OPC migration [56, 146]. Interestingly, in the context of MS, OPCs in the vicinity of blood vessels exhibit a phenomenon known as “clustering”, where the OPCs fail to migrate individually along the blood vessels to the demyelinated sites for myelin repair [56]. However, the underlying reasons for OPC clustering around blood vessels and whether a similar phenomenon of blood vessel-associated OPC clustering exists in AD remain unclear [146]. Few studies have investigated OPC migration in the context of AD. Therefore, the impact of OPC migration on AD remains uncertain, necessitating further research to fill the theoretical gap.
Under normal conditions, myelin sheath production relies on the relentless proliferation of OPCs derived from the pool of stem cells [147]. Studies in mice and rats have indicated that aging decreases the OPC proliferation efficiency [139, 148,149,150]. However, research on monkeys suggests that the efficiency of OPC proliferation remains consistent throughout their lifespan, without any significant decrease observed [151]. In AD pathology, the changes in OPC proliferation become more intricate, with different studies reporting distinct patterns of change (Table 1).
An early study has indicated that amyloid plaque deposition increases the proliferation of microglia around plaques but does not affect the proliferation of cortical OPCs in double-transgenic AD mice at different ages [152]. Recent investigations, nevertheless, have yielded divergent findings that deviate significantly from the previous study. Between 6 and 9 months of age, there was a notable rise in the population of oligodendrocyte lineage Olig2+ cells within the cortical gray matter of APP/PS1 mice, coinciding with the emergence of myelin abnormalities [153]. Furthermore, at as early as 2 months of age, an increase in OPCs was observed in the hippocampus of AD mice, concurrent with a prevalent thickening of myelin [154]. Likewise, an upregulation of NG2+ cells was reported in the temporal cortex of the 6-month-old APP/PS1 mice, which coincided with a downregulation of myelin basic protein (MBP) [155]. NG2+ cells increase in the cortex and cluster around amyloid plaques at 14 months of age [88].
Furthermore, the NG2+ OPCs can internalize and degrade Aβ in culture, supporting their potential involvement in Aβ clearance [88]. These findings suggest that the early proliferation of OPCs in animal models of AD may represent a compensatory response to myelin damage and cytotoxicity of Aβ [156]. However, the increased OPC proliferation does not hold in older AD mice, as the number of OPCs in the hippocampus of 24-month-old 3×Tg-AD mice is significantly lower than that in 6-month-old counterparts [17]. It is evident that with the progression of AD pathology, cellular senescence becomes more pronounced, leading to a significant decline in cell proliferation [18].
The studies mentioned above collectively indicate that the proliferation of OPCs in AD pathology exhibits temporal and spatial variations [17, 18]. Initially, OPC proliferation may be upregulated as a response to counteract the progression of AD pathology. In the later stages, due to the deteriorating brain environment and the declined regenerative capacity [18], the proliferative ability of OPCs diminishes [17, 18].
To summarize, the varying results of the studies mentioned above may largely be attributed to the limited specificity of the antibodies used, with many studies using NG2 or Olig2 as a marker for OPCs. It should be noted that the NG2+ cells may potentially represent differentiating OPCs or a subset of cells transitioning into other glial cell types, while Olig2 marks the entire oligodendrocyte lineage [19, 68, 157, 158]. Although further research is warranted to elucidate the impact of AD pathology on OPC proliferation and migration, OPC proliferation and migration could be a potential therapeutic target for AD prevention and treatment.
Impaired OPC differentiation in AD
OPCs serve as a reservoir of oligodendrocytes, which is crucial for myelin formation and repair [159]. In the pathological context of AD, demyelination is a significant element [160]. The first connection between neurodegeneration in AD and the susceptibility of myelin-forming cells lies in the accumulation of Aβ peptides, which induce dysfunction and death of OPCs and mature oligodendrocytes in vitro and in vivo [161]. Furthermore, emerging scientific investigations have shed light on the interplay between demyelination of neurons and impaired differentiation of OPCs [47]. Due to the prevalent oxidative and carbonyl stress in AD, differentiation of OPCs, which inherently require a high energy supply and are associated with a limited antioxidant capacity, becomes further constrained [14]. These findings imply that efficient differentiation of OPCs is equally important as proliferation in AD.
A study examining the RNA expression profile of post-mortem brain tissues from dementia cases revealed disruption of signaling pathways, contributing to the decline of OPC differentiation [162]. Similar disruptions of signaling pathways involved in OPC differentiation and migration (such as PDGF-2 A and FGF-2) have been observed in MS. These disruptions ultimately lead to a blockade of differentiation during the chronic phase [146, 163]. Additionally, extracellular myelin debris derived from damaged oligodendrocytes inhibits OPC differentiation, and AD is characterized by abundance of such debris [160, 164].
Taken together, under the pathological conditions of AD, the differentiation of OPCs is susceptible to the deterioration of the microenvironment, resulting in a reduction of OPC differentiation capability. Therefore, further research is needed to elucidate the balance between OPC proliferation and differentiation under AD conditions.
OPC-mediated demyelination and remyelination in AD
Cutting-edge neuroimaging research has greatly advanced our understanding of AD by revealing the presence of demyelination in the white matter. The demyelination process, characterized by a loss of protective myelin sheaths, directly impacts synaptic function as well as learning and memory abilities [160, 165]. In a recent study, a significant loss of pre-existing myelin and increased oligodendrogenesis and remyelination were observed in an AD mouse model, suggesting the existence of both myelin degeneration and remyelination in AD progression [166]. However, the overall levels of myelination were decreased, indicating that although the rate of myelin formation increases in AD, it is difficult to compensate for the increased myelin degeneration rate [166]. Therefore, approaches that could enhance myelination or affect the balance between myelin degeneration and formation may be promising for AD treatment. Further investigations are warranted to explore the balance between myelin degeneration and remyelination under AD.
Moreover, there is evidence for a correlation between focal demyelination and the presence of Aβ plaques, further emphasizing the link between demyelination and AD [167]. Interestingly, a recent study reported that myelin dysfunction and demyelination injury are also risk factors for Aβ plaque formation in AD (Fig. 3) [168]. Mechanistically, myelin dysfunction may exacerbate the accumulation of the Aβ-producing machinery within axonal swellings and cause increased cleavage of cortical amyloid precursor protein (APP) [168]. In addition, the microglia originally responsible for Aβ clearance are increasingly recruited to demyelination sites, reducing Aβ clearance [168]. Also, recent studies have suggested that the senescence of OPCs plays a significant role in the etiology of demyelination in the contexts of AD and MS [18, 169], suggesting OPC senescence as a potential target for alleviating AD pathology.

Myelin dysfunction drives Aβ deposition. Myelin dysfunction and demyelination injury are also upstream risk factors contributing to formation of Aβ plaques in AD. Mechanistically, myelin dysfunction may exacerbate the accumulation of the Aβ-producing machinery within axonal swellings and cause the increased cleavage of cortical amyloid precursor protein. In addition, the microglia originally responsible for Aβ clearance are increasingly drawn to demyelination sites, reducing Aβ clearance
OPC senescence
Cellular senescence refers to a state in which cells are blocked in the G1 phase, unable to proliferate and unresponsive to external stimuli, resulting in the inability to perform normal functions [170]. Cellular senescence is closely associated with the aging of the organism, contributing to cellular functional decline [171]. The senescence-associated secretory phenotype (SASP) is characterized by the extensive release of pro-inflammatory cytokines, cytotoxic mediators, matrix metalloproteinases, and reactive oxygen species. The release of these factors further affects neighboring cells, inducing cellular senescence in the surrounding microenvironment and perpetuating a vicious cycle that accelerates the AD process [172, 173].
Compared to young mice, aged mice exhibit a higher abundance of cellular senescence markers in the brain, indicating increased senescent cells [174, 175]. Previous studies have identified various forms of cellular senescence in neurodegenerative diseases, and these senescent cells have a profound impact on the transmission of signals between neurons and the maintenance of neuronal structures [176, 177].
A human study found that patients diagnosed with late-onset AD had a significantly higher average count of senescent OPCs in the inferior parietal cortex than patients with mild cognitive impairment (MCI) [18]. The number of senescent OPCs in MCI patients was higher compared to the control group without dementia, although the difference was not statistically significant [18]. The results of single-cell sequencing in the frontal cortex indicate a negative correlation between the number of OPCs and amyloid protein [178]. These findings confirmed that patients at a late stage exhibit a decline in white matter integrity and increased cellular senescence compared to the healthy control group [160]. Further investigations on an AD mouse model revealed that Olig2 and NG2 expression in Aβ plaque-associated OPCs exhibited an aging-like phenotype [18]. This change was characterized by the upregulation of p21/CDKN1A and p16/INK4/CDKN2A proteins, and increased activity of the senescence-associated β-galactosidase [18]. The secretion profile associated with cellular senescence, known as SASP, includes a substantial amount of pro-inflammatory cytokines, cytotoxic mediators, metalloproteinases, and reactive oxygen species. These substances have the potential to impact neighboring cells, triggering their senescence and initiating a vicious cycle that accelerates the aging process [173]. According to a previous study, OPCs are required for the maintenance of microglial homeostatic state, and loss of OPCs abolishes the homeostatic microglial state [157], which further promotes the transformation of more microglial cells into the pro-inflammatory type, exerting harmful effects on a more significant number of neurons [179,180,181,182]. Therefore, these findings suggest that the loss of OPC function caused by cellular senescence contributes to aging and neurodegenerative diseases. Overall, these studies emphasize the essential role of OPC senescence in forming AD-associated pathology and progression.
Adaptive myelination in AD
Adaptive myelination, often called activity-dependent myelin plasticity in adulthood, significantly contributes to cognitive function [183]. Experience-driven changes in oligodendrocyte generation are essential for memory consolidation [184], working memory [185], spatial learning [186], and contextual fear memory [187]. These mechanisms are associated with the regulation of OPC proliferation, oligodendrocyte generation, and myelin formation by neuronal activity [188, 189]. However, single-cell sequencing conducted in oligodendrocytes under AD pathology has revealed alterations in genes associated with ion channels and glutamate receptor subunit genes related to neural activity sensing and regulation [190, 191]. Furthermore, adaptive myelination is impaired in a mouse model of chemotherapy-related cognitive impairment. This impairment and associated cognitive deficits can be rescued by the action of a small-molecule TrkB agonist on OPCs [192]. Building upon the previous discussion on myelin loss in AD, it appears that in AD, the adaptive myelination is compromised, potentially linked to weakened signaling between the oligodendrocyte lineage and neuronal activity. However, further research is needed to establish the role of OPCs in this context.
Current therapeutic approaches targeting OPCs in AD
Over the past few decades, the Aβ and tau protein aggregation hypotheses have been considered the mainstream hypotheses for the pathogenesis of AD and have been extensively targeted as primary therapeutic targets in clinical trials [9, 10]. Unfortunately, to date, nearly all of the interventions targeting Aβ and tau protein hyperphosphorylation in AD have failed in phase III clinical trials [193,194,195], including immunotherapies, drugs reducing Aβ production or enhancing Aβ clearance, as well as drugs inhibiting tau phosphorylation or aggregation in these years [196,197,198]. Although Lecanemab, a humanized IgG1 monoclonal antibody targeting Aβ, is approved by the US Food and Drug Administration (FDA) for the treatment of MCI or mild dementia, its efficacy and safety remain controversial [199,200,201,202]. Therefore, developing new therapeutic targets for AD is still needed. Interestingly, an increasing body of research indicates that OPCs undergo functional dysregulation in the pathological state of AD, suggesting OPCs as a potential therapeutic target for AD [18, 148, 203]. In the subsequent discussion, we will discuss several existing OPC-based therapeutic approaches for AD (Table 2).
Senolytic and rejuvenation strategies
In recent years, emerging evidence suggests the therapeutic potential of senescent OPC clearance in AD treatment [18, 176, 204]. Senolytics are a class of compounds that can selectively clear senescent cells by inducing apoptosis of senescent cells [18, 176, 204, 205]. The administration of senolytic treatment effectively targets and eliminates senescent OPCs in the Aβ plaque microenvironment of AD mice [18].
Furthermore, rejuvenation strategies are another widely studied AD therapy [139]. In neurodegenerative diseases, the decline in the functionality of stem cells and progenitor cell populations is a significant cause of reduced tissue regeneration capacity, wherein OPCs are mainly affected [139]. Interestingly, a previous study found that the OPC microenvironment determines OPC senescence and aging [139]. For example, the transfer of aged OPCs into the stem cell pool of young rats significantly improves OPC functionality [139], suggesting that the microenvironment affects cellular senescence. Consistently, another study found that the young cerebrospinal fluid restores oligodendrogenesis and long-term memory consolidation in aged mice via Fgf17 [206]. These studies highlight the importance of mitigating OPC aging for improving AD. Therefore, some studies found that enhancing the expression of the anti-aging gene KLOTHO attenuates amyloid and tau pathology and alleviates cognitive deficits [207,208,209]. The potential underlying mechanisms include promoting OPC maturation and enhancing remyelination [210,211,212].
Strategies targeting OPC differentiation-associated signaling pathways
Previous studies have found that impaired OPC differentiation is the leading cause of remyelination failure in various neurogenerative diseases, including multiple system atrophy, amyotrophic lateral sclerosis, and MS [169, 213,214,215]. OPC differentiation-promoting therapies exhibit excellent potential in restoring CNS remyelination. For instance, treatment with metformin significantly improves remyelination in aged animals by restoring the regenerative capacity of aged OPCs. Further, metformin promotes the responsiveness of aged OPCs to pro-differentiation signals, suggesting the essential role of OPC differentiation in remyelination [148]. Similarly, several studies have focused on enhancing the signaling pathways involved in OPC differentiation in AD that contains demyelinating environment (Fig. 4). For instance, the signaling pathway of nuclear receptor retinoid X receptor (RXR) upregulates the expression of ATP-binding cassette transporter A1 (ABCA1) and apolipoprotein E (ApoE), thereby directly enhancing the maturation of OPCs and oligodendrocytes, and improving AD-related cognitive function [216, 217]. The interaction between RXR, ABCA1 and ApoE plays a crucial role in modulating lipid metabolism, cholesterol transport, and the pathogenesis of AD [218]. Moreover, RXR activation promotes Aβ clearance, inhibits Aβ generation, modulates neuronal function, and exerts anti-inflammatory actions [219, 220]. RXR agonists activate RXR/LXR and PPAR/RXR heterodimers, promote ABCA1 and ApoE mRNA expression in cells, reduce Aβ levels, and thereby reverse cognitive impairments in AD [221, 222]. Furthermore, RXR agonists enhance the differentiation of OPCs into mature oligodendrocytes, which accelerates CNS remyelination [217]. In addition, RXR activation can also promote remyelination by inducing monocytes, macrophages and microglia to clear myelin debris [223]. RXR serves as a clinically relevant target, and the FDA-approved RXR agonist, bexarotene, has been associated with OPC maturation and remyelination in stroke mice, and cognitive recovery in AD mice [216, 224].

Strategies targeting OPC differentiation-associated signaling pathways in AD. Strategies targeting OPC differentiation-associated signaling pathways include the mammalian target of rapamycin (mTOR) signaling agonist and retinoid X receptor (RXR) signaling agonist. a Streamline illustration. b Detailed molecular pathway diagrams: The mTOR inhibitor promotes OPC differentiation by downregulating p-mTOR and promoting autophagy-related genes. Elevated autophagy enhances OPC differentiation and myelination. RXR activation promotes Aβ clearance, inhibits Aβ generation, and modulates neuronal function. In addition, the interaction between RXR, ABCA1, and ApoE plays a crucial role in modulating cholesterol transport genes. The enhanced cholesterol transport gene expressions promote OPC differentiation
The PI3K/Akt/mammalian target of rapamycin (mTOR) signaling cascade is also an effective target. Donepezil, an FDA-approved acetylcholinesterase inhibitor (AChEI) used in the treatment of AD, has been shown to promote OPC differentiation into oligodendrocytes, enhance myelination, and upregulate myelin-specific proteins [204, 225]. Another compound that acts on OPCs through the mTOR pathway is Clemastine. It is also an FDA-approved antihistamine drug. Clemastine can inhibit OPC aging by downregulating p-mTOR and promoting OPC differentiation, leading to enhancement of autophagy, improved myelin integrity, and reduction in Aβ deposition over long-term treatment, thereby enhancing overall outcomes [15, 166]. Furthermore, the activation of PI3K agonists has been shown to improve peripheral muscle-nerve regeneration [226]. This effect holds promise for alleviating the decline in motor function observed in patients with AD [227]. These studies collectively indicate the significance of targeting and enhancing OPC differentiation signaling pathways in AD therapy.
Other potential therapeutic approaches
Nano-delivery systems
Nano-delivery systems possess finely-tuned physicochemical properties and can be widely used to enhance drug delivery by ameliorating unfavorable drug characteristics, enhancing permeability, and improving tissue distribution and in vivo metabolism [228, 229]. A nanoparticle-based strategy has been developed to enhance the differentiation of OPCs into mature oligodendrocytes capable of repairing myelin. This strategy involves the construction of poly(lactic-co-glycolic acid) nanoparticles with a diameter of approximately 120 nm, which are surface-functionalized with NG-2 antibodies. In a previous study, this nanoparticle was used to deliver leukemia inhibitory factor, a promoter of myelination, to promote OPC differentiation into mature oligodendrocytes and enhance myelin repair [230].
Exosomes
Exosomes are small vesicles derived from the cell membrane of various living cells that are released into body fluids and circulate throughout the body [231,232,233]. Previous research has indicated that exosomes derived from mesenchymal stem cells (MSC-Exo) have shown potentials to enhance neuroregeneration and promote functional recovery in animal models of CNS disease and injury [234,235,236,237]. MSC-Exo can cross the BBB in demyelinated mouse models and target neural cells. The MSC-Exo administration significantly increases mature OPCs, elevates the levels of MBP, reduces neuroinflammation by promoting the M2 phenotype of microglial cells while suppressing the M1 phenotype and their associated cytokines, and decreases the level of APP [238]. Currently, a specific exosome targeting OPCs contains a lentivirus with a PDGFRα ligand capable of anchoring to the membrane [239, 240]. This targeted approach towards OPCs in the affected areas significantly enhances the protective capacity of myelin sheaths and promotes OPC differentiation [239, 240].
AMPK activators
Several AMPK activators also hold therapeutic potential for AD [241]. For example, disrupted OPC differentiation has been observed in the aging brain [242]. Interventions such as the administration of metformin or dietary restrictions have shown the ability to alleviate the pathological changes of OPCs and restore the regenerative capacity [148]. These changes presumably result from enhanced mitochondrial function and remyelination in aged demyelinated animals [148]. Moreover, as mentioned previously, metformin has been shown to improve myelin recovery in animal models of demyelination induced by cuprizone and alleviate the antioxidant response [243]. Curcumin, another AMPK activator administered by loading into dendrimer nanoparticles, can penetrate the BBB and exert neuroprotective effects by enhancing OPC proliferation and migration [244].
Stem cell therapy
Increasing OPCs in patients with AD can represent increased oligodendrocyte production to compensate for the loss of oligodendrocytes in the disease [245]. It has been suggested that stem cell therapy is a direct approach to increasing OPCs [246]. As early as 2005, researchers utilized neural sphere culture to manually select and generate OPCs, which were then transplanted into mice, resulting in increased differentiation into oligodendrocytes and denser myelin sheaths [246]. Subsequent studies have focused on using growth factors such as Sox10, Olig2, and insulin-like growth factor-I for induction [247, 248]. Research on ischemic stroke has revealed that transplantation of OPCs can contribute to the protection of the BBB and the inhibition of brain damage [249]. However, the distribution of OPCs is complex, and when considering transplantation, the regional specificity and the effects require serious consideration [68].
Exercise
As a “non-invasive” intervention, physical exercise has gained widespread recognition for its role in preventing and treating various diseases [250, 251]. While the effects of exercise on OPCs do not target a specific pathway, it is cost-effective and provides multiple benefits [106, 252]. In addition, innovative motor tasks, such as the skilled reaching task or complex running wheel, have been found to stimulate the proliferation, migration, and differentiation of OPCs in rodents, leading to oligodendrocyte generation and remyelination [253,254,255,256]. In the brains of AD patients, the activities of antioxidant enzymes SOD2 and CAT are significantly reduced [257]. However, long-term exercise can dramatically increase the contents of SOD2 and CAT, enhancing antioxidative capacity [106, 257]. Exercise can regulate the release of irisin from muscles, which regulates the release of brain-derived neurotrophic factors and promotes the ability of synaptic mitochondria to transport glucose and enhance respiratory coupling efficiency. Furthermore, exercise regulates mitochondrial regeneration by increasing the expression of PGC-1α, thus influencing the number of mitochondria [258]. More importantly, exercise is essential in maintaining the dynamic balance of mitochondrial fission and fusion and the structural integrity of mitochondria [259]. Therefore, exercise regulation on OPCs may be achieved by improving the brain microenvironment [260].
Photobiomodulation (PBM)
Similarly, a growing body of research has demonstrated that PBM by applying low-level laser (light) exerts beneficial effects in various neurodegenerative disorders [261,262,263]. In animal models of demyelinating diseases, PBM has been shown to stimulate OPC proliferation and promote myelin repair, thereby enhancing neuroplasticity and facilitating disease recovery [264, 265]. A recent study revealed that in a rat model of early-life adversity, there is a decrease of OPC proliferation and differentiation, an increase of myelin loss, and elevated levels of oxidative damage. However, these changes can be reversed through early PBM treatment [266]. Unfortunately, no reported evidence regarding PBM therapy has been shown to specifically target OPCs in AD. However, it is worth noting that studies have investigated the involvement of myelin sheath repair in AD [11, 267]. In particular, research in transgenic AD rodent models has shown promising results regarding the effects of PBM therapy on MBP+ myelin-related changes [11]. PBM has been widely studied to enhance the activity of mitochondrial cytochrome c oxidase, reduce oxidative damage, and improve antioxidant enzyme activity [10]. Therefore, mitochondrial protection, in turn, can improve the microenvironment associated with AD [11, 268, 269]. These studies suggest that PBM may promote the recovery of various brain disorders by regulating OPC differentiation and myelin formation. However, additional research is still needed to investigate the precise targets of PBM.
Conclusion and future perspectives
In conclusion, OPCs play critical roles in the CNS, contributing to myelination, intercellular signaling, phagocytosis, and BBB formation and repair [47, 79]. However, in the context of AD, the microenvironment in the brain deteriorates, leading to alterations in OPC-related events [137,138,139]. Under pathological conditions, the impaired migration and proliferation of OPCs compromise the differentiation of OPCs into mature oligodendrocytes [47]. The disrupted OPC differentiation contributes to myelin degeneration and pathological change in neurodegenerative diseases.
Additionally, OPC senescence contributes to demyelination in AD [176]. Given the significance of OPCs in AD, current therapeutic approaches target OPC senescence and function. Strategies focusing on improving OPC senescence, rejuvenating OPC functions, enhancing myelin repair processes, promoting OPC differentiation, facilitating remyelination, and restoring neuronal circuitry exhibit great potential for AD treatment. Moreover, there are other potential therapeutic approaches beyond OPC targeting. Combining strategies targeting OPCs or microenvironments may provide synergistic effects and enhance therapeutic outcomes. Therefore, further research is warranted to deepen our understanding of the intricate mechanisms underlying OPC-related events in AD pathology. Unraveling the specific signaling pathways, molecular players, and cellular interactions involved will pave the way for more precise and effective therapeutic interventions. Additionally, exploring the potential of emerging technologies, such as gene editing, holds promise in harnessing the regenerative capacity of OPCs and promoting myelin repair in AD [270]. In addition, considering the association between AD and the pathological loss of functional synapses [271,272,273], it is crucial to ascertain the potential role of OPCs in synaptic phagocytosis, which may underlie the neurobiological deficits observed in AD and related disorders.
In summary, elucidating the complex interplay between OPC, the microenvironment, and AD pathology is crucial for developing targeted therapeutic strategies. Focusing on rejuvenating senescent OPCs, modulating differentiation pathways, and addressing the altered microenvironment may help mitigate myelin abnormalities and restore cognitive function in AD patients.
Availability of data and materials
Not applicable.
Abbreviations
- OPC:
-
Oligodendrocyte progenitor cell
- nOPC:
-
Neonatal oligodendrocyte progenitor cell
- aOPC:
-
Adult oligodendrocyte progenitor cell
- BBB:
-
Blood–brain barrier
- AD:
-
Alzheimer’s disease
- CNS:
-
Central nervous system
- IFNγ:
-
Interferon-gamma
- IL:
-
Interleukin
- MHC:
-
Major histocompatibility complex
- LRP1:
-
Low-density lipoprotein receptor-related protein 1
- DAM:
-
Disease-associated microglia
- DAA:
-
Disease-associated astrocyte
- PI3K:
-
Phosphatidylinositol-3 kinase
- LTP:
-
Long-term potentiation
- Aβ:
-
Amyloid-beta
- NFT:
-
Neurofibrillary tangle
- mtDNA:
-
Mitochondrial DNA
- DAMP:
-
Damage-associated molecular pattern
- SASP:
-
Senescence-associated secretory phenotype
- MCI:
-
Mild cognitive impairment
- RXR:
-
Receptor retinoid X receptor
- ABCA1:
-
ATP-binding cassette transporter A1
- ApoE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- PBM:
-
Photobiomodulation
References
Nishiyama A, Polydendrocytes. NG2 cells with many roles in development and repair of the CNS. Neuroscientist. 2007;13:62–76.
Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007;14:1324–35.
Nampoothiri S, Nogueiras R, Schwaninger M, Prevot V. Glial cells as integrators of peripheral and central signals in the regulation of energy homeostasis. Nat Metab. 2022;4:813–25.
Healy LM, Stratton JA, Kuhlmann T, Antel J. The role of glial cells in multiple sclerosis disease progression. Nat Rev Neurol. 2022;18:237–48.
Yun W, Choi KA, Hwang I, Zheng J, Park M, Hong W, et al. OCT4-induced oligodendrocyte progenitor cells promote remyelination and ameliorate disease. NPJ Regen Med. 2022;7:4.
Galichet C, Clayton RW, Lovell-Badge R. Novel tools and investigative approaches for the study of oligodendrocyte precursor cells (NG2-Glia) in CNS development and disease. Front Cell Neurosci. 2021;15:673132.
Duncan ID, Radcliff AB, Heidari M, Kidd G, August BK, Wierenga LA. The adult oligodendrocyte can participate in remyelination. Proc Natl Acad Sci USA. 2018;115:E11807-16.
Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22.
Feng S, Wu C, Zou P, Deng Q, Chen Z, Li M, et al. High-intensity interval training ameliorates Alzheimer’s disease-like pathology by regulating astrocyte phenotype-associated AQP4 polarization. Theranostics. 2023;13:3434–50.
Wu C, Yang L, Feng S, Zhu L, Yang L, Liu TC, et al. Therapeutic non-invasive brain treatments in Alzheimer’s disease: recent advances and challenges. Inflamm Regen. 2022;42:31.
Yang L, Wu C, Parker E, Li Y, Dong Y, Tucker L, et al. Non-invasive photobiomodulation treatment in an Alzheimer disease-like transgenic rat model. Theranostics. 2022;12:2205–31.
Psenicka MW, Smith BC, Tinkey RA, Williams JL. Connecting neuroinflammation and neurodegeneration in multiple sclerosis: Are oligodendrocyte precursor cells a Nexus of disease? Front Cell Neurosci. 2021;15:654284.
Clayton BLL, Tesar PJ. Oligodendrocyte progenitor cell fate and function in development and disease. Curr Opin Cell Biol. 2021;73:35–40.
Spaas J, van Veggel L, Schepers M, Tiane A, van Horssen J, Wilson DM, et al. Oxidative stress and impaired oligodendrocyte precursor cell differentiation in neurological disorders. Cell Mol Life Sci. 2021;78:4615–37. 3rd.
Xie YY, Pan TT, Xu DE, Huang X, Tang Y, Huang W, et al. Clemastine ameliorates myelin deficits via preventing senescence of oligodendrocytes precursor cells in Alzheimer’s disease model mouse. Front Cell Dev Biol. 2021;9:733945.
Chacon-De-La-Rocha I, Fryatt G, Rivera AD, Verkhratsky A, Raineteau O, Gomez-Nicola D, et al. Accelerated dystrophy and decay of oligodendrocyte precursor cells in the APP/PS1 model of Alzheimer’s-like pathology. Front Cell Neurosci. 2020;14:575082.
Vanzulli I, Papanikolaou M, De-La-Rocha IC, Pieropan F, Rivera AD, Gomez-Nicola D, et al. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2020;94:130–9.
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, et al. Senolytic therapy alleviates abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22:719–28.
Kuhn S, Gritti L, Crooks D, Dombrowski Y. Oligodendrocytes in development, myelin generation and beyond. Cells. 2019;8:1424.
Richardson WD, Kessaris N, Pringle N. Oligodendrocyte wars. Nat Rev Neurosci. 2006;7:11–8.
Lecca D, Raffaele S, Abbracchio MP, Fumagalli M. Regulation and signaling of the GPR17 receptor in oligodendroglial cells. Glia. 2020;68:1957–67.
Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, et al. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–82.
Pringle NP, Richardson WD. A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development. 1993;117:525–33.
Yeh HJ, Ruit KG, Wang YX, Parks WC, Snider WD, Deuel TF. PDGF A-chain gene is expressed by mammalian neurons during development and in maturity. Cell. 1991;64:209–16.
Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature. 1988;333:560–2.
Marx M, Perlmutter RA, Madri JA. Modulation of platelet-derived growth factor receptor expression in microvascular endothelial cells during in vitro angiogenesis. J Clin Invest. 1994;93:131–9.
Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–82.
Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res. 2010;88:758–63.
Auguste YSS, Ferro A, Kahng JA, Xavier AM, Dixon JR, Vrudhula U, et al. Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat Neurosci. 2022;25:1273–8.
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–401.
Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71.
Qin G, Wang Y, Liu Z, Mana L, Huang S, Wang P. Shenzhiling oral solution promotes myelin repair through PI3K/Akt-mTOR pathway in STZ-induced SAD mice. 3 Biotech. 2021;11:361.
Chomiak T, Hu B. What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PLoS One. 2009;4:e7754.
York EN, Martin SJ, Meijboom R, Thrippleton MJ, Bastin ME, Carter E, et al. MRI-derived g-ratio and lesion severity in newly diagnosed multiple sclerosis. Brain Commun. 2021;3:fcab249.
Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927.
Ong WY, Levine JM. A light and electron microscopic study of NG2 chondroitin sulfate proteoglycan-positive oligodendrocyte precursor cells in the normal and kainate-lesioned rat hippocampus. Neuroscience. 1999;92:83–95.
Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351:379–84.
Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, et al. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–83.
Robinson S, Tani M, Strieter RM, Ransohoff RM, Miller RH. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J Neurosci. 1998;18:10457–63.
Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology. 2010;35:147–68.
Rhodes KE, Raivich G, Fawcett JW. The injury response of oligodendrocyte precursor cells is induced by platelets, macrophages and inflammation-associated cytokines. Neuroscience. 2006;140:87–100.
Tiane A, Schepers M, Rombaut B, Hupperts R, Prickaerts J, Hellings N, et al. From OPC to oligodendrocyte: an epigenetic journey. Cells. 2019;8:1236.
Sock E, Wegner M. Transcriptional control of myelination and remyelination. Glia. 2019;67:2153–65.
Kuypers NJ, Bankston AN, Howard RM, Beare JE, Whittemore SR. Remyelinating oligodendrocyte precursor cell miRNAs from the Sfmbt2 cluster promote cell cycle arrest and differentiation. J Neurosci. 2016;36:1698–710.
Magri L, Gacias M, Wu M, Swiss VA, Janssen WG, Casaccia P. c-Myc-dependent transcriptional regulation of cell cycle and nucleosomal histones during oligodendrocyte differentiation. Neuroscience. 2014;276:72–86.
Magri L, Swiss VA, Jablonska B, Lei L, Pedre X, Walsh M, et al. E2F1 coregulates cell cycle genes and chromatin components during the transition of oligodendrocyte progenitors from proliferation to differentiation. J Neurosci. 2014;34:1481–93.
Skaper SD. Oligodendrocyte precursor cells as a therapeutic target for demyelinating diseases. Prog Brain Res. 2019;245:119–44.
Fields RD. Myelin formation and remodeling. Cell. 2014;156:15–7.
Gouvea-Junqueira D, Falvella ACB, Antunes A, Seabra G, Brandao-Teles C, Martins-de-Souza D, et al. Novel treatment strategies targeting myelin and oligodendrocyte dysfunction in Schizophrenia. Front Psychiatry. 2020;11:379.
Serrano-Regal MP, Luengas-Escuza I, Bayon-Cordero L, Ibarra-Aizpurua N, Alberdi E, Perez-Samartin A, et al. Oligodendrocyte differentiation and myelination is potentiated via GABA(B) receptor activation. Neuroscience. 2020;439:163–80.
Sobottka B, Ziegler U, Kaech A, Becher B, Goebels N. CNS live imaging reveals a new mechanism of myelination: the liquid croissant model. Glia. 2011;59:1841–9.
Snaidero N, Mobius W, Czopka T, Hekking LH, Mathisen C, Verkleij D, et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell. 2014;156:277–90.
de Faria O Jr., Pivonkova H, Varga B, Timmler S, Evans KA, Karadottir RT. Periods of synchronized myelin changes shape brain function and plasticity. Nat Neurosci. 2021;24:1508–21.
Hughes AN, Appel B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci. 2020;23:1055–66.
Beiter RM, Rivet-Noor C, Merchak AR, Bai R, Johanson DM, Slogar E, et al. Evidence for oligodendrocyte progenitor cell heterogeneity in the adult mouse brain. Sci Rep. 2022;12:12921.
Niu J, Tsai HH, Hoi KK, Huang N, Yu G, Kim K, et al. Aberrant oligodendroglial-vascular interactions disrupt the blood–brain barrier, triggering CNS inflammation. Nat Neurosci. 2019;22:709–18.
Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol. 2005;252(Suppl 5):v3–9.
Miyamoto Y, Yamauchi J, Tanoue A. Cdk5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase fyn. J Neurosci. 2008;28:8326–37.
Franklin RJM, Ffrench-Constant C. Regenerating CNS myelin—from mechanisms to experimental medicines. Nat Rev Neurosci. 2017;18:753–69.
Blakemore WF. Pattern of remyelination in the CNS. Nature. 1974;249:577–8.
Raff M, Apperly J, Kondo T, Tokumoto Y, Tang D. Timing cell-cycle exit and differentiation in oligodendrocyte development. Novartis Found Symp. 2001;237:100–7 discussion 107–112, 158–163.
Monaghan TK, Mackenzie CJ, Plevin R, Lutz EM. PACAP-38 induces neuronal differentiation of human SH-SY5Y neuroblastoma cells via cAMP-mediated activation of ERK and p38 MAP kinases. J Neurochem. 2008;104:74–88.
Baron W, Colognato H, ffrench-Constant C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia. 2005;49:467–79.
Blumer KJ, Johnson GL. Diversity in function and regulation of MAP kinase pathways. Trends Biochem Sci. 1994;19:236–40.
Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–92.
Wu C, Duan R, Yang L. Astrocyte-mediated oligodendrocyte precursor cells detachment from vessels. Cell Biochem Biophys. 2023;81:379–81.
Su Y, Wang X, Yang Y, Chen L, Xia W, Hoi KK, et al. Astrocyte endfoot formation controls the termination of oligodendrocyte precursor cell perivascular migration during development. Neuron. 2023;111:190–201.
Suzuki N, Sekimoto K, Hayashi C, Mabuchi Y, Nakamura T, Akazawa C. Differentiation of oligodendrocyte precursor cells from Sox10-Venus mice to oligodendrocytes and astrocytes. Sci Rep. 2017;7:14133.
Buchanan J, Elabbady L, Collman F, Jorstad NL, Bakken TE, Ott C, et al. Oligodendrocyte precursor cells ingest axons in the mouse neocortex. Proc Natl Acad Sci USA. 2022;119:e2202580119.
Zhang X, Liu Y, Hong X, Li X, Meshul CK, Moore C, et al. NG2 glia-derived GABA release tunes inhibitory synapses and contributes to stress-induced anxiety. Nat Commun. 2021;12:5740.
Matejuk A, Vandenbark AA, Offner H. Cross-talk of the CNS with immune cells and functions in health and disease. Front Neurol. 2021;12:672455.
Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–91.
Ge WP, Yang XJ, Zhang Z, Wang HK, Shen W, Deng QD, et al. Long-term potentiation of neuron-glia synapses mediated by Ca2+-permeable AMPA receptors. Science. 2006;312:1533–7.
Nakano M, Tamura Y, Yamato M, Kume S, Eguchi A, Takata K, et al. NG2 glial cells regulate neuroimmunological responses to maintain neuronal function and survival. Sci Rep. 2017;7:42041.
Desu HL, Illiano P, Choi JS, Ascona MC, Gao H, Lee JK, et al. TNFR2 signaling regulates the immunomodulatory function of oligodendrocyte precursor cells. Cells. 2021;10:1785.
Clemente D, Ortega MC, Melero-Jerez C, de Castro F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front Cell Neurosci. 2013;7:268.
Sozmen EG, DiTullio DJ, Rosenzweig S, Hinman JD, Bridges SP, Marin MA, et al. White matter stroke induces a unique oligo-astrocyte niche that inhibits recovery. J Neurosci. 2019;39:9343–59.
Xu D, Liu Z, Wang S, Peng Y, Sun X. Astrocytes regulate the expression of Sp1R3 on oligodendrocyte progenitor cells through Cx47 and promote their proliferation. Biochem Biophys Res Commun. 2017;490:670–5.
Akay LA, Effenberger AH, Tsai LH. Cell of all trades: oligodendrocyte precursor cells in synaptic, vascular, and immune function. Genes Dev. 2021;35:180–98.
Fernandez-Castaneda A, Chappell MS, Rosen DA, Seki SM, Beiter RM, Johanson DM, et al. The active contribution of OPCs to neuroinflammation is mediated by LRP1. Acta Neuropathol. 2020;139:365–82.
Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2 + glial cells. Nat Neurosci. 2013;16:1401–8.
Falcao AM, van Bruggen D, Marques S, Meijer M, Jakel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24:1837–44.
Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. 2019;10:3887.
Moyon S, Dubessy AL, Aigrot MS, Trotter M, Huang JK, Dauphinot L, et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci. 2015;35:4–20.
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705.
Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527.
Irfan M, Evonuk KS, DeSilva TM. Microglia phagocytose oligodendrocyte progenitor cells and synapses during early postnatal development: implications for white versus gray matter maturation. FEBS J. 2022;289:2110–27.
Li W, Tang Y, Fan Z, Meng Y, Yang G, Luo J, et al. Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol Neurodegener. 2013;8:27.
Zhao WD, Liu DX, Wei JY, Miao ZW, Zhang K, Su ZK, et al. Caspr1 is a host receptor for meningitis-causing Escherichia coli. Nat Commun. 2018;9:2296.
Maki T. Novel roles of oligodendrocyte precursor cells in the developing and damaged brain. Clin Exp Neuroimmunol. 2017; 8:33–42.
Kishida N, Maki T, Takagi Y, Yasuda K, Kinoshita H, Ayaki T, et al. Role of perivascular oligodendrocyte precursor cells in angiogenesis after brain ischemia. J Am Heart Assoc. 2019;8:e011824.
Seo JH, Maki T, Maeda M, Miyamoto N, Liang AC, Hayakawa K, et al. Oligodendrocyte precursor cells support blood–brain barrier integrity via TGF-beta signaling. PLoS One. 2014;9:e103174.
Maki T, Maeda M, Uemura M, Lo EK, Terasaki Y, Liang AC, et al. Potential interactions between pericytes and oligodendrocyte precursor cells in perivascular regions of cerebral white matter. Neurosci Lett. 2015;597:164–9.
Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–5.
Hayakawa K, Seo JH, Pham LD, Miyamoto N, Som AT, Guo S, et al. Cerebral endothelial derived vascular endothelial growth factor promotes the migration but not the proliferation of oligodendrocyte precursor cells in vitro. Neurosci Lett. 2012;513:42–6.
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 2012;2012:756357.
Nissim NR, Pham DVH, Poddar T, Blutt E, Hamilton RH. The impact of gamma transcranial alternating current stimulation (tACS) on cognitive and memory processes in patients with mild cognitive impairment or Alzheimer’s Disease: a literature review. Brain Stimul. 2023;16:748–55.
Li YD, Luo YJ, Xie L, Tart DS, Sheehy RN, Zhang L, et al. Activation of hypothalamic-enhanced adult-born neurons restores cognitive and affective function in Alzheimer’s Disease. Cell Stem Cell. 2023;30:415–432e416.
Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell. 2020;19:e13081.
Laurent C, Buee L, Blum D. Tau and neuroinflammation: What impact for Alzheimer’s disease and tauopathies? Biomed J. 2018;41:21–33.
d’Errico P, Meyer-Luehmann M. Mechanisms of pathogenic tau and abeta protein spreading in Alzheimer’s disease. Front Aging Neurosci. 2020;12:265.
Barthes J, Ozcelik H, Hindie M, Ndreu-Halili A, Hasan A, Vrana NE. Cell microenvironment engineering and monitoring for tissue engineering and regenerative medicine: the recent advances. Biomed Res Int. 2014;2014:921905.
Song T, Song X, Zhu C, Patrick R, Skurla M, Santangelo I, et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: a meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res Rev. 2021;72:101503.
Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–40.
Ehrnhoefer DE, Wong BK, Hayden MR. Convergent pathogenic pathways in Alzheimer’s and Huntington’s diseases: shared targets for drug development. Nat Rev Drug Discov. 2011;10:853–67.
Wu C, Yang L, Tucker D, Dong Y, Zhu L, Duan R, et al. Beneficial effects of Exercise pretreatment in a sporadic Alzheimer’s rat model. Med Sci Sports Exerc. 2018;50:945–56.
Wang Y, Yang Y, Zhang S, Li C, Zhang L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: implications for cerebral ischemia and neurodegenerative diseases. Neurobiol Aging. 2020;87:1–10.
Ano Y, Ikado K, Uchida K, Nakayama H. Amyloid beta-induced mesenteric inflammation in an Alzheimer’s disease transgenic mouse model. Curr Alzheimer Res. 2020;17:52–9.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol. 2021;17:157–72.
Popko K, Gorska E, Stelmaszczyk-Emmel A, Plywaczewski R, Stoklosa A, Gorecka D, et al. Proinflammatory cytokines Il-6 and TNF-alpha and the development of inflammation in obese subjects. Eur J Med Res. 2010;15(Suppl 2):120–2.
Sinyor B, Mineo J, Ochner C. Alzheimer’s disease, inflammation, and the role of antioxidants. J Alzheimers Dis Rep. 2020;4:175–83.
Luque-Contreras D, Carvajal K, Toral-Rios D, Franco-Bocanegra D, Campos-Pena V. Oxidative stress and metabolic syndrome: Cause or consequence of Alzheimer’s disease? Oxid Med Cell Longev. 2014;2014:497802.
Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova NN, et al. Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. FASEB J. 2011;25:4063–72.
Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523.
Kim DK, Mook-Jung I. The role of cell type-specific mitochondrial dysfunction in the pathogenesis of Alzheimer’s disease. BMB Rep. 2019;52:679–88.
Yang L, Tucker D, Dong Y, Wu C, Lu Y, Li Y, et al. Photobiomodulation therapy promotes neurogenesis by improving post-stroke local microenvironment and stimulating neuroprogenitor cells. Exp Neurol. 2018;299:86–96.
Xiong Y, Cheng Q, Li Y, Han Y, Sun X, Liu L. Vimar/RAP1GDS1 promotes acceleration of brain aging after flies and mice reach middle age. Commun Biol. 2023;6:420.
Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30.
Flannery PJ, Trushina E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci. 2019;98:109–20.
Adav SS, Park JE, Sze SK. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol Brain. 2019;12:8.
Bajwa E, Pointer CB, Klegeris A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediat Inflamm. 2019;2019:4050796.
Han M, Cao Y, Guo X, Chu X, Li T, Xue H, et al. Mesenchymal stem cell-derived extracellular vesicles promote microglial M2 polarization after subarachnoid Hemorrhage in rats and involve the AMPK/NF-kappaB signaling pathway. Biomed Pharmacother. 2021;133:111048.
Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. 2018;115:E1896-905.
Plemel JR, Stratton JA, Michaels NJ, Rawji KS, Zhang E, Sinha S, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv. 2020;6:eaay6324.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:253–71.
Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110:3458–83.
Healy LM, Zia S, Plemel JR. Towards a definition of microglia heterogeneity. Commun Biol. 2022;5:1114.
Song WM, Colonna M. The identity and function of microglia in neurodegeneration. Nat Immunol. 2018;19:1048–58.
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–6.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–90.
Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener. 2018;13:24.
Udeochu JC, Amin S, Huang Y, Fan L, Torres ERS, Carling GK, et al. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. 2023;26:737–50.
Cheng KK, Yeung CF, Ho SW, Chow SF, Chow AH, Baum L. Highly stabilized curcumin nanoparticles tested in an in vitro blood–brain barrier model and in Alzheimer’s disease Tg2576 mice. AAPS J. 2013;15:324–36.
Zenaro E, Piacentino G, Constantin G. The blood–brain barrier in Alzheimer’s disease. Neurobiol Dis. 2017;107:41–56.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–20.
Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediat Inflamm. 2018;2018:9076485.
Masaldan S, Belaidi AA, Ayton S, Bush AI. Cellular senescence and iron dyshomeostasis in Alzheimer’s disease. Pharmaceuticals. 2019;12:93.
Killilea DW, Wong SL, Cahaya HS, Atamna H, Ames BN. Iron accumulation during cellular senescence. Ann N Y Acad Sci. 2004;1019:365–7.
Segel M, Neumann B, Hill MFE, Weber IP, Viscomi C, Zhao C, et al. Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature. 2019;573:130–4.
Nalivaeva NN, Rybnikova EA, Editorial. Brain hypoxia and ischemia: new insights into neurodegeneration and neuroprotection. Front Neurosci. 2019;13:770.
Lau V, Ramer L, Tremblay ME. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. 2023;14:1670.
Boyd A, Zhang H, Williams A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013;125:841–59.
Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F, et al. Single-cell Transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell. 2019;24:707–23.
Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Varemo L, et al. Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev Cell. 2018;46:504-517e507.
Esteve D, Molina-Navarro MM, Giraldo E, Martinez-Varea N, Blanco-Gandia MC, Rodriguez-Arias M, et al. Adult neural stem cell migration is impaired in a mouse model of Alzheimer’s disease. Mol Neurobiol. 2022;59:1168–82.
Musella A, Gentile A, Rizzo FR, De Vito F, Fresegna D, Bullitta S, et al. Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front Aging Neurosci. 2018;10:238.
Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–55.
Neumann B, Baror R, Zhao C, Segel M, Dietmann S, Rawji KS, et al. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell. 2019;25:473–485e478.
Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10:96–103.
Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22:2451–9.
Dimovasili C, Fair AE, Garza IR, Batterman KV, Mortazavi F, Moore TL, et al. Aging compromises oligodendrocyte precursor cell maturation and efficient remyelination in the monkey brain. Geroscience. 2023;45:249–64.
Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia. 2012;60:615–29.
Behrendt G, Baer K, Buffo A, Curtis MA, Faull RL, Rees MI, et al. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia. 2013;61:273–86.
Wu Y, Ma Y, Liu Z, Geng Q, Chen Z, Zhang Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. Neurosci Lett. 2017;642:102–6.
Dong YX, Zhang HY, Li HY, Liu PH, Sui Y, Sun XH. Association between Alzheimer’s disease pathogenesis and early demyelination and oligodendrocyte dysfunction. Neural Regen Res. 2018;13:908–14.
Ferreira S, Pitman KA, Wang S, Summers BS, Bye N, Young KM, et al. Amyloidosis is associated with thicker myelin and increased oligodendrogenesis in the adult mouse brain. J Neurosci Res. 2020;98:1905–32.
Liu Y, Aguzzi A. NG2 glia are required for maintaining microglia homeostatic state. Glia. 2020;68:345–55.
Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–57.
Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun. 2015;6:8518.
Papuc E, Rejdak K. The role of myelin damage in Alzheimer’s disease pathology. Arch Med Sci. 2020;16:345–51.
Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–31.
Pietrzak M, Papp A, Curtis A, Handelman SK, Kataki M, Scharre DW, et al. Gene expression profiling of brain samples from patients with Lewy body dementia. Biochem Biophys Res Commun. 2016;479:875–80.
Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–58.
Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006;26:328–32.
Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22.
Chen JF, Liu K, Hu B, Li RR, Xin W, Chen H, et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron. 2021;109:2292-2307e2295.
Mitew S, Kirkcaldie MT, Halliday GM, Shepherd CE, Vickers JC, Dickson TC. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 2010;119:567–77.
Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T, et al. Myelin dysfunction drives amyloid-beta deposition in models of Alzheimer’s disease. Nature. 2023;618:349–57.
Nicaise AM, Wagstaff LJ, Willis CM, Paisie C, Chandok H, Robson P, et al. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc Natl Acad Sci USA. 2019;116:9030–9.
Di Micco R, Krizhanovsky V, Baker D. d’Adda Di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95.
Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. 2019;99:1047–78.
Wang J, Zheng B, Yang S, Zhou D, Wang J. Olmesartan prevents oligomerized amyloid beta (abeta)-induced cellular senescence in neuronal cells. ACS Chem Neurosci. 2021;12:1162–9.
Papadopoulos D, Magliozzi R, Mitsikostas DD, Gorgoulis VG, Nicholas RS. Aging, Cellular Senescence, and Progressive multiple sclerosis. Front Cell Neurosci. 2020;14:178.
Zhang X, Pearsall VM, Carver CM, Atkinson EJ, Clarkson BDS, Grund EM, et al. Rejuvenation of the aged brain immune cell landscape in mice through p16-positive senescent cell clearance. Nat Commun. 2022;13:5671.
Ogrodnik M, Evans SA, Fielder E, Victorelli S, Kruger P, Salmonowicz H, et al. Whole-body senescent cell clearance alleviates age-related Brain Inflammation and cognitive impairment in mice. Aging Cell. 2021;20:e13296.
Saez-Atienzar S, Masliah E. Cellular senescence and Alzheimer Disease: the egg and the chicken scenario. Nat Rev Neurosci. 2020;21:433–44.
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–82.
Jian C, Wei L, Mo R, Li R, Liang L, Chen L, et al. Microglia mediate the occurrence and development of Alzheimer’s Disease through ligand-receptor Axis Communication. Front Aging Neurosci. 2021;13:731180.
Raghunatha P, Vosoughi A, Kauppinen TM, Jackson MF. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia. 2020;68:1421–34.
Zhang SZ, Wang QQ, Yang QQ, Gu HY, Yin YQ, Li YD, et al. NG2 glia regulate brain innate immunity via TGF-beta2/TGFBR2 axis. BMC Med. 2019;17:204.
Ahmed ME, Iyer S, Thangavel R, Kempuraj D, Selvakumar GP, Raikwar SP, et al. Co-localization of Glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J Alzheimers Dis. 2017;60:1143–60.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42:318–33.
Knowles JK, Batra A, Xu H, Monje M. Adaptive and maladaptive myelination in health and disease. Nat Rev Neurol. 2022;18:735–46.
Steadman PE, Xia F, Ahmed M, Mocle AJ, Penning ARA, Geraghty AC, et al. Disruption of oligodendrogenesis impairs memory consolidation in adult mice. Neuron. 2020;105:150-164e156.
Schiller RM, Madderom HIJ, van Rosmalen MJ, van Heijst J, Smits AFJ. Training-induced white matter microstructure changes in survivors of neonatal critical Illness: a randomized controlled trial. Dev Cogn Neurosci. 2019;38:100678.
Hofstetter S, Tavor I, Tzur Moryosef S, Assaf Y. Short-term learning induces white matter plasticity in the fornix. J Neurosci. 2013;33:12844–50.
Pan S, Mayoral SR, Choi HS, Chan JR, Kheirbek MA. Preservation of a remote fear memory requires new myelin formation. Nat Neurosci. 2020;23:487–99.
Mitew S, Gobius I, Fenlon LR, McDougall SJ, Hawkes D, Xing YL, et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat Commun. 2018;9:306.
Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344:1252304.
Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087–97.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7.
Geraghty AC, Gibson EM, Ghanem RA, Greene JJ, Ocampo A, Goldstein AK, et al. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron. 2019;103:250–65.
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378:321–30.
Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8:18.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33.
Xie J, Liang R, Wang Y, Huang J, Cao X, Niu B. Progress in target drug molecules for Alzheimer’s disease. Curr Top Med Chem. 2020;20:4–36.
Loera-Valencia R, Cedazo-Minguez A, Kenigsberg PA, Page G, Duarte AI, Giusti P, et al. Current and emerging avenues for Alzheimer’s disease drug targets. J Intern Med. 2019;286:398–437.
Barage SH, Sonawane KD. Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides. 2015;52:1–18.
Mahase E. Alzheimer’s disease: lecanemab gets full FDA approval and black box safety warning. BMJ. 2023;382:p1580.
Thambisetty M, Howard R. Lecanemab trial in AD brings hope but requires greater clarity. Nat Rev Neurol. 2023;19:132–3.
Reish NJ, Jamshidi P, Stamm B, Flanagan ME, Sugg E, Tang M, et al. Multiple cerebral hemorrhages in a patient receiving Lecanemab and treated with t-PA for Stroke. N Engl J Med. 2023;388:478–9.
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9–21.
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24:1246–56.
Cui X, Guo YE, Fang JH, Shi CJ, Suo N, Zhang R, et al. Donepezil, a drug for Alzheimer’s disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol Sin. 2019;40:1386–93.
Kirkland JL, Tchkonia T. Senolytic Drugs: from discovery to translation. J Intern Med. 2020;288:518–36.
Iram T, Kern F, Kaur A, Myneni S, Morningstar AR, Shin H, et al. Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature. 2022;605:509–15.
Neitzel J, Franzmeier N, Rubinski A, Dichgans M, Brendel M, Alzheimer’s Disease Neuroimaging Initiative (ADNI), et al. KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer’s Disease. Nat Commun. 2021;12:3825.
Belloy ME, Eger SJ, Le Guen Y, Napolioni V, Deters KD, Yang HS, et al. KL *VS heterozygosity reduces brain amyloid in asymptomatic at-risk APOE *4 carriers. Neurobiol Aging. 2021;101:123–9.
Dubal DB, Zhu L, Sanchez PE, Worden K, Broestl L, Johnson E, et al. Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J Neurosci. 2015;35:2358–71.
Zeldich E, Chen CD, Avila R, Medicetty S, Abraham CR. The anti-aging protein klotho enhances remyelination following cuprizone-induced demyelination. J Mol Neurosci. 2015;57:185–96.
Chen CD, Li H, Liang J, Hixson K, Zeldich E, Abraham CR. The anti-aging and tumor suppressor protein klotho enhances differentiation of a human oligodendrocytic hybrid cell line. J Mol Neurosci. 2015;55:76–90.
Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, et al. The antiaging protein klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci. 2013;33:1927–39.
Tepavcevic V, Lubetzki C. Oligodendrocyte progenitor cell recruitment and remyelination in multiple sclerosis: The more, the merrier? Brain. 2022;145:4178–92.
Raffaele S, Boccazzi M, Fumagalli M. Oligodendrocyte dysfunction in amyotrophic lateral sclerosis: mechanisms and therapeutic perspectives. Cells. 2021;10:565.
May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E, et al. Alpha-synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging. 2014;35:2357–68.
Santos-Gil DF, Arboleda G, Sandoval-Hernandez AG. Retinoid X receptor activation promotes re-myelination in a very old triple transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2021;750:135764.
Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14:45–53.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18.
Mariani MM, Malm T, Lamb R, Jay TR, Neilson L, Casali B, et al. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci Rep. 2017;7:42270.
Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, et al. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res Ther. 2016;8:4.
Wang W, Nakashima KI, Hirai T, Inoue M. Anti-inflammatory effects of naturally occurring retinoid X receptor agonists isolated from Sophora Tonkinensis Gagnep. Via retinoid X receptor/liver X receptor heterodimers. J Nat Med. 2019;73:419–30.
Wang W, Nakashima KI, Hirai T, Inoue M. Neuroprotective effect of naturally occurring RXR agonists isolated from Sophora Tonkinensis Gagnep. On amyloid-beta-induced cytotoxicity in PC12 cells. J Nat Med. 2019;73:154–62.
Keough MB, Yong VW. Remyelination therapy for multiple sclerosis. Neurotherapeutics. 2013;10:44–54.
Song S, Yu L, Hasan MN, Paruchuri SS, Mullett SJ, Sullivan MLG, et al. Elevated microglial oxidative phosphorylation and phagocytosis stimulate post-stroke brain remodeling and cognitive function recovery in mice. Commun Biol. 2022;5:35.
Imamura O, Arai M, Dateki M, Takishima K. Donepezil promotes differentiation of neural stem cells into mature oligodendrocytes at the expense of astrogenesis. J Neurochem. 2017;140:231–44.
Gong GQ, Bilanges B, Allsop B, Masson GR, Roberton V, Askwith T, et al. A small-molecule PI3Kalpha activator for cardioprotection and neuroregeneration. Nature. 2023;618:159–68.
Pettersson AF, Olsson E, Wahlund LO. Motor function in subjects with mild cognitive impairment and early Alzheimer’s disease. Dement Geriatr Cogn Disord. 2005;19:299–304.
Dong P, Rakesh KP, Manukumar HM, Mohammed YHE, Karthik CS, Sumathi S, et al. Innovative nano-carriers in anticancer drug delivery-a comprehensive review. Bioorg Chem. 2019;85:325–36.
Dadwal A, Baldi A, Kumar Narang R. Nanoparticles as carriers for drug delivery in cancer. Artif Cells Nanomed Biotechnol. 2018;46:295–305.
Rittchen S, Boyd A, Burns A, Park J, Fahmy TM, Metcalfe S, et al. Myelin repair in vivo is increased by targeting oligodendrocyte precursor cells with nanoparticles encapsulating Leukaemia inhibitory factor (LIF). Biomaterials. 2015;56:78–85.
Barteneva NS, Maltsev N, Vorobjev IA. Microvesicles and intercellular communication in the context of parasitism. Front Cell Infect Microbiol. 2013;3:49.
Wang Y, Han ZB, Song YP, Han ZC. Safety of mesenchymal stem cells for clinical application. Stem Cells Int. 2012; 2012:652034.
Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol. 2012;83:1484–94.
Gorabi AM, Kiaie N, Barreto GE, Read MI, Tafti HA, Sahebkar A. The therapeutic potential of mesenchymal stem cell-derived exosomes in treatment of neurodegenerative Diseases. Mol Neurobiol. 2019;56:8157–67.
Zhang ZG, Buller B, Chopp M. Exosomes - beyond stem cells for restorative therapy in Stroke and neurological injury. Nat Rev Neurol. 2019;15:193–203.
Reza-Zaldivar EE, Hernandez-Sapiens MA, Minjarez B, Gutierrez-Mercado YK, Marquez-Aguirre AL, Canales-Aguirre AA. Potential effects of MSC-derived exosomes in neuroplasticity in Alzheimer’s disease. Front Cell Neurosci. 2018;12:317.
Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells. 2017;35:851–8.
Zhang J, Buller BA, Zhang ZG, Zhang Y, Lu M, Rosene DL, et al. Exosomes derived from bone marrow mesenchymal stromal cells promote remyelination and reduce neuroinflammation in the demyelinating central nervous system. Exp Neurol. 2022;347:113895.
Xiao Y, Zhang Y, Gao YH, Zhao ZH, He J, Gao R, et al. A targeted extracellular vesicles loaded with montelukast in the treatment of demyelinating Diseases. Biochem Biophys Res Commun. 2022;594:31–7.
Wu XY, Liao BY, Xiao D, Wu WC, Xiao Y, Alexander T, et al. Encapsulation of bryostatin-1 by targeted exosomes enhances remyelination and neuroprotection effects in the cuprizone-induced demyelinating animal model of multiple sclerosis. Biomater Sci. 2022;10:714–27.
Yang L, Jiang Y, Shi L, Zhong D, Li Y, Li J, et al. AMPK: potential therapeutic target for Alzheimer’s disease. Curr Protein Pept Sci. 2020;21:66–77.
Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, et al. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–34.
Sanadgol N, Barati M, Houshmand F, Hassani S, Clarner T, Shahlaei M, et al. Metformin accelerates myelin recovery and ameliorates behavioral deficits in the animal model of multiple sclerosis via adjustment of AMPK/Nrf2/mTOR signaling and maintenance of endogenous oligodendrogenesis during brain self-repairing period. Pharmacol Rep. 2020;72:641–58.
Motavaf M, Sadeghizadeh M, Babashah S, Zare L, Javan M. Protective effects of a nano-formulation of curcumin against cuprizone-induced demyelination in the mouse corpus callosum. Iran J Pharm Res. 2020;19:310–20.
Nielsen HM, Ek D, Avdic U, Orbjorn C, Hansson O, Netherlands Brain B, et al. NG2 cells, a new trail for Alzheimer’s Disease mechanisms? Acta Neuropathol Commun. 2013;1:7.
Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005;49:385–96.
Li P, Li M, Tang X, Wang S, Zhang YA, Chen Z. Accelerated generation of oligodendrocyte progenitor cells from human induced pluripotent stem cells by forced expression of Sox10 and Olig2. Sci China Life Sci. 2016;59:1131–8.
Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–22.
Wang L, Geng J, Qu M, Yuan F, Wang Y, Pan J, et al. Oligodendrocyte precursor cells transplantation protects blood–brain barrier in a mouse model of brain ischemia via Wnt/beta-catenin signaling. Cell Death Dis. 2020;11:9.
Wu C, Yang L, Li Y, Dong Y, Yang B, Tucker LD, et al. Effects of exercise training on anxious-depressive-like behavior in Alzheimer rat. Med Sci Sports Exerc. 2020;52:1456–69.
Zou L, Loprinzi PD, Yeung AS, Zeng N, Huang T. The Beneficial effects of mind-body exercises for people with mild cognitive impairment: a systematic review with Meta-analysis. Arch Phys Med Rehabil. 2019;100:1556–73.
Yang L, Wu C, Li Y, Dong Y, Wu CY, Lee RH, et al. Long-term exercise pre-training attenuates Alzheimer’s disease-related pathology in a transgenic rat model of Alzheimer’s disease. Geroscience. 2022;44:1457–77.
Bacmeister CM, Barr HJ, McClain CR, Thornton MA, Nettles D, Welle CG, et al. Motor learning promotes remyelination via new and surviving oligodendrocytes. Nat Neurosci. 2020;23:819–31.
Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, et al. Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci. 2016;19:1210–7.
McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, et al. Motor skill learning requires active central myelination. Science. 2014;346:318–22.
Sampaio-Baptista C, Khrapitchev AA, Foxley S, Schlagheck T, Scholz J, Jbabdi S, et al. Motor skill learning induces changes in white matter microstructure and myelination. J Neurosci. 2013;33:19499–503.
Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932.
Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, et al. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250.
Yan QW, Zhao N, Xia J, Li BX, Yin LY. Effects of treadmill exercise on mitochondrial fusion and fission in the hippocampus of APP/PS1 mice. Neurosci Lett. 2019;701:84–91.
Zhang X, Ashcraft KA, Betof Warner A, Nair SK, Dewhirst MW. Can exercise-induced modulation of the tumor physiologic microenvironment improve antitumor immunity? Cancer Res. 2019;79:2447–56.
Nizamutdinov D, Ezeudu C, Wu E, Huang JH, Yi SS. Transcranial near-infrared light in treatment of neurodegenerative diseases. Front Pharmacol. 2022;13:965788.
Berman MH, Nichols TW. Treatment of neurodegeneration: integrating photobiomodulation and neurofeedback in Alzheimer’s dementia and Parkinson’s: a review. Photobiomodul Photomed Laser Surg. 2019;37:623–34.
Hong N. Photobiomodulation as a treatment for neurodegenerative disorders: current and future trends. Biomed Eng Lett. 2019;9:359–66.
Duarte KCN, Soares TT, Magri AMP, Garcia LA, Le Sueur-Maluf L, Renno ACM, et al. Low-level laser therapy modulates demyelination in mice. J Photochem Photobiol B. 2018;189:55–65.
Goncalves ED, Souza PS, Lieberknecht V, Fidelis GS, Barbosa RI, Silveira PC, et al. Low-level laser therapy ameliorates disease progression in a mouse model of multiple sclerosis. Autoimmunity. 2016;49:132–42.
Huang Z, Zhang Y, Ma X, Feng Y, Zong X, Jordan JD, et al. Photobiomodulation attenuates oligodendrocyte dysfunction and prevents adverse neurological consequences in a rat model of early life adversity. Theranostics. 2023;13:913–30.
Yang L, Wu C, Tucker L, Dong Y, Li Y, Xu P, et al. Photobiomodulation therapy attenuates anxious-depressive-like behavior in the TgF344 rat model. J Alzheimers Dis. 2021;83:1415–29.
De Marchi T, Ferlito JV, Ferlito MV, Salvador M, Leal-Junior ECP. Can photobiomodulation therapy (PBMT) minimize exercise-induced oxidative stress? A systematic review and meta-analysis. Antioxid. 2022;11:1671.
Ramezani F, Neshasteh-Riz A, Ghadaksaz A, Fazeli SM, Janzadeh A, Hamblin MR. Mechanistic aspects of photobiomodulation therapy in the nervous system. Lasers Med Sci. 2022;37:11–8.
Meneghini V, Peviani M, Luciani M, Zambonini G, Gritti A. Delivery platforms for CRISPR/Cas9 genome editing of glial cells in the central nervous system. Front Genome Ed. 2021;3:644319.
Colom-Cadena M, Spires-Jones T, Zetterberg H, Blennow K, Caggiano A, DeKosky ST, et al. The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:21.
Jackson J, Jambrina E, Li J, Marston H, Menzies F, Phillips K, et al. Targeting the synapse in Alzheimer’s disease. Front Neurosci. 2019;13:735.
Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (31971096 and 32100918), the China Postdoctoral Science Foundation (2021M690060 and 2022T150227), the Sigma Xi Grants in Aid of Research (GIAR) program (G03152021115804390), Guangzhou Scientific Research Grant (SL2022B04J00013 and SL2024A04J00578) and the SCNU Young Faculty Development Program (22KJ04).
Author information
Authors and Affiliations
Contributions
PZ reviewed the literature and drafted the manuscript. PZ, CW, and LY prepared the figures. LY revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zou, P., Wu, C., Liu, TY. et al. Oligodendrocyte progenitor cells in Alzheimer’s disease: from physiology to pathology. Transl Neurodegener 12, 52 (2023). https://doi.org/10.1186/s40035-023-00385-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-023-00385-7