
Abstract
Colorectal cancer (CRC) is the third most common cancer worldwide. One of the main causes of colorectal cancer is inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD). Intestinal epithelial cells (IECs), intestinal mesenchymal cells (IMCs), immune cells, and gut microbiota construct the main body of the colon and maintain colon homeostasis. In the development of colitis and colitis-associated carcinogenesis, the damage, disorder or excessive recruitment of different cells such as IECs, IMCs, immune cells and intestinal microbiota play different roles during these processes. This review aims to discuss the various roles of different cells and the crosstalk of these cells in transforming intestinal inflammation to cancer, which provides new therapeutic methods for chemotherapy, targeted therapy, immunotherapy and microbial therapy.
Similar content being viewed by others

Introduction
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), causes long-term immune-mediated colitis-associated colorectal cancer (CAC) [1]. Patients with IBD have an increased risk of developing colorectal cancer (CRC), the third most common cancer worldwide [2, 3]. An extended meta-analysis demonstrated that the risk of CRC is approximately 2% after 10 years and up to 18% at 30 years in UC patients [4]. Therefore, chronic inflammation in IBD patients leads to a significant increase in CRC risk.
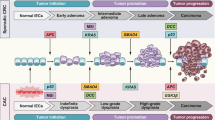
Currently, our understanding of the mechanisms leading to a high risk of intestinal cancer in IBD patients has improved [5]. In patients with IBD, the pathogenesis of CRC involves both genes and environmental factors, such as genetic mutations, epigenetic changes and alterations in immune response factors [6,7,8,9]. The molecular alterations in colorectal cancer with IBD varied significantly from those in sporadic CRC [10, 11]. First, the timing of gene alterations in colitis-associated colorectal cancer is different from that in sporadic CRC [12, 13]. The APC gene is usually lost at a later stage in CAC, whereas it always occurs at an earlier date in sporadic colorectal cancer [14]. And p53 gene mutation is an early event in CAC, whereas it appears to be a late event in sporadic disease [15]. Second, there are some differences in the frequency of gene mutations between CAC and CRC. Compared with sporadic mutations, APC and KRAS gene mutations are at a relatively low level in colitis-related cancer [16]. This evidence indicates that gene expression and pathway alterations are closely related to CAC progression in specific cells.
Several types of cells, including intestinal epithelial cells (IECs), intestinal mesenchymal cells (IMCs), immune cells and gut microbiota construct the main body of the colon. A few genes with barrier functions in the intestinal epithelium play a significant role in protecting the gastrointestinal tract from pathogen invasion [17,18,19]. In colitis and colitis-associated carcinogenesis, these cells are disrupted, resulting in damaged IECs, disorganized IMCs and excessive recruitment of immune cells [20, 21]. Patients with IBD and colon cancer have been associated with aberrant function of the epithelial barrier [22, 23]. In addition, IMCs can regulate the development of colon tumors, including intestinal inflammation regulation, epithelial proliferation, stem cell maintenance, angiogenesis, extracellular matrix remodeling and immune responses [24, 25]. Different immune cells such as neutrophils, macrophages and dendritic cells, are activated by chronic accumulation and are recognized as major contributors to gene alterations [26]. Furthermore, the gut microbiota plays a vital role in the modulation of the immune system in chronic inflammatory diseases of the intestine [27, 28]. Therefore, intestinal epithelial cells, mesenchymal cells, immune cells, and gut microflora play a pivotal role in colitis and CAC. In this review, we discuss the various roles of different cells in the transformation of intestinal inflammation to cancer and provide new therapeutic ideas for IBD and colitis-associated colorectal cancer.
IECs and colitis-associated colorectal cancer
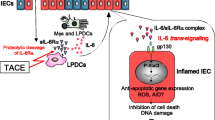
The epithelium is a single-cell layer consisting of various subtypes of particular IECs, such as cup cells, tuft cells, absorptive cells, enteroendocrine cells, M cells, Paneth cells and goblet cells [29]. These cells have pivotal and distinctive functions in maintaining intestinal homeostasis [30]. Paneth cells stay in the small intestine with the function of secreting antimicrobial peptides and maintaining the niche of stem cells in the intestine [31, 32]. In contrast, goblet cells are stayed in the large intestines and produce abundant glycosylated proteins, such as Muc2 [33]. Intestinal epithelium cells (IECs) preferentially absorb nutrients and have a protective barrier effect and strong defense ability against a harmful intestinal microenvironment [34, 35]. Disruption of the intestinal epithelium is a hallmark of IBD. Moreover, the process of gene alterations and pathway changes in intestinal epithelial cells has been demonstrated during the formation of IBD [36,37,38,39]. Intestinal tumors originate from gut epithelial cells and develop from gene mutations in a few signaling pathways, such as NF-κB, Wnt, STAT, endoplasmic reticulum (ER) stress and transforming growth factor (TGF)-β (Fig. 1) [40,41,42].

IECs are involved in CAC progression. Many signaling pathways are involved in the occurrence and development of colorectal cancer, such as the NF-kB, Wnt, STAT3 and TGF-β pathways. A few genes, such as ASAP3, promote tumor growth by binding to Nemo, while others, such as TRF9, regulate the binding of NF-kB to the promoter region by affecting target genes. Immune cells such as myeloid cells, Tregs, and DCs can regulate the phosphorylation of STAT3 to affect tumor growth. In addition, the Wnt signaling pathway also affects tumor progression. JMJD2D protein can regulate the transcription of Wnt-related genes by binding to β-catenin. Similarly, TRIB3 can also regulate the occurrence and development of tumors in intestinal stem cells by affecting Wnt-related genes
The NF-κB pathway in the intestinal epithelium is the first pathway in our discussion. Colonic organoid experiments can assess the epithelial responses to inflammatory cytokines in an in vitro model. In colonic organoids, NF-κB signaling is activated in patients with chronic inflammation, which leads to the occurrence of colitis-associated colorectal cancer [43]. In CD patients, NOD2 is a dominant genetic risk factor, accompanied by many rare variants and three main risk-conferring variants [44, 45]. NOD2 regulates TLR signaling and NF-κB pathways, which increases the risk of IBD [35]. Recent studies have suggested that P65 (an important component of NF-κB) binds to the N-terminus of ITF2 to inhibit ubiquitination, promotes the stability of ITF2 and reduces colitis-associated colorectal cancer[46]. Moreover, NFKBIZ (also known as IκBζ) gene mutations were abundant in colitis samples, but the incidence and severity of colorectal tumors decreased significantly in NFKBIZ-deficient mice [47]. The gene deficiency of Nemo (NF-κB essential modifier) in the epithelium causes epithelial cell apoptosis and affects the expression of antimicrobial peptides, leading to bacterial migration to the mucosa. In addition, ASAP3 interacts with Nemo to regulate the expression of NF-κB, which is associated with poor prognosis and plays an oncogenic role in colorectal carcinogenesis [48]. Moreover, chemotherapy can activate NF-κB and IRAK4 by increasing the transcription of TLR9. Meanwhile, the expression of TLR9 is also inhibited by IRAK4 or IKK inhibitors, which can protect colorectal cancer cells from chemotherapy drugs through a feedforward pathway [49].
The activation of the Janus kinase (JAK)/signal transducer and activation of the transcription (STAT) pathway is important in IEC disorders [50, 51]. STAT3 plays a crucial role in maintaining intestinal homeostasis and strongly protects against chemically induced colitis [52]. Mice with reduced STAT3 activity were highly susceptible to colitis by regulating the IL-6ST/gp130 cytokine receptor, which plays a key role in promoting intestinal barrier function and epithelial regeneration [53, 54]. Conditional knockout mice with a specific STAT3 or ATG16L1 deficiency in IECs can affect the secretion of IL-22, which is associated with wound healing and has a high risk of developing colitis [55,56,57]. However, abnormal STAT3 activation is also closely related to the malignant progression and pathogenesis of solid tumors such as CAC [58]. The activation and translocation of STAT3 into the nucleus promote the transcription of target genes related to cell proliferation, metastasis and inflammatory response. In colon epithelial cells, BMI1 and MEL18 can promote proliferation and reduce apoptosis to accelerate the development of CAC by regulating the secretion of IL-6/11, which is a regulator of STAT3 [59]. During CAC development, overexpression of the CAMK2γ gene facilitates the activation of STAT3 in the epithelium, thereby promoting the survival and proliferation of IECs [60]. Additionally, the oncogene RXRα is an effective regulator of the inflammatory response that promotes colorectal tumorigenesis by activating the NF-κB-IL-6-STAT3 signaling cascade [61]. Moreover, mTORC1 can activate COX-2 transcription by phosphorylating STAT3 and enhancing the interaction with COX-2 promoter in the colonic epithelium, thus recruiting T helper-17 (Th17) cells and promoting tumor growth [62]. Blockade of IL-6 levels in IECs can inhibit the activation of STAT3 and abnormal cell proliferation, which provides new therapeutic potential [63, 64]. Thus, these studies indicate that STAT3 plays a protective role by maintaining epithelial cell proliferation during acute colitis, while abnormally activated STAT3 promotes the progression of CAC.
The third pathway in our discussion is the Wnt/β-Catenin signaling pathway, which is essential for the pathogenesis of CAC [65]. Whole-exome sequencing analyses have indicated that Wnt pathways play a predominant role in IBD-associated colon tumorigenesis [66]. There is an important correlation between the activation of Wnt and the expression levels of a few genes in intestinal epithelial homeostasis [67,68,69]. In the CAC mouse model, MUC1-C can form a transcription complex with MYC and act on the LGR5 promoter region, thereby activating LGR5 expression and tumor growth [70, 71]. In addition, histone demethylase JMJD2D is highly expressed in tumors, regulates a few signaling pathways (including Wnt/β-Catenin and Hedgehog) and activates the transcription of downstream target genes related to proliferation, migration, and invasion, which leads to the formation of CAC [72, 73]. In colorectal cancer stem cells, TRIB3 can interact with β-catenin and Tcf4 to increase AOM/DSS-induced colorectal tumor formation and xenograft tumor growth in mice [74]. Currently, researchers are exploring drugs that can inhibit key signaling pathways. For example, Zeng et al. found that scutellarin improved colitis-related colorectal cancer by reducing Wnt/β-Catenin signaling [75]. Researchers also found that synbiotics can significantly inhibit abnormal activation of the Wnt signaling pathway in an AOM/DSS-induced mouse model and alleviate the progression of CAC [76].
Changes in gene expression may affect the development of inflammatory bowel disease [77,78,79]. The MEP1A gene, which encodes the α subunit of meprins, has a strong relationship with UC patients. In IECs, the subunit of meprins is associated with the transmembrane β subunit and cleaves different substrates, which suppresses the development of UC. Mep1A-deficient mouse models are more susceptible to chemically induced colitis, in accordance with the decreased expression of MEP1A in UC patients [80]. In addition, it has been reported that hepatocyte nuclear factor 4α (HNF4α), a nuclear transcription factor encoded by the gene HNF4α, is crucial for epithelial tight junctions and intestinal permeability by regulating several cytokines and signaling pathways. In IBD patients, HNF4α has low expression, which is consistent with the results in mice lacking HNF4α in IECs, indicating that IECs are more susceptible to drug mediated colitis in mouse models [81]. Moreover, FAM3D (a cytokine-like molecule) is highly expressed in the gastrointestinal tissues and is associated with colonic mucosal integrity, epithelial cell proliferation, antibacterial effects and the development of inflammatory bowel disease [82]. A few genes, such as BRG1 and SETD2, attenuate inflammation in CRC by modulating oxidative stress [83, 84]. E-cadherin, a cell adhesion molecule expressed in epithelial cells encoded by the gene CDH1, plays an important role in cell growth, proliferation, and epithelial differentiation. In addition, a typical pathological characteristic of IBD patients is the loss or disorder of E-cadherin accompanied by increased epithelial permeability [85]. These differentially expressed genes and signaling pathways have the potential to become targets for the diagnosis and treatment of IBD.
IMCs in CAC progression
Intestinal mesenchymal cells (IMCs) are major components of the normal intestinal tract and intestinal tumors [86]. They include numerous cell types with a similar origin, function and molecular markers, such as intestinal fibroblasts, myofibroblasts and pericytes [87] (Fig. 2). Recent studies used unbiased single-cell profiling involving over 16,500 colonic mesenchymal cells and revealed four subsets of fibroblasts, including TNF superfamily member 14 (TNFSF14), fibroblastic reticular cell-associated genes, IL-33, and lysyl oxidases, which express different transcriptional regulators and signaling pathways [88, 89]. Activated IMCs can promote inflammation and tumor progression by directly affecting the growth of neoplasms and changing the microenvironment of the surrounding tumors. Cancer-associated fibroblasts (CAFs) consist of a population of cells from different origins that lead to tumor initiation, progression, metastasis and poor outcomes of patients via interactions and changes in the microenvironment [90, 91].

The main function of IMCs in intestinal homeostasis. Intestinal mesenchymal cells play an important role in maintaining intestinal homeostasis. The main functions of fibroblasts and pericytes are maintaining epithelial homeostasis, stem cell niche, vascular function and ECM. Meanwhile, CAFs and myofibroblasts can promote inflammation, cell proliferation, angiogenesis, invasion and migration
Fibroblasts
Fibroblasts reside in the lamina propria with α-smooth muscle actin (α-SMA)-negative characteristics, are adjacent to the intestinal epithelium and remain in the quiescent phase with poor transcriptomic and metabolic activities in the normal colon [92]. Fibroblasts have different characteristics than epithelial and immune cells and possibly originate from a mesenchymal lineage. They are connective tissues that can synthesize collagen components [93]. The main functions of fibroblasts include the accumulation and maintenance of the extracellular matrix (ECM), stabilization of adjacent epithelia and regulation of inflammation [92, 94]. Many studies have shown that fibroblasts can be activated by multiple factors, such as growth factors, inflammatory cytokines and chemokines, mechanical stress and reactive oxygen species in colitis and colorectal cancer [95, 96]. The secretion of periostin in fibroblasts can promote the occurrence and development of colorectal cancer by activating FAK-Src kinase, the Yap/TAZ pathway and IL-6 expression in tumor cells [97]. In addition, IL-11+ fibroblasts can activate tumor cells and fibroblasts by producing a large amount of IL-11 and promote tumorigenesis at the same time [98]. Furthermore, fibroblasts in tumors include activated fibroblasts (myofibroblasts), cancer-associated fibroblasts (CAFs) and cancer-associated mesenchymal stem cells (MSCs) [99].
Myofibroblasts
Myofibroblasts are stellate shaped, proliferate and are usually more active, which makes them different from quiescent fibroblasts in terms of morphology and metabolism [100]. The typical characteristics of intestinal myofibroblasts are the expression of α-SMA, CD90, and vimentin. Among these markers, α-SMA is considered the most typical intestinal myofibroblast marker. However, α-SMA is also expressed in pericytes, partial smooth muscle cells and bone marrow-derived mesenchymal stromal cells [101]. Myofibroblasts can perform various functions by regulating signaling pathways and cytokines. MyD88 signaling in myofibroblasts promotes CAC progression by activating osteopontin secretion, macrophage M2 polarization and the STAT3/PPARγ pathway [102]. Activated fibroblasts can produce MMPs, which are ECM-degrading proteases and promote cancer cell invasion. For example, the overexpression of MMP1 induces invasiveness, and MMP3 expression promotes epithelial-to-mesenchymal transition (EMT) progression and the invasion of cancer cells into adjacent tissues [103]. Studies conducted in TnfΔARE/+ mice have shown an increase in the expression of MMP9 and ICAM1, thereby inducing ECM remodeling and adaptive immune responses [104, 105]. Activated fibroblasts play a role in regulating immune homeostasis, including immune cell recruitment and modulation by secreting chemokines and infection- or injury-related cytokines. Additionally, myofibroblasts can maintain epithelial homeostasis by sensing the inflammatory environment created by the tissue or bacteria and mediate epithelial regeneration through the activation of the Cox-2 signaling pathway [106,107,108].
Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) have a specific definition that includes all fibroblastic, non-vascular, non-neoplastic, non-inflammatory and non-epithelial features in tumorigenesis [109,110,111]. Usually, the markers of CAFs are α-SMA, fibroblast activation protein-α (FAP-α), fibroblast-specific protein-1 and platelet-derived growth factor receptor-β (PDGFR-β) [112]. In cancer-associated fibroblasts, lncRNA-H19 is highly expressed and promotes stemness and chemoresistance through exosomal transmission. And H19 promotes CRC progression by competitively binding miR-141 and increasing the expression of β-catenin [113]. In recent years, the importance of CAFs in the progression of colitis and CAC has been recognized.
In CAFs, TGF-β signaling is necessary for metastasis by promoting the activation of STAT3 signaling and the secretion of CCL2 and CCL8 during the development of CAC disease [114]. Additionally, Smad7 and Smurf1 (negative regulators in the TGF-β signaling pathway) are decreased in Ikkβ-deficient fibroblasts, which can increase the secretion of hepatocyte growth factor (HGF) and activate CAC progression [115, 116]. In addition, CAFs are associated with ECM remodeling, which can produce ECM constituents and rebuild enzymes, such as TIMPs, MMPs and other proteases [117, 118]. Moreover, another study found that inhibiting the activation of STAT3 in COL1+ fibroblasts can reduce tumor growth, while the activation of STAT3 can accelerate the progression of CAC. Thus, reducing the activation of STAT3 in COL1+ fibroblasts has the potential to become a therapeutic target for CAC [119].
Additionally, CAFs have been proposed to regulate the tumor microenvironment [120, 121]. CAFs produce various cytokines and chemokines to regulate tumor proliferation, migration and adhesion [122, 123]. In CAC patients, a high level of CCL2 produced by CAFs leads to the formation of macrophages with a tumor-suppressive function [124]. In addition, researchers have found that MCAM is a specific marker for colorectal cancer stromal cells in the CAC mouse model. In MCAM+ cancer-associated fibroblasts, MCAM interacts with IL-1 receptor 1 to enhance the NF-κB-IL34/CCL8 signaling pathway and promotes the recruitment of tumor-associated macrophages [125]. The co-expression of CAFs and macrophages has become an important marker of malignant tumors [126, 127].
Various types of immune cells are correlated with CAC
Several studies have recently shown that colitis-associated colorectal cancer is accompanied by many adaptive immune cells, including T and B lymphocytes, and innate immune cells that contain myeloid-derived suppressor cells (MDSCs), macrophages, dendritic cells (DCs), neutrophils, and NK cells (Fig. 3) [128,129,130,131].

Immune cells regulate the progression of colon cancer by affecting the secretion of cytokines and chemokines. Under inflammatory conditions, immune cells including T cells, dendritic cells, MDSCs, macrophages and neutrophils can secrete cytokines and chemokines to change the tumor microenvironment, thus causing DNA damage and activating tumor-related genes
T lymphocytes, such as regulatory T cells (Tregs) and helper T cells (Th) are associated with CAC [132, 133]. The removal of CD4+ T cells and blocking CCL4 or IL-17 reduced the formation of tumorigenesis caused by myeloid cells [134, 135]. G-CSF/G-CSFR increased the secretion of FoxP3-expressing CD4+ and CD8+ T cells, while G-CSFR deficiency in T cells increased cytotoxic activity in the tumor microenvironment by producing IL-17A which improved resistance to anti-PD-1 therapy [136,137,138]. In IBD patients, the blockade of IL-7R reduced colonic inflammation by inhibiting T-cell homing to the gut and altering the activation of effector T cells [139]. Additionally, S1PR4 affected the expression of PIK3AP1 and LTA4H, which is associated with the proliferation and survival of CD8+ T cells [140]. In recent years, immunotherapy has played an important role in the treatment of IBD patients [141,142,143]. FK228 is a histone deacetylase inhibitor that can upregulate the expression of PD-L1 in tumor cells and enhance the antitumor effect by affecting the activity of CD4+ and CD8+ T cells [144]. In addition, the recruitment of CD4+ Th lymphocytes can play an important role in maintaining chronic enteritis in UC and CD patients and promoting colitis-associated colon cancer. Detection in patients with colonic CD showed that high levels of Th1 cytokines lead to an increased risk of CAC in the intestine, whereas the Th2 immune response is directly involved in colitis-associated tumorigenesis by inducing DNA mutations caused by Th2-associated cytokines (i.e., IL-4 and IL-13) in cultured colonic epithelial cells [145, 146]. The expression of FAM64A regulates the differentiation of Th17 but not Th1 cells by modulating the IL-6/STAT3 axis in CAC [147, 148]. Moreover, regulatory T cells are CD4+ T cells that express the master transcription factors Foxp3 and CD25, showing immunosuppressive effects by direct cell communication and the release of the cytokines TGF-β and IL-10 [149, 150]. A few components are necessary for the appropriate differentiation and function of Tregs [151, 152]. The analysis of single-cell RNA sequencing data of human colorectal cancer tissues showed that MondoA-thioredoxin-TXNIP axis maintains Tregs and regulates glucose uptake [153]. Erdman et al. suggested that Tregs could develop pathological changes and at the same time reduce tumor formation, which is consistent with a recent study that Tregs have antitumor activity in colitis-associated colon cancer [154].
Myeloid-derived suppressor cells (MDSCs) originate from the bone marrow, which participates in the activation of specific elements and continues multiplication in the pathologic environment [155, 156]. MDSCs are involved in suppressing T-cell immunity via numerous mechanisms, including disturbing T-cell function by reducing necessary nutrients, destroying the normal response of effector T cells, indirectly depressing T-cell function by activating the expansion of regulatory T cells (Treg), and inhibiting the proper expression of L-selectin in naïve T cells [157, 158]. In addition to their immunosuppressive activity, MDSCs play an important role in enhancing angiogenesis [159, 160]. In B16 and RENCA mouse models, STAT3 signaling is considered a fundamental factor in increasing angiogenesis activity in tumor cells. MDSCs and macrophages and the expression of related proteins, such as β-FGF and VEGF, are controlled by STAT3 signaling, which is activated in endothelial cells, facilitates angiogenesis and can be interrupted by STAT3 inhibitors [161]. The pace of colitis development slows down with the treatment of resveratrol in IL-10-deficient mice accompanied by MDSC multiplication and reduced colitis-associated cytokine production [162]. In the development of CAC, MyD88 signaling plays an important role in colonic myeloid cells by regulating the production of pro-inflammatory cytokines and by increasing proliferation and reducing apoptosis in epithelial cells [163, 164]. Recent studies indicate that suppressing the activity of EZH2 promotes MDSC production and ameliorates CAC [165]. Moreover, IL-27 plays an important role in the accumulation of MDSCs and increases tumor cell proliferation, which contributes to CAC development in a murine model [166]. In the AOM/DSS model, there were increased tumor loads and MDSCs in Card9-deficient mice compared with WT mice. In addition, Card9−/− macrophages caused changes in the composition of the intestinal flora, such as a significant increase in C. tropicalis [167]. The gut microbiota was associated with the increased CXCL1, CXCL2, CXCL5 expression and MDSC accumulation in tumor tissues [168].
DCs (dendritic cells) play a vital role in the development of colitis because DC deficiency has a strong correlation with the ease of DSS-induced colitis in a mouse model [169, 170]. DCs exhibit a protective effect by activating the rehabilitation of the intestinal epithelium instead of regulating the immune reaction [171]. Another experiment ablated DCs before DSS treatment and inflammation was aggravated, indicating that DCs play a protective role in the initiation of colitis, except for pathogenicity during the progression of tumors [172]. An observation in TGF-β-deficient and IL-10-deficient mice suggested that IL-10 and TGF-β act as determinant factors of DC function in the intestine [173, 174]. In IBD patients, the depletion of DCs in their peripheral blood exacerbates the development of diseases, which regulate the infiltration of MDSCs and cause an increasing number of chemokines, such as CCL20 or MAdCAM-1 (mucosal vascular address in cell adhesion molecule-1) [175,176,177]. In acute IBD, immature DCs are dramatically decreased, showing that several DC subsets may be absent during disease recurrence. In addition, M-DC8+ DCs exist in the subepithelial dome ileum of CD patients, which secrete abundant TNF-α in the treatment of lipopolysaccharide (LPS), contributing to the tumorigenesis of IBD [178]. Moreover, p38α deficiency in DCs influences the activation of Tr1 cells by regulating IL-27 and IL-22 secretion, which plays a pivotal role in the intestinal inflammatory response and tumorigenesis [179]. Additionaly, lymphotoxin signaling induces the expression of IL22BP through the activation of NF-κB in human colorectal tumors and cultured human dendritic cells [180].
Intestinal macrophages are located under the epithelial layer and are considered to originate from classical blood monocytes activated by CCL2/CCR2 [181, 182]. These macrophages express many innate receptors, including the scavenger receptors CD36/CD163, the triggering receptor expressed on myeloid cells (TREM)-2, the C-type lectin receptor CD209 and the FcgR CD64, which promotes the chemotaxis and phagocytosis of bacteria [183, 184]. In a colitis environment, monocytes are moved to the LP and transition to inflammatory macrophages in respond to TLR stimulation and secrete pro-inflammatory cytokines, such as IL-23 [185, 186]. In the human body with Crohn’s disease, CD14+ macrophages are most abundant in inflamed tissues, accompanied by the secretion of pro-inflammatory cytokines stimulated by TLR, whereas resident macrophages do not respond to TLR [187,188,189]. IL-10-deficient macrophages produce higher amounts of prostaglandin E2 after LPS stimulation to impede bacterial killing [190, 191]. Moreover, the deletion of EPRAP in macrophages increased the levels of p105, MEK, and ERK phosphorylation, which led to the activation of stromal macrophages in DSS-induced colitis [192]. CX3CR1-deficient mice showed significantly lower expression of HOMX-1 (an antioxidant and anti-inflammatory enzyme) in adenomatous colon tissue by mediating the CX3CR1 receptor [193]. In primary human and mouse colorectal cancer samples, mTORC2 is only expressed in the adjacent area of macrophages, but not in tumor cells, as mTORC2-deficient macrophages stimulate tumor growth through the cytokine SPP1/osteopontin [194]. BATF2 attenuated inflammation and protected intestinal epithelial cells by inhibiting the transcriptional activation of STAT1/CCL2 and reducing the recruitment of macrophages in colon tissues [195].
Neutrophils accumulate in specific tissues with acute inflammation, acting as the first line of defense, and play an important role in resisting pathogenic microorganisms [196]. In various mouse colitis-associated models, the depletion of circulating neutrophils accelerates inflammation, which suggested that neutrophils are a protective factor in the progression of inflammation [197]. Activated neutrophils produce numerous pro-inflammatory cytokines, including TNF-α, IL-1, TGF-β and IL-6, as well as chemokines, such as CXCL1, CCL2, CCL3, CXCL8 and CXCL9, which result in further recruitment of leukocytes and activation of inflammatory pathways [198]. Additionally, neutrophils increase the production of pro-inflammatory microRNAs (miR-31 and miR-155), which affect genomic instability by modulating replication fork collapse and inhibiting homologous recombination [199, 200]. Moreover, Zhou et al. found that CD177+ neutrophils suppress tumorigenesis of epithelial cells and are markedly increased in tumor tissues compared with controls in colorectal cancer [201]. Lin et al. found that the expression of BATF3 was correlated with the poor prognosis of colitis-associated colorectal cancer and promoted the recruitment of neutrophils by modulating the CXCL5/CXCR2 axis [202]. Recently, Zhang et al. identified IRAK-M as an innate suppressor of neutrophils that regulates the activation of STAT1/3/5 and promotes tumor growth [203].
In addition, a few innate immune cells, including basophils and γδ T cells, also play an important role in the pathogenesis of IBD and CAC. The inhibition of AKR1B8 activates innate immunity (including excessive infiltration of basophils and neutrophils) and promotes the occurrence of IBD [204]. γδ T cells, a subset of T cells, are abundant in the intestinal mucosa and maintain epithelial homeostasis [205]. AKR1B8 deficiency leads to increased infiltration of neutrophils and mast cells, as well as a decrease in the number of γδ T cells, thereby disrupting the self-renewal of intestinal epithelium and promoting the progression of inflammation-related colorectal cancer [206].
Impact of the intestinal microbiome on CAC
Gut bacteria play an essential role in regulating gut homeostasis by affecting immunity [207,208,209]. Dysbiosis of the intestinal microbiome is strongly associated with many intestinal diseases, such as inflammatory bowel diseases (IBD) and colitis-associated colorectal cancer (CAC) [210,211,212]. With the development of next-generation sequencing technology, an unprecedented view of the intestinal microbiome has been recognized as being involved in intestinal disorders in IBD and CAC patients [213, 214]. Several studies have demonstrated that bacteria promote CAC progression by recruiting macrophages and activating T helper cells [215]. The interaction between the gut and different microorganisms such as pathogenic bacteria, probiotics and fungi will exert or reduce tumorigenic factors in the host [216,217,218] (Fig. 4).

Intestinal flora affects tumor progression. In the presence of probiotics in the intestinal tract, NK cells and macrophages phagocytize the abnormal flora due to the normal state of the body's immune system. At the same time, the secretion of cytokines and chemokines inhibits the abnormal proliferation of intestinal epithelium. In CAC, E.coli and other harmful bacteria enter intestinal epithelial cells, regulate various immune cells, and activate tumor-associated transcription factors (such as TLR4, NF-κB and PI3K/AKT), thus aggravating the tumor progression
Harmful microbiota, including F. nucleatum and E. coli can promote the development of inflammation-related colorectal cancer. F. nucleatum was first known as a common anaerobic gram-negative bacterium in the oral cavity and was recently associated with preterm birth, rheumatoid arthritis and colorectal cancer [219]. Recent studies have found that F. nucleatum is closely related to the occurrence and development of CAC. Adherence and invasion are important mechanisms in the induction of host defense and host responses. CAC has a strong correlation with the invasiveness of F. nucleatum [220, 221]. And F. nucleatum was detected in 39.5% of human CAC samples [222]. In addition, F. nucleatum increased the expression of tumorigenic genes in CAC by regulating the TLR4-PI3K-AKT-NF-κB pathway [223]. Specifically, F. nucleatum increased the expression of miR-21, which represents an increased risk of poor outcomes and may regulate the levels of RAS GTPase by activating the TLR4 signaling pathway and causing the activation of NF-κB signaling in CAC progression [224]. Moreover, F. nucleatum can promote the EMT process by activating the expression of the EGFR signaling pathway, thereby accelerating the progression of CAC [221]. Additionally, Rubinstein et al. suggested that F. nucleatum promotes the progression of CAC development via its special adhesin FadA and regulation of the E-cadherin/β-catenin signaling pathway [225].
E. coli is another commensal bacterium in the human gastrointestinal tract and belongs to the gram-negative and aero-anaerobic bacteria [226]. Various studies have indicated that there is a specific link between E. coli and CAC [227, 228]. Adherent invasive E. coli (AIEC) is an important pathotype, and the amount of AIEC has increased in colitis-associated colorectal cancer compared with normal tissues [229, 230]. FimH adhesin variants of AIEC can more easily bind to intestinal epithelial cells, which causes the recruitment of dendritic cells (DCs) and macrophages to prevent infection by modulating the secretion of the pro-inflammatory cytokines IL-8 and CCL20 in intestinal epithelial cells [231]. In addition, increased oxygenation of colon epithelium and proliferation of E. coli in chemically induced CAC lead to the production of colibactin (an oncogenic factor produced by E. coli) [232]. In the CAC mouse model, researchers have found that restricting the proliferation of E. coli can alleviate intestinal inflammation and reduce colorectal tumors [27]. Thus, inhibiting the proliferation of Escherichia coli is proposed as a preventive strategy for alleviating inflammation-related colorectal cancer.
Probiotics are living microorganisms that have a strong association with diverse health benefits, such as regulating gut microflora, suppressing inflammation and exerting antitumor effects [233,234,235]. Lactobacillus and Bifidobacterium are two species of probiotics with tumor-suppressive effects indicated in colorectal cancer cell lines and mouse models [237, 238]. And there are three main mechanisms by which probiotic bacteria prevent colorectal cancer: modulating the immune response, inducing cell apoptosis, and exerting antioxidant activity [239, 240]. The combination of probiotics and prebiotics inhibits the secretion of pro-inflammatory cytokines and inflammation-associated enzymes, such as TNF-α, IL-1β, iNOS and COX-2, and simultaneously upregulates the expression of anti-inflammatory cytokines and pro-apoptotic factors, such as IL-4, IL-10, p53 and p21 [240,241,242]. The probiotic strain Lactobacillus casei Shirota can suppress tumor growth by enhancing the cytotoxicity of natural killer (NK) cells and IL-12 produced by dendritic cells [244, 245]. A recent study revealed that L. casei BL23 has an immunomodulatory role in CAC by downregulating the cytokine IL-22 and has an anti-proliferative effect by upregulating caspase-7, caspase-9, and Bik [245]. Moreover, MSCs can migrate into the colon and maintain the dynamic balance of intestinal microorganisms by inhibiting chronic inflammation, thereby alleviating CAC progression [246]. Recently, microbiota therapy has been involved in preventing and treating intestinal dysfunction, including IBD, CAC, pathogenic bacterial or viral infection, irritable bowel syndrome (IBS), and antibiotic-associated diarrhea [248, 249]. Oral treatment with zerumbone inhibits the progression of colitis-associated colorectal cancer by reducing the harmful bacteria enterotoxigenic Bacteroides fragilis [249].
Although fungi account for only 0.02–0.03% of the intestinal microbiota, the increase in the number of fungi in the intestinal microbiota is an important factor in the development of IBD and IBD-associated colorectal cancer [251, 252]. The fungi in the intestine mainly include Saccharomyces, Candida, Penicillium and Kluyveromyces. Compared with healthy individuals, the diversity of fungal species increased in IBD patients [252]. Notably, the abundances of C. albicans and Cryptococcus neoformans were increased, while Malassezia sympodialis and S. cerevisiae were decreased in IBD [252]. The use of a single antibiotic, such as cefoperazone, will induce fungal infections (especially C. albicans), which affect the composition of bacterial microbiota in the intestine [253]. The immune response against intestinal fungi may affect intestinal inflammation in patients with IBD. Mice with IL-22 deficiency are more likely to be infected by C. albicans in the gastrointestinal tract [254]. The secretion of the anti-inflammatory cytokine IL-10 increased significantly under S.cerevisiae stimulation, indicating that S.cerevisiae plays an anti-inflammatory role. And the amount of S.cerevisiae was negatively correlated with a CARD9 SNP allele (rs10781499, ‘A’ allele) in IBD patients [255]. The SYK-CARD9 signaling axis promotes inflammasome activation mediated by commensal gut fungi and thereby inhibits colitis and CAC. In the AOM/DSS-induced mouse model, treatment of mice with antifungal drugs aggravated colitis and CAC [256]. In addition, Malassezia restricta exists on the surface of mammalian skin, which can aggravate colitis in mice and trigger the innate inflammatory response through CARD9 [257]. Moreover, using fungal ITS sequencing, researchers found that some mucosa-related fungi were more abundant in CD patients, and Malassezia restricta was especially present in patients with the IBD CARD9 risk allele [258]. These results indicated that targeting specific fungi may become a therapeutic strategy for colitis-related colorectal cancer.
Therapies for IBD-associated CRC
Currently, surgical resection is the most commonly used treatment for patients with early-stage (stage 0 to II) colorectal cancer. However, chemotherapy drugs such as 5-fluorouracil (5-FU), folinic acid, oxaliplatin and capecitabine are usually used for patients with stage II colorectal cancer. And patients with stage III and IV colorectal cancer are usually treated with chemotherapy and targeted therapy [260, 261]. For IBD-associated CRC, anti-inflammatory therapy in IBD patients may be an effective way to prevent CAC [261].
Chemotherapy has been widely used in colorectal cancer. And 5-fluorouracil is the preferred anticancer drug for the clinical therapy of colorectal cancer. Although 5-FU has therapeutic effects on advanced CRC, the development of drug resistance limits its antitumor effect. And researchers have conducted in-depth research on the molecular mechanism of 5-FU resistance in recent years, hoping to alleviate the development of resistance caused by 5-FU [262]. The inhibition of METTL3 can enhance DNA damage and induce apoptosis in CRC cells by regulating the expression of RAD51AP1, thereby promoting the therapeutic sensitivity of 5-FU. Targeting the METTL3/RAD51AP1 axis has the potential to become a new adjuvant therapy strategy in 5-FU-resistant CRC patients [263]. In addition, the activation of the PI3K/Akt and Wnt/β-catenin signaling pathways can lead to the upregulation of HIF-1α in 5-FU-resistant CRC cells. And the inhibition of HIF-1α combined with 5-FU treatment may enhance the sensitivity of colorectal cancer to 5-FU [264].
The epidermal growth factor receptor (EGFR) gene and its proteins play a key role in promoting CRC tumor growth [265]. EGFR-targeted monoclonal antibodies such as cetuximab and panitumumab have been widely used to treat advanced colorectal cancer [267, 268]. KRAS/NRAS (RAS) wild-type, as well as BRAF/HER2 and MAP2K1 (MEK) mutated CRC patients, are sensitive to anti-EGFR therapy [268]. The expression of EGFR increased in tumor-related myeloid cells and was associated with the outcomes of CRC patients [269]. Anti-angiogenic therapy is also an effective treatment for CRC that targets the vascular endothelial growth factor (VEGF) protein and affects the development of blood vessels during tumor growth. In CAC, inflammation leads to colorectal tumors that are unresponsive to anti-VEGF therapy [138]. To improve the therapeutic effect, multi-target combination therapies are used to treat CAC. The combination of a VEGF inhibitor and a C3b/C4b blocking agent can effectively inhibit angiogenesis and tumor immunity in a mouse CAC model [270].
Immunotherapy has become an effective approach for the treatment of various types of cancers [271]. A growing number of studies indicate that immune checkpoint therapy plays an important role in mediating the immune response mediated by antitumor T cells in the tumor microenvironment [272]. Antibodies targeting PD-1 (programmed cell death 1, also known as PDCD1) and PD-L1 (PDCD1 ligand 1, also known as B7H1 and CD274) are promising strategies in many cancer types, including colorectal cancer. PD-1 is normally seen on the surface of immune cells, such as activated T cells, and is especially overexpressed in inflammatory and tumor conditions [274, 275]. Recently, it has been shown that the loss of PD-L1 in human colorectal cancer cells can lead to chemoresistance [275]. Moreover, CTLA-4 is a co-inhibitory protein usually seen on tumor cells that plays a vital role in immune checkpoint therapy by downregulating the activation and expansion of tumor reactive T cells. Additionally, anti-CTLA-4 promotes antitumor activity by selectively reducing Tregs and simultaneously activating Teffs in tumors [276]. Recent studies have shown that treating mice with therapeutic TNF inhibitors combined with PD-1 and CTLA-4 immunotherapy can improve colitis and antitumor efficacy [278, 279]. In addition, the gene expression of the immune checkpoint molecules Tim-3, LAG-3, Galectin-9, PTPN2 and BTLA is significantly upregulated in patients with colorectal cancer and may become a potential therapeutic target [279,280,281].
Anti-cytokine therapy, such as anti-TNF and IL-6 therapy, is used in the treatment of IBD, which improves the therapeutic efficiency and prevents the occurrence of CAC (Table 1) [282,283,284]. Anti-TNFα treatment can block the activation of TNFα receptors and reduce the apoptosis of intestinal epithelial cells, as well as inhibit intestinal permeability. Infliximab, an anti-TNFα drug for IBD treatment, was recently found to facilitate restoration of the colonic barrier of microbiota in Crohn’s disease [285]. As an effective pro-inflammatory cytokine, IL-6 plays an important role in regulating the immune system, such as regulating T-cell activation to control the balance between Th cells and immunosuppressive regulatory T cells in IBD [286]. Also, IL-22 is closely related to mucosal immunity and can directly participate in regulating fungal function. It has been found in humans and mice that IL-22 induces anti-bacterial related reactions, promotes epithelial regeneration, coordinates the endoplasmic reticulum (ER) stress response, and has potential clinical application as a mucosal healing therapy for IBD [40, 288]. In addition, the downstream signaling targets of inflammatory cytokines are also new therapeutic strategies for IBD. Small molecule JAK inhibitors repress the expression of a large variety of pro-inflammatory cytokines, including IL-6, IL-12 and IFN-γ, in the process of IBD [288]. And in the early stage of Crohn’s disease, Smad7 is expressed in a large number of cells in the epithelium and lamina propria of the new terminal ileal mucosa, and the use of Smad7 blocker is helpful to prevent postoperative recurrence [290, 291]. Moreover, a clinical study showed that bone marrow mesenchymal stem cells can be widely used in fistula treatment of patients with Crohn’s disease [291]. And human mesenchymal stem cell-derived exosomes (MSC-Exos) have similar functions as bone marrow mesenchymal stem cells in immune regulation and tissue repair, which can protect against experimental colitis and play a potential role in the treatment of IBD [293 294]. T-cell trafficking disruption and transcription factor inhibition are new therapeutic strategies that have recently been carried out in clinical trials. Specifically, targeting β7 integrins and the endothelial adhesion molecule MAdCAM-1 can effectively inhibit the migration of lymphocytes [294]. Recent studies have indicated that α4β7− and α4β7+ T cells may upregulate αEβ7 in the intestinal mucosa by activating TGF-β signaling [295]. Another trafficking modulators sphingosine-1-phosphate receptors (S1PRs) is dysregulated on intestinal vascular endothelial cells in patients with IBD, and is involved in the growth, angiogenesis, migration and barrier homeostasis of vascular endothelial cells [296]. In addition, the transcription factor GATA3, whose expression is correlated with the secretion of Th2- and Th9-related cytokines, has been found in UC patients, and the GATA3 DNAzyme may play a role in the treatment of UC patients [297]. Additionally, RORγt (a transcription factor in Th17 cells) can be regulated by a specific strain that induces Th17 cells in IBD patients and promotes the process of colitis [298]. Therefore, new targets of chemotherapy, targeted therapy and immunotherapy have the potential to become effective methods for the treatment and prevention of colitis-associated colorectal cancer.
Conclusion
In this study, we reviewed the important functions and crosstalk among different cells, such as IECs, IMCs, immune cells and gut microbiota, in the progression of colitis-associated colorectal cancer. These cells can regulate the occurrence and development of CAC in many ways. Wnt, NF-κB, STAT and other signaling pathways regulate the carcinogenesis of intestinal epithelial cells and stromal cells. Furthermore, various immune cells affect tumor progression by secreting cytokines and chemokines, and the gut microbiota regulates tumorigenesis by influencing the immune response. The interactions among microorganisms, immune cells, and epithelial cells regulate the occurrence and development of tumors. However, the regulatory mechanisms between these cells are still unclear.
In recent years, a few studies have shown that chemotherapy, targeted therapy and immunotherapy have been carried out in mouse models of CAC and have had therapeutic effects. However, clinical research on CAC is rare and requires further in-depth investigation. In addition, many researchers have edited harmful bacteria and used probiotics to alleviate tumor progression and chemoresistance in CAC [300]. However, the target cells and specific molecular mechanisms of these drugs and therapies still need to be further studied. Therefore, in future studies, we should focus on regulating specific signaling pathways in various cell types and clarifying the regulatory mechanisms between different cell types. It is important to investigate the crosstalk between various cells and to design drugs for chemotherapy, targeted therapy and immunotherapy, which provide new approaches for CAC patients.
Availability of data and materials
Not applicable.
Abbreviations
- CRC:
-
Colorectal cancer
- IBD:
-
Inflammatory bowel disease
- UC:
-
Ulcerative colitis
- CD:
-
Crohn’s disease
- IECs:
-
Intestinal epithelial cells
- IMCs:
-
Intestinal mesenchymal cells
- CAC:
-
Colitis-associated colorectal cancer
- ER:
-
Endoplasmic reticulum
- JAK/STAT:
-
Janus kinase/signal transducer and activation of the transcription
- MSCs:
-
Mesenchymal stem cells
- CAFs:
-
Cancer-associated fibroblasts
- α-SMA:
-
α-Smooth muscle actin
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-to-mesenchymal transition
- FAP-α:
-
Fibroblast activation protein-α
- PDGFR-β:
-
Platelet-derived growth factor receptor-β
- EREG:
-
Epiregulin
- Bmps:
-
Bone morphogenetic proteins
- HGF:
-
Hepatocyte growth factor
- FGF:
-
Fibroblast growth factors
- CTLA-4:
-
Cytotoxic T lymphocyte antigen-4
- EGF:
-
Epidermal growth factor
- VEGF:
-
Vascular endothelial growth factor
- MDSC:
-
Myeloid-derived suppressor cell
- DC:
-
Dendritic cell
- Treg:
-
Regulatory T cell
- Th:
-
Helper T cell
- pDC:
-
Plasmacytoid DC
- LPS:
-
Lipopolysaccharide
- TCS:
-
Triclosan
- RA:
-
Rosmarinic acid
- AIEC:
-
Adherent invasive E. coli
- ETBF:
-
Enterotoxigenic Bacteroides fragilis
- NK cell:
-
Natural killer cell
- IBS:
-
Irritable bowel syndrome
- EGFR:
-
Epidermal growth factor receptor
- PD-1:
-
Programmed cell death 1
- CCL2::
-
C–C motif chemokine ligand 2
- CCL8::
-
C–C motif chemokine ligand 8
References:
Sun J, Halfvarson J, Bergman D, et al. Statin use and risk of colorectal cancer in patients with inflammatory bowel disease. EClinicalMedicine. 2023;63:102182.
Siegel RL, Miller KD, Wagle NS, et al. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Waldum H, Fossmark R. Inflammation and digestive cancer. Int J Mol Sci. 2023;24(17):13503.
Eaden J. Review article: colorectal carcinoma and inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20(4):24–30.
Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51(1):27–41.
Porter RJ, Arends MJ, Churchhouse A, et al. Inflammatory bowel disease-associated colorectal cancer: translational risks from mechanisms to medicines. J Crohn’s Colitis. 2021. https://doi.org/10.1093/ecco-jcc/jjab102.
Soomro S, Venkateswaran S, Vanarsa K, et al. Predicting disease course in ulcerative colitis using stool proteins identified through an aptamer-based screen. Nat Commun. 2021;12(1):1–11.
Gasparetto M, Payne F, Nayak K, et al. Transcription and DNA methylation patterns of blood-derived CD8+ T cells are associated with age and inflammatory bowel disease but do not predict prognosis. Gastroenterology. 2021;160(1):232-44.e7.
Fazio A, Bordoni D, Kuiper JW, et al. DNA methyltransferase 3A controls intestinal epithelial barrier function and regeneration in the colon. Nat Commun. 2022;13(1):1–19.
Hirsch D, Hardt J, Sauer C, et al. Molecular characterization of ulcerative colitis-associated colorectal carcinomas. Mod Pathol. 2021;34(6):1153–66.
Rajamäki K, Taira A, Katainen R, et al. Genetic and epigenetic characteristics of inflammatory bowel disease associated colorectal cancer. Gastroenterology. 2021. https://doi.org/10.1053/j.gastro.2021.04.042.
Li J, Ma X, Chakravarti D, et al. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021;35(11–12):787–820.
Matsumoto K, Urabe Y, Oka S, et al. Genomic landscape of early-stage colorectal neoplasia developing from the ulcerative colitis mucosa in the Japanese population. Inflamm Bowel Dis. 2021;27(5):686–96.
Huang D, Sun W, Zhou Y, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018;37(1):173–87.
Brentnall TA, Crispin DA, Rabinovitch PS, et al. Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology. 1994;107(2):369.
Aust DE, Terdiman JP, Willenbucher RF, et al. The APC/beta-catenin pathway in ulcerative colitis-related colorectal carcinomas: a mutational analysis. Cancer. 2002;94(5):1421–7.
Wei M, Ma Y, Shen L, et al. NDRG2 regulates adherens junction integrity to restrict colitis and tumourigenesis. EBioMedicine. 2020;61:103068.
Kumar A, Priyamvada S, Ge Y, et al. A novel role of SLC26A3 in the maintenance of intestinal epithelial barrier integrity. Gastroenterology. 2021;160(4):1240-55.e3.
Grosheva I, Zheng D, Levy M, et al. High-throughput screen identifies host and microbiota regulators of intestinal barrier function. Gastroenterology. 2020;159(5):1807–23.
Spalinger MR, Sayoc-Becerra A, Santos AN, et al. PTPN2 regulates interactions between macrophages and intestinal epithelial cells to promote intestinal barrier function. Gastroenterology. 2020;159(5):1763-77.e14.
Sahoo D, Swanson L, Sayed IM, et al. Artificial intelligence guided discovery of a barrier-protective therapy in inflammatory bowel disease. Nat Commun. 2021;12(1):1–14.
Grivennikov S, Karin E, Terzic J, et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis associated cancer. Cancer Cell. 2009;15(2):103–13.
Meir M, Burkard N, Ungewiß H, et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J Clin Investig. 2019;129(7):2824–40.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392.
Hidalgo García L, Molina Tijeras JA, Huertas Peña FJ, et al. Intestinal mesenchymal cells regulate immune responses and promote epithelial regeneration in vitro and in dextran sulfate sodium-induced experimental colitis in mice. Acta Physiol. 2021. https://doi.org/10.1111/apha.13699.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539.
Zhu W, Miyata N, Winter MG, et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J Exp Med. 2019;216(10):2378–93.
Cohen LJ, Cho JH, Gevers D, et al. Genetic factors and the intestinal microbiome guide development of microbe-based therapies for inflammatory bowel diseases. Gastroenterology. 2019;156(8):2174–89.
López-Posadas R, Neurath MF, Atreya I. Molecular pathways driving disease-specific alterations of intestinal epithelial cells. Cell Mol Life Sci. 2017;74(5):803–26.
Hu S, Venema WTU, Westra H-J, et al. Inflammation status modulates the effect of host genetic variation on intestinal gene expression in inflammatory bowel disease. Nat Commun. 2021;12(1):1–10.
Xiao L, Li X-X, Chung HK, et al. RNA-binding protein HuR regulates Paneth cell function by altering membrane localization of TLR2 via post-transcriptional control of CNPY3. Gastroenterology. 2019;157(3):731–43.
Yu S, Balasubramanian I, Laubitz D, et al. Paneth cell-derived lysozyme defines the composition of mucolytic microbiota and the inflammatory tone of the intestine. Immunity. 2020;53(2):398-416.e8.
Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2015;14(1):20–32.
Banerjee A, Herring CA, Chen B, et al. Succinate produced by intestinal microbes promotes specification of tuft cells to suppress ileal inflammation. Gastroenterology. 2020;159(6):2101-155.e5.
Balasubramanian I, Gao N. From sensing to shaping microbiota: insights into the role of NOD2 in intestinal homeostasis and progression of Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2017;313(1):G7–13.
Landi MT, Bishop DT, MacGregor S, et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet. 2020;52(5):494–504.
Liu Z-Y, Zheng M, Li Y-M, et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics. 2019;9(12):3659.
Zhou M, He J, Shi Y, et al. ABIN3 negatively regulates necroptosis-induced intestinal inflammation through recruiting A20 and restricting the ubiquitination of RIPK3 in inflammatory bowel disease. J Crohns Colitis. 2021;15(1):99–114.
Kosinsky RL, Saul D, Ammer-Herrmenau C, et al. USP22 suppresses sparc expression in acute colitis and inflammation-associated colorectal cancer. Cancers. 2021;13(8):1817.
Powell N, Pantazi E, Pavlidis P, et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut. 2020;69(3):578–90.
Perez LG, Kempski J, McGee HM, et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat Commun. 2020;11(1):1–14.
Sheng YH, Giri R, Davies J, et al. A nucleotide analog prevents colitis-associated cancer via beta-catenin independently of inflammation and autophagy. Cell Mol Gastroenterol Hepatol. 2021;11(1):33–53.
Hibiya S, Tsuchiya K, Hayashi R, et al. Long-term inflammation transforms intestinal epithelial cells of colonic organoids. J Crohns Colitis. 2017;11(5):621–30.
De Salvo C, Buela K-A, Creyns B, et al. NOD2 drives early IL-33–dependent expansion of group 2 innate lymphoid cells during Crohn’s disease–like ileitis. J Clin Invest. 2021;131(5):e140624.
Solà-Tapias N, Vergnolle N, Denadai-Souza A, et al. The interplay between genetic risk factors and proteolytic dysregulation in the pathophysiology of inflammatory bowel disease. J Crohns Colitis. 2020;14(8):1149–61.
Lee M, Kim Y-S, Lim S, et al. Protein stabilization of ITF2 by NF-κB prevents colitis-associated cancer development. Nat Commun. 2023;14(1):2363.
Kakiuchi N, Yoshida K, Uchino M, et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature. 2020;577(7789):260–5.
Tian H, Qian J, Ai L, et al. Upregulation of ASAP 3 contributes to colorectal carcinogenesis and indicates poor survival outcome. Cancer Sci. 2017;108(8):1544–55.
Li Q, Chen Y, Zhang D, et al. IRAK4 mediates colitis-induced tumorigenesis and chemoresistance in colorectal cancer. JCI insight. 2019;4(19):e130867.
Salas A, Hernandez-Rocha C, Duijvestein M, et al. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(6):323–37.
Lamichhane S, Mo J-S, Sharma G, et al. MicroRNA 452 regulates IL20RA-mediated JAK1/STAT3 pathway in inflammatory colitis and colorectal cancer. Inflamm Res. 2021;70(8):903–14.
Willson TA, Jurickova I, Collins M, et al. Deletion of intestinal epithelial cell STAT3 promotes T-lymphocyte STAT3 activation and chronic colitis following acute dextran sodium sulfate injury in mice. Inflamm Bowel Dis. 2013;19(3):512.
Pang L, Huynh J, Alorro MG, et al. STAT3 signalling via the IL-6ST/gp130 cytokine receptor promotes epithelial integrity and intestinal barrier function during DSS-induced colitis. Biomedicines. 2021;9(2):187.
Josa V, Ferenczi S, Szalai R, et al. Thrombocytosis and effects of IL-6 knock-out in a colitis-associated cancer model. Int J Mol Sci. 2020;21(17):6218.
Backert I, Koralov SB, Wirtz S, et al. STAT3 activation in Th17 and Th22 cells controls IL-22-mediated epithelial host defense during infectious colitis. J Immunol. 2014;193(7):3779.
Keir ME, Yi T, Lu TT, et al. The role of IL-22 in intestinal health and disease. J Exp Med. 2020;217(3):e20192195.
Aden K, Tran F, Ito G, et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS–STING. J Exp Med. 2018;215(11):2868–86.
Delgado-Ramirez Y, Baltazar-Perez I, Martinez Y, et al. STAT1 is required for decreasing accumulation of granulocytic cells via IL-17 during initial steps of colitis-associated cancer. Int J Mol Sci. 2021;22(14):7695.
Liu X, Wei W, Li X, et al. BMI1 and MEL18 promote colitis-associated cancer in mice via REG3B and STAT3. Gastroenterology. 2017;153(6):1607.
Ma X, Meng Z, Jin L, et al. CAMK2γ in intestinal epithelial cells modulates colitis-associated colorectal carcinogenesis via enhancing STAT3 activation. Oncogene. 2017;36(28):4060–71.
Ye X, Wu H, Sheng L, et al. Oncogenic potential of truncated RXRα during colitis-associated colorectal tumorigenesis by promoting IL-6-STAT3 signaling. Nat Commun. 2019;10(1):1–15.
Lin X, Sun Q, Zhou L, et al. Colonic epithelial mTORC1 promotes ulcerative colitis through COX-2-mediated Th17 responses. Mucosal Immunol. 2018;11(6):1663–73.
Rizzo A, Di Giovangiulio M, Stolfi C, et al. RORγt-expressing Tregs drive the growth of colitis-associated colorectal cancer by controlling IL6 in dendritic cells. Cancer Immunol Res. 2018;6(9):1082–92.
Schreiber S, Aden K, Bernardes JP, et al. Therapeutic Interleukin-6 Trans-signaling Inhibition by Olamkicept (sgp130Fc) in patients with active inflammatory bowel disease. Gastroenterology. 2021;160(7):2354-66.e11.
Network TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Robles AI, Traverso G, Zhang M, et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology. 2016;150(4):931.
Sakai K, De Velasco MA, Kura Y, et al. Transcriptome profiling and metagenomic analysis help to elucidate interactions in an inflammation-associated cancer mouse model. Cancers. 2021;13(15):3683.
Biscaglia G, Latiano A, Castellana S, et al. Germline alterations in patients with IBD-associated colorectal cancer. Inflamm Bowel Dis. 2021. https://doi.org/10.1093/ibd/izab195.
Mäki-Nevala S, Ukwattage S, Olkinuora A, et al. Somatic mutation profiles as molecular classifiers of ulcerative colitis-associated colorectal cancer. Int J Cancer. 2021;148(12):2997–3007.
Kesari MV, Gaopande VL, Joshi AR, et al. Immunohistochemical study of MUC1, MUC2 and MUC5AC in colorectal carcinoma and review of literature. Indian J Gastroenterol Off J Indian Soc Gastroenterol. 2015;34(1):63–7.
Li W, Zhang N, Jin C, et al. MUC1-C drives stemness in progression of colitis to colorectal cancer. JCI insight. 2020;5(12):e137112.
Peng K, Kou L, Yu L, et al. Histone demethylase JMJD2D interacts with β-catenin to induce transcription and activate colorectal cancer cell proliferation and tumor growth in mice. Gastroenterology. 2019;156(4):1112–26.
Zhuo M, Chen W, Shang S, et al. Inflammation-induced JMJD2D promotes colitis recovery and colon tumorigenesis by activating Hedgehog signaling. Oncogene. 2020;39(16):3336–53.
Hua F, Shang S, Yang Y-w, et al. TRIB3 interacts with β-catenin and TCF4 to increase stem cell features of colorectal cancer stem cells and tumorigenesis. Gastroenterology. 2019;156(3):708-21.e15.
Zeng S, Chen L, Sun Q, et al. Scutellarin ameliorates colitis-associated colorectal cancer by suppressing Wnt/β-catenin signaling cascade. Eur J Pharmacol. 2021;906:174253.
Wu H, Wu Z, Qiu Y, et al. Supplementing a specific synbiotic suppressed the incidence of AOM/DSS-induced colorectal cancer in mice. iScience. 2023. https://doi.org/10.1016/j.isci.2023.106979.
Južnić L, Peuker K, Strigli A, et al. SETDB1 is required for intestinal epithelial differentiation and the prevention of intestinal inflammation. Gut. 2021;70(3):485–98.
Wang Q, Wang Z, Zhang Z, et al. Landscape of cell heterogeneity and evolutionary trajectory in ulcerative colitis-associated colon cancer revealed by single-cell RNA sequencing. Chin J Cancer Res. 2021;33(2):271.
Hirano T, Hirayama D, Wagatsuma K, et al. Immunological mechanisms in inflammation-associated colon carcinogenesis. Int J Mol Sci. 2020;21(9):3062.
Coskun M. TNF-a-induced down-regulation of CDX2 suppresses MEP1A expression in colitis. Biochimica Biophysica Acta Molecular Basis Dis. 2014;1822(6):843–51.
Ahn SH, Shah YM, Junko Inoue MS, et al. Hepatocyte nuclear factor 4α in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm Bowel Dis. 2008;14(7):908–20.
Liang W, Peng X, Li Q, et al. FAM3D is essential for colon homeostasis and host defense against inflammation associated carcinogenesis. Nat Commun. 2020;11(1):1–16.
Liu M, Sun T, Li N, et al. BRG1 attenuates colonic inflammation and tumorigenesis through autophagy-dependent oxidative stress sequestration. Nat Commun. 2019;10(1):1–15.
Liu M, Rao H, Liu J, et al. The histone methyltransferase SETD2 modulates oxidative stress to attenuate experimental colitis. Redox Biol. 2021;43:102004.
Muise AM, Walters TD, Glowacka WK, et al. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut. 2009;58(8):1121–7.
Henriques A, Koliaraki V, Kollias G. Mesenchymal MAPKAPK2/HSP27 drives intestinal carcinogenesis. Proc Natl Acad Sci. 2018;115(24):E5546–55.
Powell DW, Pinchuk IV, Saada JI, et al. Mesenchymal cells of the intestinal lamina propria. Annu Rev Physiol. 2011;73(1):213–37.
Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175(2):372-86.e17.
Kienzl M, Hasenoehrl C, Valadez-Cosmes P, et al. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology. 2020;9(1):1776059.
Cortez E, Roswall P, Pietras K. Functional subsets of mesenchymal cell types in the tumor microenvironment. Semin Cancer Biol. 2014;25(2):3–9.
Abolarinwa BA, Ibrahim RB, Huang Y-H. Conceptual development of immunotherapeutic approaches to gastrointestinal cancer. Int J Mol Sci. 2019;20(18):4624.
Tommelein J, Verset L, Boterberg T, et al. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front Oncol. 2015;5:63.
Darby IA, Laverdet B, Bonté F, et al. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol. 2014;7:301.
Gomes RN, Manuel F, Nascimento DS. The bright side of fibroblasts: molecular signature and regenerative cues in major organs. NPJ Regen Med. 2021;6(1):1–12.
Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316(17):2713–22.
Boesch M, Baty F, Rumpold H, et al. Fibroblasts in cancer: defining target structures for therapeutic intervention. Biochimica Biophysica Acta Rev Cancer. 2019;1872(1):111–21.
Ma H, Wang J, Zhao X, et al. Periostin promotes colorectal tumorigenesis through integrin-FAK-Src pathway-mediated YAP/TAZ activation. Cell reports. 2020;30(3):793-806.e6.
Nishina T, Deguchi Y, Ohshima D, et al. Interleukin-11-expressing fibroblasts have a unique gene signature correlated with poor prognosis of colorectal cancer. Nat Commun. 2021;12(1):1–20.
Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochimica Biophysica Acta Molecular Basis Dis. 2013;1832(7):1070–8.
Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol. 2013;229(2):198–207.
Mifflin RC, Pinchuk IV, Saada JI, et al. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G684–96.
Yuan Q, Gu J, Zhang J, et al. MyD88 in myofibroblasts enhances colitis-associated tumorigenesis via promoting macrophage M2 polarization. Cell Rep. 2021;34(5):108724.
Boire A, Covic L, Agarwal A, et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120(3):303–13.
Roulis M, Armaka M, Manoloukos M, et al. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc Natl Acad Sci. 2011;108(13):5396–401.
Diez-Obrero V, Moratalla-Navarro F, Ibañez-Sanz G, et al. Transcriptome-wide association study for inflammatory bowel disease reveals novel candidate susceptibility genes in specific colon subsites and tissue categories. J Crohn’s Colitis. 2021;16(2):275–85.
Kim Y-G, Kamada N, Shaw MH, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34(5):769–80.
Lin J-D, Devlin JC, Yeung F, et al. Rewilding Nod2 and Atg16l1 mutant mice uncovers genetic and environmental contributions to microbial responses and immune cell composition. Cell host & microbe. 2020;27(5):830-40.e4.
Roulis M, Nikolaou C, Kotsaki E, et al. Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis. Proc Natl Acad Sci. 2014;111(43):E4658–67.
Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211(8):1503–23.
Gregorieff A, Pinto D, Begthel H, et al. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology. 2005;129(2):626–38.
McCarthy N, Manieri E, Storm EE, et al. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell. 2020;26(3):391-402.e5.
Rupp C, Scherzer M, Rudisch A, et al. IGFBP7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor–stroma interaction. Oncogene. 2015;34(7):815.
Ren J, Ding L, Zhang D, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8(14):3932.
Hawinkels LJ, Paauwe M, Verspaget HW, et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene. 2014;33(1):97–107.
Koliaraki V, Pasparakis M, Kollias G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J Exp Med. 2015;212(13):2235–51.
Pallangyo CK, Ziegler PK, Greten FR. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J Exp Med. 2015;212(13):2253–66.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801.
Levi-Galibov O, Lavon H, Wassermann-Dozorets R, et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat Commun. 2020;11(1):1–19.
Heichler C, Schmied A, Enderle K, et al. Targeting STAT3 signaling in COL1+ fibroblasts controls colitis-associated cancer in mice. Cancers. 2022;14(6):1472.
Gao L, Yu Q, Zhang H, et al. A resident stromal cell population actively restrains innate immune response in the propagation phase of colitis pathogenesis in mice. Sci Transl Med. 2021;13(603):eabb5071.
Thomson CA, Nibbs RJ, McCoy KD, et al. Immunological roles of intestinal mesenchymal cells. Immunology. 2020;160(4):313–24.
Walton KL, Holt L, Sartor RB. Lipopolysaccharide activates innate immune responses in murine intestinal myofibroblasts through multiple signaling pathways. Am J Physiol Gastroint Liver Physiol. 2009;296(3):601–11.
Horiguchi H, Kadomatsu T, Miyata K, et al. Stroma-derived ANGPTL2 establishes an anti-tumor microenvironment during intestinal tumorigenesis. Oncogene. 2021;40(1):55–67.
Stadler M, Pudelko K, Biermeier A, et al. Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett. 2021. https://doi.org/10.1016/j.canlet.2021.07.006.
Kobayashi H, Gieniec KA, Lannagan TR, et al. The origin and contribution of cancer-associated fibroblasts in colorectal carcinogenesis. Gastroenterology. 2022;162(3):890–906.
Herrera M, Herrera A, DomãNguez G, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Science. 2013;104(4):437–44.
Torres S, Bartolomé RA, Mendes M, et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res. 2013;19(21):6006–19.
Geremia A, Arancibia-Cárcamo CV. Innate lymphoid cells in intestinal inflammation. Front Immunol. 2017;8:1296.
Mitsialis V, Wall S, Liu P, et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and Crohn’s disease. Gastroenterology. 2020;159(2):591-608.e10.
Li Z-W, Sun B, Gong T, et al. GNAI1 and GNAI3 reduce colitis-associated tumorigenesis in mice by blocking IL6 signaling and down-regulating expression of GNAI2. Gastroenterology. 2019;156(8):2297–312.
Na YR, Stakenborg M, Seok SH, et al. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16(9):531–43.
Galon J, Costes A, Sanchezcabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4.
Leite CA, Mota JM, de Lima KA, et al. Paradoxical interaction between cancer and long-term postsepsis disorder: impairment of de novo carcinogenesis versus favoring the growth of established tumors. J Immunother Cancer. 2020;8(1):e000129.
Ortiz ML, Kumar V, Martner A, et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17–producing CD4+ T cells. J Exp Med. 2015;212(3):351–67.
Liao Y, Zhao J, Bulek K, et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat Commun. 2020;11(1):1–15.
Karagiannidis I, Jerman SJ, Jacenik D, et al. G-CSF and G-CSFR modulate CD4 and CD8 T cell responses to promote colon tumor growth and are potential therapeutic targets. Front Immunol. 2020;11:1885.
Liu C, Liu R, Wang B, et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J Immunother Cancer. 2021;9(1):e001895.
Itatani Y, Yamamoto T, Zhong C, et al. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc Natl Acad Sci. 2020;117(35):21598–608.
Belarif L, Danger R, Kermarrec L, et al. IL-7 receptor influences anti-TNF responsiveness and T cell gut homing in inflammatory bowel disease. J Clin Investig. 2019;129(5):1910–25.
Olesch C, Sirait-Fischer E, Berkefeld M, et al. S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J Clin Investig. 2020;130(10):5461–76.
Abu-Sbeih H, Faleck DM, Ricciuti B, et al. Immune checkpoint inhibitor therapy in patients with preexisting inflammatory bowel disease. J Clin Oncol. 2020;38(6):576.
Zundler S, Becker E, Schulze LL, et al. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut. 2019;68(9):1688–700.
Sasson SC, Slevin SM, Cheung VT, et al. IFNγ-producing CD8+ tissue resident memory T cells are a targetable hallmark of immune checkpoint inhibitor-colitis. Gastroenterology. 2021. https://doi.org/10.1053/j.gastro.2021.06.025.
Shi Y, Fu Y, Zhang X, et al. Romidepsin (FK228) regulates the expression of the immune checkpoint ligand PD-L1 and suppresses cellular immune functions in colon cancer. Cancer Immunol Immunother. 2021;70(1):61–73.
Endo Y, Marusawa H, Kou T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135(3):1–3.
Canavan C, Abrams K, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther. 2006;23(8):1097–104.
Xu Z-S, Zhang H-X, Li W-W, et al. FAM64A positively regulates STAT3 activity to promote Th17 differentiation and colitis-associated carcinogenesis. Proc Natl Acad Sci. 2019;116(21):10447–52.
Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. 2019;20(8):970–9.
Sakaguchi S. Regulatory T cells. Springer Semin Immunopathol. 2006;28(1):1–2.
Clough JN, Omer OS, Tasker S, et al. Regulatory T-cell therapy in Crohn’s disease: challenges and advances. Gut. 2020;69(5):942–52.
Phuong NNT, Palmieri V, Adamczyk A, et al. IL-33 drives expansion of type 2 innate lymphoid cells and regulatory T cells and protects mice from severe, acute colitis. Front Immunol. 2021;12:669787.
Huang L-j, Mao X-t, Li Y-y, et al. Multiomics analyses reveal a critical role of selenium in controlling T cell differentiation in Crohn’s disease. Immunity. 2021. https://doi.org/10.1016/j.immuni.2021.07.004.
Lu Y, Li Y, Liu Q, et al. MondoA-TXNIP axis maintains regulatory T cell identity and function in colorectal cancer microenvironment. Gastroenterology. 2021. https://doi.org/10.1053/j.gastro.2021.04.041.
Erdman SE, Sohn JJ, Rao VP, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ Mice. Can Res. 2005;65(10):3998–4004.
Ibrahim ML, Klement JD, Lu C, et al. Myeloid-derived suppressor cells produce IL-10 to elicit DNMT3b-dependent IRF8 silencing to promote colitis-associated colon tumorigenesis. Cell Rep. 2018;25(11):3036-46.e6.
Wang Y, Ding Y, Deng Y, et al. Role of myeloid-derived suppressor cells in the promotion and immunotherapy of colitis-associated cancer. J Immunother Cancer. 2020;8(2):e000609.
Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Pan PY, Ma G, Weber KJ, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Can Res. 2010;70(1):99–108.
Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res. 2019;25(18):5449–57.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8.
Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Investig. 2008;118(10):3367–77.
Singh UP, Singh NP, Singh B, et al. Role of resveratrol-induced CD11b(+) Gr-1(+) myeloid derived suppressor cells (MDSCs) in the reduction of CXCR3(+) T cells and amelioration of chronic colitis in IL-10(−/−) mice. Brain Behav Immun. 2012;26(1):72–82.
Song J, Chen Z, Geng T, et al. Deleting MyD88 signaling in myeloid cells promotes development of adenocarcinomas of the colon. Cancer Lett. 2018;433:65–75.
Eftychi C, Schwarzer R, Vlantis K, et al. Temporally distinct functions of the cytokines IL-12 and IL-23 drive chronic colon inflammation in response to intestinal barrier impairment. Immunity. 2019;51(2):367-80.e4.
Zhou J, Huang S, Wang Z, et al. Targeting EZH2 histone methyltransferase activity alleviates experimental intestinal inflammation. Nat Commun. 2019;10(1):1–11.
Cui B, Lu S, Lai L, et al. Protective function of interleukin 27 in colitis-associated cancer via suppression of inflammatory cytokines in intestinal epithelial cells. Oncoimmunology. 2017;6(2):e1268309.
Wang T, Fan C, Yao A, et al. The adaptor protein CARD9 protects against colon cancer by restricting mycobiota-mediated expansion of myeloid-derived suppressor cells. Immunity. 2018;49(3):504-14. e4.
Harusato A, Viennois E, Etienne-Mesmin L, et al. Early-life microbiota exposure restricts myeloid-derived suppressor cell–driven colonic tumorigenesis. Cancer Immunol Res. 2019;7(4):544–51.
Dou D, Liang J, Zhai X, et al. Oxytocin signalling in dendritic cells regulates immune tolerance in the intestine and alleviates DSS-induced colitis. Clin Sci. 2021;135(4):597–611.
Abbasi-Kenarsari H, Heidari N, Baghaei K, et al. Synergistic therapeutic effect of mesenchymal stem cells and tolerogenic dendritic cells in an acute colitis mouse model. Int Immunopharmacol. 2020;88:107006.
Berndt BE, Zhang M, Chen G-H, et al. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol. 2007;179(9):6255–62.
Abe K, Nguyen KP, Fine SD, et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc Natl Acad Sci USA. 2007;104(43):17022–7.
Murai M, Turovskaya O, Kim G, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10(11):1178–84.
Wei H-X, Wang B, Li B. IL-10 and IL-22 in mucosal immunity: driving protection and Pathology. Front Immunol. 2020;11:1315.
Kaser A, Ludwiczek O, Holzmann S, et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24(1):74–85.
Souza HS, Elia CC, Spencer J, et al. Expression of lymphocyte-endothelial receptor-ligand pairs, alpha4beta7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut. 1999;45(6):856–63.
Ko H-J, Hong E-H, Cho J, et al. Plasmacytoid dendritic cells regulate colitis-associated tumorigenesis by controlling myeloid-derived suppressor cell infiltration. Cancer Lett. 2020;493:102–12.
De BA, Mende I, Baretton G, et al. A subset of human dendritic cells in the T cell area of mucosa-associated lymphoid tissue with a high potential to produce TNF-alpha. J Immunol. 2003;170(10):5089–94.
Zheng T, Zhang B, Chen C, et al. Protein kinase p38α signaling in dendritic cells regulates colon inflammation and tumorigenesis. Proc Natl Acad Sci. 2018;115(52):E12313–22.
Kempski J, Giannou AD, Riecken K, et al. IL22BP mediates the antitumor effects of lymphotoxin against colorectal tumors in mice and humans. Gastroenterology. 2020;159(4):1417-303.e3.
Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(11):929–37.
He J, Song Y, Li G, et al. Fbxw7 increases CCL2/7 in CX3CR1 hi macrophages to promote intestinal inflammation. J Clin Investig. 2019;129(9):3877–93.
Bain CC, Scott CL, Uronenhansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498–510.
Smith P, Smythies L, Shen R, et al. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol. 2011;4(1):31–42.
Platt AM, Bain CC, Bordon Y, et al. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J Immunol. 2010;184(12):6843–54.
Aschenbrenner D, Quaranta M, Banerjee S, et al. Deconvolution of monocyte responses in inflammatory bowel disease reveals an IL-1 cytokine network that regulates IL-23 in genetic and acquired IL-10 resistance. Gut. 2021;70(6):1023–36.
Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Investig. 2008;118(6):2269–80.
Deng F, He S, Cui S, et al. A molecular targeted immunotherapeutic strategy for ulcerative colitis via dual-targeting nanoparticles delivering miR-146b to intestinal macrophages. J Crohns Colitis. 2019;13(4):482–94.
Wunderlich CM, Ackermann PJ, Ostermann AL, et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat Commun. 2018;9(1):1–16.
Mukhopadhyay S, Heinz E, Porreca I, et al. Loss of IL-10 signaling in macrophages limits bacterial killing driven by prostaglandin E2. J Exp Med. 2020;217(2):e20180649.
Mola S, Pandolfo C, Sica A, et al. The macrophages-microbiota interplay in colorectal cancer (CRC)-related inflammation: prognostic and therapeutic significance. Int J Mol Sci. 2020;21(18):6866.
Masato N, Manabu M, Hiroshi S, et al. EP4 receptor-associated protein in macrophages ameliorates colitis and colitis-associated tumorigenesis. PLoS Genet. 2015;11(10):e1005542.
Marelli G, Erreni M, Anselmo A, et al. Heme-oxygenase-1 production by intestinal CX3CR1+ macrophages helps to resolve inflammation and prevents carcinogenesis. Can Res. 2017;77(16):4472–85.
Katholnig K, Schütz B, Fritsch SD, et al. Inactivation of mTORC2 in macrophages is a signature of colorectal cancer that promotes tumorigenesis. JCI Insight. 2019;4(20):e124164.
Dai L, Liu Y, Cheng L, et al. SARI attenuates colon inflammation by promoting STAT1 degradation in intestinal epithelial cells. Mucosal Immunol. 2019;12(5):1130–40.
Triner D, Devenport SN, Ramakrishnan SK, et al. Neutrophils restrict tumor-associated microbiota to reduce growth and invasion of colon tumors in mice. Gastroenterology. 2019;156(5):1467–82.
Kühl AA, Kakirman H, Janotta M, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133(6):1882–92.
Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. 2014;5:508.
Butin-Israeli V, Bui TM, Wiesolek HL, et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J Clin Investig. 2019;129(2):712–26.
Tian Y, Xu J, Li Y, et al. MicroRNA-31 reduces inflammatory signaling and promotes regeneration in colon epithelium, and delivery of mimics in microspheres reduces colitis in mice. Gastroenterology. 2019;156(8):2281-96.e6.
Zhou G, Peng K, Song Y, et al. CD177+ neutrophils suppress epithelial cell tumourigenesis in colitis-associated cancer and predict good prognosis in colorectal cancer. Carcinogenesis. 2017;39(2):272–82.
Lin Y, Cheng L, Liu Y, et al. Intestinal epithelium-derived BATF3 promotes colitis-associated colon cancer through facilitating CXCL5-mediated neutrophils recruitment. Mucosal Immunol. 2020;14(D1):1–12.
Zhang Y, Diao N, Lee CK, et al. Neutrophils deficient in innate suppressor IRAK-M enhances anti-tumor immune responses. Mol Ther. 2020;28(1):89–99.
Deng Q, Yao Y, Yang J, et al. AKR1B8 deficiency drives severe DSS-induced acute colitis through invasion of luminal bacteria and activation of innate immunity. Front Immunol. 2022;13:1042549.
Li G-Q, Xia J, Zeng W, et al. The intestinal γδ T cells: functions in the gut and in the distant organs. Front Immunol. 2023;14:1206299.
Wang X, Khoshaba R, Shen Y, et al. Impaired barrier function and immunity in the colon of aldo-keto reductase 1B8 deficient mice. Front Cell Dev Biol. 2021;9:632805.
Plovier H, Everard A, Druart C, et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat Med. 2017;23(1):107–13.
Bhattacharya N, Yuan R, Prestwood TR, et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8+ T cell-mediated immunity in colorectal cancer. Immunity. 2016;45(3):641–55.
Ryzhakov G, West NR, Franchini F, et al. Alpha kinase 1 controls intestinal inflammation by suppressing the IL-12/Th1 axis. Nat Commun. 2018;9(1):1–13.
Nasef NA, Mehta S. Role of inflammation in pathophysiology of colonic disease: an update. Int J Mol Sci. 2020;21(13):4748.
Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223–37.
Lee JG, Lee Y-R, Lee A-r, et al. Role of the global gut microbial community in the development of colitis-associated cancer in a murine model. Biomed Pharmacother. 2021;135:111206.
Hu S, Vila AV, Gacesa R, et al. Whole exome sequencing analyses reveal gene–microbiota interactions in the context of IBD. Gut. 2021;70(2):285–96.
Vila AV, Imhann F, Collij V, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med. 2018;10(472):eaap8914.
Yang Y, Li L, Xu C, et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut. 2021;70(8):1495–506.
Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134(2):577–94.
Richard ML, Sokol H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2019;16(6):331–45.
Kudelka MR, Stowell SR, Cummings RD, et al. Intestinal epithelial glycosylation in homeostasis and gut microbiota interactions in IBD. Nat Rev Gastroenterol Hepatol. 2020;17(10):597–617.
Kostic AD, Gevers D, Pedamallu CS, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22(2):292–8.
Strauss J, Kaplan GG, Beck PL, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17(9):1971–8.
Yu MR, Kim HJ, Park HR. Fusobacterium nucleatum accelerates the progression of colitis-associated colorectal cancer by promoting EMT. Cancers. 2020;12(10):2728.
Dregelies T, Haumaier F, Sterlacci W, et al. Detection of Fusobacterium nucleatum in patients with colitis-associated colorectal cancer. Curr Microbiol. 2023;80(9):293.
Han JX, Tao ZH, Qian Y, et al. ZFP90 drives the initiation of colitis-associated colorectal cancer via a microbiota-dependent strategy. Gut Microbes. 2021;13(1):1–20.
Yang Y, Weng W, Peng J, et al. Fusobacterium nucleatum Increases proliferation of colorectal cancer cells and tumor development in mice by activating TLR4 signaling to NFκB, upregulating expression of microRNA-21. Gastroenterology. 2016;152(4):851.
Rubinstein MR, Wang X, Liu W, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/Î2-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206.
Tenaillon O, Skurnik D, Picard B, et al. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. 2010;8(3):207–17.
Arthur JC, Perezchanona E, Mühlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Gut Microbes. 2013;338(3):120–3.
Mirsepasi-Lauridsen HC, Vallance BA, Krogfelt KA, et al. Escherichia coli pathobionts associated with inflammatory bowel disease. Clin Microbiol Rev. 2019;32(2):e00060-e118.
Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–21.
Viladomiu M, Metz ML, Lima SF, et al. Adherent-invasive E. coli metabolism of propanediol in Crohn’s disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe. 2021;29(4):607-19.e8.
Eavespyles T, Allen CA, Taormina J, et al. Escherichia coli isolated from a Crohn’s disease patient adheres, invades, and induces inflammatory responses in polarized intestinal epithelial cells. Int J Med Microbiol. 2008;298(5–6):397–409.
Cevallos SA, Lee J-Y, Tiffany CR, et al. Increased epithelial oxygenation links colitis to an expansion of tumorigenic bacteria. MBio. 2019. https://doi.org/10.1128/mbio.
Isolauri E, Salminen S. Probiotics: use in allergic disorders: a nutrition, allergy, mucosal immunology, and intestinal microbiota (NAMI) research group report. J Clin Gastroenterol. 2008;42:S91–6.
Silveira DSC, Veronez LC, Lopes-Júnior LC, et al. Lactobacillus bulgaricus inhibits colitis-associated cancer via a negative regulation of intestinal inflammation in azoxymethane/dextran sodium sulfate model. World J Gastroenterol. 2020;26(43):6782.
Bedada TL, Feto TK, Awoke KS, et al. Probiotics for cancer alternative prevention and treatment. Biomed Pharmacother. 2020;129:110409.
Cousin FJ, Jouanlanhouet S, Corcos L, et al. Milk fermented by Propionibacterium freudenreichii induces apoptosis of HGT-1 human gastric cancer cells. PLoS ONE. 2012;7(3):e31892.
Iwama T, Fujiya M, Konishi H, et al. Bacteria-derived ferrichrome inhibits tumor progression in sporadic colorectal neoplasms and colitis-associated cancer. Cancer Cell Int. 2021;21(1):1–15.
Zhong XZ, Covasa M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J Gastroenterol. 2014;20(24):7878–86.
Zhao Y, Jiang Q. Roles of the polyphenol-gut microbiota interaction in alleviating colitis and preventing colitis-associated colorectal cancer. Adv Nutr. 2021;12(2):546–65.
Oh NS, Lee JY, Kim Y-T, et al. Cancer-protective effect of a synbiotic combination between Lactobacillus gasseri 505 and a Cudrania tricuspidata leaf extract on colitis-associated colorectal cancer. Gut microbes. 2020;12(1):1785803.
van der Giessen J, Binyamin D, Belogolovski A, et al. Modulation of cytokine patterns and microbiome during pregnancy in IBD. Gut. 2020;69(3):473–86.
Choi JH, Moon CM, Shin T-S, et al. Lactobacillus paracasei-derived extracellular vesicles attenuate the intestinal inflammatory response by augmenting the endoplasmic reticulum stress pathway. Exp Mol Med. 2020;52(3):423–37.
Takagi A, Ikemura H, Matsuzaki T, et al. Relationship between the in vitro response of dendritic cells to Lactobacillus and prevention of tumorigenesis in the mouse. J Gastroenterol. 2008;43(9):661–9.
Greving CNA, Towne JE. A role for IL-12 in IBD after all? Immunity. 2019;51(2):209–11.
Jacouton E, Chain F, Sokol H, et al. Probiotic strain Lactobacillus casei BL23 prevents colitis-associated colorectal cancer. Front Immunol. 2017;8:1553.
He R, Han C, Li Y, et al. Cancer-preventive role of bone marrow-derived mesenchymal stem cells on colitis-associated colorectal cancer: roles of gut microbiota involved. Front Cell Dev Biol. 2021;9:610.
Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA Cancer J Clin. 2017;67(4):326–44.
Helmink BA, Khan MW, Hermann A, et al. The microbiome, cancer, and cancer therapy. Nat Med. 2019;25(3):377–88.
Hwang S, Jo M, Hong JE, et al. Protective effects of zerumbone on colonic tumorigenesis in enterotoxigenic bacteroides fragilis (ETBF)-colonized AOM/DSS BALB/c mice. Int J Mol Sci. 2020;21(3):857.
Akutko K, Stawarski A. Probiotics, prebiotics and synbiotics in inflammatory bowel diseases. J Clin Med. 2021;10(11):2466.
Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. 2017;152(2):327-39.e4.
Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66(6):1039–48.
Downward JRE, Falkowski NR, Mason KL, et al. Modulation of post-antibiotic bacterial community reassembly and host response by Candida albicans. Sci Rep. 2013;3(1):1–11.
De Luca A, Zelante T, D’angelo C, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010;3(4):361–73.
Jostins L, Ripke S, Weersma RK, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24.
Malik A, Sharma D, Malireddi RS, et al. SYK-CARD9 signaling axis promotes gut fungi-mediated inflammasome activation to restrict colitis and colon cancer. Immunity. 2018;49(3):515-30.e5.
Limon JJ, Tang J, Li D, et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25(3):377-88.e6.
Tang J, Iliev ID, Brown J, et al. Mycobiome: approaches to analysis of intestinal fungi. J Immunol Methods. 2015;421:112–21.
Ohnmacht AJ, Stahler A, Stintzing S, et al. The oncology biomarker discovery framework reveals cetuximab and bevacizumab response patterns in metastatic colorectal cancer. Nat Commun. 2023;14(1):5391.
Gang W, Wang JJ, Guan R, et al. Strategy to targeting the immune resistance and novel therapy in colorectal cancer. Cancer Med. 2018;7(5):1578–603.
Nardone OM, Zammarchi I, Santacroce G, et al. Inflammation-driven colorectal cancer associated with colitis: from pathogenesis to changing therapy. Cancers. 2023;15(8):2389.
Yang C, Song J, Hwang S, et al. Apigenin enhances apoptosis induction by 5-fluorouracil through regulation of thymidylate synthase in colorectal cancer cells. Redox Biol. 2021;47:102144.
Li M, Xia M, Zhang Z, et al. METTL3 antagonizes 5-FU chemotherapy and confers drug resistance in colorectal carcinoma. Int J Oncol. 2022;61(3):1–13.
Dong S, Liang S, Cheng Z, et al. ROS/PI3K/Akt and Wnt/β-catenin signalings activate HIF-1α-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res. 2022;41(1):1–27.
Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Long non coding RNA Biology. Berlin: Springer; 2017. p. 1–46.
Kim N, Cho D, Kim H, et al. Colorectal adenocarcinoma-derived EGFR mutants are oncogenic and sensitive to EGFR-targeted monoclonal antibodies, cetuximab and panitumumab. Int J Cancer. 2020;146(8):2194–200.
Martinelli E, Ciardiello D, Martini G, et al. Implementing anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer: challenges and future perspectives. Ann Oncol. 2020;31(1):30–40.
Parseghian C, Loree J, Morris V, et al. Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol. 2019;30(2):243–9.
Srivatsa S, Paul MC, Cardone C, et al. EGFR in tumor-associated myeloid cells promotes development of colorectal cancer in mice and associates with outcomes of patients. Gastroenterology. 2017;153(1):178-90.e10.
Wang H, Li Y, Shi G, et al. A novel antitumor strategy: simultaneously inhibiting angiogenesis and complement by targeting VEGFA/PIGF and C3b/C4b. Mol Ther Oncolytics. 2020;16:20–9.
Zhu H, Liu X. Advances of tumorigenesis, diagnosis at early stage, and cellular immunotherapy in gastrointestinal malignancies. Front Oncol. 2021;11:666340.
Shek D, Akhuba L, Carlino MS, et al. Immune-checkpoint inhibitors for metastatic colorectal cancer: a systematic review of clinical outcomes. Cancers. 2021;13(17):4345.
Payandeh Z, Khalili S, Somi MH, et al. PD-1/PD-L1-dependent immune response in colorectal cancer. J Cell Physiol. 2020;235(7–8):5461–75.
Choucair K, Radford M, Bansal A, et al. Advances in immune therapies for the treatment of microsatellite instability-high/deficient mismatch repair metastatic colorectal cancer. Int J Oncol. 2021;59(3):1–17.
Sun L, Patai ÁV, Hogenson TL, et al. Irreversible JNK blockade overcomes PD-L1-mediated resistance to chemotherapy in colorectal cancer. Oncogene. 2021;40(32):5105–15.
Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42.
Perez-Ruiz E, Minute L, Otano I, et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature. 2019;569(7756):428–32.
Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38(1):1–12.
Kamal AM, Wasfey EF, Elghamry WR, et al. Genetic signature of CTLA-4, BTLA, TIM-3 and LAG-3 molecular expression in colorectal cancer patients: Implications in diagnosis and survival outcomes. Clin Biochem. 2021;96:13–8.
Sasidharan Nair V, Toor SM, Taha RZ, et al. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin Epigenetics. 2018;10(1):1–9.
Zaravinos A, Roufas C, Nagara M, et al. Cytolytic activity correlates with the mutational burden and deregulated expression of immune checkpoints in colorectal cancer. J Exp Clin Cancer Res. 2019;38(1):1–18.
Koelink PJ, Bloemendaal FM, Li B, et al. Anti-TNF therapy in IBD exerts its therapeutic effect through macrophage IL-10 signalling. Gut. 2020;69(6):1053–63.
Aden K, Rehman A, Waschina S, et al. Metabolic functions of gut microbes associate with efficacy of tumor necrosis factor antagonists in patients with inflammatory bowel diseases. Gastroenterology. 2019;157(5):1279-9211.e11.
Lee JWJ, Plichta D, Hogstrom L, et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe. 2021;29(8):1294-1304.e4.
Yakymenko O, Schoultz I, Gullberg E, et al. Infliximab restores colonic barrier to adherent-invasive E. coli in Crohn’s disease via effects on epithelial lipid rafts. Scand J Gastroenterol. 2018;53(6):677–84.
Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin Immunol. 2014;26(1):75–9.
Patnaude L, Mayo M, Mario R, et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021;271:119195.
Danese S, Grisham M, Hodge J, et al. JAK inhibition using tofacitinib for inflammatory bowel disease treatment a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310(3):G155–62.
Zorzi F, Calabrese E, Di Fusco D, et al. High Smad7 in the early post-operative recurrence of Crohn’s disease. J Transl Med. 2020;18(1):1–8.
Troncone E, Marafini I, Stolfi C, et al. Involvement of Smad7 in inflammatory diseases of the gut and colon cancer. Int J Mol Sci. 2021;22(8):3922.
Garcia-Arranz M, Herreros MD, Gonzalez-Gomez C, et al. Treatment of Crohn’s-related rectovaginal fistula with allogeneic expanded-adipose derived stem cells: a phase I-IIa clinical trial. Stem Cells Transl Med. 2016;5(11):1441–6.
Shen Z, Huang W, Liu J, et al. Effects of mesenchymal stem cell-derived exosomes on autoimmune diseases. Front Immunol. 2021;12:749192.
Liu H, Liang Z, Wang F, et al. Exosomes from mesenchymal stromal cells reduce murine colonic inflammation via a macrophage-dependent mechanism. JCI Insight. 2019;4(24):e131273.
Tyler CJ, Guzman M, Lundborg LR, et al. Antibody secreting cells are critically dependent on integrin α4β7/MAdCAM-1 for intestinal recruitment and control of the microbiota during chronic colitis. Mucosal Immunol. 2021;15(1):109–19.
Keir ME, Fuh F, Ichikawa R, et al. Regulation and Role of αE integrin and gut homing Integrins in migration and retention of intestinal lymphocytes during inflammatory bowel disease. J Immunol. 2021;207(9):2245–54.
Wang X, Chen S, Xiang H, et al. Role of sphingosine-1-phosphate receptors in vascular injury of inflammatory bowel disease. J Cell Mol Med. 2021;25(6):2740–9.
Scarozza P, Schmitt H, Monteleone G, et al. Oligonucleotides—a novel promising therapeutic option for IBD. Front Pharmacol. 2019;10:314.
Britton GJ, Contijoch EJ, Spindler MP, et al. Defined microbiota transplant restores Th17/RORγt+ regulatory T cell balance in mice colonized with inflammatory bowel disease microbiotas. Proc Natl Acad Sci. 2020;117(35):21536–45.
Chung Y, Ryu Y, An BC, et al. A synthetic probiotic engineered for colorectal cancer therapy modulates gut microbiota. Microbiome. 2021;9(1):1–17.
Uchiyama K, Naito Y, Takagi T. Intestinal microbiome as a novel therapeutic target for local and systemic inflammation. Pharmacol Ther. 2019;199:164–72.
Acknowledgements
We thank Wencheng Zhou, Gang Shi and Lin Cheng for a critical reading of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China Program grant (No. 81972607), the 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (No. ZYGD20003), and the Basic Research for Application of Sichuan Province, China (No. 2020YJ0066).
Author information
Authors and Affiliations
Contributions
JL wrote the manuscript and drew the figures and tables. YJ and NC provided the introduction and discussion of the manuscript. HD and LD conceptualized the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, J., Ji, Y., Chen, N. et al. Colitis-associated carcinogenesis: crosstalk between tumors, immune cells and gut microbiota. Cell Biosci 13, 194 (2023). https://doi.org/10.1186/s13578-023-01139-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-023-01139-8