
Abstract
The etiology of pulmonary arterial hypertension (PAH) is complex, especially the investigation of rare pathogeny is difficult. Congenital portosystemic venous shunt (CPSS) is a rare congenital anomaly in which the portal blood completely or partially bypasses the liver through a congenital portosystemic shunt, and drains directly into the inferior vena cava (IVC) (Howard and Davenport in J Pediatr Surg 32:494–497, 1997).CPSS is an uncommon cause of PAH (Christiane et al. in J Pediatr Gastroenterol Nutr 56:675–681, 2013), and often covered by other pathogenic factors. The clinical manifestations of CPSS-associated PAH are not specific, thus making it difficult to distinguish from PAH caused by other pathogenetic factors based on clinical presentations alone. This is a retrospective analysis of data from six patients with CPSS at a single center. Of these, five were diagnosed as PAH: four were also associated with other predisposing factors of pulmonary hypertension (PH). All patients had high serum bile concentration and high cardiac output. The aim of this retrospective study was to investigate the clinical recognition of PAH secondary to CPSS. The concentration of serum bile acid and cardiac output can be used as two important non-invasive indicators in clinical practice. Thus far, few studies have reported the clinical outcomes of CPSS-associated PAH specifically (Anna et al. in Hepatology 71:658–669, 2020;Franchi-Abella et al. in J Pediatr Gastroenterol Nutr 51:322–330, 2010;Uike et al. in Pediatr Pulmonol 53:505–511, 2018;). In the current study, such patients carried a poor prognosis if left untreated, or treated with pulmonary vasodilators alone.
Similar content being viewed by others


Case report
The data of patients who were admitted in the Pulmonary Hypertension Center of our hospital from May 2018 to July 2021 were reviewed and a total of six cases were diagnosed with CPSS:three males and three females. All of the six patients were admitted to our center because echocardiography indicated tricuspid regurgitation and the elevation of estimated pulmonary arterial systolic pressure. The main clinical manifestations of six patients included physical decline, shortness of breath, palpitation and cough, two of them had syncope history. The general conditions of the six patients were presented in Table 1. Right heart catheterization (RHC) revealed five cases of diagnosed PAH, and four of them also complicated with other possible pathogenic factors (despite one case combined with patent ductus arteriosus (PDA), PAH was not considered due to its small diameter and flow rate). All six patients had high cardiac output: cardiac index (CI) and cardiac output (CO) were increased. The results of RHC were showed in Table 2.
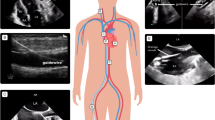
Five patients diagnosed with CPSS-associated PAH had varying degrees of liver dysfunction, including elevated alanine aminotransferase, aspartate aminotransferase and bilirubin, which were considered to be caused by liver congestion and poor function. It is noteworthy that different from the patients with PAH treated in the past, the bile acids of six patients were significantly increased. The laboratory examinations of six patients were exhibited in Table 3. Their chest radiographs indicated all had bulging pulmonary artery segments and increased pulmonary blood volume. Ultrasonic cardiogram revealed widened pulmonary artery, enlarged right atrium, tricuspid regurgitation and elevation of estimated pulmonary arterial systolic pressure as shown in Table 2. Computed tomography angiography (CTA) confirmed the fact that they had CPSS: two patients had congenital intrahepatic portosystemic shunt (CIPS) and the other patients had congenital extrahepatic portosystemic shunt (CEPS) (one type I and three type II) (Fig. 1). Five CPSS-associated PAH sufferers were followed up: Two patients underwent trans-catheter closure to block the portosystemic shunt and the pulmonary arterial pressure returned to normal. Two were managed conservatively to take pulmonary vasodilators for a long time and there were no significant changes in pulmonary artery pressure during follow-up. One patient who was recommended for surgery to close the portosystemic shunt lost follow-up.

SV/SPV splenic vein, IVC inferior vena cava, PV portal vein, SMV superior mesenteric vein, LRV left renal vein. Patient 1a Superior mesenteric vein and splenic vein converge into portal vein, and portal vein directly flows into inferior vena cava. Patient 2b Superior mesenteric vein, splenic vein and left renal vein converge into portal vein, most of the portal vein flows into inferior vena cava, and another slender branch enters the liver. Patient 3c Most of the portal vein flows into the inferior vena cava through the left lobe of the liver, and a few into the liver. Patient 4d The branch of portal vein drains into inferior vena cava. Patient 5e Part of the portal vein flows into the inferior vena cava through the iliac vein. Patient 6f: Superior mesenteric vein and splenic vein converge into portal vein, the left branch of portal vein enters inferior vena cava and the right branch of portal vein enters liver
Discussion
PAH refers to the clinical and pathophysiological syndrome that causes pulmonary vascular resistance and elevated pulmonary artery pressure as a result of the structural or functional changes in the pulmonary vessels caused by multiple etiologies and different pathogenesis, thereby developing into right heart failure and even death. There are many factors leading to PAH, congenital heart disease (CHD)-related PAH, heritable pulmonary arterial hypertension (HPAH) and drug‐induced pulmonary arterial hypertension (DPAH) are frequently encountered. A patient with PAH may run the risks of two or more pathogenic factors at the same time.
CPSS is an uncommon congenital portal vascular malformation which can be divided into CIPS and CEPS according to the location of abnormal vessels. CEPS is also known as Abernethy malformation (AM) as John Abernethy first reported this deformity in 1793.In 1994, Morgan and Superina [6] classified CEPS into two types in light of the absence or presence of an intrahepatic portal venous supply: Type I was congenital portal vein atresia, that was, the blood flow failed to enter the portal vein but flew into the vena cava through an abnormal channel. Based on the confluence of the superior mesenteric and splenic veins, it was sub-grouped into type Ia and type Ib. Type II was congenital portal vein dysplasia, only partial blood flow entered the portal vein and the rest flew into the vena cava through side-to-side extrahepatic communication [7,8,9]. Due to the diverse anatomical changes of CPSS, the clinical manifestations were also diverse and complex. The literature on CPSS combined with PAH is not common, and most of them are based on case reports [10], the pathogenesis is not completely clear. The current point has highlighted that the blood flow in the lungs increases due to the shunt of the portal venous system blood through the abnormal pathway [11]. Meanwhile, vasoactive substances (including 5-hydroxytryptamine, glucagon, histamine, estrogen, and endotoxin) produced by the visceral vascular bed enter the pulmonary circulation directly without being inactivated by the liver [12,13,14]. Consequently, vasodilators and vasoconstrictors cannot maintain balance in the lungs, thereby causing pulmonary vasoconstriction, remodeling and the generation of in situ thrombosis, which ultimately leads to PH [15,16,17]. Furthermore, some reports have revealed that heredity or genetic mutations also play a role in the occurrence and development of diseases [18].
As PAH patients with CPSS present no special clinical manifestations, it is easily to be misdiagnosed or neglected. Angiography is the gold standard for the diagnosis of CPSS. Unfortunately, it cannot be applied as a routine screening method because of its invasiveness. As the non-invasive cross-sectional imaging techniques, ultrasound, CTA or magnetic resonance imaging(MRI) can show shunts and intrahepatic portal vein branches, they can be adopted as the main means of cause screening for clinical PAH patients. Moreover, we reviewed the clinical data of six CPSS patients and found all patients had high cardiac output which was consistent with previous literature reports [19]. We also found that although some patients had varying degrees of elevated liver enzymes due to liver congestion, it is worth noting that bile acid levels were significantly elevated in all patients, whereas the patients previously diagnosed with PAH were not. There is no clear research on this by consulting the previous literature, but the consistent result was found in the data shown in an article on PAH in children with AM [20] .
The mechanism of increased bile acid in CPSS-related PAH patients is rarely reported in the literature. It may be associated with bile acid reabsorption and abnormal metabolism. Bile acid is a product metabolized from cholesterol in the liver. When blood runs through the liver, hepatocytes can uptake 60–80% of bile acids, and this is the reason why the concentration of total bile acid (TBA) in the blood of healthy individuals is very low. The increase of serum TBA concentration is the consequence of decreased liver intake and excretion (hepatocyte damage) or the result of portal system shunt. Aspartate transaminase (AST), alanine transaminase (ALT) and alkaline phosphatase (ALP) exist in cytoplasm and mitochondria, and they are enzymes reflecting liver parenchymal cell injury. In patients with CPSS-PAH, especially those with less severe elevation of pulmonary hypertension, hepatic parenchymal cells are not seriously damaged and liver enzymes may not be elevated markedly. Nevertheless, portal vein blood flows directly into the IVC completely or partially through the congenital abnormal channel, resulting in a significant increase in TBA. Therefore, we put forward this view for the first time: as a simple, non-invasive and sensitive laboratory index, bile acid is helpful in performing etiological screening of patients with PAH. When PAH patients present a remarkable increase in bile acid level, it should be alert to the existence of CPSS .
Conclusions
Because CPSS is a rare cause of PAH, and PAH patients with CPSS are easily covered by other pathogenic factors, or be misdiagnosed as IPAH, all PAH patients need to exclude the presence or absence of CPSS. CPSS and PAH require two-way screening to avoid delays in diagnosis and treatment. As a non-invasive, fast, convenient and economical laboratory index, the level of bile acid reveals the possibility of CPSS in PAH patients. High cardiac output also serves as a reminder. Unfortunately, no unified experience or consensus has been reached on the treatment of such patients [4]. The current treatment plan rely on the type of shunt and individual circumstances. In addition to the application of standardized targeted drugs in treating PAH, a comprehensive clinical evaluation remains necessary, and even surgical management are recommended when necessary. As deeper understanding of the disease and more promising novel treatments emerge in future, the CPSS-associated PAH patients will benefit more with better therapies .
Availability of data and materials
Not applicable.
Abbreviations
- PAH:
-
Pulmonary arterial hypertension
- CPSS:
-
Congenital portosystemic venous shunt
- IVC:
-
Inferior vena cava
- PH:
-
Pulmonary hypertension
- RHC:
-
Right heart catheterization
- PDA:
-
Patent ductus arteriosus
- CI:
-
Cardiac index
- CO:
-
Cardiac output
- CTA:
-
Computed tomography angiography
- CIPS:
-
Congenital intrahepatic portosystemic shunt
- CEPS:
-
Congenital extrahepatic portosystemic shunt
- CHD:
-
Congenital heart disease
- HPAH:
-
Heritable pulmonary arterial hypertension
- DPAH:
-
Drug‐induced pulmonary arterial hypertension
- AM:
-
Abernethy malformation
- MRI:
-
Magnetic resonance imaging
- TBA:
-
Total bile acid
- AST:
-
Aspartate transaminase
- ALT:
-
Alanine transaminase
- ALP:
-
Alkaline phosphatase
References
Howard ER, Davenport M. Congenital extrahepatic portocaval shunts-the Abernethy malformation. J Pediatr Surg. 1997;32(3):494–7.
Christiane S, Bandsma Robert HJ, Gana Juan C, et al. Congenital portosystemic shunt: characterization of a multisystem disease. J Pediatr Gastroenterol Nutr. 2013;56:675–81.
Anna B, Fanny T, Macarena S-T, et al. Congenital extrahepatic portosystemic shunts (Abernethy malformation): an international observational study. Hepatology. 2020;71:658–69.
Franchi-Abella S, Branchereau S, Lambert V, et al. Complications of congenital portosystemic shunts in children:therapeutic options and outcomes. J Pediatr Gastroenterol Nutr. 2010;51:322–30.
Uike K, Nagata H, Hirata Y, et al. Effective shunt closure for pulmonary hypertension and liver dysfunction in congenital portosystemic venous shunt. Pediatr Pulmonol. 2018;53:505–11.
Morgan G, Superina R. Congenital absence of the portal vein:two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatr Surg. 1994;29(9):1239–41.
Ávila LF, Luis AL, et al. Shunt porto cava congénito.Malformación de Abernethy. Cir Pediatr. 2006;19:204–9.
Alonso-Gamarra E, et al. Clinical and radiologic manifestations of congenital extrahepatic portosystemic shunts:a comprehensive review. Radiographics. 2011;31:707–22.
Witters P, Maleux G, et al. Congenital veno-venous malformations of the liver: widely variable clinical presentations. J Gastroenterol Hepatol. 2008;23:e390–4.
Ohio T, Muneuchi J, Ihara K, et al. Pulmonary hypertension in patients with congenital portosystemic venous shunt:a previously unrecognized association. Pediatrics. 2008;121:e892–9.
Benjaminov FS, Prentice M, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut. 2003;52:1355–62.
OlingS OlsonR. Congenital absence of portal venous system in a 50-year-old woman. Acta Med Scand. 1974;196(4):343–5.
Carolina S, Adelia M, et al. Pulmonary arterial hypertension in a patient with a portosystemic shunt: diagnostic challenge. Case. 2020;4(2):93–6.
Galie N, Humbert M, Vachiery JL, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The task force on diagnosis and treatment of pulmonary arterial hypertension of the European society of cardiology. Eur Heart J. 2004;25:2243–78.
Simonneau G, Gatzoulis MA, et al. Updated clinical classification of pulmonary hypertension. Turk Kardiyol Dern Ars. 2014. https://doi.org/10.1016/j.jacc.2013.10.028.
Dumbar I, Abman S, Barst R, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol. 2013;62:D117–26.
Henriksen JH, Moller S. Cardiac and systemic haemodynamic complications of liver cirrhosis. Scand Cardiovasc J. 2009;43:218–25.
Law YM, Mack CL, Sokol RJ, et al. Cardio pulmonary manifestations of portovenous shunts from congenital absence of theportalvein:pulmonary hypertension and pulmonary vascular dilation. PediatrTransplant. 2011;15(8):e162-168.
Spruijt OA, Bogaard H-J, Vonk-Noordegraaf A. Pulmonary arterial hypertension combined with a high cardiac output state: Three remarkable cases. Pulm Circ. 2013;3(2):440–3.
Yunbin,ZENG Yunhong,XIAO Zhenghui et al.Clinical analysis of pulmonary arterial hypertension associated with congenital portosystemic shunt in children.J Clin Pediatr.2019,37(12).
Acknowledgements
We would like to thank all patients and their families participating in the article.We also would like to thank all colleagues who helped in designing and performing the survey.
Funding
Hubei Province Health and Family Planning Scientific Research Project, Grant/Award Number: WJ2019H237.
Author information
Authors and Affiliations
Contributions
YLand XD designed the study. QX, HZ, XZ, and GZcollected data. YL and XD wrote the manuscript. YL reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
We have obtained the written consent from the patients to submit this case series for publication.
Competing interests
The author(s) declare that there is no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Deng, X., Zhou, H. et al. Bile acid predicts congenital portosystemic venous shunt in patients with pulmonary arterial hypertension. Eur J Med Res 28, 74 (2023). https://doi.org/10.1186/s40001-023-01039-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-023-01039-0