
Abstract
Background
Gaucher disease is one of the most common lipid-storage disorders, affecting approximately 1 in 75,000 births. Enzyme replacement therapy with recombinant glucocerebrosidase is currently considered the first-line treatment choice for patients with symptomatic Gaucher disease type 1. Oral substrate reduction therapy is generally considered a second-line treatment option for adult patients with mild to moderate Gaucher disease type 1 who are unable or unwilling to receive lifelong intravenous enzyme infusions. The efficacy and safety of the oral substrate reduction therapy miglustat (Zavesca®) in patients with Gaucher disease type 1 have been established in both short-term clinical trials and long-term, open-label extension studies. Published data indicate that miglustat can be used as maintenance therapy in patients with stable Gaucher disease type 1 switched from previous enzyme replacement therapy.
Case presentation
We report a case of a 44-year-old Caucasian man with Gaucher disease type 1 who was initially treated with enzyme replacement therapy but, owing to repeated cutaneous allergic reactions, had to be switched to miglustat after several attempts with enzyme replacement therapy. Despite many attempts, desensitization treatment did not result in improved toleration of imiglucerase infusions, and the patient became unwilling to continue with any intravenous enzyme replacement therapy. He subsequently agreed to switch to oral substrate reduction therapy with miglustat 100 mg twice daily titrated up to 100 mg three times daily over a short period. Long-term miglustat treatment maintained both hemoglobin and platelet levels within acceptable ranges over 8 years. The patient’s spleen volume decreased, his plasma chitotriosidase levels stayed at reduced levels, and his bone mineral density findings have remained stable throughout follow-up. The patient’s quality of life has remained satisfactory. Miglustat showed good gastrointestinal tolerability in this patient, and no adverse events have been reported.
Conclusions
Oral miglustat therapy proved to be a valid alternative treatment to intravenous enzyme replacement therapy for long-term maintenance in this patient with Gaucher disease type 1, who showed persistent allergic intolerance to imiglucerase infusions. This report exemplifies the type of patient with Gaucher disease type 1 who can benefit from switching from enzyme replacement therapy to substrate reduction therapy.
Similar content being viewed by others

Background
Gaucher disease (GD) is an autosomal recessive inherited lysosomal storage disease caused by a deficiency of the enzyme β-glucocerebrosidase (also called β-glucosidase [GBA]), which catalyzes the degradation of the sphingolipid glucocerebroside (glucosylceramide) into ceramide and glucose [1–3]. This deficiency, caused by mutations on chromosome 1q21 [4], leads to the accumulation of glucocerebroside in the lysosomes of macrophages that collect in the liver, spleen, bone marrow, and skeleton [5]. Subsequent progressive visceral involvement has a large impact on both quality of life and life expectancy.
Traditionally, on the basis of the presence or absence of neurological symptoms and signs, GD is categorized into three subtypes [6, 7]. The most common form, Gaucher disease type 1 (GD1), is generally considered to be nonneuronopathic in nature and is characterized by hepatosplenomegaly, hematological abnormalities (anemia and thrombocytopenia), and skeletal manifestations such as osteopenia, fragility fractures, and bone pain. Primarily neuronopathic forms are less common. GD type 2 is an acute neuronopathic variant associated with early-onset and rapidly progressive neurological deterioration as well as visceral manifestations. GD type 3 is a chronic neuronopathic form characterized by later onset and more protracted neurological and visceral involvement. However, a number of published reports indicate that a significant proportion of patients with GD1 have neurological abnormalities [8–10]. The different types of GD are therefore perhaps better viewed as occurring across a phenotypic continuum [11, 12]. In light of the weight of published evidence, it seems appropriate to drop the terms type 2 and type 3 GD and more representative to apply the terms acute neuronopathic and chronic neuronopathic GD [13].
Enzyme replacement therapy (ERT) with recombinant glucocerebrosidase is currently considered the first-line treatment choice for patients with symptomatic GD1, providing proven beneficial effects on hematological and visceral manifestations and improvements in some skeletal indices [14, 15]. Oral substrate reduction therapy (SRT) is generally considered a second-line treatment option for adult patients with mild to moderate GD1 who are unable or unwilling to receive lifelong intravenous enzyme infusions [16].
The iminosugar miglustat (N-butyldeoxynojirimycin, Zavesca®; Actelion Pharmaceuticals, Allschwil, Switzerland) is an inhibitor of glucosylceramide synthase (ceramide-specific glucosyltransferase), which is the enzyme that catalyzes the first committed step in the biosynthesis of glycosphingolipids [17]. The efficacy and safety of miglustat in patients with GD1 have been established in both short-term clinical trials and long-term, open-label extension studies [16, 18, 19]. Further studies have indicated that miglustat can be used as a maintenance therapy in patients with stable GD1 switched from previous ERT [20].
We report a case of a man with GD1 who was initially treated with ERT but, owing to repeated cutaneous allergic reactions, had to be switched to miglustat after several attempts with ERT. The patient’s clinical details are discussed in relation to the safety profile of miglustat.
Case presentation
A 44-year-old Caucasian man was admitted to our department in March 2002 with thrombocytopenia and leukocytosis. The same signs had appeared 2 years before in another hospital, without a definitive diagnosis. At admission, he had a platelet count of 123,000/mm3 and a white blood cell count of 8750/mm3. Physical and neurological examinations did not reveal any physical symptoms.
The patient’s clinical history revealed a spontaneous right lung collapse in 1987 (treated by drainage for 1 week) and an abdominal cardiac valve defect with gastroesophageal reflux since 1989. Liver and spleen hemangioma were detected by abdominal ultrasound. A bone marrow biopsy showed 70 % cellularity, with approximately half of the cell population comprising well-differentiated histiocytes. The cell cytoplasm was distended and closely packed with fine granules staining blue with a May-Grünwald-Giemsa preparation, and centrally located nuclei in some cells were displaced toward the periphery in others. These features were consistent with sea-blue histiocytes.
During follow-up, the patient’s initial hemoglobin (Hb) concentration and platelet count were 15.4 g/dl and 154,000/mm3, respectively, and his plasma chitotriosidase activity was 2150 nmol/h/ml. In May 2002, a fluorometric assay showed a β-glucosidase activity of 0.30 nmol/h/mg (range 1.7–4.3 nmol/h/mg), which is compatible with GD. This diagnosis was confirmed by molecular genetic testing, showing homozygous N370S mutations. At this point, the patient’s spleen volume (calculated on the basis of polar diameter) was 12.5 cm3. In addition, bone mineral density (BMD) measurement by dual-energy X-ray absorptiometry revealed severe osteoporosis (T-score greater than −2.5).
In April 2003, several months after admission, ERT with imiglucerase (Cerezyme®; Genzyme, Cambridge, MA, USA) was started with a dosage of 60 IU/kg of body weight every other week. Approximately 1 year later (March 2004), the patient underwent upper-right lobectomy with mediastinal lymphadenectomy for treatment of lung cancer (clear-cell adenocarcinoma). In follow-up approximately 2.5 years later (December 2005), a clear improvement in BMD (T-score −1 and less than or equal to −2.5) (Table 1) and a substantial (> 70 %) decrease in plasma chitotriosidase to 616 nmol/h/ml were observed. The patient’s standard hematological parameters remained stable (Hb 15.4 g/dl, platelet count 126,000/mm3). In July 2004, a cutaneous allergic reaction appeared on the patient’s abdomen and shoulder during the ninth infusion of imiglucerase, which led to stoppage of the infusion and subsequent treatment with methylprednisolone 20 mg. Similar adverse reactions occurred at the 10th and 11th infusions. Thereafter the patient was prepared 30 minutes before starting ERT with intravenous chlorpheniramine maleate 5 mg to prevent allergic manifestations. No further infusion reactions occurred up to the 20th administration, at which point the patient developed urticarial, pruritus, diarrhea, and chest discomfort.
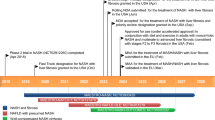
The patient became unwilling to continue with any intravenous ERT because of his recurrent allergic manifestations, which were poorly prevented by antihistamine therapy. However, he later agreed to switch to oral therapy with miglustat (Zavesca®, Actelion Pharmaceuticals), which was started at a dosage of 100 mg twice daily and titrated up to 100 mg three times daily over a short period according to the manufacturer’s instructions [21]. He has since remained on miglustat therapy with good compliance and has not reported any adverse events. His core hematological parameters and further BMD findings have remained stable throughout follow-up (Fig. 1a–c). His spleen volume decreased from 12.0 cm3 at miglustat initiation to 11.1 cm3 in December 2015, after a total of 12 years of ERT and SRT. The patient has never had hepatosplenomegaly, and his liver and renal parameters were always normal before, during, and after ERT. To date, no bisphosphonate therapy for osteoporosis has ever been applied. Last follow-up was in December 2015.

Hematological parameters during initial enzyme replacement therapy and subsequent miglustat treatment. Black, white, and blue triangles represent start of enzyme replacement therapy, cessation of enzyme replacement therapy, and start of miglustat therapy. a) platelet count during treatment: Black triangle represent start of enzyme replacement therapy, white triangle represent cessation of enzyme replacement therapy and Blue triangle represent start of miglustat therapy. b) hemoglobin concentration during treatment: black triangle represent start of enzyme replacement therapy, white triangle represent cessation of enzyme replacement therapy and blue triangle represent start of miglustat therapy. c) plasma chitotriosidase activity: Black triangle represent start of enzyme replacement therapy, white triangle represent cessation of enzyme replacement therapy and blue triangle represent start of miglustat therapy
Discussion
GD is one of the most common lipid-storage disorders, affecting approximately 1 in 75,000 births [22]. The homozygous N370S mutation on the chromosome 1q21 is the most common genotype observed in affected patients. Our patient, who was homozygous for N370S, was treated in accordance with current treatment recommendations and other literature reports.
The standard of care for the treatment of the systemic, nonneuronopathic manifestations of GD is ERT with a recombinant analogue of human GBA. Intravenous ERT infusions for GD have been shown to improve hematological parameters (increased Hb concentrations and platelet counts), reduce organomegaly, and decrease bone involvement [14, 15]. In most cases, the therapeutic response to ERT reaches a plateau within the first 2–5 years [14, 15].
The response of our patient to ERT was initially good, including improvements in Hb and platelets, decreased bone involvement, and reduced chitotriosidase levels that were in line with findings reported in the literature [14, 15]. However, repeated infusion reactions curtailed the continued use of ERT in this case. The development of immunoglobulin G antibodies in response to intravenous infusions has been reported in approximately 15 % of ERT-treated patients, with many of those affected experiencing a range of immune reactions, including pruritus, flushing, tachycardia, and hypotension [23–26]. These side effects are usually mild [26, 27], allowing continuation of therapy after slowing of infusion rates or the use of pharmacological support with corticosteroids and antihistamines. In patients who respond poorly to these treatments, a desensitization protocol has successfully been implemented in both pediatric and adult patients [28, 29]. Unfortunately, despite many attempts, premedication with methylprednisolone and chlorpheniramine maleate did not result in improved toleration of imiglucerase infusions in our patient. ERT was therefore stopped with the patient’s consent, and it was decided that miglustat therapy should be commenced.
Miglustat acts as a competitive inhibitor of glucosylceramide synthase, thereby decreasing the synthesis and accumulation of glucosylceramide and related glycosphingolipids in reticuloendothelial cell lysosomes [30]. The efficacy of miglustat has previously been demonstrated over the short term and long term in patients with GD regarding both reduced visceral involvement (sustained reductions in both liver and spleen volumes) and improved hematological parameters [16, 18, 19, 31, 32]. Data also indicate that miglustat is effective as maintenance therapy in patients with stable GD switched from previous ERT [33–35]. In particular, this agent has shown beneficial effects on BMD indices in cortical and trabecular bone, bone marrow burden scores, and bone pain, which are possibly related to its physicochemical properties that enable a wide distribution throughout body tissues [32, 36, 37]. Published data derived from a long-term safety surveillance program for miglustat have demonstrated its safety in patients with GD treated for up to 9 years [38, 39].
Overall, the long-term effects of miglustat in this case are in agreement with previous findings. Both Hb and platelet count remained within acceptable ranges throughout 8 years of follow-up on miglustat since the patient was switched from previous ERT. His spleen volume decreased, and his plasma chitotriosidase levels were maintained at reduced levels. The patient’s quality of life has remained satisfactory.
It is well known on the basis of early clinical trials in patients with GD that miglustat can lead to gastrointestinal disturbances, mainly during the initial weeks or months of treatment [40]. This is due to reversible inhibition by orally administered miglustat of intestinal disaccharidases, including sucrose and maltase, and to a lesser extent lactase [40, 41]. As a result of this activity, the amount of undigested oligosaccharides in the gastrointestinal lumen increases, which leads to osmotic diarrhea and related symptoms [40]. However, in recent years, the management of these side effects in miglustat-treated patients has improved. In particular, simple dietary modifications such as restriction of dietary disaccharide intake, ideally commenced before initiation of treatment, has proved useful in reducing gastrointestinal side effects and in maintaining body-weight gain within normal limits in pediatric patients [40, 42, 43]. Notably, probiotics rich in Saccharomyces boulardii, combined with miglustat dose escalation, have been reported to confer further possible benefits in this respect [44]. We suggested restriction of disaccharides for our patient, and we observed acceptable gastrointestinal tolerability based on the patient’s self-reports.
Conclusions
We report a case of a patient with GD who underwent long-term SRT treatment with miglustat. He showed improvements in visceral and hematological parameters during initial ERT, but he had to stop ERT because of an unacceptable level of adverse infusion reactions. After he was switched to miglustat, his core disease parameters, particularly bone indices, remained stable or improved, and his quality of life was maintained. In terms of tolerability, dietary modifications and general patient education regarding gastrointestinal disturbances that can occur during initial miglustat treatment appeared to result in acceptable compliance with treatment. While reports of previous open-label trials have indicated that miglustat can, in general, act as a maintenance therapy in patients switched from previous intravenous treatment with ERT, this case provides a specific example of the type of patient with GD1 in whom switching to oral SRT can be of particular benefit.
Abbreviations
- BMD:
-
Bone mineral density
- ERT:
-
Enzyme replacement therapy
- GBA:
-
β-glucocerebrosidase
- GD:
-
Gaucher disease
- GD1:
-
Gaucher disease type 1
- Hb:
-
Hemoglobin
- SRT:
-
Substrate reduction therapy
References
Wanders JA, Poorthuis BJHM, Rodenburg RJT. Enzymes, metabolic pathways, flux control analysis, and the enzymology of specific groups of inherited metabolic diseases. In: Hoffmann GF, Zschocke J, Nyhan WL, editors. Inherited metabolic diseases: a clinical approach. Berlin: Springer-Verlag; 2010. p. 283–303.
Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005;129:178–88.
Nilsson O, Svennerholm L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J Neurochem. 1982;39:709–18.
Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66:1777–86.
Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A. Gaucher’s disease. Lancet. 2001;358:324–7.
Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Pediatr Health Med Ther. 2011;2:59–73.
Chen M, Wang J. Gaucher disease: review of the literature. Arch Pathol Lab Med. 2008;132:851–3.
Cherin P, Rose C, de Roux-Serratrice C, Tardy D, Dobbelaere D, Grosbois B, et al. The neurological manifestations of Gaucher disease type 1: the French Observatoire on Gaucher disease (FROG). J Inherit Metab Dis. 2010;33:331–8.
Biegstraaten M, van Schaik IN, Aerts JM, Hollak CE. Non-neuronopathic’ Gaucher disease reconsidered: prevalence of neurological manifestations in a Dutch cohort of type I Gaucher disease patients and a systematic review of the literature. J Inherit Metab Dis. 2008;31:337–49.
Pastores GM, Barnett NL, Bathan P, Kolodny EH. A neurological symptom survey of patients with type I Gaucher disease. J Inherit Metab Dis. 2003;26:641–5.
Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr. 2003;143:273–6.
Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83:6–15.
Vellodi A, Tylki-Szymanska A, Davies EH, Kolodny E, Bembi B, Collin-Histed T, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009;32:660–4.
Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–9.
Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-Cheng A, Zimran A, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22:119–26.
Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–5.
Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269:8362–5.
Heitner R, Elstein D, Aerts J, Weely S, Zimran A. Low-dose N-butyldeoxynojirimycin (OGT 918) for type I Gaucher disease. Blood Cells Mol Dis. 2002;28:127–33.
Elstein D, Hollak C, Aerts JM, van Weely S, Maas M, Cox TM, et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J Inherit Metab Dis. 2004;27:757–66.
Cox TM, Amato D, Hollak CE, Luzy C, Silkey M, Giorgino R, et al. Evaluation of miglustat as maintenance therapy after enzyme therapy in adults with stable type 1 Gaucher disease: a prospective, open-label non-inferiority study. Orphanet J Rare Dis. 2012;7:102.
Zavesca (miglustat). Summary of product characteristics 2016. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000435/human_med_001171.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001d125. Accessed 17 June 2016.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–54.
Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C, Weinreb NJ, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev. 2015;3:CD010324.
Aviner S, Levy Y, Yaniv I, Cohen IJ. Anaphylactoid reaction to imiglucerase, but not to alglucerase, in a type I Gaucher patient. Blood Cells Mol Dis. 1999;25:92–4.
Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122:33–9.
Starzyk K, Richards S, Yee J, Smith SE, Kingma W. The long-term international safety experience of imiglucerase therapy for Gaucher disease. Mol Genet Metab. 2007;90:157–63.
Elstein D, Zimran A. Review of the safety and efficacy of imiglucerase treatment of Gaucher disease. Biologics. 2009;3:407–17.
Erdogdu D, Gelincik A, Canbaz B, Colakoglu B, Buyukozturk S, Tanakol R. Successful desensitization to imiglucerase of an adult patient diagnosed with type I Gaucher disease. Int Arch Allerg Immunol. 2013;160:215–7.
Peroni DG, Pescollderungg L, Piacentini GL, Cassar W, Boner AL. Effective desensitization to imiglucerase in a patient with type I Gaucher disease. J Pediatr. 2009;155:940–1.
Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. Substrate reduction therapy of glycosphingolipid storage disorders. J Inherit Metab Dis. 2006;29:449–56.
Giraldo P, Latre P, Alfonso P, Acedo A, Alonso D, Barez A, et al. Short-term effect of miglustat in every day clinical use in treatment-naive or previously treated patients with type 1 Gaucher’s disease. Haematologica. 2006;91:703–6.
Giraldo P, Alfonso P, Atutxa K, Fernandez-Galan MA, Barez A, Franco R, et al. Real-world clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL project. Haematologica. 2009;94:1771–5.
Kuter DJ, Mehta A, Hollak CE, Giraldo P, Hughes D, Belmatoug N, et al. Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study. Blood Cells Mol Dis. 2013;51:116–24.
Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 2007;110:2296–301.
Serratrice C, Swiader L, Serratrice J. Switching from imiglucerase to miglustat for the treatment of French patients with Gaucher disease type 1: a case series. J Med Case Rep. 2015;9:146.
Treiber A, Morand O, Clozel M. The pharmacokinetics and tissue distribution of the glucosylceramide synthase inhibitor miglustat in the rat. Xenobiotica. 2007;37:298–314.
Pastores GM, Elstein D, Hrebicek M, Zimran A. Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin Ther. 2007;29:1645–54.
Hollak CE, Hughes D, van Schaik IN, Schwierin B, Bembi B. Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf. 2009;18:770–7.
Brand M, Muller A, Alsop J, van Schaik IN, Bembi B, Hughes D. Results from a 9-year Intensive Safety Surveillance Scheme (IS3) in miglustat (Zavesca®)-treated patients. Pharmacoepidemiol Drug Saf. 2015;24:329–33.
Belmatoug N, Burlina A, Giraldo P, Hendriksz CJ, Kuter DJ, Mengel E, et al. Gastrointestinal disturbances and their management in miglustat-treated patients. J Inherit Metab Dis. 2011;34:991–1001.
Amiri M, Naim HY. Miglustat-induced intestinal carbohydrate malabsorption is due to the inhibition of α-glucosidases, but not β-galactosidases. J Inherit Metab Dis. 2012;35:949–54.
Déchelotte P. Type 1 Gaucher’s disease in the adult. Nutritional management during initiation of treatment with miglustat [in French]. Presse Med. 2004;33:494–6.
Champion H, Ramaswami U, Imrie J, Lachmann RH, Gallagher J, Cox TM, et al. Dietary modifications in patients receiving miglustat. J Inherit Metab Dis. 2010;33 Suppl 3:S379–83.
Remenova T, Morand O, Amato D, Chadha-Boreham H, Tsurutani S, Marquardt T. A double-blind, randomized, placebo-controlled trial studying the effects of Saccharomyces boulardii on the gastrointestinal tolerability, safety, and pharmacokinetics of miglustat. Orphanet J Rare Dis. 2015;10:81.
Acknowledgements
Matthew Reilly, PhD, at InTouch Medical Ltd provided editorial assistance in the preparation of this manuscript for submission, paid for by Actelion Pharmaceuticals Ltd.
Funding
No funding was received for the conduct of this case analysis. Actelion Pharmaceuticals Ltd paid for editorial assistance for the preparation of this manuscript.
Availability of data and materials
Not applicable.
Authors’ contributions
GG coordinated the specialist clinical care of the patient, wrote the first draft of the manuscript, and was involved at all stages of writing the manuscript. RL, EDF, LP, AF and FDR contributed to the clinical assessments and reviewed and provided critical input into the manuscript content. All authors read and approved the final manuscript.
Competing interests
We received support from Actelion Pharmaceuticals to pay for manuscript preparation.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Ethics approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the 1975 Declaration of Helsinki as revised in 2000.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Giuffrida, G., Lombardo, R., Di Francesco, E. et al. Successful switch from enzyme replacement therapy to miglustat in an adult patient with type 1 Gaucher disease: a case report. J Med Case Reports 10, 315 (2016). https://doi.org/10.1186/s13256-016-1060-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-016-1060-y