
Abstract
Background
Cytokine-induced killer (CIK) cells are a novel subgroup of immune effectors, classified as one of the modified T cell-mediated arms for immunotherapy. These cells exert MHC‐unrestricted cytotoxicity against both hematological and solid malignancies with low incidence of treatment‐related severe complications. This study reviews the application of CIK cells in treating cases with hematologic malignancies.
Main body
CIK cells consist of CD3+/CD56+ natural killer (NK) T cells, CD3−/CD56+ NK cells, and CD3+/CD56− cytotoxic T cells. In this regard, the CD3+/CD56+ NK T cells are the primary effectors. Compared with the previously reported antitumor immune cells, CIK cells are characterized by improved in vitro proliferation and amplification, enhanced migration and invasive capacity to tumor region, more significant antitumor activity, and a broader antitumor spectrum. CIK cells can also induce death in tumor cells via numerous pathways and mechanisms. Hence, CIKs-based therapy has been used in various clinical trials and has shown efficacy with a very low graft versus host disease (GVHD) against several cancers, such as hematologic malignancies, even in relapsing cases, or cases not responding to other therapies. Despite the high content of T cells, CIK cells induce low alloreactivity and, thus, pose a restricted threat of GVHD induction even in MHC-mismatched transplantation cases. Phase 1 and 2 clinical trials of CIK cell therapy have also highlighted satisfactory therapeutic advantages against hematologic cancers, indicating the safety of CIK cells even in haploidentical transplantation settings.
Conclusion
CIK cells have shown promising results in the treatment of hematologic malignancies, especially in combination with other antitumor strategies. However, the existing controversies in achieving desired clinical responses underscore the importance of future studies.
Similar content being viewed by others

Background
Hematological malignancies are a specific category of tumors that impact the normal function of blood cells, bone marrow, and lymph nodes [1]. In this regard, several well-researched hematopoietic cancers are leukemia, non‑Hodgkin lymphoma (NHL), Hodgkin lymphoma (HL), multiple myeloma (MM), myelodysplastic syndromes (MDS) and myeloproliferative disorders [2, 3]. In addition to other blood disorders, hematologic cancers drastically increase worldwide death and morbidity rates [4]. These malignancies are the fifth most prevalent form of cancer and the 2nd highest etiology of cancer-induced death [5, 6]. Although the disease etiology is not entirely understood, several factors have been recognized that elevate the risk of these conditions, including immunosuppression or immunodeficient states [7, 8], viral infections such as human immunodeficiency virus (HIV), Epstein Barr virus (EBV), human T- lymphotropic virus (HTLV) and chronic bacterial infections like helicobacter pylori [8]. Manifestations of these malignancies are generally non-specific and impacted by several factors, among which are the awareness level of the patients, the type of cancer, and the presence of underlying comorbidities [9]. Early diagnosis of the disease plays a pivotal role in the improved prognosis of the patients [10]. However, an early diagnosis is quite complicated and is only accomplished using histologic, cytologic, cytogenetic, immunophenotypic, and radiologic studies [11]. It is worth mentioning that the constantly developing genetic methods have contributed to additional techniques that improve the diagnosis of patients [12]. Furthermore, the therapeutic strategy alters from patient to patient and is determined based on factors such as disease stage, pathologic findings, serum biomarkers, and the patient’s clinical status.
Diffuse large B cell lymphoma (DLBCL) and human leukemia (HL) are among the most common hematologic malignancies that are potentially curable with currently available chemotherapies; however, patients with failed chemotherapy usually experience unfavorable outcomes [13]. On the other hand, follicular lymphoma (FL) and MM are the only treatable malignancies. Nevertheless, consolidation therapy with initial autologous stem-cell transplantation (ASCT) and chemotherapy is a favorable option for eligible individuals with MM. The key problem in treating hematologic malignancies is recurrence following the primary therapy. In such conditions, patients usually undergo high-dose chemotherapy plus ASCT if not already done. However, more potent therapies are still required, particularly for individuals ineligible for stem-cell transplantation (SCT) or those who experience relapse following SCT [13].
The concept of exploiting immunity to protect against malignancies commenced in the late twentieth century when Prof. Coley W. suggested immunotherapy of malignant tumors with bacterial toxins [14]. Recent developments in molecular biology and an enhanced understanding of tumor-immunity interactions have contributed to the design of novel immunotherapies that are effective in various cancers [15, 16]. Among these immunotherapeutic strategies are bispecific T-cell engagers (BiTEs), immune checkpoint inhibitors [17], monoclonal antibodies [18], immunomodulatory drugs [19, 20], immunotherapy vaccines [21, 22] and adoptive immunotherapy including CAR-T cell [23], NK cells [24] and CIK cells which demonstrate remarkable efficacy in hematopoietic malignancies [25, 26].
CIK cells belong to a new subgroup of immune regulatory cells, categorized as a member of the adoptive T cell-mediated immunotherapy methods, which exploit MHC‐unrestricted cytolytic activity toward both hematologic and solid malignancies. A part of the growing penchant for the use of CIK cells in the treatment of cancers is due to the lack of life-threatening side effects [27]. Schmidt Wolf et al. [28] were the first to suggest a practical protocol for developing CIK cells, which was later shown to have antitumoral effects in both in vitro and in vivo settings. In summary, peripheral blood mononuclear cells (PBMCs) obtained from either healthy or unhealthy donors are treated with cytokines, such as interferon-γ (IFN‐γ), anti‐CD3 monoclonal antibody (mAb), and interleukin (IL)‐2 in a specific order, readily leading to the synthesis of CIK cells [27]. CD3+ CD56+ NK T cells are the main cellular subgroup of CIK cells, which show the properties of both NK and T cells. CD3−/CD56+ NK cells and CD3+/CD56− cytotoxic T cells are the other effector cells of this cellular population [28, 29].
Compared with conventional antitumor immune cells, CIK is marked by a more remarkable ability for in vitro expansion and consolidation, increased ability to migrate and invade the tumor zone, superior antitumor capacity, and broader antitumor spectrum. It has overcome the conventional problems such as gradual migration, limitation of cell source, and insufficient antitumor activity displayed in the lymphokine-activated killer cell (LAK), tumor-infiltrating lymphocyte (TIL), anti-CD3 mAb activated killer cell, and cytotoxic T lymphocyte (CTL) [30]. The CIK cells exhibit potent antitumor activity toward various tumor cells, particularly hematologic malignancies, mediated through multiple pathways [27, 31]. This review aimed to summarize the clinical administration of CIK cells and their novel combinations with other therapies in hematologic malignancies.
Hematologic malignancies: an overview
Acute lymphoblastic leukemia (ALL)
ALL is a heterogeneous hematopoietic neoplasm marked by the multiplication of immature lymphoid cells in the bone marrow, peripheral blood, and other tissues [32]. ALL has an age-adjusted prevalence rate of 1.8 per 100,000 individuals annually in the United States, with an average diagnosis age of 17 years [33]. ALL is the most common form of pediatric leukemia, accounting for 75–80% of cases in this age; however, ALL accounts for only about 20% of acute leukemia in adolescents [32, 34]. On the other hand, adolescent and young adult (AYA) patients are less likely to have favorable cytogenetic subtypes, such as ETV6-RUNX1 and hyperdiploidy, than children. In contrast, the occurrence of ALLs with unfavorable outcomes, such as BCR-ABL (Philadelphia positive (Ph+) ALL) or Ph-like ALL, is higher among AYA patients [35]. Risk factors for developing ALL consist of aging (> 70 years), chemotherapy, radiotherapy exposure, and genetic abnormalities, especially Down’s syndrome [36]. Induction therapy with multiple systemic chemotherapy agents, including glucocorticoids, asparaginase, vincristine, anthracyclines, and intrathecal chemotherapy, is the typical choice for pediatric B- and T-ALL treatment [37]. For high-risk patients with properly matched donors and appropriate performance status, allogeneic stem cell transplantation could be replaced with or added to the initial phases of chemotherapy, ameliorating long-term consequences [38]. Clinical trials using pediatric-inspired regimens in 50–60-year-old adults have shown 5-year overall survival (OS) rates of 50–60% [39]. Moreover, various studies have demonstrated that cellular immunotherapy is a useful therapeutic option for patients with relapsed or refractory B-lineage ALL [40, 41]. More recent treatments comprising blinatumomab, inotuzumab, Chimeric antigen receptor (CAR) T-cell therapy, nelarabine, and CIK cells have shown promising results in managing lymphoblastic leukemia [42, 43].
Acute myeloid leukemia (AML)
AML is characterized by increased immature myeloid progenitor cells in bone marrow and peripheral blood circulation. AML incidence is reported in all age groups; however, it is typically diagnosed after the sixth decade of life, indicating greater occurrence rates in the elderly, unlike ALL [44]. Somatic mutations in hematopoietic stem and progenitor cells (HSPCs) are the leading causes of AML development. Aging is accompanied by the accumulation of mutated HSPCs, a process called clonal hematopoiesis, eventually leading to cancer [45]. FLT3 mutation is the most known genetic alteration in AML patients, seen in approximately 30% of cases. This mutation results in the ligand-independent activation of tyrosine kinase, leading to cellular survival enhancement. The use of BiTE anti-FLT3/CD3 (AMG-427) has been considered by various clinical trials [46]. Despite the recent advancements in AML treatment, the success rate of cure in patients over 60 years old is only 35–40% [47]. The heterogeneity of AML cells makes it difficult for the immune system to identify specific tumor biomarkers, making it challenging to develop practical immunotherapies for AML [48]. Furthermore, AML cells are capable of scaping immunity by downregulating the expression of particular receptors or overexpression of inhibitory immune checkpoints, such as PD-L1, PD-L2, CD47, and CD70 [49]. Thus, numerous clinical studies have been attempting to improve the outcome of these patients by developing novel immunotherapeutic strategies or enhancing the existing ones by combining them with other treatments. For example, Liao et al. showed that administration of anti-PD1 or anti-CTLA4 in AML patients is accompanied by an improved T cells’ reaction, resulting in suppressed measurable residual disease (MRD) [50]. Anti-CD33/CD3 (AMG330) [51] and anti-CD123/CD3 [52] are also potential therapeutic agents in AML patients. Moreover, studies have demonstrated the ability of CIK cells to destroy autologous AML malignant cells [53].
Chronic lymphocytic leukemia (CLL)
CLL is a commonly occurring form of leukemia with an incidence rate of about 0.6%, slightly more prevalent among men (1.2:1–1.7:1) [54]. CLL is considered an aging-related disease with an average diagnosis age of 70 [55]. CLL is identified by the clonal multiplication and building up of mature, typically CD5-positive B-cells within the bone marrow, lymph nodes, spleen, and blood [56]. Almost 80% of all cases with CLL have at least 25% of the most prevalent chromosomal mutations, including deletion in 13q14.3 (del13q), del11q, del17p, and trisomy 12 [57]. Del13q is the most prevalent mutation, seen in up to two third of cases. An isolated del13q14 is accompanied by a slow cancer progression. Besides, del(17p) is reported in 5–8% of chemotherapy-naïve cases, which usually involves 17p13, where the most critical tumor suppressor gene, TP53, is situated. CLL cases with del17p demonstrate considerable genotoxic chemoresistance [58]. Therapeutic options for CLL patients have developed over the past years. The innovative addition of immunotherapy to chemotherapy has shown marked enhancements in the patients’ overall response (OR) and OS rates [58, 59]. In this regard, the combination of venetoclax (a B-cell lymphoma 2 (BCL2) inhibitor) with obinutuzumab, monotherapy with Bruton tyrosine kinase (BTK) inhibitors such as ibrutinib and acalabrutinib, CAR-T cell, CIK or chemoimmunotherapy have shown promising outcomes [60, 61].
Myeloproliferative neoplasm (MPN)
MPNs are hematopoietic neoplasms that emerge in the bone marrow and are categorized into different subgroups, including (I) chronic myeloid leukemia (CML), (II) chronic neutrophilic leukemia, (III) chronic eosinophilic leukemia-not otherwise specified, (IV) primary myelofibrosis (PMF), (V) polycythemia vera (PV), (VI) essential thrombocythemia (ET), and (VII) MPN unclassifiable. MPNs are characterized by an excessive proliferation of blood cell lines, leading to the thickening of blood and disrupted bone marrow function. [62]. CML is typically identified by BCR-ABL1 mutation and is usually managed with imatinib, a kinase inhibitor. Nonetheless, JAK2-mutated clones can be identified in some CML cases ensuing therapy [63]. Moreover, most CML patients experience remission and long-term disease-free survival following intensive chemotherapy and BCR-ABL1-targeting compounds [64]. However, nonresponse and remission are still challenging issues in treating many patients. Recently, novel targeted treatment strategies, such as antibody-based therapies, checkpoint inhibitors, CIK cells, CAR-T, and CAR-NK cells, have been proposed for CML [65, 66]. Among these treatments, CIK cells were shown to be able to inhibit the growth of CML-positive colonies both in vitro and in Severe Combined Immunodeficiency (SCID) mice transplanted with autologous CML cells; however, controversies are undeniable [67, 68].
Lymphoma
Hodgkin lymphoma
HL usually develops sporadically and constitutes about 10% of all lymphoma cases. HL emerges from lymph nodes, and the underlying etiology could be related to the EBV or HIV/AIDS infection [69]. The diagnostic hallmark of HL is the presence of malignant Reed–Sternberg cells in the lymph nodes of the patients. Notably, an abnormal increase in chromosome 9p24.1 in HL individuals, a locus containing JAK2, PD-L1, and PD-L2, is involved in the pathophysiology of HL [70]. This abnormal amplification directly increases the expression of PD-L1 and PD-L2. Besides, PD-L1 transcription is also enhanced via gene dose-dependent JAK-STAT pathway in HL. Moreover, EBV is another factor that induces PD-L1 overexpression in EBV-positive cancerous cells [71]. In contrast to non-Hodgkin lymphoma, in classical HL, the tumor microenvironment contains numerous ineffective immune cells and a small number of malignant cells [72].
Non-Hodgkin lymphoma
NHL typically involves lymph nodes and spleen and can potentially damage the bone marrow. However, leukemia generally implicates the bone marrow and blood. The classification of NHL is based on the type of lymphocytes involved and the disease severity [73]. There are five prevalent forms of NHL, including (I) DLBCL, II) FL, (III) small lymphocytic lymphoma (also known as SLL), (IV) marginal zone B cell lymphoma (MZL), and (V) mantle cell lymphoma (MCL). While DLBCL is the most prevalent aggressive lymphoma, FL and CLL have slow progression [74]. Malignant lymphoid cells have restricted tumor antigen presentation and impair immune response by releasing immunosuppressive cytokines or recruiting immunoregulatory immune cells [75, 76]. In this regard, immunotherapeutic strategies have been developed to overcome these inefficient immune responses, among which are the novel immune checkpoint inhibitors affecting the CD47-SIRPα pathway, leading to the regulation of innate immunity [16, 77]. Moreover, antibody-based immunotherapeutic strategies, such as antibody–drug conjugates and bispecific T-cell or NK-cell engagers, are rapidly developed with favorable clinical outcomes to overcome the limited antigen presentation in malignant cells [78, 79].
Multiple myeloma
MM is the second most common blood cancer and the most prevalent form of myeloma [80]. Abnormal plasmacytes in the bone marrow result in excessive production of monoclonal immunoglobulins, known as M proteins, which are the hallmarks of MM. These proteins can cause end-organ injury by attacking organs such as kidneys and bone. Whereas MM has remained an incurable disease, novel therapeutic strategies such as immunotherapies using mAbs [81], CAR-T cells, and therapies combining chemotherapeutics and DC/CIK cells have recently been exploited with promising results [82, 83].
Cytokine induced killer cells
CIK fundamentals
Immunotherapies are novel and promising strategies that are potential alternatives to conventional treatments such as surgery, chemotherapy, or radiotherapy, particularly in patients with recurrent or advanced stages of malignancies. The main goal of immunotherapy is to enhance the native immune response against tumor cells, potentializing it to eradicate them [84]. Currently, there are different immunotherapies to treat malignancies, such as vaccines, cytokine or antibody-based, and adoptive cell‐based strategies. Adoptive cell‐based immunotherapy is a therapeutic method where the immune cells from either the patient or donor are multiplied, activated, and even altered outside of the body to produce engineered cells with significantly increased cytotoxicity against malignant cells, which are then transfused into the patient’s body [85]. This strategy is primarily mediated by T cells such as TILs, engineered T‐cell receptor (TCR) or CAR‐T cells, CIK cells, NK cells, and natural killer T (NKT) cells [28, 86, 87]. CIK cells consist of T cells (CD3+CD56−), NK‐T cells (CD3+CD56+), and NK cells (CD3−CD56+) [88]. Among these three distinct types of cells, a higher fraction of CD3+CD56+ NK‐T cells are CD8+ T cells, possess increased granzymes, and show enhanced effector phenotype. As a result, these cells have the highest cytotoxicity and antitumor potential [89]. Interestingly, CIK cells have considerable proliferative capacity and MHC-unrestricted cytotoxicity against both hematopoietic and solid tumors. In addition, they identify and eliminate neoplastic cells without requiring preceding exposure to malignant components or priming [28, 89].
CIK design and biology
CIK cells are obtained by ex vivo incubation of cells derived from various lymphocyte sources such as PBMCs, umbilical cord blood precursors, and bone marrow [90] in a time-sensitive sequence using IFN-γ, anti‐CD3 mAb, and IL-2 (Fig. 1). The first protocol for generating CIK cells was introduced by Schmidt‐Wolf et al. [28] in 1991. According to this protocol, PBMCs were isolated from whole blood and then exposed to 1000 IU/ml IFN‐γ. The next day, 50 ng/ml of anti‐CD3 mAb, 100 IU/ml rIL‐1, and 300 IU/ml IL‐2 were added to the medium, and then IL-2 was added during the culture process [28]. However, today, the induction protocol of CIK cells is not limited to this method, and different studies use various strategies to obtain these valuable cells (Table 1) [91].

The overview of CIK cell expansion from PBMN. This figure illustrates how CIK cells are expanded from PBMN in autologous or allogenic settings and delivered to the patients after preparation. A typical expansion protocol of CIK cells consists of incubation with IFN-γ, CD3 mAbs, and IL-2. However, there is no unique protocol for producing CIK cells, and various modifications and combination therapeutic strategies (such as PD-1 mABs mentioned in the figure) are usually exploited to enhance the antitumor efficacy and cytotoxicity of CIK cells against hematologic malignancies
Each factor used in the process of CIK cell induction is used with a particular goal. Anti-CD3 Ab is a mitogenic factor, and specific levels of IL-2 primarily enhance the expression of natural killer group 2 member D (NKG2D) and transmembrane adapter protein DAP10, which are pivotal for cytolysis (Fig. 2) [92]. While IL-2 has a considerable role in lymphocyte proliferation, IFN-γ activates CIK cells and induces CD58 expression, also known as lymphocyte function-associated antigen (LFA)-3 [93]. Various studies have also reported that activating or transfecting CIK cells by cytokines such as IL‐6, IL‐7, IL‐12, IL‐15, IL‐21, or thymoglobulin can lead to phenotypic variation, increased multiplication, and improved antitumor activity [94,95,96,97]. According to Lin et al. [94], using IL-6 every two to three days in CIK cell expansion could result in a higher fraction of CD3+CD56+ effector cells and a decreased proportion of regulatory T cells (Tregs), significantly enhancing these cells' in vitro cytolytic activity.

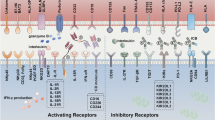
Double T/NK cell specificity of CIK cells and mechanism of action. CIK cells are effector memory T (T-EMRA) lymphocytes with TCR specificity that also show the cytotoxicity of NK cells. The antitumor activity of CIK cells is regulated through DNAM-1 and LFA-1, besides NKG2D, NKp30, and CD56
Using IL-15 in CIK cell expansion in combination with IL-12 or as an alternative to IL-2 has demonstrated significant benefits [98]. Accordingly, Rettinger et al. have reported that IL-15-activated CIK cells exhibit an increased antitumor efficacy against ALL and lymphoma targets [96]. Another study highlighted that using IL-15 during CIK cell induction results in an increased proportion of CD3+CD56+cells, a decreased immunosuppressive activity of Tregs, and suppressed IL-35 levels, enhancing antileukemic efficacy [99]. Moreover, the expansion of CIK cells using IL-21 has resulted in increased levels of perforin, granzyme B, Fas ligand, IFN-γ, tumor necrosis factor-α, and IL-21 receptors. Besides, IL-21 was able to increase the fraction of CD3+CD56+ effector cells, although the general proliferation rate remained unchanged [100].
Mature CIK cells express elevated levels of anti-malignant cell components, such as perforin, granzyme, Fas ligand (FasL), and CD40 ligand (CD40L), compared to early-stage CIK cells. These cells also have altered expression of immune checkpoints so that PD‐1, CD28, CD137, and VSIR are decreased, and LAG3, CTLA4, and TIM‐3 are upregulated [101]. Although the exact mechanisms behind the CIK cells’ cytolysis activity are still unclear, some important molecules and pathways have been recognized. For example, a considerable cytolysis suppression after blocking LFA-1 and intracellular cell adhesion molecule (ICAM)-1 indicates that cytotoxicity relies on cell-to-cell contact. On the other hand, failure in inhibition of the cytotoxic activity of CIK cells using antibodies against CD2, CD3, CD8, CD28, CD56, very late antigen (VLA)-4, TCR, and MHC class I and II molecules suggests that these cells exploit an MHC-independent method for recognizing targets [29].
NKG2D is a conserved member of the c-type lectin-activating receptor family found within the NK gene complex on human chromosome 12p12-p13, which is a pivotal molecule in identifying tumors by CIK cells [102]. NK cells express NKG2D receptors that are able to recognize at least six counter-ligands, including the MHC-class I-like molecules, MHC class I polypeptide-related sequence A (MICA), MHC class I polypeptide-related sequence B (MICB), and some members of the UL16 binding protein family (ULPB1-4) [103]. It has been established that the expression of NKG2D is increased in all CIK cells without being limited to the CD3+CD56+ subgroup. Recent findings based on NKG2D-targeting antibodies, small interfering RNA, and redirected cytotoxicity have shown that the CIK cells’ cytolysis activity is mainly associated with the NKG2D interaction rather than TCR engagement. Activation of the NKG2D pathway seems to be directly related to disulfide adaptor protein 10 upregulation, which is induced by the high levels of recombinant human IL-2 (rhIL-2) in the culture medium of CIK cells [93]. Interestingly, NKG2D has also been found to play a vital role in target recognition by CIK cells that are expanded with IL-15 instead of IL-2 [96]. Besides NKG2D, CIK cells have antitumor effects through their TCR‐mediated lytic manner, indicating the dual‐functional capability of these cells [104].
Moreover, a transcriptomic study illustrated the contribution of CD8 and Lck kinase, a member of the Src kinase family, in the antitumor activity of CIK cells [101]. CIK and NK cells have many characteristics in common, including the large granular lymphocyte morphology, the ability to eradicate the K562 cell line (an HLA (Human leukocyte antigen) class I negative cell line), and the high densities of CD56 and NKG2D in the cell surface. CIK cells have lower levels of NKp30 than NK cells and do not express NKp44, NKp46, inhibitory killer immunoglobulin-like receptors, NKG2A, and CD94 [88]. Even though NKp30 is expressed on CIK cells at low density, it has been revealed that DNAM-1 and NKp30 are involved in CIK cell-mediated cytolysis in malignant tumors [104]. Besides, it has been previously shown that the CD3+CD56+CD16+ subset of CIK cells displays remarkable cytotoxicity against tumors through antibody-dependent cell‐mediated cytotoxicity (ADCC), which is exclusively medicated by the CD16 receptor and can be augmented by using monoclonal antibodies. The same study reported that the fraction of the CD3+CD56+CD16+ subset of CIK cells differs from donor to donor [105].
CIK and cancer immunotherapy
CIK cell therapy is used to treat both hematologic and solid malignancies. Interestingly, the expansion of CIK cells is achievable using autologous sources and can even be administrated in combination with various allo-hematopoietic stem cell transplantation (HSCT) strategies [106]. The earliest clinical trial of autologous CIK cells was performed on cases with relapsed lymphomas following autologous HSCT. Nine patients (7 with advanced HD and 2 with NHL) were administered autologous CIK cells (1 × 109 to 10 × 109 cells per cycle) during an average of two cycles (range 1–3). CD3+CD56+CIK cells reflected 8–58% (median 22%) of the overall cellular population. While two patients achieved disease stabilization for over 18 months, and two others experienced a partial response, the other five patients did not show a favorable response. Only one of the patients experienced mild hypotension, and another had a low-grade fever, indicating that the adverse effects were minor [107]. Likewise, Hoyle et al. [68] established the feasibility of generating autologous CIK cells from patients with CML. They also demonstrated that CIK cells have cytolytic activity against OCI-LY8 and K562 tumor cell lines.
Zhang et al. [27] reviewed 106 CIK clinical trials from 1999 to 2019, conducted on a total number of 10,225 patients with over 30 distinct cancers. Among the studied patients, 4,889 received CIK cells as their treatment, either alone or in combination with other therapeutics. They concluded that using CIK cells in the treatment of various malignancies notably ameliorates patients’ median progression‐free survival (mPFS), median overall survival (mOS), and overall response rate (ORR). A multicenter clinical trial on 230 patients with hepatocellular carcinoma showed that CIK cell therapy could extend recurrence‐free survival by about 50% compared to the control group [108]. In another study, CIK cell therapy showed considerably prolonged mOS (36 months vs. 16 months) and significantly enhanced 3‐year PFS and OS compared to the control subgroup [109]. Chen et al. studied the efficacy of combination therapy using CIK cells and chemotherapy on 136 patients with non-small cell lung cancer (NSCLC) following surgery. They reported a higher mOS and 3-year survival median in the combination therapy group than in the chemotherapy group (P = 0.032, P = 0.036, respectively) [110]. Other clinical trials have shown that combination therapy using CIK cells and chemotherapy is more effective strategy compared to only chemotherapy in patients with advanced pancreatic cancer [111] and postoperative epithelial ovarian cancer based on their mOS and favorable PFS [112].
Clinical application of CIK cells in hematologic malignancies
Numerous clinical studies have utilized CIK cells to treat various cancers, such as hematologic malignancies, and reported high efficacy even in relapsing patients who failed to respond to conventional treatments (Table 2) [26, 113]. Despite having a substantial number of T cells, CIK cells exhibit minimal alloreactive reactions, resulting in a low incidence of GVHD even in MHC-mismatched transplantation settings [114, 115]. The following sections will discuss the findings of different clinical trials on patients with hematological malignancies undergoing CIK cell therapy.
Monotherapy
Allogenic
Allogeneic HSCT is the standard therapy for numerous advanced hematologic cancers, although molecular relapse is the primary etiology of treatment failure. As one of the novel cell-based immunotherapy strategies, CIK cells show promising results in resolving molecular and overt relapse following allogeneic HSCT in these patients [25, 26]. Merker et al. [106] appraised the efficacy of using donor lymphocyte infusion (DLI) and CIK cells in 91 cases with hematopoietic neoplasms following allogeneic HSCT. Among the studied cases, 31 had AML, two had CML, 53 had ALL, one had biphenotypic leukemia, and four had T-NHL. IL-15 was used in the expansion protocol of CIK cells, readily producing CD3+CD56+CD25+ CIK cells. Despite the more severe overall condition of patients receiving CIK cells compared to patients receiving DLI, patients in the CIK subgroup experienced a limited 6-month cumulative incidence of relapse (CIR) and higher complete remission (CR) [25].
In another clinical trial, allogeneic CIK cells were transfused as consolidation therapy following non-myeloablative allogeneic transplant in high-risk patients with myeloid malignancies, including MDS, MPN, therapy-related myeloid neoplasms (t-MN) and secondary acute myeloid leukemia (sAML). While patients in the intervention group received a single dose of CD3+CIK cells (1 × 108/kg) between the 21st and 35th day after total lymphoid irradiation and anti-thymocyte globulin (TLI-ATG)-based allo-HCT, cases in the control group only received TLI-ATG-based allo-HCT. CIK-treated patients had lower 1-year II–IV grade aGVHD and higher CIR compared to the control patients, indicating the safety and feasibility of CIK cell therapy as an early consolidation treatment in the ambulatory setting. However, the two groups had no significant difference regarding two-year non-relapse mortality, event-free survival rates, two-year OS, and full donor chimerism (FDC) rate by the 90th day. Such failure could be a result of using only a single dose of CIK cells, adverse effects of cyclosporine and mycophenolate mofetil on the function, proliferation, or survival of CIK cells, which were used with GVHD immunosuppressive goals [116].
Post-transplant lymphoproliferative disorders (PTLD) can be a fatal complication following allogeneic HSCT [117]. Generally, there is a low chance of PTLD after stem cell transplantation; however, weakened immunosurveillance can raise the risk of developing PTLD during the early post-transplantation phase [118]. In most patients, PTLD incidence is correlated with EBV-induced atypical lymphoproliferative disorders that occur due to viral reactivation or primary EBV infection following HSCT, which can usually be controlled by cellular immunity in an immunocompetent population [119]. To overcome such complications, Pfeffermann et al. [120] obtained CIK cells from EBV-seropositive donors using IFN-γ, anti-CD3, IL-2, IL-15, and EBV-antigen pulsing as stimulants. EBV-antigen was added to the expansion protocol to target the generation of CIK cells with EBV-reactive T cells to treat patients with EBV-associated PTLD rapidly progressed to severe DLBCL. 10 × 106 T cells/kg of EBV-specific CIK cells were infused, which resulted in the eradication of DLBCL without recurrence after 1 week of transfusion with no acute toxicities. Moreover, CIK cells were still detectable after 32 days of administration. The in vitro evaluations highlighted improved cytotoxicity against EBV-positive targets and elevated expansion of CD8 + T cells and T-NK cells compared to conventional CIK protocol.
Autologous
A pioneering clinical study using autologous CIK therapy in refractory APL patients [121], which administered three cycles of CIK (1 × 109 cells/cycle) in each patient, reported that CIK cell therapy improved outcomes in these patients. Similarly, Zhou et al. [122] studied autologous CIK cells in 20 patients with high-risk DLBCL with an IPI ≥ 3. On average, patients received two CIK cycles (median of 55.12 ± 14.63 × 108 cells) every three months, each followed by 100 mU of subcutaneous IL-2 for ten consecutive days. The univariate analysis demonstrated an elevation in 5-year disease-free survival (DFS) and OS of CIK-treated patients compared to the control subgroup; however, after adjusting the results, only the DFS statistically remained significant. Besides, although one patient experienced a mild flu-like syndrome, which was naturally resolved without any intervention, no severe complication was reported.
In another clinical trial study, autologous CIK cells were used to treat 24 patients with hematologic malignancies (13 with AML and 11 with CML). On average, AML patients received two cycles of CIK (median of 12.72 × 109 cells), and CML patients received four cycles of CIK (median of 25.72 × 109 cells). It was reported that the survival and relapse of AML patients and the BCR–ABL transcript level in CML patients treated with CIK did not differ from those not treated with CIK cells, indicating that CIK therapy had no superiority over conventional antitumor therapies. However, the advantage of no serious side effects following CIK cell therapy was also established in this study [67].
Combination therapy
As mentioned above, CIKs are crucial components of adoptive cellular immunotherapy and have been identified as major cytotoxic immunologic effector cells, which can be exploited to treat various malignancies. In the last decade, CIK cells have been used in combination with other therapeutic strategies such as CAR, DC, mAbs, and PD-1/PD-L1 inhibitors to enhance their antitumor efficacy against hematologic malignancies (Fig. 1). Following sections summarize the recent improvements in CIK cell combinational therapeutic strategies.
CAR + CIK therapy
CAR T-cells have revolutionized the treatment of hematological cancers. However, there are severe adverse effects related to this therapeutic strategy, such as cytokine storm syndrome, immunity-induced neural toxicity, and significant risk of GVHD in allogeneic settings, making it challenging to apply it on a wide scale [123]. CIK cells have a low risk of GVHD and are readily expanded platforms for CAR engineering, similar to the conventional CAR T-cells. Besides, the high expenses of CAR T-cell therapy and the cost-effectiveness of CIK cells make the combination of these two strategies a matching therapeutic choice [124]. Moreover, the preliminary clinical studies in both adult and pediatric patients have revealed that CAR T-cell and CIK therapies have synergic antitumor effects and even show higher specificity against certain malignancies [125,126,127]. Oelsner et al. [42] designed specific CIK cells using IL-15 and transduced them to generate CAR.CIK cells specific for CD19 and carrying CD28 endodomain, briefly called CAR.CIK-63.28.z. In vitro studies showed that CAR.CIK-63.28.z had selective cytotoxicity against B-ALL cells previously resistant to CIK therapy.
The in vitro efficacy of CD44v6-cCAR.CIK, the engineered CIK cells obtained from fully donor chimeric individuals redirected against CD44v6 with the CD28 costimulatory endodomain, was investigated by Circosta et al. [123]. CD44v6–fcCAR.CIK demonstrated notably increased in vitro cytolysis against malignant cells compared to the non-transduced (NTD) CIK cells (fcNTD.CIK). However, the cytotoxic efficacy of CD44v6–fcCAR.CIK was not superior to the cytotoxic potential of conventional CD44v6-fcCAR.T cells. By the way, CD44v6–fcCAR.CIK showed nonenhanced alloreactivity across HLA barriers, indicating an essential advantage for CD44v6–fcCAR.CIK against CD44v6-fcCAR.T. Besides, CD44v6–fcCAR.CIK had higher secretion of the IL-1β and IL6 following coculture with CD44v6 + targets compared to CD44v6-fcCAR-T cells. Similarly, Magnani et al. [128] investigated the feasibility of engineered CD19-CAR.CIK in ALL. They showed that CD19-CAR.CIK could extend the durability in a patient‐derived xenograft (PDX) with the ph‐like ALL PAX5/AUTS2 translocation and showed a dose‐dependent antitumor activity, with 15 × 106 per cycle being the optimal cellular dose. Moreover, in the lymphoma survival model, complete elimination of disseminated neoplasms was achieved. Overall, the safety and tolerability of CARCIK‐CD19 administration were verified in a bio‐distribution and toxicity model [128].
In vivo efficacy of engineered CD33-CAR.CIK was investigated in another preclinical study [129]. CD33-CAR.CIK cells were infused weekly for 3 weeks into patient-derived AML PDX mice (107 cells/cycle). In the early approach, CIK cells were transfused on 3rd or 5th days following the AML induction in the mice. CD33-CAR.CIK cells displayed considerable antitumor activity toward AML targets in vitro and the PDX mice and significantly delayed the progression of AML in the treated mice. They also highlighted that CD33-CAR.CIK cells are capable of harnessing relapsed and refractory AML following the administration of a xenograft chemotherapy approach. Notably, CD33-CAR.CIK cells were well tolerated, and after a maximum of three cycles, no CD33-negative AML cells were detectable.
Despite the anti-AML efficacy of CAR.CIK cells in various in vitro studies, CIK therapy is not accompanied by favorable findings in the treatment of AML patients when compared to the success achieved in treating B-cell neoplasms. Among the obstacles to the use of CAR.CIK therapy in AML patients are the cellular heterogeneity, the lack of AML-specific cellular markers, and the leukemia-friendly tumor microenvironment (TME) [67, 130]. In this regard, Alberti et al. [131] designed bispecific Tandem CAR.CIK cells targeting CD33 on malignant cells and CD146 on mesenchymal stromal cells (CD33-CD146-CAR.CIK), aiming to modify the TME alongside the cancerous cells themselves. CD146 is known to enhance malignant myeloid cell growth and survival, and blocking it would hypothetically suppress tumor invasion. However, they reported that the long-term coculture of CD33-CD146-CAR.CIK with CD146 + mesenchymal stromal cells suppressed the function of CD33-CD146-CAR.CIK based on the reduced synthesis of IFN-ɣ, IL-2, and ki-67 and decreased cellular proliferation. These discoveries imply that the CD146 + mesenchymal stromal cells present in the TME play an essential role in the resistance of malignancies against therapies, and manipulating them could be a promising target for future studies. By the way, despite the suppressive role of CD146 + stromal cells on CD33-CD146-CAR.CIK, these engineered cells demonstrated improved cytolysis activity against the KG-1 myelogenous cell line compared to NTD.CIK cells.
Homing of leukemic blasts in bone marrow is another obstacle in treating AML, primarily due to the increased level of CXCR4 in AML blasts. The CXCL12/CXCR4 axis plays a pivotal role in AML pathogenesis, as it regulates blast adhesion into the protective BM surroundings, accommodation to the hypoxia, and cellular migration and durability [132]. On the other hand, ex vivo culture conditions impair the expression of chemokine receptors in human lymphocytes, which hinders the ability of CIK cells to migrate into the BM [133, 134]. Biondi et al. [135] designed novel CAR.CIK cells, using CD33 and wild-type CXCR4 to overcome such problems. CXCR4-CD33-CAR.CIK cells exhibited a promoted ability to migrate towards the CXCL12 chemokine, thereby facilitating them to effectively eliminate myelogenous cell lines. In addition, CXCR4-CD33-CAR.CIK cells showed improved homing to bone marrow in a mouse model of AML and superior control over AML progression, leading to increased survival time in treated mice compared to conventional CD33-CAR.CIK cells.
Dendritic cells (DCs) + CIK therapy
Combining DCs and CIK cell therapy is another emerging strategy in the treatment of various malignancies such as ALL, CML, and lymphoma, and its safety and efficacy have been established by multiple studies [67, 136]. Cao et al. [137] demonstrated that in vitro incubation of CIK cells beside DCs can enhance the replication rate and tumor suppressive efficacy of CIK cells. Another survey reported that the combination of CIK cells with DCs alters the expressing markers of both populations and improves the secretion capacity of IL-12 by CIK cells, enhancing their cytotoxicity against malignancies [138]. Furthermore, investigations have revealed that DCs can reduce the number of Treg cells present in the CIK cellular population, improving the antitumor efficacy of CIK cells [139].
Zhang et al. [140] administrated autologous DC-CIK and allogeneic NK cells in 85 AML cases. Among these, 16 patients received only autologous DC-CIK, 9 received only allogeneic NK cells, and the remaining alternately received DC-CIK and NK cells in two to four cycles without any severe adverse effects or treatment-related mortality. The average number of DC-CIK or NK cells was 5.12 × 109/cycle. The results revealed an increase in OS and relapse-free survival (RFS) in patients who alternately received DC-CIK and NK cells compared to those patients who received either DC-CIK or NK cells. In another study, Zhao et al. [82] investigated the efficacy of DC + CIK immunotherapy in combination with bortezomib and dexamethasone chemotherapy in MM patients. DC + CIK was administered daily to the patients 15–20 days after chemotherapy for six cycles (2.0–5.0 × 109 cells/cycle). Results revealed that DC + CIK combination therapy is superior to only chemotherapy strategy based on the performance status (PS) scores. Furthermore, the serum proportion of MM cells, β2-microglobulin (β2-MG) level, M protein level, creatinine level, and 24-h urine light chain level were lower among patients who received combination therapy, indicating the better prognosis of these patients.
In another study, Xiao et al. [141] conducted DC-CIK cell therapy in a 52-year-old patient with Ph− B-precursor ALL who experienced complete remission following a standard VDCLP regimen including vincristine, daunorubicin, cyclophosphamide, l-asparaginase, and prednisone. However, the patient declined to continue the therapy due to some serious complications, leading to disease relapse. Thus, the therapeutic plan was changed to DC-CIK. Following the first infusion, DC-CIK therapy was continued for seven more cycles (4–6 × 109 cells/cycle) besides chemotherapy, which resulted in the long-lasting complete remission of the patient.
Dong et al. [142] found that the proportions of CD4+PD-1+ and CD8+PD-1+ cells are significantly higher in AML mice models than in the control group, a trend that is mitigated by applying DC-CIK cells. Furthermore, they observed that MMP9- and CCL1-mediated silencing of DC-CIK cells effectively enhances the reduction in CD4+ PD-1+ and CD8+ PD-1+ cells. MMP9- and CCL1-silencing could probably exert their role by increasing T-cell activation and reducing T-cell exhaustion, which was evident in both their in vivo and ex vivo experiments.
Monoclonal antibodies + CIK therapy
Over the past 20 years, immunotherapy utilizing targeted antibodies has emerged as a highly effective treatment approach. Various research has demonstrated that combining CIK cells with antibodies can enhance their antitumor efficacy. A study showed that the in vitro combination of rituximab or Obinutuzumab (OBI) with CIK is accompanied by a significant increase in the CIK cells' antitumor activity toward B-cell lymphoma [143]. Esser et al. [144] found that combining CIK cell therapy with anti-CD30 mAb, Brentuximab Vedotin, substantially improves treatment efficacy against three distinct CD30+ lymphoma targets (Daudi, KI-JK, and L-540) without disrupting CIK cells’ function.
In another study, Pietà AD et al. [145] reported the combination of OBI and CIK cells as a novel and effective therapeutic strategy in B cell cancers. In this study, the CIK cells were obtained using a novel strategy that included blinatumomab during CIK cell expansion. They compared the in vitro cytotoxicity of CIK and OBI combination with CIK and RTX combination toward various B-cell lines expressing CD20. In general, redirected CIK cell therapy with mAB showed substantial antitumor activity against all malignant cell lines. However, the OBI-CIK combination therapy demonstrated superiority against the RTX-CIK method. Besides, in vivo investigations highlighted that daily administration of 107 CIK cells besides OBI in mice models of MCL for seven sequential days suppresses cancer progression, improves immune infiltration, and extends overall survival [145]. Frank et al. [146] introduced a new method for directly conjugating antibodies to surface proteins of CIK cells, including rituximab and daratumumab, which notably improved CIK cells' cytotoxicity compared to soluble antibodies. Surprisingly, direct conjugation of rituximab to cellular surface resulted in improved antitumor efficacy in all CD3−CD56+ (NK), CD3+CD56+ (CIK), and CD3+CD56− (T) cell lines. This strategy showed increased intracellular signaling following interaction with tumor targets, which improved the cytotoxicity of CIK cells. Moreover, Interdonato et al. [147] developed a new tetravalent IgG1-like bispecific (bs)Ab called BL-01 that contains two distinct binding sites for CD5 and CD20 proteins. It was demonstrated that BL-01 can concomitantly bind to CD20 and CD5, redirect CIK cells toward CD20 + targets, and enhance their cytotoxicity up to 3 folds. Besides, BL-01-CIK cells were able to prolong the survival rate in rituximab-resistant PDX DLBCL mice more potently compared to exposure to bsAb or CIK cells alone.
In another study, Gloy et al. [148] exploited banked cryopreserved cord blood units to expand CIK cells. The costimulatory and inhibitory/exhaustion levels in cord blood (CB)-CIKs and peripheral blood (PB)-CIKs did not differ; however, PB-CIK cells with positive CD8 had higher CD28 expression. CB-CIKs and PB-CIKs also had similar in vitro efficacy. Furthermore, the comparison between only CB-CIK (20 × 106/cycle for five cycles) treatment and CB-CIK plus blinatumomab (100 ng) treatment revealed that the efficacy of combination therapy is superior to the prior method based on the higher survival rate among PDX Ph + ALL mice models. Tita-Nwa et al. [149] also described increased CIK cell efficacy against B-lymphoma cells after combining with bsAb against CD19 and CD5, which are present on effector T cells and malignant target cells, respectively.
Immune checkpoint inhibitors + CIK Cells
Immune checkpoint inhibitors, such as anti-PD-1 agents, have been identified as an efficacious therapeutic approach in various high-grade cancers like lymphoma in both monotherapy [150] and combination therapy forms [151, 152]. Moreover, recent studies have shown that combining immune checkpoint inhibitors with CIK cells can considerably improve the anti-tumor characteristics of these cells [153, 154].
In a combination therapy, Li et al. [155] added immune checkpoint inhibitors against PD-1, TIM-3, and LAG-3 to CIK cells to investigate the cumulative cytotoxicity of this therapeutic approach. In their study, CIK cells were harvested from PBMCs of patients with AML, ALL, or MM, and their anti-tumor efficacy was investigated against ALL and MM samples and U937 and Raji cell lines. The results favored the improved cytotoxicity of CIK cells against primary AML blasts and U937 cell lines. However, compared to PD-1 and LAG-3, the blockade of TIM3 resulted in the highest anti-tumor characteristics. Despite increased efficacy against AML and U937, not only ALL and MM targets were resistant to CIK-mediated cytotoxicity, but the treatment with anti-immune checkpoints (D-1, TIM-3, and LAG-3) did not significantly enhance its cytotoxicity. It was also demonstrated that the expression of these receptors is increased during CIK cell culture (TIM-3 had the highest increase in expression, followed by LAG-3 and PD-1).
In another study, Li et al. [156] investigated the effect of PD-1/PD-L1 inhibitors on CIK-mediated cytotoxicity in vitro. The CIK cells were derived from PBMCs of healthy donors. They reported that the simultaneous administration of CIK cells and PD-1/PD-L1 inhibitors on the DAUDI cell line of B-NHL significantly improves CIK efficacy based on the reduced viability of DAUDI cells. However, the same therapy did not enhance the cytotoxicity of CIK cells when administered on the SU-DHL-4 cell line of B-NHL.
Although the number of studies investigating the combined efficacy of CIK cell and immune checkpoint inhibitors against hematologic malignancies is limited, several studies have reported promising results in the management of non-hematologic cancers [152, 157]. Zhang et al. [158] reported an increase in the DC-CIK effectiveness when combined with pembrolizumab, a PD-1 inhibitor, among individuals with hepatocellular carcinoma. Another study found an increase in CIK cells’ proliferation following exposure to anti-PD-1 and anti-CTLA-4 antibodies while treating patients with renal cell carcinoma [153]. In a similar study, it was demonstrated that combining CIK cells with Crizotinib (a receptor tyrosine kinase inhibitor) and Nivolumab (PD-1 inhibitor) could notably enhance the in vitro anti-tumor immune response against NSCL cell lines [159]. Taking into account the promising potential of CIK + immune checkpoint inhibitors against non-hematologic malignancies, it is essential to further investigate the efficacy of this combination therapy against hematologic malignancies in future studies.
Other combination therapies with CIK cells
Previous studies have shown that activating cannabinoid receptor 2 (CB2), which is highly expressed in the hematopoietic system, has immunomodulatory effects. Besides, cannabinoids can selectively induce apoptosis in MM cell lines and plasma cells in patients with MM through caspase activation (mainly caspase-2) without affecting regular cells [160]. A recent study [161] reported that CB2 receptors are highly expressed on both CIK and MM cells. They also highlighted that combining cannabidiol with CIK cell therapy in MM patients not only enhances the cytotoxicity of CIK cells against MM cells but also has protective effects on CIK cells. However, the protective effect on CIK cells was replaced with cytotoxic effects after increasing the cannabidiol concentration to more than one µM, indicating that combining cannabinoids with CIK therapy requires additional considerations in the future. Innovatively, Introna et al. [26] investigated the protective role of CIK therapy on the incidence of GVHD following conventional DLI therapy in patients with various hematological malignancies, including AML, ALL, MM, HL, NHL, MDS, and MPNs. Patients were administered two cycles of unmanipulated DLI (1 × 106 /kg) every 3 weeks, followed by three cycles of donor CIK therapy (1–10 × 106/kg) every 3 weeks. The incidence rate of severe GVHD was remarkably reduced in this method compared to only DLI treatment. Furthermore, the antitumor efficacy of both CIK and DLI therapy was satisfactory.
Limitations of CIK cells
CIK cell therapies present numerous advantages in the treatment of cancer patients; however, they also come with limitations. Some clinical trials applying CIK cell therapy on various malignancies have reported unsatisfactory results, especially when using CIK cells as monotherapy. However, unfavorable outcomes have been more common in the treatment of solid tumors than hematologic cancer. One explanation for such variations in the efficacy of CIK cell therapy is a tumor microenvironment with diverse immunosuppressive characteristics created by some tumors [162].
Furthermore, CIK cells consist of a heterogeneous cellular population. Different proportions of these cells within each ex vivo expanded CIK population cause a significant challenge while treating malignant tumors. It has been demonstrated that the altered proportion of CD4+ T, Treg, and CD3+CD56+ T cells in the population of CIK cells significantly impacts the efficacy of this therapeutic strategy [163]. Moreover, the rates of CD3+CD56+ subset cells, as potent effector cells of expanded CIK populations, substantially vary in a range of 6% to over 60% in different studies [67, 107].
In another study, Liu et al. [163] divided the CIK cells based on their CD4 markers. They demonstrated that CD4-CIK cells show diminished cytotoxic activity within tumor tissues compared to the general CIK population. Besides, CD4- CIK cells were accompanied by a reduced number of CD8+ T cells and a higher proportion of PD-1+Tim-3+ CD8+ T cells, which are commonly recognized as terminally exhausted T cells. They also found that the functional exhaustion of PD-1+Tim-3+ CIKs in tumor tissues of a mouse model of NSCLC can be restored through combination therapy with rIL-17A. On the other hand, CD4+ T cells, particularly Th1/Th17 cell subsets, were shown to have a greater suppressive impact on the inhibitory receptors on CIKs, improving the tumor-killing function, migration, and locomotion of these cells. A general look at these findings implies that the different rates of CD4+ cells in the CIK population may play a significant role in how patients respond to the treatment protocol. Future studies should focus on finding ways to overcome this limitation by obtaining a relatively predetermined proportion of cells after CIK induction.
Treg cells are other components of CIK cells that are considered a major drawback for the induction of immunity and cytotoxicity against cancerous cells. These cells induce CD8 + cytotoxic T lymphocyte exhaustion in the tumor microenvironment. It has been established that IL-2, an indispensable cytokine in the induction of CIK cells, has a drastic role in developing these cells [99]. Therefore, developing novel induction methods is required to mitigate the number and negative impact of Treg cells on the cytotoxicity of CIKs. However, it is worth mentioning that the availability of diverse protocols to expand CIK cells imposes another limitation to this therapeutic strategy. Various CIK expansion protocols differ in many aspects, such as the concentration and timing of stimulating elements and the type of media, making the comprehension of findings reported by different researchers difficult [164].
Conclusion
Immunotherapy is a powerful tool for treating various malignancies, especially hematologic cancers, and it has great potential to improve patients' OS and prognosis. In recent years, among different types of immunotherapies, CIK cells have gotten special attention due to their unique characteristics, including the mixed T–NK phenotype, MHC unrestricted function, expansion in vitro in both allogeneic and autologous settings, low GVHD rate, lack of severe side effects report, and high safety. Another major advantage of CIK cell therapy is its potential to be combined with other antitumor therapeutic strategies, which have shown promising results. However, additional studies are essential to increase the therapeutic efficacy of these cells in cancer patients, especially in ones with high-grade disease. Thus, the current research has focused on improving the antitumor potential of CIK cells in various malignancies, including hematologic ones.
Availability of data and materials
Not applicable.
Abbreviations
- ADCC:
-
Antibody-dependent cell‐mediated cytotoxicity
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- ASCT:
-
Autologous stem-cell transplantation
- AYA:
-
Adolescent and young adult
- BCL2:
-
B-cell lymphoma 2
- BiTEs:
-
Bispecific T-cell engagers
- bs:
-
Bispecific
- BTK:
-
Bruton tyrosine kinase
- CAR:
-
Chimeric antigen receptor
- CB:
-
Cord blood
- CB2:
-
Cannabinoid receptor 2
- CD40L:
-
CD40 ligand
- CIK:
-
Cytokine-induced killer
- CIR:
-
Cumulative incidence of relapse
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- CTL:
-
Cytotoxic T lymphocyte
- DC:
-
Dendritic cell
- DFS:
-
Disease-free survival
- DLBCL:
-
Diffuse large B cell lymphoma
- DLI:
-
Donor lymphocyte infusion
- EBV:
-
Epstein Barr virus
- FasL:
-
Fas ligand
- FDC:
-
Full donor chimerism
- FL:
-
Follicular lymphoma
- GVHD:
-
Graft versus host disease
- HIV:
-
Human immunodeficiency virus
- HL:
-
Hodgkin lymphoma
- HL:
-
Human leukemia
- HLA:
-
Human leukocyte antigen
- HSCT:
-
Hematopoietic stem cell transplantation
- HSPCs:
-
Hematopoietic stem and progenitor cells
- HTLV:
-
Human T-lymphotropic virus
- ICAM:
-
Intracellular cell adhesion molecule
- IL:
-
Interleukin
- LAK:
-
Lymphokine-activated killer cell
- LFA:
-
Lymphocyte function-associated antigen
- mAb:
-
Monoclonal antibody
- MCL:
-
Mantle cell lymphoma
- MDS:
-
Myelodysplastic syndromes
- MM:
-
Multiple myeloma
- mOS:
-
Median overall survival
- mPFS:
-
Median progression‐free survival
- MPN:
-
Myeloproliferative Neoplasm
- MRD:
-
Measurable residual disease
- MZL:
-
Marginal zone B cell lymphoma
- NHL:
-
Non‑Hodgkin lymphoma
- NK:
-
Natural killer
- NKG2D:
-
Natural killer group 2 member D
- NSCLC:
-
Non-small cell lung cancer
- NTD:
-
Non-transduced
- OBI:
-
Obinutuzumab
- OR:
-
Overall response
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PB:
-
Peripheral blood
- PBMCs:
-
Peripheral blood mononuclear cells
- PDX:
-
Patient‐derived xenograft
- Ph+:
-
Philadelphia positive
- PMF:
-
Primary myelofibrosis
- PTLD:
-
Post-transplant lymphoproliferative disorders
- PV:
-
Polycythemia vera
- rhIL:
-
Recombinant human IL
- sAML:
-
Secondary acute myeloid leukemia
- SCID:
-
Severe Combined Immunodeficiency
- SCT:
-
Stem-cell transplantation
- SLL:
-
Small lymphocytic lymphoma
- TCR:
-
T‐cell receptor
- TIL:
-
Tumor-infiltrating lymphocyte
- TLI-ATG:
-
Total lymphoid irradiation and anti-thymocyte globulin
- TME:
-
Tumor microenvironment
- t-MN:
-
Therapy-related myeloid neoplasms
- Tregs:
-
Regulatory T cells
- VLA:
-
Very late antigen
- β2-MG:
-
β2-Microglobulin
References
Pearce L. Haematological cancers. Nurs Stand. 2016;30(48):15.
Pérez GB, Calaf GM, Villalba MTM, Prieto KS, Burgos FC. Frequency of hematologic malignancies in the population of Arica. Chile Oncol Lett. 2019;18(5):5637–43.
Taylor J, Xiao W, Abdel-Wahab O. Diagnosis and classification of hematologic malignancies on the basis of genetics. Blood. 2017;130(4):410–23.
The LH. The global burden of haematological diseases. Lancet Haematol. 2018;5(1): e1.
Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3(4):524–48.
Burns R, Leal J, Sullivan R, Luengo-Fernandez R. Economic burden of malignant blood disorders across Europe: a population-based cost analysis. Lancet Haematol. 2016;3(8):e362–70.
Lehmann L, El-Haddad A, Barr RD. Global approach to hematologic malignancies. Hematol Oncol Clin N Am. 2016;30(2):417–32.
Park LS, Tate JP, Rodriguez-Barradas MC, Rimland D, Goetz MB, Gibert C, et al. Cancer incidence in HIV-infected versus uninfected veterans: comparison of cancer registry and ICD-9 code diagnoses. J AIDS Clin Res. 2014;5(7):1000318.
Savage DG, Szydlo RM, Goldman JM. Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period. Br J Haematol. 1997;96(1):111–6.
Vedsted P, Olesen F. Early diagnosis of cancer–the role of general practice. Scand J Prim Health Care. 2009;27(4):193–4.
Li J, Smith A, Crouch S, Oliver S, Roman E. Estimating the prevalence of hematological malignancies and precursor conditions using data from haematological malignancy research network (HMRN). Cancer Causes Control. 2016;27(8):1019–26.
Prakash G, Kaur A, Malhotra P, Khadwal A, Sharma P, Suri V, et al. Current role of genetics in hematologic malignancies. Indian J Hematol Blood Transfus. 2016;32(1):18–31.
Kansara RR, Speziali C. Immunotherapy in hematologic malignancies. Curr Oncol. 2020;27(Suppl 2):S124–31.
Brouckaert PGG, Fiers W. Coley’s vaccine and TNF therapy. Nature. 1992;358(6388):630.
Yang Z-Z, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Investig. 2012;122(4):1271–82.
Barclay AN, Berg TKVD. The Interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Ann Rev Immunol. 2014;32(1):25–50.
Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for relapsed/refractory classic hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol. 2018;36(14):1428–39.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–10.
Jackson GH, Davies FE, Pawlyn C, Cairns DA, Striha A, Collett C, et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019;20(1):57–73.
Facon T, Dimopoulos MA, Dispenzieri A, Catalano JV, Belch A, Cavo M, et al. Final analysis of survival outcomes in the phase 3 FIRST trial of up-front treatment for multiple myeloma. Blood. 2018;131(3):301–10.
Shimizu Y, Yoshikawa T, Kojima T, Shoda K, Nosaka K, Mizuno S, et al. Heat shock protein 105 peptide vaccine could induce antitumor immune reactions in a phase I clinical trial. Cancer Sci. 2019;110(10):3049–60.
Shah NJ, Najibi AJ, Shih TY, Mao AS, Sharda A, Scadden DT, et al. A biomaterial-based vaccine eliciting durable tumour-specific responses against acute myeloid leukaemia. Nat Biomed Eng. 2020;4(1):40–51.
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–9.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Merker M, Salzmann-Manrique E, Katzki V, Huenecke S, Bremm M, Bakhtiar S, et al. Clearance of hematologic malignancies by allogeneic cytokine-induced killer cell or donor lymphocyte infusions. Biol Blood Marrow Transplant. 2019;25(7):1281–92.
Introna M, Lussana F, Algarotti A, Gotti E, Valgardsdottir R, Micò C, et al. Phase II study of sequential infusion of donor lymphocyte infusion and cytokine-induced killer cells for patients relapsed after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2017;23(12):2070–8.
Zhang Y, Schmidt-Wolf IGH. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J Cell Physiol. 2020;235(12):9291–303.
Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–49.
Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, et al. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol. 1993;21(13):1673–9.
Guo Y, Han W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin J Cancer. 2015;34(3):1–9.
Schmidt-Wolf GD, Negrin RS, Schmidt-Wolf IG. Activated T cells and cytokine-induced CD3+CD56+ killer cells. Ann Hematol. 1997;74(2):51–6.
Jabbour EJ, Faderl S, Kantarjian HM. Adult acute lymphoblastic leukemia. Mayo Clin Proc. 2005;80(11):1517–27.
Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, et al. SEER cancer statistics review, 1975–2018. Bethesda: National Cancer Institute; 2021.
Esparza SD, Sakamoto KM. Topics in pediatric leukemia–acute lymphoblastic leukemia. MedGenMed. 2005;7(1):23.
Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017;130(19):2064–72.
Whitlock JA. Down syndrome and acute lymphoblastic leukaemia. Br J Haematol. 2006;135(5):595–602.
Siegel SE, Stock W, Johnson RH, Advani A, Muffly L, Douer D, et al. Pediatric-inspired treatment regimens for adolescents and young adults with philadelphia chromosome-negative acute lymphoblastic leukemia: a review. JAMA Oncol. 2018;4(5):725–34.
Vey N, Thomas X, Picard C, Kovascovicz T, Charin C, Cayuela JM, et al. Allogeneic stem cell transplantation improves the outcome of adults with t(1;19)/E2A-PBX1 and t(4;11)/MLL-AF4 positive B-cell acute lymphoblastic leukemia: results of the prospective multicenter LALA-94 study. Leukemia. 2006;20(12):2155–61.
DeAngelo DJ, Stevenson KE, Dahlberg SE, Silverman LB, Couban S, Supko JG, et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18–50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia. 2015;29(3):526–34.
Xiong Y, Bensoussan D, Decot V. Adoptive immunotherapies after allogeneic hematopoietic stem cell transplantation in patients with hematologic malignancies. Transfus Med Rev. 2015;29(4):259–67.
Blair A, Goulden NJ, Libri NA, Oakhill A, Pamphilon DH. Immunotherapeutic strategies in acute lymphoblastic leukaemia relapsing after stem cell transplantation. Blood Rev. 2005;19(6):289–300.
Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genßler S, Rettinger E, et al. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int J Cancer. 2016;139(8):1799–809.
Rafei H, Kantarjian HM, Jabbour EJ. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma. 2019;60(11):2606–21.
Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010.
Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–4.
Einsele H, Borghaei H, Orlowski RZ, Subklewe M, Roboz GJ, Zugmaier G, et al. The BiTE (bispecific T-cell engager) platform: development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer. 2020;126(14):3192–201.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. 2019;25(4):603–11.
Noh JY, Seo H, Lee J, Jung H. Immunotherapy in hematologic malignancies: emerging therapies and novel approaches. Int J Mol Sci. 2020;21(21):8000.
Liao D, Wang M, Liao Y, Li J, Niu T. A review of efficacy and safety of checkpoint inhibitor for the treatment of acute myeloid leukemia. Front Pharmacol. 2019;10:609.
Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood J Am Soc Hematol. 2012;119(26):6198–208.
Jordan C, Upchurch D, Szilvassy S, Guzman M, Howard D, Pettigrew A, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–84.
Linn YC, Lau LC, Hui KM. Generation of cytokine-induced killer cells from leukaemic samples with in vitro cytotoxicity against autologous and allogeneic leukaemic blasts. Br J Haematol. 2002;116(1):78–86.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Smith A, Howell D, Patmore R, Jack A, Roman E. Incidence of haematological malignancy by sub-type: a report from the haematological malignancy research network. Br J Cancer. 2011;105(11):1684–92.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–60.
Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–74.
Freeman CL, Gribben JG. Immunotherapy in chronic lymphocytic leukaemia (CLL). Curr Hematol Malig Rep. 2016;11(1):29–36.
Kornacker M, Moldenhauer G, Herbst M, Weilguni E, Tita-Nwa F, Harter C, et al. Cytokine-induced killer cells against autologous CLL: direct cytotoxic effects and induction of immune accessory molecules by interferon-gamma. Int J Cancer. 2006;119(6):1377–82.
Wiedmeier-Nutor J, Leis J. Chronic lymphocytic leukemia: chemotherapy free and other novel therapies including CAR T. Curr Treat Options Oncol. 2022;23(6):904–19.
Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15.
Hussein K, Bock O, Seegers A, Flasshove M, Henneke F, Buesche G, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood. 2007;109(9):4106–7.
Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17(1):49.
Valent P, Sadovnik I, Eisenwort G, Bauer K, Herrmann H, Gleixner KV, et al. Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML. Int J Mol Sci. 2019;20(17):4233.
Scheffold C, Brandt K, Johnston V, Lefterova P, Degen B, Schöntube M, et al. Potential of autologous immunologic effector cells for bone marrow purging in patients with chronic myeloid leukemia. Bone Marrow Transplant. 1995;15(1):33–9.
Linn YC, Yong HX, Niam M, Lim TJ, Chu S, Choong A, et al. A phase I/II clinical trial of autologous cytokine-induced killer cells as adjuvant immunotherapy for acute and chronic myeloid leukemia in clinical remission. Cytotherapy. 2012;14(7):851–9.
Hoyle C, Bangs CD, Chang P, Kamel O, Mehta B, Negrin RS. Expansion of Philadelphia chromosome-negative CD3(+)CD56(+) cytotoxic cells from chronic myeloid leukemia patients: in vitro and in vivo efficacy in severe combined immunodeficiency disease mice. Blood. 1998;92(9):3318–27.
Grewal R, Irimie A, Naidoo N, Mohamed N, Petrushev B, Chetty M, et al. Hodgkin’s lymphoma and its association with EBV and HIV infection. Crit Rev Clin Lab Sci. 2018;55(2):102–14.
Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. 2018;131(1):68–83.
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18(6):1611–8.
Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14(8):517–34.
Perry AM, Diebold J, Nathwani BN, MacLennan KA, Müller-Hermelink HK, Bast M, et al. Non-Hodgkin lymphoma in the developing world: review of 4539 cases from the International non-Hodgkin lymphoma classification project. Haematologica. 2016;101(10):1244–50.
Karabon L, Partyka A, Ciszak L, Pawlak-Adamska E, Tomkiewicz A, Bojarska-Junak A, et al. Abnormal expression of BTLA and CTLA-4 immune checkpoint molecules in chronic lymphocytic leukemia patients. J Immunol Res. 2020;2020:6545921.
Maruhashi T, Okazaki I-M, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol. 2018;19(12):1415–26.
Roemer MGM, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34(23):2690–7.
Tun AM, Ansell SM. Immunotherapy in Hodgkin and non-Hodgkin lymphoma: innate, adaptive and targeted immunological strategies. Cancer Treat Rev. 2020;88:102042.
Herrera AF, Moskowitz AJ, Bartlett NL, Vose JM, Ramchandren R, Feldman TA, et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood J Am Soc Hematol. 2018;131(11):1183–94.
Viardot A, Goebeler M-E, Hess G, Neumann S, Pfreundschuh M, Adrian N, et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016;127(11):1410–6.
Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos M-V, et al. Multiple myeloma. Nat Rev Dis Primers. 2017;3(1):17046.
Mateos M-V, Orlowski RZ, Ocio EM, Rodríguez-Otero P, Reece D, Moreau P, et al. Pembrolizumab combined with lenalidomide and low-dose dexamethasone for relapsed or refractory multiple myeloma: phase I KEYNOTE-023 study. Br J Haematol. 2019;186(5):e117–21.
Zhao X, Ji C-Y, Liu G-Q, Ma D-X, Ding H-F, Xu M, et al. Immunomodulatory effect of DC/CIK combined with chemotherapy in multiple myeloma and the clinical efficacy. Int J Clin Exp Pathol. 2015;8(10):13146.
Quach H, Ritchie D, Stewart A, Neeson P, Harrison S, Smyth M, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32.
Wang M, Yin B, Wang HY, Wang RF. Current advances in T-cell-based cancer immunotherapy. Immunotherapy. 2014;6(12):1265–78.
Cappuzzello E, Sommaggio R, Zanovello P, Rosato A. Cytokines for the induction of antitumor effectors: the paradigm of cytokine-Induced Killer (CIK) cells. Cytokine Growth Factor Rev. 2017;36:99–105.
Rohaan MW, Wilgenhof S, Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch. 2019;474(4):449–61.
Shahrabi S, Zayeri ZD, Ansari N, Hadad EH, Rajaei E. Flip-flops of natural killer cells in autoimmune diseases versus cancers: immunologic axis. J Cell Physiol. 2019;234(10):16998–7010.
Franceschetti M, Pievani A, Borleri G, Vago L, Fleischhauer K, Golay J, et al. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp Hematol. 2009;37(5):616-28.e2.
Linn YC, Lau SK, Liu BH, Ng LH, Yong HX, Hui KM. Characterization of the recognition and functional heterogeneity exhibited by cytokine-induced killer cell subsets against acute myeloid leukaemia target cell. Immunology. 2009;126(3):423–35.
Introna M, Franceschetti M, Ciocca A, Borleri G, Conti E, Golay J, et al. Rapid and massive expansion of cord blood-derived cytokine-induced killer cells: an innovative proposal for the treatment of leukemia relapse after cord blood transplantation. Bone Marrow Transplant. 2006;38(9):621–7.
Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IG. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol. 2015;141(5):839–49.
Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci USA. 1999;96(12):6879–84.
Verneris MR, Karami M, Baker J, Jayaswal A, Negrin RS. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103(8):3065–72.
Lin G, Wang J, Lao X, Wang J, Li L, Li S, et al. Interleukin-6 inhibits regulatory T cells and improves the proliferation and cytotoxic activity of cytokine-induced killer cells. J Immunother. 2012;35(4):337–43.
Zoll B, Lefterova P, Ebert O, Huhn D, Von Ruecker A, Schmidt-Wolf IG. Modulation of cell surface markers on NK-like T lymphocytes by using IL-2, IL-7 or IL-12 in vitro stimulation. Cytokine. 2000;12(9):1385–90.
Rettinger E, Kuçi S, Naumann I, Becker P, Kreyenberg H, Anzaghe M, et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy. 2012;14(1):91–103.
Finke S, Trojaneck B, Lefterova P, Csipai M, Wagner E, Kircheis R, et al. Increase of proliferation rate and enhancement of antitumor cytotoxicity of expanded human CD3+ CD56+ immunologic effector cells by receptor-mediated transfection with the interleukin-7 gene. Gene Ther. 1998;5(1):31–9.
Pfirrmann V, Oelsner S, Rettinger E, Huenecke S, Bonig H, Merker M, et al. Cytomegalovirus-specific cytokine-induced killer cells: concurrent targeting of leukemia and cytomegalovirus. Cytotherapy. 2015;17(8):1139–51.
Tao Q, Chen T, Tao L, Wang H, Pan Y, Xiong S, et al. IL-15 improves the cytotoxicity of cytokine-induced killer cells against leukemia cells by upregulating CD3+ CD56+ cells and downregulating regulatory T cells as well as IL-35. J Immunother. 2013;36(9):462–7.
Zhao N, Zhao M, Rajbhandary S, Lu W, Zhu H, Xiao X, et al. Effects of humanized interleukin 21 on anti-leukemic activity of cytokine induced killer cells and the mechanism. Zhonghua xue ye xue za zhi= Zhonghua xueyexue zazhi. 2012;33(10):823–8.
Meng M, Li L, Li R, Wang W, Chen Y, Xie Y, et al. A dynamic transcriptomic atlas of cytokine-induced killer cells. J Biol Chem. 2018;293(51):19600–12.
Houchins JP, Yabe T, McSherry C, Bach FH. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med. 1991;173(4):1017–20.
Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002;17(1):19–29.
Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+ CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood J Am Soc Hematol. 2011;118(12):3301–10.
Cappuzzello E, Tosi A, Zanovello P, Sommaggio R, Rosato A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology. 2016;5(8):e1199311.
Rettinger E, Huenecke S, Bonig H, Merker M, Jarisch A, Soerensen J, et al. Interleukin-15-activated cytokine-induced killer cells may sustain remission in leukemia patients after allogeneic stem cell transplantation: feasibility, safety and first insights on efficacy. Haematologica. 2016;101(4):e153–6.
Leemhuis T, Wells S, Scheffold C, Edinger M, Negrin RS. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. Biol Blood Marrow Transplant. 2005;11(3):181–7.
Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148(7):1383-91.e6.
Zhao H, Wang Y, Yu J, Wei F, Cao S, Zhang X, et al. Autologous cytokine-induced killer cells improves overall survival of metastatic colorectal cancer patients: results from a phase II clinical trial. Clin Colorectal Cancer. 2016;15(3):228–35.
Chen D, Sha H, Hu T, Dong S, Zhang J, Liu S, et al. Cytokine-induced killer cells as a feasible adoptive immunotherapy for the treatment of lung cancer. Cell Death Dis. 2018;9(3):366.
Wang Z, Liu Y, Re Li, Shang Y, Zhang Y, Zhao L, et al. Autologous cytokine-induced killer cell transfusion increases overall survival in advanced pancreatic cancer. J Hematol Oncol. 2016;9:1–7.
Zhou Y, Chen C-l, Jiang S-W, Feng Y, Yuan L, Chen P, et al. Retrospective analysis of the efficacy of adjuvant CIK cell therapy in epithelial ovarian cancer patients who received postoperative chemotherapy. Oncoimmunology. 2019;8(2):e1528411.
Li X, Zhou H, Huang W, Wang X, Meng M, Hou Z, et al. Retrospective analysis of the efficacy of adjuvant cytokine-induced killer cell immunotherapy combined with chemotherapy in colorectal cancer patients after surgery. Clin Transl Immunol. 2022;11(1):e1368.
Linn YC, Niam M, Chu S, Choong A, Yong HX, Heng KK, et al. The anti-tumour activity of allogeneic cytokine-induced killer cells in patients who relapse after allogeneic transplant for haematological malignancies. Bone Marrow Transplant. 2012;47(7):957–66.
Martino I, Gianmaria B, Elena C, Marta F, Anna Maria B, Raewyn B, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study. Haematologica. 2007;92(7):952–9.
Narayan R, Benjamin JE, Shah O, Tian L, Tate K, Armstrong R, et al. Donor-derived cytokine-induced killer cell infusion as consolidation after nonmyeloablative allogeneic transplantation for myeloid neoplasms. Biol Blood Marrow Transplant. 2019;25(7):1293–303.
Rasche L, Kapp M, Einsele H, Mielke S. EBV-induced post transplant lymphoproliferative disorders: a persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant. 2014;49(2):163–7.
van Esser JWJ, van der Holt B, Meijer E, Niesters HGM, Trenschel R, Thijsen SFT, et al. Epstein-Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV-lymphoproliferative disease following T-cell–depleted SCT. Blood. 2001;98(4):972–8.
Styczynski J, Gil L, Tridello G, Ljungman P, Donnelly JP, van der Velden W, et al. Response to rituximab-based therapy and risk factor analysis in Epstein Barr virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: a study from the infectious diseases working party of the european group for blood and marrow transplantation. Clin Infect Dis. 2013;57(6):794–802.
Pfeffermann L-M, Pfirrmann V, Huenecke S, Bremm M, Bonig H, Kvasnicka H-M, et al. Epstein-Barr virus–specific cytokine-induced killer cells for treatment of Epstein-Barr virus–related malignant lymphoma. Cytotherapy. 2018;20(6):839–50.
Wang H, Cao F, Li J, Li Y, Liu X, Wang L, et al. Homing of cytokine-induced killer cells during the treatment of acute promyelocytic leukemia. Int J Hematol. 2014;100(2):165–70.
Zhou M, Wang J, Li C-P, Xu J-Y, Chen B. Autologous cytokine-induced killer cell immunotherapy for patients with high-risk diffuse large B cell lymphoma after the first complete remission. Onco Targets Ther. 2020;13:5879.
Circosta P, Donini C, Gallo S, Giraudo L, Gammaitoni L, Rotolo R, et al. Full chimaeric CAR.CIK from patients engrafted after allogeneic haematopoietic cell transplant: feasibility, anti-leukaemic potential and alloreactivity across major human leukocyte antigen barriers. Br J Haematol. 2023;200(1):64–9.
Leuci V, Donini C, Grignani G, Rotolo R, Mesiano G, Fiorino E, et al. CSPG4-Specific CAR.CIK lymphocytes as a novel therapy for the treatment of multiple soft-tissue sarcoma histotypes. Clin Cancer Res. 2020;26(23):6321–34.
Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. 2020;130(11):6021–33.
Zuo S, Wen Y, Panha H, Dai G, Wang L, Ren X, et al. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol Immunol. 2017;85:293–304.
Marin V, Dander E, Biagi E, Introna M, Fazio G, Biondi A, et al. Characterization of in vitro migratory properties of anti-CD19 chimeric receptor-redirected CIK cells for their potential use in B-ALL immunotherapy. Exp Hematol. 2006;34(9):1219–29.
Magnani CF, Mezzanotte C, Cappuzzello C, Bardini M, Tettamanti S, Fazio G, et al. Preclinical efficacy and safety of CD19CAR cytokine-induced killer cells transfected with sleeping beauty transposon for the treatment of acute lymphoblastic leukemia. Hum Gene Ther. 2018;29(5):602–13.
Rotiroti MC, Buracchi C, Arcangeli S, Galimberti S, Valsecchi MG, Perriello VM, et al. Targeting CD33 in chemoresistant AML patient-derived xenografts by CAR-CIK cells modified with an improved SB transposon system. Mol Ther. 2020;28(9):1974–86.
Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. 2017;32(4):506-19.e5.
Alberti G, Arsuffi C, Pievani A, Salerno D, Mantegazza F, Dazzi F, et al. Engineering tandem CD33xCD146 CAR CIK (cytokine-induced killer) cells to target the acute myeloid leukemia niche. Front Immunol. 2023;14:1192333.
Ladikou EE, Chevassut T, Pepper CJ, Pepper AG. Dissecting the role of the CXCL12/CXCR4 axis in acute myeloid leukaemia. Br J Haematol. 2020;189(5):815–25.
Zou Y, Li F, Hou W, Sampath P, Zhang Y, Thorne SH. Manipulating the expression of chemokine receptors enhances delivery and activity of cytokine-induced killer cells. Br J Cancer. 2014;110(8):1992–9.
Zou Y, Liang J, Li D, Fang J, Wang L, Wang J, et al. Application of the chemokine-chemokine receptor axis increases the tumor-targeted migration ability of cytokine-induced killer cells in patients with colorectal cancer. Oncol Lett. 2020;20(1):123–34.
Biondi M, Tettamanti S, Galimberti S, Cerina B, Tomasoni C, Piazza R, et al. Selective homing of CAR-CIK cells to the bone marrow niche enhances control of the acute myeloid leukemia burden. Blood. 2023;141(21):2587–98.
Boeck CL, Amberger DC, Doraneh-Gard F, Sutanto W, Guenther T, Schmohl J, et al. Significance of frequencies, compositions, and/or antileukemic activity of (DC-stimulated) invariant NKT, NK and CIK cells on the outcome of patients with AML, ALL and CLL. J Immunother. 2017;40(6):224–48.
Cao J, Chen C, Wang Y, Chen X, Chen Z, Luo X. Influence of autologous dendritic cells on cytokine-induced killer cell proliferation, cell phenotype and antitumor activity in vitro. Oncol Lett. 2016;12(3):2033–7.
Märten A, Ziske C, Schöttker B, Renoth S, Weineck S, Buttgereit P, et al. Interactions between dendritic cells and cytokine-induced killer cells lead to an activation of both populations. J Immunother. 2001;24(6):502–10.
Pan Y, Tao Q, Wang H, Xiong S, Zhang R, Chen T, et al. Dendritic cells decreased the concomitant expanded Tregs and Tregs related IL-35 in cytokine-induced killer cells and increased their cytotoxicity against leukemia cells. PLoS ONE. 2014;9(4):e93591.
Zhang X, Yang J, Zhang G, Song L, Su Y, Shi Y, et al. 5 years of clinical DC-CIK/NK cells immunotherapy for acute myeloid leukemia—a summary. Immunotherapy. 2020;12(1):63–74.
Xiao X, Ye X, Xu C, Huang J. Successful alternative treatment for relapsed adult acute lymphoblastic leukemia with dendritic cells-cytokine-induced killer cells combined with a rituximab-based regimen. OncoTargets Ther. 2018;11:7555–8.
Dong M, Zhang G, Meng J, Liu B, Jiang D, Liu F. MMP9-associated tumor stem cells, CCL1-silenced dendritic cells, and cytokine-induced killer cells have a remarkable therapeutic efficacy for acute myeloid leukemia by activating T cells. Stem Cells Int. 2023;2023:2490943.
Pievani A, Belussi C, Klein C, Rambaldi A, Golay J, Introna M. Enhanced killing of human B-cell lymphoma targets by combined use of cytokine-induced killer cell (CIK) cultures and anti-CD20 antibodies. Blood. 2011;117(2):510–8.
Esser L, Weiher H, Schmidt-Wolf I. Increased efficacy of brentuximab vedotin (SGN-35) in combination with cytokine-induced killer cells in lymphoma. Int J Mol Sci. 2016;17(7):1056.
Dalla Pietà A, Cappuzzello E, Palmerini P, Ventura A, Visentin A, Astori G, et al. Innovative therapeutic strategy for B-cell malignancies that combines obinutuzumab and cytokine-induced killer cells. J Immunother Cancer. 2021;9(7):e002475.
Frank MJ, Olsson N, Huang A, Tang SW, Negrin RS, Elias JE, et al. A novel antibody-cell conjugation method to enhance and characterize cytokine-induced killer cells. Cytotherapy. 2020;22(3):135–43.
Interdonato A, Choblet S, Sana M, Valgardsdottir R, Cribioli S, Alzani R, et al. BL-01, an Fc-bearing, tetravalent CD20 × CD5 bispecific antibody, redirects multiple immune cells to kill tumors in vitro and in vivo. Cytotherapy. 2022;24(2):161–71.
Golay J, Martinelli S, Alzani R, Cribioli S, Albanese C, Gotti E, et al. Cord blood–derived cytokine-induced killer cells combined with blinatumomab as a therapeutic strategy for CD19+ tumors. Cytotherapy. 2018;20(8):1077–88.
Tita-Nwa F, Moldenhauer G, Herbst M, Kleist C, Ho AD, Kornacker M. Cytokine-induced killer cells targeted by the novel bispecific antibody CD19xCD5 (HD37xT5.16) efficiently lyse B-lymphoma cells. Cancer Immunol Immunother. 2007;56(12):1911–20.
Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. 2016;13(1):25–40.
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21(1):28.
Liu S, Meng Y, Liu L, Lv Y, Wei F, Yu W, et al. Rational pemetrexed combined with CIK therapy plus anti-PD-1 mAbs administration sequence will effectively promote the efficacy of CIK therapy in non-small cell lung cancer. Cancer Gene Ther. 2023;30(2):277–87.
Dehno MN, Li Y, Weiher H, Schmidt-Wolf IGH. Increase in efficacy of checkpoint inhibition by cytokine-induced-killer cells as a combination immunotherapy for renal cancer. Int J Mol Sci. 2020;21(9):3078.
Chen J, Chen Y, Feng F, Chen C, Zeng H, Wen S, et al. Programmed cell death protein-1/programmed death-ligand 1 blockade enhances the antitumor efficacy of adoptive cell therapy against non-small cell lung cancer. J Thorac Dis. 2018;10(12):6711–21.
Poh SL, Linn YC. Immune checkpoint inhibitors enhance cytotoxicity of cytokine-induced killer cells against human myeloid leukaemic blasts. Cancer Immunol Immunother. 2016;65(5):525–36.
Li Y, Sharma A, Bloemendal MW, Schmidt-Wolf R, Kornek M, Schmidt-Wolf IG. PD-1 blockade enhances cytokine-induced killer cell-mediated cytotoxicity in B-cell non-Hodgkin lymphoma cell lines. Oncol Lett. 2021;22(2):1–6.
Lv Y, Zhao H, Liu S, Meng Y, Yu W, Liu T, et al. Anlotinib and anti-PD-1 mAbs perfected CIK cell therapy for lung adenocarcinoma in preclinical trials. J Leukoc Biol. 2024. https://doi.org/10.1093/jleuko/qiae037.
Zhang W, Song Z, Xiao J, Liu X, Luo Y, Yang Z, et al. Blocking the PD-1/PD-L1 axis in dendritic cell-stimulated cytokine-induced killer cells with pembrolizumab enhances their therapeutic effects against hepatocellular carcinoma. J Cancer. 2019;10(11):2578–87.
Li Y, Sharma A, Wu X, Weiher H, Skowasch D, Essler M, et al. A combination of cytokine-induced killer cells with PD-1 blockade and ALK inhibitor showed substantial intrinsic variability across non-small cell lung cancer cell lines. Front Oncol. 2022;12:713476.
Barbado MV, Medrano M, Caballero-Velázquez T, Álvarez-Laderas I, Sánchez-Abarca LI, García-Guerrero E, et al. Cannabinoid derivatives exert a potent anti-myeloma activity both in vitro and in vivo. Int J Cancer. 2017;140(3):674–85.
Garofano F, Schmidt-Wolf IGH. High expression of cannabinoid receptor 2 on cytokine-induced killer cells and multiple myeloma cells. Int J Mol Sci. 2020;21(11):3800.
Schmidt TL, Negrin RS, Contag CH. A killer choice for cancer immunotherapy. Immunol Res. 2014;58(2–3):300–6.
Liu S, Meng Y, Liu L, Lv Y, Yu W, Liu T, et al. CD4(+) T cells are required to improve the efficacy of CIK therapy in non-small cell lung cancer. Cell Death Dis. 2022;13(5):441.
Cappuzzello E, Vigolo E, D’Accardio G, Astori G, Rosato A, Sommaggio R. How can cytokine-induced killer cells overcome CAR-T cell limits. Front Immunol. 2023;14:1229540.
Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J, et al. Dual-functional capability of CD3+ CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118(12):3301–10.
Acknowledgements
Not applicable
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Morteza Raisi and Amir Mehdizadeh contributed to the study’s conception. The first draft of the manuscript was written by Faezeh Ghanbari Sevari and Seyed Sina Hejazian; all authors commented on previous versions of the manuscript. Figures were designed by Khadijeh Abbasi. The final revision and editing of the manuscript were conducted by Faezeh Ghanbari Sevari and Seyed Sina Hejazian. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghanbari Sevari, F., Mehdizadeh, A., Abbasi, K. et al. Cytokine-induced killer cells: new insights for therapy of hematologic malignancies. Stem Cell Res Ther 15, 254 (2024). https://doi.org/10.1186/s13287-024-03869-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-024-03869-z