
Abstract
Previously, the contribution of peripheral infection to cognitive decline was largely overlooked however, the past 15 years have established a key role for infectious pathogens in the progression of age-related neurodegeneration. It is now accepted that the immune privilege of the brain is not absolute, and that cells of the central nervous system are sensitive to both the inflammatory events occurring in the periphery and to the infiltration of peripheral immune cells. This is particularly relevant for the progression of Alzheimer’s disease, in which it has been demonstrated that patients are more vulnerable to infection-related cognitive changes. This can occur from typical infectious challenges such as respiratory tract infections, although a number of specific viral, bacterial, and fungal pathogens have also been associated with the development of the disease. To date, it is not clear whether these microorganisms are directly related to Alzheimer’s disease progression or if they are opportune pathogens that easily colonize those with dementia and exacerbate the ongoing inflammation observed in these individuals. This review will discuss the impact of each of these challenges, and examine the changes known to occur with age in the peripheral immune system, which may contribute to the age-related vulnerability to infection-induced cognitive decline.
Similar content being viewed by others

Background
It has been estimated that by the year 2050 the population of individuals over the age of 60 will double from 901 million in 2015 to 2.1 billion people worldwide [1]. Importantly this increase in life expectancy will go hand-in-hand with an increase in age-related diseases, with the elderly currently expected to spend more of their later years in overall ill-health [2]. Indeed dementia, one of the principle causes of disability in the elderly, currently affects 44 million people globally, with this figure expected to increase to over 135 million people by the year 2050 [3]. As the annual cost of dementia care is expected to increase from $600 billion to $1 trillion over the next 15 years [3], finding a way to prevent disease progression is vital. This review will summarize the impact of inflammation on the progression of neurodegeneration, with a focus on the role of infection-related neuroinflammation in dementia, which is a rapidly growing area of interest in the field.
Review
Infection in the elderly and in Alzheimer’s disease
The immune system undergoes many changes with age that leaves the elderly more susceptible to infection [4], indeed older individuals are more vulnerable to bacterial or viral infections of the urinary or respiratory tract, with influenza-related morbidity also increased in this group [5, 6]. Sepsis, which is caused by severe infection, can also lead to permanent cognitive dysfunction, particularly in older individuals [7]. Importantly, infectious burden in the elderly is associated with mini-mental state examination (MMSE) scores below 24, which indicate dementia [8]. This is in line with a previous study that linked infection with lower MMSE scores, however Hodgson and colleagues also observed that 36% of elderly subjects with dementia had an infection which was formerly undiagnosed [9]. Unfortunately, the symptoms of infection often present atypically in this group [10] and, as dementia patients are often unable to communicate their symptoms [11], diagnosis is difficult. To further complicate matters, bacterial resistance is often increased in older patients [12].
Individuals with Alzheimer’s disease (AD) are even more vulnerable to the effects of peripheral infection. In a 10-year follow-up study, delirium (which is often caused by infection) correlated with an eightfold increase in dementia development [13]. Furthermore, the cognitive capabilities of AD patients worsened significantly after an episode of delirium, which has been confirmed by others [14]. Indeed poor health [15] and viral burden [16] have been linked with cognitive impairment and AD development in the elderly. Natalwala and colleagues found that the incidence of many infectious conditions such as pneumonia, lower respiratory tract, or urinary tract infections is higher in AD patients than healthy, age-matched controls [17]. Previous studies have demonstrated that numerous infections over a 4-year period doubled the risk of AD development [18]. Indeed cognitive decline has been observed just 2 or 6 months after a resolved peripheral infection, with an association between cognitive impairment and circulating proinflammatory cytokines [19, 20]. Pneumonia is a frequent, if not the most common, cause of death in AD [21, 22], conversely, vaccination against influenza and other infections significantly reduced the risk of AD development [23, 24] and antibiotic treatment has been observed to slow cognitive decline in patients [25]. Many specific viral, bacterial, and fungal pathogens are suspected to play a role in the progression of neurodegeneration including herpes simplex virus type 1 (HSV-1), Chlamydia pneumonia, spirochetes, and Candida [26,27,28] thus, the contribution of each pathogenic group will be examined in detail.
Viral infections
Chronic infection with HSV-1 and cytomegalovirus (CMV) has been implicated in neurodegeneration. HSV-1 is typically a lifelong, latent infection of the central nervous system (CNS) and, while the virus has been found in the brains of control and AD subjects, viral deoxyribonucleic acid (DNA) was located in regions such as the hippocampus, which are particularly affected in AD [29]. HSV-1 is a risk factor for AD in people carrying the apolipoprotein E epsilon 4 (APOE4) allele, indeed the allele frequency is much higher in the HSV-1-infected than non-infected AD population [28, 30]. In vitro studies have demonstrated that HSV-1 triggers amyloid precursor protein (APP) processing, resulting in the production of amyloid β (Aβ) via β- and γ-secretases [31], and murine studies have shown that APOE4-expressing mice have a significantly enhanced viral burden after infection with HSV-1 [32]. It is believed that HSV-1 outcompetes ApoE4 in binding to heparan sulfate proteoglycans (HSPG) on the cell surface, thus facilitating viral internalization and infection of the host cell in APOE4 carriers in particular [26].
CMV is another lifelong, latent infection that, along with HSV-1, was associated with lower MMSE scores in the elderly [16]. In a 5-year follow-up study, CMV was linked with faster cognitive decline and development of AD [33], which supports two other reports that found an association between CMV seropositivity and AD development [34, 35]. Interestingly, Westman and colleagues observed that the peripheral blood mononuclear cells (PBMCs) from CMV+ AD patients were more reactive after stimulation than non-infected patients, suggesting that CMV is an inflammatory promoter in AD [36].
Bacterial infections
A number of bacterial pathogens have also been associated with the development of AD. Chlamydia pneumonia is an obligate intracellular, Gram-negative bacteria that was first observed in the postmortem AD brain by Balin and colleagues in 1998 [37], although the finding has been replicated many times since [38, 39]. Infection with C. pneumonia is associated with a fivefold increase in AD development [40], and AD patients have increased levels of C. pneumonia-specific antibodies in circulation in comparison with control subjects [34]. It is believed that the bacteria can cross the blood-brain barrier (BBB) via the olfactory route or within infected monocytes [41]. Once inside the CNS, C. pneumonia can infect microglia, astrocytes, and neurons. Importantly, Gérard and colleagues observed infected cells containing viable, metabolically active pathogens in close proximity to AD-plaque pathology [39]. Similar to HSV-1, AD patients with the APOE4 allele are more susceptible to infection with C. pneumonia as a significantly greater bacterial burden was observed in regions such as the hippocampus in comparison to those without APOE4 [38]. C. pneumonia can inhibit neuronal apoptosis in vitro, thus facilitating the maintenance of a chronic infection [42]. Interestingly, intranasal infection of mice with C. pneumonia induced Aβ deposition in the brain, which co-localized with reactive glia [43], importantly C. pneumonia remains active in the murine CNS for months after infection [44].
Heliobacter pylori is a Gram-negative bacteria that grows in the digestive tract and was recently demonstrated to have a significant association with the development of dementia [45]. In older individuals, the presence of H. pylori IgG antibodies was associated with decreased cognitive performance [46], indeed research has shown that AD patients also have increased H. pylori seropositivity in the serum and cerebrospinal fluid (CSF) [47]. Furthermore, Kountouras and colleagues have demonstrated that individuals with AD had an increased incidence of H. pylori infection of the gastric mucosa compared to controls [48]. Within AD patients, those infected with H. pylori had more severe dementia, characterized by lower MMSE scores, with increased proinflammatory cytokines and tau levels in the CSF [49]. A 20-year follow-up study also found H. pylori to be a significant risk factor for the development of dementia, although even at baseline those positive for H. pylori had lower MMSE scores [50]. It has been reported that elimination of H. pylori infection reduced the mortality rate of AD patients when examined 5 years later [51]. Furthermore, AD patients who were treated for their infection and remained H. pylori-free for 2 years had improved cognition than when they were first examined, while those who were still positive for the bacterium had further declined [52]. Animal studies have demonstrated that intraperitoneal injection of H. pylori filtrate into rats increased the concentration of Aβ42 in the cortex and hippocampus, which was associated with memory deficits and impaired spatial learning [53]. This group also reported that H. pylori filtrate significantly increased tau phosphorylation in neuronal cultures in vitro and in the rat hippocampus in vivo [54]. Conversely, infection of C57BL/6 J mice with H. pylori did not affect amyloid deposition when assessed 18 months later [55], however the effect of H. pylori on amyloid pathology in AD-transgenic mice has yet to be examined.
Periodontitis is another a risk factor for AD and it has been demonstrated that healthy elderly individuals with periodontal disease have a higher accumulation of amyloid in the CNS [56], with an association found between elevated interleukin (IL)-6 and tumor necrosis factor (TNF)α in the circulation and periodontitis in AD patients [57]. A common cause of periodontitis is spirochete infection, which is a Gram-negative, neurotropic bacterium. The periodontal spirochete pathogen Treponema has been detected in the AD brain, with co-infection of multiple Treponema species observed in some patients [58]. Many other species of periodontal pathogens have been found in the AD brain including lipopolysaccharide (LPS) from P. gingivalis [59] and Borrelia burgdorferi [58, 60, 61]. Importantly, B. burgdorferi co-localised with Aβ deposits in patients [61] and was found to induce Aβ deposition by glial and neuronal cells in vitro [62]. Indeed, it has recently been suggested that bacterial amyloid, along with host-derived Aβ, are constituents of the senile plaques observed in AD [63]. A number of studies have found that significantly more AD patients have a spirochete infection when examined in the post-mortem brain, than controls [58, 60]. In addition, AD patients have increased levels of B. burgdorferi-specific antibodies in circulation [34], and a recent study demonstrated a tenfold increase in the occurrence of AD with spirochete infection [40].
Fungal infections
There have been a number of reports over the past 3 years on the contribution of fungal infections to the progression of AD. In 2014, Alonso and colleagues first demonstrated the presence of fungal proteins and DNA in the AD brain [64]. Many different species were detected including Saccharomyces cerevisiae, Malassezia globosa, Malassezia restricta, and Penicillium. Analysis of CSF samples from patients also revealed the presence of S. cerevisiae, M. globose, and M. restricta DNA, while the CSF levels of Candida albicans and C. glabrata proteins were significantly greater in those with AD [65]. In both studies it was observed that many AD patients were co-infected with a number of fungal species, while no fungal DNA was detected in control samples. In line with this finding, AD patients also have greater seropositivity to C. albicans and C. glabrata [66]. Immunohistochemical analysis identified fungal material inside neuronal cells in the postmortem AD brain, including macromolecules from Candida glabrata, Penicillium notatum, and C. albicans [67]. Furthermore, this group have found fungal material both intra- and extra-cellularly, and in many brain regions including the frontal cortex, hippocampus, and the blood vessels of the CNS, with mixed-fungal infections observed in multiple patients [27]. It has yet to be established whether the fungal infection co-localises with Aβ or if the infection has a direct or indirect effect on amyloid production in the CNS.
Interestingly, it has been suggested that Aβ may also function as an antimicrobial peptide (AMP). In vitro studies have confirmed that Aβ has antimicrobial activity against a range of pathogens and was as effective, or even more potent, than LL-37 which is an established human AMP [68]. Importantly, C. albicans was the microbe most sensitive to synthetic Aβ, and brain homogenates from AD patients, but not controls, were also capable of inhibiting fungal growth. It was recently demonstrated that Aβ protects against C. albicans infection in glial cells in vitro and in nematodes in vivo [69]. In addition, Aβ inhibits HSV-1 viral replication in vitro, and protects mice from Salmonella Typhimurium infection in vivo, which led the authors of both studies to conclude that Aβ may have a previously unknown protective role in innate immunity, along with the pathogenic characteristics that are extremely well studied [69, 70].
Why are the elderly more susceptible to these infections?
The emerging evidence strongly indicates that infection has a significant role in the development of, and progression to, dementia, with a growing list of pathogens specifically associated with AD or Aβ deposition. This may be due in part to some of the changes that are known to occur to the immune system with age. One of the key changes in the adaptive immune system is the involution of the thymus, resulting in a dramatic decrease in the production of new T cells [71]. With age, there is an overall decrease in naive T cells, and a corresponding increase in memory T cells [72]. This is associated with a reduction in naive T cell diversity after the age of 65 [71], with clonally expanded subsets of memory T cells often observed in this age group, which can occur from chronic or repeat infections [4, 72]. Together, this can limit the capacity of the individual to induce a sufficient immune response to new infectious challenges. In adults, the remaining pool of naive T cells is maintained via proliferation [73], however over their life-span, these naive cells can be exposed to stressors such as oxidative species or changes in the availability of cell-survival factors, which can affect their function [74].
Interestingly, many studies have demonstrated that T cell activation is even further altered in AD. The population of naive T cells is significantly decreased in AD patients, with an increase in memory T cells in comparison with age-matched, healthy controls [75,76,77]. In addition, it has been reported that AD patients have T cells with shorter telomeres, and telomere length significantly correlated with AD severity [78]. AD patients have increased T cell reactivity to Aβ [79, 80], and the phenotype of the T cells in circulation is shifted, with increased CD4+IFN-γ+ and CD8+IFN-γ+ T cells observed [81, 82]. Saresella and colleagues also reported an Aβ-specific population of Th17 and Th9 cells that was increased in AD patients in comparison to healthy control subjects [75].
These changes can have a critical impact on the CNS, as activated T cells have been found in the CSF of AD patients [83, 84] and these cells have been reported in the human brain, with greater numbers reported in the brains of AD patients [85, 86]. Importantly, T cells were found in close association with microglia [86], which are known to have an antigen-presenting phenotype in AD [85, 87, 88]. Animal studies have demonstrated the presence of IFN-γ+ Th1 cells and IL-17+ Th17 cells in the CNS of aged APP/PS1 mice [89, 90]. In addition, respiratory infection can have a significant effect on the phenotype of T cells in the CNS [90]. It was observed that peripheral infection increased the deposition of Aβ in the brain, which was associated with increased T cell infiltration and microglial activation in older, but not younger, APP/PS1 mice.
It has also been reported that the innate immune system undergoes changes with age and in AD. Monocytes prepared from individuals with discrepant memory IQ had increased expression of CD11b, Toll-like receptor (TLR)2, and TLR4 [91]. Indeed the population of circulating myeloid dendritic cells (DCs) is decreased in the elderly [92] and these cells are further reduced in AD [93]. AD patients had increased levels of ICAM-1+ monocyte-derived DCs [94] and increased expression of MHC class II and CD16 on CD14+ monocytes [95]. Saresella and colleagues also found a significant increase in IL-6- and IL-23-producing CD14+ monocyte/macrophages in AD patients, while IL-10+ cells were reduced [75]. Furthermore, it was recently demonstrated that circulating NLRP3+caspase 1+ and NLRP3+caspase 8+ monocytes are increased in AD, and these cells produced significantly greater levels of IL-1β and IL-18 after LPS and Aβ treatment [96]. This is in line with our previous work demonstrating an important role for the NLRP3 and caspase-1 pathway in the progression of AD pathology, both in AD patients and APP/PS1 mice [97]. Myeloid cells from memory-impaired individuals also have a greater stimulus-induced proinflammatory response [91], which mirrors animal studies demonstrating the same effect in bone marrow-derived macrophages from APP/PS1 mice [98]. However, it has been found that DCs from AD patients have a reduced ability to stimulate T cell proliferation [94]. The innate immune system provides the first line of defense against infectious agents, thus an altered response here can have severe consequences for the individual and their ability to control, and respond to, infection. As peripheral myeloid cells have been detected in the AD brain [99], changes in the phenotype of these circulating cells suggest that those which have infiltrated the CNS are similarly altered.
Conclusion
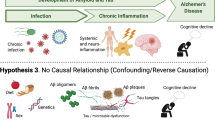
It is clear from the evidence that AD patients are more vulnerable to the effects of peripheral infection than their age-matched, healthy counterparts. Importantly, it is indisputable that many specific viral, bacterial, and fungal infections are associated with AD development, although whether these pathogens are a direct cause of dementia or instead are advantageous, infiltrating microorganisms that exacerbate the neuroinflammation already ongoing in these individuals remains to be confirmed. Importantly, the BBB of AD patients is significantly leakier than in healthy subjects, which facilitates infiltration of peripheral immune cells [100] and possibly these infectious pathogens (Fig. 1). Together, this review demonstrates the critical need for early detection and treatment of infections in the elderly and in those with dementia. As infectious diseases can present atypically in this group, frequent screening and vaccination are key to preventing infection-related deterioration of cognition until new therapies are established that can protect the elderly from these unnecessary insults.

The impact of infection on Alzheimer’s disease pathology. Healthy aging is accompanied by increased blood-brain barrier (BBB) permeability and an elevation in baseline inflammation however, Alzheimer’s disease (AD) is characterized by a significant increase in microglia activation, amyloid β (Aβ) deposition, BBB disruption and neuronal loss, far beyond that observed in non-demented, age-matched controls. It is established that microglia produce a range of cytokines in response to the growing presence of Aβ, however, with the increased number of T cells in the AD brain that have the capacity to interact with microglia, this can lead to elevated cytokine production. In addition, pathogens have been found in the AD brain, indeed many species were in close association with Aβ plaques. The presence of these microorganisms can exacerbate the ongoing neuroinflammation, thus negatively affecting nearby neuronal and glial cells leading to neurodegeneration. The situation is further confounded by inflammatory events occurring in the periphery, such as respiratory infection. This can result in increased immune cells and cytokines in circulation, which would have little influence on the healthy CNS, but the inflamed AD brain cannot efficiently handle this extra challenge. As the innate and adaptive immune cells of AD patients have altered reactivity, together they have the potential to further affect the BBB and exacerbate inflammatory changes within the CNS. This is an original figure, which was designed for this manuscript
Abbreviations
- AD:
-
Alzheimer’s disease
- AMP:
-
Antimicrobial peptide
- APOE4:
-
Apolipoprotein E epsilon 4
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid β
- BBB:
-
Blood-brain barrier
- CMV:
-
Cytomegalovirus
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DC:
-
Dendritic cell
- DNA:
-
Deoxyribonucleic acid
- HSPG:
-
Heparan sulfate proteoglycans
- HSV-1:
-
Herpes simplex virus type 1
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MMSE:
-
Mini-mental state examination
- NLRP3:
-
NOD-, LRR- and pyrin domain-containing 3
- PBMC:
-
Peripheral blood mononuclear cell
- TLR:
-
Toll-like receptor
- TNF:
-
Tumour necrosis factor
References
United Nations, D.o.E.a.S.A., Population Division, World Population Ageing 2015. 2015: (ST/ESA/SER.A/390).
Westendorp RG. What is healthy aging in the 21st century? Am J Clin Nutr. 2006;83(2):404S–9S.
Prince M, et al. World Alzheimer Report 2014: dementia and risk reduction: an analysis of protective and modifiable factors. London: Alzheimer’s Disease International (ADI); 2014.
Kovaiou RD, Herndler-Brandstetter D, Grubeck-Loebenstein B. Age-related changes in immunity: implications for vaccination in the elderly. Expert Rev Mol Med. 2007;9(3):1–17.
Castle SC. Clinical relevance of age-related immune dysfunction. Clin Infect Dis. 2000;31(2):578–85.
Engelhart ST, et al. Prospective surveillance for healthcare-associated infections in German nursing home residents. J Hosp Infect. 2005;60(1):46–50.
Widmann CN, Heneka MT. Long-term cerebral consequences of sepsis. Lancet Neurol. 2014;13(6):630–6.
Katan M, et al. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology. 2013;80(13):1209–15.
Hodgson NA, et al. Undiagnosed illness and neuropsychiatric behaviors in community residing older adults with dementia. Alzheimer Dis Assoc Disord. 2011;25(2):109–15.
Crossley KB, Peterson PK. Infections in the elderly. Clin Infect Dis. 1996;22(2):209–15.
McCloskey RM. Caring for patients with dementia in an acute care environment. Geriatr Nurs. 2004;25(3):139–44.
Linhares I, et al. Frequency and antimicrobial resistance patterns of bacteria implicated in community urinary tract infections: a ten-year surveillance study (2000–2009). BMC Infect Dis. 2013;13:19.
Davis DH, et al. Delirium is a strong risk factor for dementia in the oldest-old: a population-based cohort study. Brain. 2012;135(Pt 9):2809–16.
Fong TG, et al. Delirium accelerates cognitive decline in Alzheimer disease. Neurology. 2009;72(18):1570–5.
Tilvis RS, et al. Predictors of cognitive decline and mortality of aged people over a 10-year period. J Gerontol A Biol Sci Med Sci. 2004;59(3):268–74.
Strandberg TE, et al. Cognitive impairment and infectious burden in the elderly. Arch Gerontol Geriatr Suppl. 2004;9:419–23.
Natalwala A, et al. Reasons for hospital admissions in dementia patients in Birmingham, UK, during 2002–2007. Dement Geriatr Cogn Disord. 2008;26(6):499–505.
Dunn N, et al. Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord. 2005;19(2):91–4.
Holmes C, et al. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74(6):788–9.
Holmes C, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–74.
Foley NC, Affoo RH, Martin RE. A systematic review and meta-analysis examining pneumonia-associated mortality in dementia. Dement Geriatr Cogn Disord. 2015;39(1–2):52–67.
Magaki S, et al. Comorbidity in dementia: update of an ongoing autopsy study. J Am Geriatr Soc. 2014;62(9):1722–8.
Tyas SL, et al. Risk factors for Alzheimer’s disease: a population-based, longitudinal study in Manitoba,Canada. Int J Epidemiol. 2001;30(3):590–7.
Verreault R, et al. Past exposure to vaccines and subsequent risk of Alzheimer’s disease. CMAJ. 2001;165(11):1495–8.
Loeb MB, et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc. 2004;52(3):381–7.
Itzhaki RF, et al. Infiltration of the brain by pathogens causes Alzheimer’s disease. Neurobiol Aging. 2004;25(5):619–27.
Pisa D, et al. Different brain regions are infected with fungi in alzheimer’s disease. Scientific Reports, Published online: 15 October 2015. doi:10.1038/srep15015.
Honjo K, van Reekum R, Verhoeff NP. Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 2009;5(4):348–60.
Jamieson GA, et al. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. 1992;167(4):365–8.
Itzhaki RF, et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349(9047):241–4.
Piacentini R, et al. HSV-1 promotes Ca2 + -mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol Aging. 2011;32(12):2323.e13-26.
Burgos JS, et al. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;80(11):5383–7.
Barnes LL, et al. Cytomegalovirus infection and risk of alzheimer disease in older black and white individuals. J Infect Dis. 2015;211(2):230–37.
Bu XL, et al. A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol. 2015;22(12):1519–25.
Carbone I, et al. Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol Aging. 2014;35(1):122–9.
Westman G, et al. Increased inflammatory response in cytomegalovirus seropositive patients with Alzheimer’s disease. PLoS One. 2014;9(5):e96779.
Balin BJ, et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187(1):23–42.
Gérard HC, et al. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog. 2005;39(1-2):19–26.
Gérard HC, et al. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol. 2006;48(3):355–66.
Maheshwari P, Eslick GD. Bacterial infection and Alzheimer’s disease: a meta-analysis. J Alzheimers Dis. 2015;43(3):957–66.
Harris SA, Harris EA. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2015;48(2):319–53.
Appelt DM, et al. Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci. 2008;9:13.
Little CS, et al. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25(4):419–29.
Little CS, et al. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front Aging Neurosci. 2014;6:304.
Shindler-Itskovitch T, et al. A systematic review and meta-analysis of the association between Helicobacter pylori infection and dementia. J Alzheimers Dis. 2016;52(4):1431–42.
Beydoun MA, et al. Helicobacter pylori seropositivity and cognitive performance among US adults: evidence from a large national survey. Psychosom Med. 2013;75(5):486–96.
Kountouras J, et al. Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease. Int J Neurosci. 2009;119(6):765–77.
Kountouras J, et al. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;66(6):938–40.
Roubaud-Baudron C, et al. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: preliminary results. Neurobiol Aging. 2012;33(5):1009.e11–9.
Roubaud Baudron C, et al. Does Helicobacter pylori infection increase incidence of dementia? The Personnes Agées QUID Study. J Am Geriatr Soc. 2013;61(1):74–8.
Kountouras J, et al. Five-year survival after Helicobacter pylori eradication in Alzheimer disease patients. Cogn Behav Neurol. 2010;23(3):199–204.
Kountouras J, et al. Eradication of Helicobacter pylori may be beneficial in the management of Alzheimer’s disease. J Neurol. 2009;256(5):758–67.
Wang XL, et al. Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases β-amyloid by enhancing expression of presenilin-2. Front Aging Neurosci. 2014;6:66.
Wang XL, et al. Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3beta. J Alzheimers Dis. 2015;43(1):153–65.
Roubaud-Baudron C, et al. An eighteen-month helicobacter infection does not induce amyloid plaques or neuroinflammation in brains of wild type C57BL/6J mice. J Alzheimers Dis. 2015;45(4):1045–50.
Kamer AR, et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging. 2015;36(2):627–33.
Cestari JA, et al. Oral infections and cytokine levels in patients with Alzheimer’s Disease and mild cognitive impairment compared with controls. J Alzheimers Dis. 2016;52(4):1479–85.
Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;17(2):113–8.
Poole S, et al. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–77.
Miklossy J. Alzheimer’s disease—a spirochetosis? Neuroreport. 1993;4(7):841–8.
Miklossy J, et al. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimers Dis. 2004;6(6):639–49. discussion 673–81.
Miklossy J, et al. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging. 2006;27(2):228–36.
Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis. 2016;53(4):1459–73.
Alonso R, et al. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis. 2014;41(1):301–11.
Alonso R, et al. Cerebrospinal fluid from Alzheimer’s disease patients contains fungal proteins and DNA. J Alzheimers Dis. 2015;47(4):873–6.
Alonso R, et al. Alzheimer’s disease and disseminated mycoses. Eur J Clin Microbiol Infect Dis. 2014;33(7):1125–32.
Pisa D, et al. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis. 2015;43(2):613–24.
Soscia SJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505.
Kumar DKV, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. 2016.
Bourgade K, et al. Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis. 2016;50(4):1227–41.
Naylor K, et al. The influence of age on T cell generation and TCR diversity. J Immunol. 2005;174(11):7446–52.
Pawelec G, et al. T cells and aging, January 2002 update. Front Biosci. 2002;7:d1056–183.
Mackall CL, Gress RE. Thymic aging and T-cell regeneration. Immunol Rev. 1997;160:91–102.
Haynes L, Swain SL. Why aging T cells fail: implications for vaccination. Immunity. 2006;24(6):663–6.
Saresella M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav Immun. 2011;25(3):539–47.
Larbi A, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103.
Schindowski K, et al. Increased T-cell reactivity and elevated levels of CD8+ memory T-cells in Alzheimer’s disease-patients and T-cell hyporeactivity in an Alzheimer’s disease-mouse model: implications for immunotherapy. Neuromolecular Med. 2007;9(4):340–54.
Panossian LA, et al. Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiol Aging. 2003;24(1):77–84.
Monsonego A, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112(3):415–22.
Saresella M, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;21(3):927–38.
Fiala M, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7(3):221–32. discussion 255-62.
Baglio F, et al. Neuroinflammation and brain functional disconnection in Alzheimer’s disease. Front Aging Neurosci. 2013;5:81.
Lueg G, et al. Clinical relevance of specific T-cell activation in the blood and cerebrospinal fluid of patients with mild Alzheimer’s disease. Neurobiol Aging. 2015;36(1):81–9.
Monson NL, et al. Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab. 2014;34(1):30–3.
McGeer PL, et al. Immune system response in Alzheimer’s disease. Can J Neurol Sci. 1989;16(4 Suppl):516–27.
Togo T, et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol. 2002;124(1-2):83–92.
McGeer PL, et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79(1–2):195–200.
Togo T, et al. Expression of CD40 in the brain of Alzheimer’s disease and other neurological diseases. Brain Res. 2000;885(1):117–21.
Browne TC, et al. IFN-γ Production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol. 2013;190(5):2241–51.
McManus RM, et al. Respiratory infection promotes T cell infiltration and amyloid-beta deposition in APP/PS1 mice. Neurobiol Aging. 2014;35(1):109–21.
Downer EJ, et al. Identifying early inflammatory changes in monocyte-derived macrophages from a population with IQ-discrepant episodic memory. PLoS One. 2013;8(5):e63194.
Della Bella S, et al. Peripheral blood dendritic cells and monocytes are differently regulated in the elderly. Clin Immunol. 2007;122(2):220–8.
Ciaramella A, et al. Myeloid dendritic cells are decreased in peripheral blood of Alzheimer’s disease patients in association with disease progression and severity of depressive symptoms. J Neuroinflammation. 2016;13:18.
Ciaramella A, et al. Increased pro-inflammatory response by dendritic cells from patients with Alzheimer’s disease. J Alzheimers Dis. 2010;19(2):559–72.
Zhang R, et al. Systemic immune system alterations in early stages of Alzheimer’s disease. J Neuroimmunol. 2013;256(1-2):38–42.
Saresella M, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11:23.
Heneka MT, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8.
Barrett JP, et al. Bone marrow-derived macrophages from AbetaPP/PS1 mice are sensitized to the effects of inflammatory stimuli. J Alzheimers Dis. 2015;44(3):949–62.
Fiala M, et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32(5):360–71.
McManus RM, Mills KH, Lynch MA. T cells—protective or pathogenic in Alzheimer’s disease? J Neuroimmune Pharmacol. 2015;10(4):547–60.
Acknowledgements
Not applicable.
Funding
This is an EU Joint Programme - Neurodegenerative Disease Research (JPND) project (see www.jpnd.eu). The project is supported through the following funding organisations under the aegis of JPND: France, Agence National de la Recherche; Germany, Federal Ministry of Education and Research (BMBF; funding code 01ED1505A); Italy, Ministry of Education, Universities and Research; Netherlands, The Netherlands Organisation for Health Research and Development; Sweden, Swedish Research Council (VR).
Availability of data and materials
Not applicable.
Authors’ contributions
Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
McManus, R.M., Heneka, M.T. Role of neuroinflammation in neurodegeneration: new insights. Alz Res Therapy 9, 14 (2017). https://doi.org/10.1186/s13195-017-0241-2
Published:
DOI: https://doi.org/10.1186/s13195-017-0241-2