
Abstract
DNA methylation is a pivotal epigenetic modification that affects gene expression. Tumor immune microenvironment (TIME) comprises diverse immune cells and stromal components, creating a complex landscape that can either promote or inhibit tumor progression. In the TIME, DNA methylation has been shown to play a critical role in influencing immune cell function and tumor immune evasion. DNA methylation regulates immune cell differentiation, immune responses, and TIME composition Targeting DNA methylation in TIME offers various potential avenues for enhancing immune cytotoxicity and reducing immunosuppression. Recent studies have demonstrated that modification of DNA methylation patterns can promote immune cell infiltration and function. However, challenges persist in understanding the precise mechanisms underlying DNA methylation in the TIME, developing selective epigenetic therapies, and effectively integrating these therapies with other antitumor strategies. In conclusion, DNA methylation of both tumor cells and immune cells interacts with the TIME, and thus affects clinical efficacy. The regulation of DNA methylation within the TIME holds significant promise for the advancement of tumor immunotherapy. Addressing these challenges is crucial for harnessing the full potential of epigenetic interventions to enhance antitumor immune responses and improve patient outcomes.
Similar content being viewed by others


Introduction
Epigenetics refers to the scientific study of reversible alterations in gene expression and function that result in heritable phenotypic changes, while preserving the underlying DNA sequence. Histone modifications, non-coding RNA regulation, and alterations in DNA methylation are important epigenetic mechanisms. One of the oldest and most important epigenetic alterations is DNA methylation [1], assumes a critical role in embryonic development, X-chromosome inactivation, gene imprinting, and transposon activity control.
Furthermore, DNA methylation reprogramming, prominently observed in various diseases, particularly malignancies and immune-related disorders, is closely associated with the tumor microenvironment (TME), including the tumor immune microenvironment (TIME). Recent research has highlighted the intricate relationship between methylation remodeling and the TME, particularly the TIME. A profound understanding of the inherent mechanisms governing the interplay between DNA methylation and the TIME may reveal a novel avenue for cancer combination immunotherapy, achieved by targeted manipulation of DNA methylation [2, 3]. Figure 1 illustrates the perspectives and relevant mechanisms used in this review. This article primarily focuses on a comprehensive discourse concerning the interactions between DNA methylation regulatory networks and the TIME, along with potential collaborative therapeutic approaches.

The perspectives and relevant mechanisms of this review. Methylation-driven remodeling is intricately associated with the maintenance or alteration of the TME, particularly the TIME. The methylation of tumor suppressor genes (left) or the demethylation of proto-oncogenes (right) is closely linked to the initiation and progression of cancer. The TIME is generally classified into immune-exempt “cold tumors” and inflamed “hot tumors” phenotypes. DNA methylation reprogramming exerts multifaceted regulation on the TIME through its influence on the infiltration, differentiation, secretion of various tumor immune cells (Tregs, B cells, macrophages), vascular genesis (hypoxic microenvironment), metabolism (lipids, mitochondria), and other aspects
Molecular level of the TIME and DNA methylation reprogramming
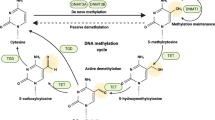
DNA methylation is an epigenetic process involving the addition of methyl groups to cytosine residues. This modification occurs at the C5 position of cytosine and is catalyzed by enzymes called DNA methyltransferases (DNMT), leading to the formation of 5-methylcytosine (5mC). In vivo, DNA methylation dynamically regulates gene modification and maintains equilibrium through methylation, recognition, and removal. Three active DNMTs have been identified in mammals and are denoted as DNMT1, DNMT3a, and DNMT3b [4,5,6]. DNA demethylation primarily involves mediation by the ten-eleven translocation (TET) family of enzymes. In the active demethylation pathway, the dioxygenase domain catalyzes the hydroxylation of 5mC to generate 5-hydroxymethylcytosine, which is further converted to 5-formylcytosine and 5-carboxylcytosine. Subsequent processes involve the dilution of DNA methylation through replication or the removal of bases through thymine DNA glycosylase-dependent base excision repair, resulting in changes in DNA methylation. TET family dioxygenases and DNA demethylation exhibit pleiotropic biological effects in both stem cells and cancer cells [7] and play significant roles in immune cell differentiation and maturation [8]. Three active mammalian TET enzymes have been identified to date, TET1, TET2, and TET3 respectively [9,10,11,12,13].
The TME is an intricate assemblage of stromal cells, tumor cells, and the extracellular matrix (ECM), encompassing immune cells, inflammatory cells from the bone marrow, fibroblasts, blood vessels, and various signaling molecules. Notably, the TIME is a vital component of the TME, comprising T cells, B cells, macrophages, dendritic cells, tumor-associated fibroblasts, neutrophils, and secreted cytokines, collectively comprising various infiltrating immune cells [14]. The composition and abundance of stromal cell types dictate TIME characteristics, further influencing tumor progression and immune responses [2, 3, 15]. The heterogeneity of the TIME results in significant differences in tumor progression between individuals. Generally, the TIME can be categorized as either immune-inflamed (“hot tumors”) or immune-excluded (“cold tumors”) [2, 3, 16].
DNA methylation and the TIME modulating
The mechanisms by which DNA methylation influences TIME can be summarized as methylation of tumor suppressor genes, which subsequently execute immune defense or immune cytotoxic functions, and demethylation of oncogenes, which perform immune suppression or immune tolerance functions. Table 1 presents the statuses of some of the relevant genes reported to date. Reversing methylation/demethylation may offer potential novel epigenetic therapeutic interventions targeting the TIME.
There is a growing consensus that the DNA methylation of tumor suppressor genes (immune defense genes) is closely associated with cancer development. In a study conducted by Li et al. [17], it was cancer patients with high levels of neurofilament medium polypeptides (NEFM) exhibited improved overall survival (OS) and recurrence-free survival (RFS). Conversely, DNA methylation in NEFM is associated with worse OS, potentially owing to reduced NEFM expression. Upon further examination, it was discovered that there was a noteworthy inverse relationship between NEFM DNA methylation levels and the infiltration levels of CD4+ T cells, CD8+ T cells, B cells, macrophages, dendritic cells, and neutrophils in tumors, with a positive correlation observed with macrophage infiltration. These results suggest that NEFM DNA methylation leads to a poorer prognosis in patients by modulating the breast TIME. One possible mechanism for this phenomenon is that the absence of NEFM in breast tumor cells causes intermediate filaments to become unstable, which in turn causes cytoskeletal disarray and dynamic cell deformation. This, in turn, facilitates tumor EMT and interferes with immune microenvironment signaling, increasing the motility of tumor cells and their capacity to colonize nearby tissues [18].
Zinc finger and SCAN domain-containing protein 18 (ZSCAN18) is a crucial member of a family of transcription factors associated with the cell cycle and glycolysis signaling pathways. It can bind to the promoter of tumor protein 53-induced nuclear protein 2 (TP53INP2) and modulate the anti-tumor response [19]. An additional study by Wang et al. discovered that whereas relatively high expression of ZSCAN18 is linked to a favorable prognosis, DNA methylation-modified ZSCAN18 is underexpressed in breast tumors. Breast tumor tissues have higher levels of ZSCAN18 DNA methylation than normal tissues. Numerous genes linked to the Wnt/β-catenin and glycolysis signaling pathways can be inhibited by ZSCAN18 upregulation. ZSCAN18 expression negatively correlated with infiltrating B cells and DC, whereas ZSCAN18 DNA methylation positively correlated with activated B cells, CD8+ and CD4+ T cells, macrophages, neutrophils, and DC. According to the study, DNA methylation modifications significantly influence the TIME through transcriptional regulation and the glycolysis signaling pathway.
STING, an essential tumor suppressor gene and vital regulator of tumor immunity, has been shown to play an important role in tumor suppression and immune control [20]. Recently, Lin et al. [21] found that both mRNA and protein expression of STING are reduced in adenocarcinoma (LUAD), and that LUAD patients with low STING expression have a poorer prognosis, which may be due to the hypermethylation of STING. Similar investigations have demonstrated a strong relationship between STING methylation and the TIME. A549 and H1975 are two LUAD cell lines whose proliferation and metastasis are inhibited by TET2 overexpression. However, these cells proliferate, migrate, and invade more readily when TET2 is knocked down. Mechanistically, LUAD carcinogenesis and metastasis were significantly influenced by the TET2-mediated DNA methylation balance in STING. [22] In order to activate STING and produce Type I IFN, which in turn restores the CD8+ T cell-dependent immune response in tumor-bearing mice, Falahat R employed DNA methylation inhibitors to reverse the methylation silent of STING in mouse melanoma cells [23]. According to a previously described study, one potential new avenue for the therapy of certain malignancies is to reverse the methylation status of STING via epigenetic reprogramming, which would modify the TIME.
DNA demethylation and the TIME modulating
Even though certain immunological tolerance-related genes or proto-oncogenes that are demethylated have been strongly linked to tumor formation, as well as the intrinsic roles of these genes, they have received relatively little research attention [24, 25]. Initially considered a Tregs-specific expression molecule, Forkhead box protein 3 (FOXP3) was believed to be involved in regulating immune-suppressive functions. Recent studies have revealed FOXP3 expression in various cancers, including gastric [26], pancreatic [27], liver [28], and breast [29] cancer [29]. Although its functional role may vary [30,31,32], numerous cancer studies have suggested that FOXP3 is highly expressed in tumor cells or T-cells, and that this high expression is associated with FOXP3 demethylation.
Ke et al. [33] observed that compared to healthy individuals, individuals with non-small cell lung cancer (NSCLC) have an increased prevalence of CD4+ Treg cells in the bloodstream. Moreover, individuals with NSCLC were found to have demethylation at eight CpG sites in the foxp3 promoter, with methylation levels showing a negative correlation with the proportion of CD4+ CD25+ Foxp3+ T cells. In vitro studies showed that tumor cells impacted the function of CD4+ Tregs, leading to the secretion of IL-10 and TGF-β1. Furthermore, a decrease in DNA methyltransferase activity was observed in CD4+ T cells, leading to demethylation of eight CpG sites in the foxp3 promoter. These findings indicate that the increased expression of FOXP3 in CD4+ T cells may be a result of demethylation of the promoter region. Tregs exhibit strong immunosuppressive effects and significantly inhibit the proliferation of naïve CD4+ T cells. This study confirmed that tumor cells in patients with NSCLC downregulate immune responses promoted tumor progression by affecting foxp3 promoter demethylation in T cells. In line with the demethylation of T-cell FOXP3, Schultze et al. [32] examined the correlation between the demethylation of FOXP3 in tumor cells and the TIME. The results showed that the average frequency of cells with demethylated FOXP3 in normal tissues was significantly lower than that in tumor tissues from both patients with colorectal cancer (CRC) and intrahepatic cholangiocarcinoma (ICC) rats. This suggests that FOXP3, a Treg biomarker, may play an intriguing role in immune evasion induced by tumor cells.
Thrombospondin-2 (THBS2) is a glycoprotein of the extracellular matrix that effectively prevents tumor development and angiogenesis. The expression of THBS2 is dramatically elevated in colon adenocarcinoma (COAD), as shown by Liu et al. [33]. It is negatively correlated with DNA replication, repair, and the cell cycle, and favorably correlated with angiogenesis and epithelial-mesenchymal transition. The expression of THBS2 has a favorable correlation with microsatellite instability and a substantial relationship with the progression-free interval in COAD. THBS2 methylation levels in COAD tissues were markedly lower than those in healthy tissues. Nuclear translocation of HIF1 is greatly increased by the high exogenous expression of THBS2 in CT26 cells, which enhances intracellular lactate metabolism. Additional studies conducted both in vitro and in vivo suggest that lactate generated by tumor cells stimulates macrophage M2 polarization, which in turn prevents T cell proliferation and destruction. By mediating DNA methylation modifications, THBS2 functions as a mediator between the tumor extracellular matrix and immune infiltration, thereby influencing biological processes, including immune cell infiltration, immune regulation, cell death, migration, epithelial-mesenchymal transition, and angiogenesis [34].
Tenascin-C (TNC), which was first discovered in the 1980s, is a multidomain extracellular matrix glycoprotein that is highly expressed during multicellular organisms [35]. TNC levels are generally undetectable in most adult tissues, likely because of epigenetic silencing during embryonic development. Sustained TNC expression is associated with chronic inflammation and many malignant tumors, including prostate cancer, glioblastoma, and nasopharyngeal carcinoma (NPC) [25, 26, 36]. Through interactions with their receptor integrins and numerous other binding components, TNC trigger environmental and cell type-dependent functions to regulate cell adhesion, migration, proliferation, and angiogenesis. Additionally, it plays a role in tumor epithelial-mesenchymal transition (EMT) and TME modulation [37]. TNC, an endogenous TLR4 activator, enhances inflammatory responses by increasing the production of pro-inflammatory cytokines in innate immune cells such as macrophages and microglia. Furthermore, TNC promotes macrophage differentiation and polarization toward an M1-like phenotype, whereas TNC exhibits immunosuppressive functions in T cells. In glioblastomas, TNC is expressed in the tumor and stromal cells, and its high expression is associated with tumor progression and poor prognosis. In addition to promoting glioblastoma invasion and angiogenesis, TNC affect the morphology and function of tumor-associated microglia/macrophages, suggesting that TNC contribute to glioblastoma progression by influencing EMT and TIME [36].
In a recent study conducted by our team [25], we found that TNC underwent significant demethylation in radioresistant NPC cell strains with high TNC expression. The traditional Chinese medicine Shengmai Yin is radiosensitive and partially restores the demethylated state of radioresistant NPC cell strains, suggesting that TNC demethylation is involved in the remodeling of the NPC radiation microenvironment. During this process, the radiation microenvironment may directly or indirectly interact with the immune microenvironment, thereby collectively influencing the TME.
In addition to their direct effects on the immune-related genes, DNA methylation and demethylation may indirectly shape the TIME through various mechanisms, including the EMT [38,39,40]. The delicate equilibrium between DNA methylation and demethylation [41] is crucial for the normal functioning of cells and development of mammals. DNMTs, TETs, and related enzymes collectively regulate this physiological balance and have significant implications for the interactions between cancerous tumors and the immune system. Disruption of this balance in the TME may be an important potential avenue for DNA methylation-related tumor immunotherapy or adjunctive therapy.
DNA methylation remodeling and the cellular level of tumor immunotherapy
Reversing this process, which involves the remodeling of DNA methylation and disruption of the TIME, has become an important avenue in tumor immunotherapy. The distribution and proportions of various functionally distinct immune cells within the TIME determine whether a tumor is “hot” or “cold” [3]. DNA methylation is strongly linked to the maturation, polarization, differentiation, and function of immune cells, and contributes to tumor evasion from the immune system [2, 3]. The following sections elaborate on the current research status of enhancing immune cytotoxicity and reducing immune escape/suppression through DNA methylation remodeling to regulate the TIME and immunotherapy.
Regulating the TIME by enhancing immune cytotoxicity through DNA methylation remodeling
The epigenetic regulation of CD8+ T cells plays a crucial role in acquiring and maintaining immune cytotoxicity as well as in mounting rapid and robust responses to antigen re-challenges. CD8+ T-cells of various cells exhibit dynamic methylation patterns at various stages of differentiation For example, in response to severe and frequent antigen stimulation, transcription factors (TFs) associated with effector T cells bind to particular demethylated sites in genes such as IFNG and GZMB, resulting in effector phenotypic differentiation [42]. Since that increases the methylation of the promoter of T cell-specific transcription factor 7 (TCF7), DNMT3A is required for CD8+ effector T-cell development [43]. TCF7 encodes TCF1, a transcription factor that is upregulated in naïve T cells and central memory T cells but downregulated in effector memory T cells. TCF7 silencing impairs stem cell-like T cell renewal and central memory CD8+ T cell memory response [44]. Despite the complexity of the epigenetic regulation of CD8+ T cells in tumor formation [45], various studies have shown that suppressing DNMTs may successfully boost the antitumor effects of CD8+ effector T cells and alter the TIME [46].
Tumor-infiltrating lymphocytes (TILs), particularly CD8+ TILs, are closely linked to the TME immune landscape. These markers serve as valuable prognostic indicators of responsiveness to immunotherapy and patient survival. In a study by Zou et al. [47] that focused on CRC, three CD8+ T cell-specific differentially methylated regions were identified, which enabled the establishment of a CD8+ MeTIL feature score. These findings revealed that lower CD8+ MeTIL scores, indicating enriched CD8+ TILs, correlated with favorable prognoses in patients with CRC. Ovarian cancer (OCs), a notably lethal gynecological malignancy, is a subset of diseases with a limited response to prevailing immunotherapies. This phenomenon can be attributed to modulation of the TIME and inadequate recruitment and activation of immune cells, which are essential for the elimination of cancer cells. Gomez et al. [48] observed that DNA methyltransferase inhibitors (DNMTis) activate the immune-suppressed TME in OC. This activation mechanism involves the restructuring of methylation patterns, subsequently leading to the recruitment and activation of more CD8+ T cells. Moufarrij et al. [49] found that combining DNMTis and HDAC6 inhibitors enhanced the Type I interferon response, leading to an increased production of cytokines, chemokines, and MHC I antigen presentation complex components in OC cells. This alteration in the TIME is characterized by a rise in tumor-killing cells including interferon (IFN)γ+ CD8, NK, and NKT-cells, accompanied by a reduction in MDSCs and PD-1hi CD4 T-cells, ultimately reversing the immune-suppressive TME.
CD4+ T-cells, under the supervision of several TFs, differentiate into various T-helper cell subsets. Epigenetic processes play integral roles in the development and function of Th1 and Th2 cells. The role of Th1 cells in immunological cytotoxicity in the TIME is discussed here. Travers et al.[13] illustrated that medicine with the combination of 5-azacytidine (5AZA-C) and α-difluoromethylornithine (DFMO) can induce the recruiting process of activated CD8+ T-cells, IFNγ+ CD4+ T-cells (Th1), and NK cells, while significantly reducing immunosuppressive cells like M2-polarized macrophages and increasing tumor-killing M1 macrophages, in the TIME of patients with OC. Li et al. [50] proposed that epigenetic modifications might lead to the abnormal regulation of angiogenesis and the TME. They found that the prognosis of patients with NSCLC treated with chemotherapy combined with bevacizumab differed based on their DNA methylation patterns. Significant enrichment of differentially methylated regions in genes related to the VEGFA-VEGFR2 signaling pathway, neutrophil-mediated immunity, neutrophil degranulation, and other biological processes was observed. Peng et al. [51] discovered that DNMT1 mediates DNA methylation in OC, decreasing the synthesis of Th1-secreted chemokines CXCL9 and CXCL10, and increasing effector T-cell infiltration. Furthermore, the expression levels of tumor DNMT1 were negatively associated with infiltrating CD8+ T cells and patient prognosis. According to this study, targeted epigenetic remodeling alters T-cell distribution and may enhance the clinical efficacy of cancer therapies. This also suggests that the epigenetic suppression of Th1-type chemokines represents a distinct mechanism of cancer immune evasion.
Regulating the TIME by reducing immune suppression through DNA methylation remodeling
Compared to antitumor immune cells, the TIME harbors a greater proportion of immune-inhibitory cells that are associated with tumor immune evasion. These immune-inhibitory cells progressively develop immune escape mechanisms during tumor development. Major players in this category include Tregs, MDSC, and TAM-2s [52]. CD4+ CD25+ T cells are a subset of suppressive T cells that play a dominant role in downregulating or inhibiting the induction and proliferation of effector T cells and mediating peripheral immune tolerance [53]. Within the TIME, Tregs are considered the primary immune suppressive factors, and they employ various mechanisms to dampen immune responses [54,55,56]. One of the key challenges in Treg-based therapy is finding effective strategies to inhibit Tregs while activating TILs without compromising the body's immune functions [57, 58].
The role of Foxp3 in the development and suppression of Tregs makes it a prime target for epigenetic therapies that target Tregs. Sabir et al. [59] discovered a strong negative association between the efficacy of imatinib therapy in patients with late-stage and optimal-response chronic myeloid leukemia, Treg demethylation percentage, and the Foxp3 Treg-specific demethylation region (TSDR). According to Ma et al. [60], increased STAT5 expression in CRC CD4+ T cells can attract additional TET2 to FOXP3-TSDR, resulting in elevated FOXP3 expression via DNA demethylation. This highlights the mechanism underlying low methylation of FOXP3-TSDR in tumor-infiltrating CD4+ T cells in patients with CRC. Other studies on FOXP3/TSDR have provided similar insights [61, 62]. Changes in cell-permeable ketoglutarate (KG) modify the DNA methylation profile of initial CD4+ T-cells stimulated under Treg polarization conditions, considerably decreasing FOXP3+ Treg differentiation and enhancing inflammatory cytokine production, according to Matias et al. [63]. This research suggests that altering αKG, mitochondrial metabolism, and lipid homeostasis represents a novel approach to inhibiting Tregs and enhanceing immune therapy in the TIME.
Within the TIME, MDSCs, which originate from immature bone marrow precursors, play a crucial role in facilitating tumor immune evasion [64, 65]. They activate the immunosuppressive activity of Tregs by modulating the production of interleukin (IL)-10 and IFN- and enhance the conversion of initial CD4+ T cells into Tregs by secreting retinoic acid and TGF- [66]. Furthermore, MDSCs can directly affect the activities of CD8+ T cells and NK cells in the TIME via cell–cell interactions and soluble factor release. Furthermore, MDSCs can establish T cell tolerance by expressing inhibitory receptors such as PD-L1 and cytotoxic T lymphocyte-associated antigen 4 (CTLA4), MDSCs can establish T-cell tolerance [67]. Numerous studies have found that DNA methylation influences MDSC development, maturation, and recruitment to the TME [68, 69]. Smith et al. [70] reported that the DNMTi decitabine (DAC) reduces the accumulation of MDSCs in mice with CRC, facilitating the activation of antigen-specific cytotoxic T lymphocytes. This mechanism may be attributed to the modulation of TNF promoter demethylation by DAC, which decreases DNMT expression and subsequently blocks RIP1-dependent necrotic target methylation, enhancing cell death and reducing MDSC accumulation. Guadecitabine was demonstrated in another trial by Luker et al. [71] to considerably reduce tumor burden by blocking excessive bone marrow proliferation and systemic MDSC accumulation in a T cell-dependent manner. The remaining MDSCs transitioned to the antigen-presenting phenotype. The findings of this study suggest that guadecitabine has the potential to enhance the therapeutic efficacy of adoptive transfer of APLs, leading to a decrease in tumor growth and an improvement in overall survival rates. Collectively, these findings suggest that epigenetic remodeling could serve as an effective strategy to inhibit MDSCs and regulate the TIME.
TAMs are derived from monocytes in the blood and migrate to solid tumor tissues. They are divided into two separate subgroups, M1 and M2, each with dramatically different roles [72]. TAM-1s are primarily activated by factors such as IFN-γ, IL-2, and TNF-γ. They play a role in polarizing Th1 responses, recruiting cytotoxic T lymphocytes (CTLs), and promoting antitumor immune responses. In contrast, TAM-2s are stimulated by IL-4 and IL-13, leading to the development of an immunosuppressive TME. They secrete factors like TGF-β, suppress NK cell activity, and upregulate the expression of PD-L1 and CTLA4. This ultimately hinders the infiltration of effector T cells by blocking immune checkpoints [73]. Moreover, TAM-2s can dampen immune responses by producing anti-inflammatory cytokines such as TGF-β, IL-10, IL-13, and IL-4, which inhibit CTL functions and upregulate Tregs, thereby weakening the immune response [74, 75]. Differentiation of TAMs and their transition between the M1 and M2 phenotypes are also subject to epigenetic regulation [76]. Travers et al. [13] previously reported that combination therapy with 5AZA-C and DFMO in patients with OC resulted in a substantial decrease in immunosuppressive cells, such as M2-polarized macrophages, and an increase in TAM-1s. This study suggests that altering macrophage polarization in the TME and recruiting TAM-1s can extend the survival of patients with OC. Furthermore, research by Zhang et al. [77] revealed that during the development of pancreatic ductal adenocarcinoma (PDA), tumor cells induce selective Nqo-1 DNA methylation in TAM-1s through direct cell-to-cell contact mediated by GARP and integrins αV/β8. This leads to suppression of the glycolytic state in TAM-1s, ultimately reprogramming them to become TAM-2s. These findings suggested that PDA cells can reprogram TAM-1s through DNA methylation-mediated mechanisms of metabolism and function. Epigenetic reprogramming, through either epigenetic modifications or drug interventions, has the potential to reverse the transition from M2 to M1, thereby offering a novel approach for cancer therapy.
Currently, immunotherapies that rely on reprogramming DNA methylation, aimed at either enhancing immune cytotoxicity or reducing immune tolerance, remain limited (Table 2). To date, research has primarily focused on non-solid tumors and the demethylation of tumor suppressor genes (Table 3). Therapies that actually target oncogene methylation remain relatively unknown, and progress in this area depends on further in-depth research into the relevant mechanisms. There will be significant advancements in treatments targeting oncogene methylation in the near future.
Prospects and challenges
Regulation of DNA methylation and immunotherapy are becoming increasingly prominent research fields. Convincing evidence suggests that epigenetic modifications influence the interactions between cancer, immune cells, and stromal cells, ultimately regulating the state of the TIME. Therefore, alterations in DNA methylation have the potential to open new avenues of cancer therapy. Although drugs targeting DNA methylation modifications have been explored for clinical applications, they are still in relatively early stages of development and will face significant challenges in the future.
Further development and maturation are imperative for basic research. There remains a need for an in-depth investigation into the mechanisms linking DNA methylation and the TIME. The depth of our understanding of this relationship directly affects the precision of practical applications. Key aspects that require further elucidation include the mechanisms governing the selective action of methylation enzymes on target cells and the intricate association between oncogene demethylation and tumorigenesis. This will depend on the emergence of new epigenetic technologies. For instance, novel detection technologies like next-generation sequencing enable high-throughput methylation site detection and accurate identification of various DNA/RNA methylation patterns, such as m5c, m6a, m7g [78,79,80]; these technologies also function as tumor biomarkers to inform precise disease subtyping and customized treatment [81]; concerning modification technologies, CRISPR-based epigenomic editing tools precisely manipulate DNA methylation at specific genomic sites, modifying methylation at selected biomarker locations to provide more targeted therapy[82]; concerning analysis technologies, these include the integrated analysis of multi-omics data and the simulation of treatment plans and outcomes using machine learning [83].
Second, the use of DNMT-based drugs poses significant challenges. DNA methylation modifications are pervasive, occurring in both normal and neoplastic cells, and these drugs exhibit varying degrees of function depending on the cell type. The conundrum lies in the precise targeting of tumor cells by broad-spectrum DNMTis during therapeutic interventions, while minimizing their impact on epigenetic modifications in normal cells. This predicament, which has been previously documented [84], underscores the importance of formulating DNMTis with tumor cell selectivity. The close relationship between demethylation and tumorigenesis highlights the importance of developing tailored TET inhibitors.
Third, the strategic integration of methylation-targeting drugs with alternative antitumor agents and methodologies warrants rigorous consideration. Monotherapeutic modalities are often associated with hurdles such as drug resistance and substantial side effects. Resolving these challenges hinges on the synergy engendered by the co-administration of methylation-targeting drugs along with therapeutic modalities, such as radiotherapy, chemotherapy, vaccines, immune checkpoint inhibitors (ICIs), oncolytic viruses, CAR-T/NK cells, and other technological approaches. Attaining a synergistic therapeutic effect coupled with the minimization of adverse effects via the judicious amalgamation of methylation-targeting drugs and other therapeutic strategies remains a formidable challenge in the future.
In conclusion, although the study of the regulation of DNA methylation in the context of immunotherapy holds immense promise, this landscape is framed by various challenges and opportunities. We anticipate advances at the exciting intersection of science and medicine in the coming years.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- RNA:
-
Ribonucleic acid
- 5mC:
-
5-Methylcytosine
- TME:
-
Tumor microenvironment
- TIME:
-
Tumor immune microenvironment
- DNMT:
-
DNA methyltransferase
- TET:
-
Ten–Eleven translocation
- CTL:
-
Cytotoxic T lymphocytes
- Tregs:
-
Regulatory T-cells
- TAMs:
-
Tumor-associated macrophages
- NK:
-
Natural killer
- TGF-β:
-
Transforming growth factor-β
- DCs:
-
Dendritic cells
- ECM:
-
Extracellular matrix
- CRC:
-
Colorectal cancer
- NSCLC:
-
Non-small cell lung cancer
- NEFM:
-
Neurofilament medium polypeptide
- IFN:
-
Interferon
- MDSCs:
-
Myeloid-derived suppressor cells
- PD-L1:
-
Programmed death-ligand 1
- TNC:
-
Tenascin-C
- NPC:
-
Nasopharyngeal carcinoma
- EMT:
-
Epithelial–mesenchymal transition
- TF:
-
Transcription factor
- 5AZA-C:
-
5-Azacytidine
- DFMO:
-
α-Difluoromethylornithine
- VEGFA:
-
Vascular endothelial growth factor A
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- TSDR:
-
Treg-specific demethylation region
- KG:
-
Ketoglutarate
- IL:
-
Interleukin
- IFNG:
-
Interferon gamma
- GZMB:
-
Granzyme B
- TNF:
-
Tumor necrosis factor
- RIP1:
-
Receptor-interacting protein kinase 1
- TAM-1s:
-
Tumor-associated macrophage type 1
- TAM-2s:
-
Tumor-associated macrophage type 2
- M1:
-
M1 macrophages
- M2:
-
M2 macrophages
- CAR-T:
-
Chimeric antigen receptor T-cell
- CAR-NK:
-
Chimeric antigen receptor natural killer cells
- ICIs:
-
Immune checkpoint inhibitors
- Th2:
-
T helper type 2
- Th1:
-
T helper type 1
- CpG:
-
Cytosine–phosphate–Guanine
- CTLs:
-
Cytotoxic T lymphocytes
- IFN-γ:
-
Interferon-gamma
- PDA:
-
Pancreatic ductal adenocarcinoma
- MHC:
-
Major histocompatibility complex
- FOXP3:
-
Forkhead box protein 3
- STAT5:
-
Signal transducer and activator of transcription 5
- MeTIL:
-
Methylation of TILs
- DAC:
-
Decitabine
- ZSCAN18:
-
Zinc finger and SCAN domain-containing protein 18
References
Hattori N, Liu Y-Y, Ushijima T. DNA methylation analysis. Methods Mol Biol. 2023;2691:165–83.
Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20:171.
Zhong F, Lin Y, Zhao L, Yang C, Ye Y, Shen Z. Reshaping the tumour immune microenvironment in solid tumors via tumour cell and immune cell DNA methylation: from mechanisms to therapeutics. Br J Cancer. 2023;129:24–37.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92.
Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56.
Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37:1012–27.
An J, Rao A, Ko M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp Mol Med. 2017;49:e323.
Tsiouplis NJ, Bailey DW, Chiou LF, Wissink FJ, Tsagaratou A. TET-mediated epigenetic regulation in immune cell development and disease. Front Cell Dev Biol. 2020;8:623948.
Liu W, Wu G, Xiong F, Chen Y. Advances in the DNA methylation hydroxylase TET1. Biomark Res. 2021;9:76.
Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–50.
Bray JK, Dawlaty MM, Verma A, Maitra A. Roles and regulations of TET enzymes in solid tumors. Trends Cancer. 2021;7:635–46.
Joshi K, Liu S, Breslin SJP, Zhang J. Mechanisms that regulate the activities of TET proteins. Cell Mol Life Sci. 2022;79:363.
Travers M, Brown SM, Dunworth M, Holbert CE, Wiehagen KR, Bachman KE, et al. DFMO and 5-azacytidine increase M1 macrophages in the tumor microenvironment of murine ovarian cancer. Cancer Res. 2019;79:3445–54.
Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat Rev Clin Oncol. 2020;17:251–66.
Wu B, Shi X, Jiang M, Liu H. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer. 2023;22:38.
Kumar V, Bauer C, Stewart JH. Targeting cGAS/STING signaling-mediated myeloid immune cell dysfunction in TIME. J Biomed Sci. 2023;30:48.
Li D, Zhao W, Zhang X, Lv H, Li C, Sun L. NEFM DNA methylation correlates with immune infiltration and survival in breast cancer. Clin Epigenetics. 2021;13:112.
Epigenetic silencing of neurofilament genes promotes an aggressive phenotype in breast cancer: PubMed. https://pubmed.ncbi.nlm.nih.gov/25985363/. Accessed 8 Dec 2023.
Inactivation of ZSCAN18 by promoter hypermethylation drives the proliferation via attenuating TP53INP2-mediated autophagy in gastric cancer cells: PubMed. https://pubmed.ncbi.nlm.nih.gov/36650573/. Accessed 8 2023.
Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X. STING: a master regulator in the cancer-immunity cycle. Mol Cancer. 2019;18:152.
Lin Z, Liu Y, Lin P, Li J, Gan J. Clinical significance of STING expression and methylation in lung adenocarcinoma based on bioinformatics analysis. Sci Rep. 2022;12:13951.
TET2 inhibits the proliferation and metastasis of lung adenocarcinoma cells via activation of the cGAS-STING signalling pathway: PubMed. https://pubmed.ncbi.nlm.nih.gov/37667220/. Accessed 8 Dec 2023.
Epigenetic state determines the in vivo efficacy of STING agonist therapy: PubMed. https://pubmed.ncbi.nlm.nih.gov/36949064/. Accessed 8 Dec 2023.
Zhu D, Li A, Lv Y, Fan Q. Traditional chinese medicine: a class of potentially reliable epigenetic drugs. Front Pharmacol. 2022;13:907031.
Wang Z-T, Peng Y, Lou D-D, Zeng S-Y, Zhu Y-C, Li A-W, et al. Effect of Shengmai Yin on epithelial–mesenchymal transition of nasopharyngeal carcinoma radioresistant cells. Chin J Integr Med. 2023;29:691–8.
Kim HK, Won KY, Han S-A. The antioncogenic effect of Beclin-1 and FOXP3 is associated with SKP2 expression in gastric adenocarcinoma. Medicine (Baltimore). 2021;100:e26951.
Hinz S, Pagerols-Raluy L, Oberg H-H, Ammerpohl O, Grüssel S, Sipos B, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–50.
Jubran MR, Rubinstein AM, Cojocari I, Adejumobi IA, Mogilevsky M, Tibi S, et al. Dissecting the role of crosstalk between glioblastoma subpopulations in tumor cell spreading. Oncogenesis. 2020;9:11.
Liu R, Liu C, Chen D, Yang W-H, Liu X, Liu C-G, et al. FOXP3 controls an miR-146/NF-κB Negative feedback loop that inhibits apoptosis in breast cancer cells. Cancer Res. 2015;75:1703–13.
Wang J, Gong R, Zhao C, Lei K, Sun X, Ren H. Human FOXP3 and tumour microenvironment. Immunology. 2023;168:248–55.
Yang S, Liu Y, Li M-Y, Ng CSH, Yang S-L, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16:124.
Schultze FC, Andag R, Alwahsh SM, Toncheva D, Maslyankov S, Yaramov N, et al. FoxP3 demethylation is increased in human colorectal cancer and rat cholangiocarcinoma tissue. Clin Biochem. 2014;47:201–5.
Systematic analysis of integrated bioinformatics to identify upregulated THBS2 expression in colorectal cancer cells inhibiting tumour immunity through the HIF1A/Lactic Acid/GPR132 pathway: PubMed. https://pubmed.ncbi.nlm.nih.gov/37884956/. Accessed 8 Dec 2023.
Thrombospondin-2 acts as a bridge between tumor extracellular matrix and immune infiltration in pancreatic and stomach adenocarcinomas: an integrative pan-cancer analysis: PubMed. https://pubmed.ncbi.nlm.nih.gov/35701829/. Accessed 8 Dec 2023.
Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a glance. J Cell Sci. 2016;129:4321–7.
Yalcin F, Dzaye O, Xia S. Tenascin-C function in glioma: immunomodulation and beyond. Adv Exp Med Biol. 2020;1272:149–72.
Kang X, Xu E, Wang X, Qian L, Yang Z, Yu H, et al. Tenascin-c knockdown suppresses vasculogenic mimicry of gastric cancer by inhibiting ERK- triggered EMT. Cell Death Dis. 2021;12:890.
Ye W, Siwko S, Tsai RYL. Sex and race-related DNA methylation changes in hepatocellular carcinoma. Int J Mol Sci. 2021;22:3820.
Hernandez-Meza G, von Felden J, Gonzalez-Kozlova EE, Garcia-Lezana T, Peix J, Portela A, et al. DNA methylation profiling of human hepatocarcinogenesis. Hepatology. 2021;74:183–99.
Yi J, Wu M, Zheng Z, Zhou Q, Li X, Lu Y, et al. Integrated analysis of DNA methylome and transcriptome reveals SFRP1 and LIPG as potential drivers of ovarian cancer metastasis. J Gynecol Oncol. 2023. https://doi.org/10.3802/jgo.2023.34.e71.
Jiang S. Tet2 at the interface between cancer and immunity. Commun Biol. 2020;3:667.
Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol. 2018;18:340–56.
Ladle BH, Li K-P, Phillips MJ, Pucsek AB, Haile A, Powell JD, et al. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016;113:10631–6.
Zhao X, Shan Q, Xue H-H. TCF1 in T cell immunity: a broadened frontier. Nat Rev Immunol. 2022;22:147–57.
Lee M, Li J, Li J, Fang S, Zhang J, Vo ATT, et al. Tet2 inactivation enhances the antitumor activity of tumor-infiltrating lymphocytes. Cancer Res. 2021;81:1965–76.
Dan H, Zhang S, Zhou Y, Guan Q. DNA methyltransferase inhibitors: catalysts for antitumour immune responses. Onco Targets Ther. 2019;12:10903–16.
Zou Q, Wang X, Ren D, Hu B, Tang G, Zhang Y, et al. DNA methylation-based signature of CD8+ tumor-infiltrating lymphocytes enables evaluation of immune response and prognosis in colorectal cancer. J Immunother Cancer. 2021;9:e002671.
Gomez S, Cox OL, Walker RR, Rentia U, Hadley M, Arthofer E, et al. Inhibiting DNA methylation and RNA editing upregulates immunogenic RNA to transform the tumor microenvironment and prolong survival in ovarian cancer. J Immunother Cancer. 2022;10:e004974.
Moufarrij S, Srivastava A, Gomez S, Hadley M, Palmer E, Austin PT, et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci Rep. 2020;10:3470.
Li B, Jiang C, Xu Y, Fan X, Yang L, Zou B, et al. Genome-wide DNA methylation signature predict clinical benefit of bevacizumab in non-small cell lung cancer. BMC Cancer. 2022;22:828.
D P, I K, N N, L Z, S W, W W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. https://pubmed.ncbi.nlm.nih.gov/26503055/. Accessed 5 Aug 2023.
Smith K. Tobacco Cessation Services. The CBHSQ Report [Internet]. Rockville (MD): Substance Abuse and Mental Health Services Administration (US); 2013. http://www.ncbi.nlm.nih.gov/books/NBK384677/. Accessed 23 Aug 2023.
Deng G, Song X, Fujimoto S, Piccirillo CA, Nagai Y, Greene MI. Foxp3 post-translational modifications and treg suppressive activity. Front Immunol. 2019;10:2486.
Shimazu Y, Shimazu Y, Hishizawa M, Hamaguchi M, Nagai Y, Sugino N, et al. Hypomethylation of the treg-specific demethylated region in FOXP3 Is a hallmark of the regulatory T-cell subtype in Adult T-cell leukemia. Cancer Immunol Res. 2016;4:136–45.
Goel PN, Grover P, Greene MI. PRMT5 and Tip60 modify FOXP3 function in tumor immunity. Crit Rev Immunol. 2020;40:283–95.
Bellanti JA, Li D. Treg cells and epigenetic regulation. Adv Exp Med Biol. 2021;1278:95–114.
Tanaka A, Sakaguchi S. Targeting treg cells in cancer immunotherapy. Eur J Immunol. 2019;49:1140–6.
Landman S, Cruijsen M, Urbano PCM, Huls G, van Erp PEJ, van Rijssen E, et al. DNA methyltransferase inhibition promotes Th1 polarization in Human CD4+CD25high FOXP3+ regulatory T cells but does not affect their suppressive capacity. J Immunol Res. 2018;2018:4973964.
Sabir SF, Matti BF, Alwatar WMA. Assessment of regulatory T cells (Tregs) and Foxp3 methylation level in chronic myeloid leukemia patients on tyrosine kinase inhibitor therapy. Immunogenetics. 2023;75:145–53.
Ma H, Gao W, Sun X, Wang W. STAT5 and TET2 cooperate to regulate FOXP3-TSDR demethylation in CD4+ T cells of patients with colorectal cancer. J Immunol Res. 2018;2018:6985031.
Lopez-Pastrana J, Shao Y, Chernaya V, Wang H, Yang X-F. Epigenetic enzymes are the therapeutic targets for CD4(+)CD25(+/high)Foxp3(+) regulatory T cells. Transl Res. 2015;165:221–40.
Suh KJ, Kim JW, Kim JE, Sung JH, Koh J, Kim K-J, et al. Correlation between tumor infiltrating immune cells and peripheral regulatory T cell determined using methylation analyses and its prognostic significance in resected gastric cancer. PLoS ONE. 2021;16:e0252480.
Matias MI, Yong CS, Foroushani A, Goldsmith C, Mongellaz C, Sezgin E, et al. Regulatory T cell differentiation is controlled by αKG-induced alterations in mitochondrial metabolism and lipid homeostasis. Cell Rep. 2021;37:109911.
Hegde S, Leader AM, Merad M. MDSC: markers, development, states, and unaddressed complexity. Immunity. 2021;54:875–84.
Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci. 2019;40:4–7.
Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, et al. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021;359:104254.
Cassetta L, Bruderek K, Skrzeczynska-Moncznik J, Osiecka O, Hu X, Rundgren IM, et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J Immunother Cancer. 2020;8:e001223.
Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenet. 2020;12:13.
Daurkin I, Eruslanov E, Vieweg J, Kusmartsev S. Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2′-deoxycytidine. Cancer Immunol Immunother. 2010;59:697–706.
Smith AD, Lu C, Payne D, Paschall AV, Klement JD, Redd PS, et al. Autocrine IL6-mediated activation of the STAT3-DNMT axis silences the TNFα-RIP1 necroptosis pathway to sustain survival and accumulation of myeloid-derived suppressor cells. Cancer Res. 2020;80:3145–56.
Luker AJ, Graham LJ, Smith TM, Camarena C, Zellner MP, Gilmer J-JS, et al. The DNA methyltransferase inhibitor, guadecitabine, targets tumor-induced myelopoiesis and recovers T cell activity to slow tumor growth in combination with adoptive immunotherapy in a mouse model of breast cancer. BMC Immunol. 2020;21:8.
Kadomoto S, Izumi K, Mizokami A. Macrophage polarity and disease control. Int J Mol Sci. 2021;23:144.
Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021;81:1201–8.
He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S. IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience. 2020;437:161–71.
Baxter EW, Graham AE, Re NA, Carr IM, Robinson JI, Mackie SL, et al. Standardized protocols for differentiation of THP-1 cells to macrophages with distinct M(IFNγ+LPS), M(IL-4) and M(IL-10) phenotypes. J Immunol Methods. 2020;478:112721.
Dekkers KF, Neele AE, Jukema JW, Heijmans BT, de Winther MPJ. Human monocyte-to-macrophage differentiation involves highly localized gain and loss of DNA methylation at transcription factor binding sites. Epigenet Chromatin. 2019;12:34.
Zhang M, Pan X, Fujiwara K, Jurcak N, Muth S, Zhou J, et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct Target Ther. 2021;6:366.
Integrated Analysis of Ovarian Juvenile Granulosa Cell Tumors Reveals Distinct Epigenetic Signatures and Recurrent TERT Rearrangements: PubMed. https://pubmed.ncbi.nlm.nih.gov/35031544/. Accessed 8 Dec 2023.
Stabilization of ERK-Phosphorylated METTL3 by USP5 increases m6A Methylation: PubMed. https://pubmed.ncbi.nlm.nih.gov/33217317/. Accessed 8 Dec 2023.
METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation: PubMed. https://pubmed.ncbi.nlm.nih.gov/31031083/. Accessed 8 Dec 2023.
Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma: PubMed. https://pubmed.ncbi.nlm.nih.gov/29035356/. Accessed 8 Dec 2023.
Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing: PubMed. https://pubmed.ncbi.nlm.nih.gov/33838111/. Accessed 8 Dec 2023.
Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning: PubMed. https://pubmed.ncbi.nlm.nih.gov/34131323/. Accessed 8 Dec 2023.
Traynor S, Terp MG, Nielsen AY, Guldberg P, Jakobsen M, Pedersen PG, et al. DNA methyltransferase inhibition promotes recruitment of myeloid-derived suppressor cells to the tumor microenvironment through induction of tumor cell-intrinsic interleukin-1. Cancer Lett. 2023;552:215982.
Acknowledgements
We express our gratitude to the research team members for their dedicated work. We would like to thank Editage (www.editage.cn) for the English language editing.
Funding
This research was funded by the National Natural Science Foundation of China (82074132), Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (20221258), Natural Science Foundation of Guangdong Province of China (2023A1515010017), Project of General Hospital of Southern Theater Command (2022NZB004), Science and Technology Program of Guangzhou (2023A04J1858), and 2022 “Young Science and Technology Talent Recruitment Project” of the Guangzhou Science and Technology Association (QT20220101163).
Author information
Authors and Affiliations
Contributions
DZ contributed to Conceptualization; Writing—original draft; literature review; funding acquisition; CS and SZ contributed to Writing—review and editing; JL contributed to Writing—review and editing, compilation of references; YX and YL contributed to Writing—review and editing, overall structure and organization; EX contributed to Writing—review and Editing, final proofreading, funding acquisition; QF contributed to Supervision, project administration, funding acquisition.
Corresponding authors
Ethics declarations
Informed consent
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, D., Zeng, S., Su, C. et al. The interaction between DNA methylation and tumor immune microenvironment: from the laboratory to clinical applications. Clin Epigenet 16, 24 (2024). https://doi.org/10.1186/s13148-024-01633-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01633-x