
Abstract
The aberrant appearance of DNA in the cytoplasm triggers the activation of cGAS-cGAMP-STING signaling and induces the production of type I interferons, which play critical roles in activating both innate and adaptive immune responses. Recently, numerous studies have shown that the activation of STING and the stimulation of type I IFN production are critical for the anticancer immune response. However, emerging evidence suggests that STING also regulates anticancer immunity in a type I IFN-independent manner. For instance, STING has been shown to induce cell death and facilitate the release of cancer cell antigens. Moreover, STING activation has been demonstrated to enhance cancer antigen presentation, contribute to the priming and activation of T cells, facilitate the trafficking and infiltration of T cells into tumors and promote the recognition and killing of cancer cells by T cells. In this review, we focus on STING and the cancer immune response, with particular attention to the roles of STING activation in the cancer-immunity cycle. Additionally, the negative effects of STING activation on the cancer immune response and non-immune roles of STING in cancer have also been discussed.
Similar content being viewed by others


Introduction
William Coley, the father of immunotherapy, began using Streptococcus pyogenes to treat patients with unresectable tumors in 1891 when chemotherapy and radiotherapy were not available [1]. Ultimately, Coley used a mixture of heat-inactivated Streptococcus pyogenes and Serratia marcescens, known as Coley’stoxin, to treat his cancer patients. For 40 years, Coley used his toxin to treat more than a thousand cancer patients, of which several hundred achieved near complete regression [2]. However, Coley did not know how toxins worked and did not figure out how inflammation treated tumors.
The discovery of phagocytosis by Mechnikov (a Nobel Prize winner) in 1883, led to the crucial understanding of the concept of innate immunity, and many great discoveries followed. Notably, innate immunity entered a new phase in the 1990s when Janeway proposed the concept of pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors (PRRs) [3]. It is now widely accepted that innate immunity plays a critical role in the host defense against microbial infection by recognizing different microbial PAMPs via various PRRs in immune cells and initiating the production and secretion of interferons (IFNs) and cytokines, which then stimulate and activate the adaptive immune response [4].Toll-like receptors (TLRs) on the surface of immune cells are one of the well-known PRRs, and different TLRs recognize different PAMPs. For instance, TLR3, TLR7 and TLR9 recognize dsRNA, ssRNA and CpG DNA, whereas TLR1, TLR2, TLR4 and TLR5 recognize bacterial lipopeptides, peptidoglycan, lipopolysacchride (LPS) and flagellin, respectively (reviewed in ref. [5]). There are also some PPRs within the cytosol of immune cells, such as the NOD-like receptor (NLR), which recognizes bacterial cell-wall lipids and products from damaged host cells, and the RIG-like receptor (RLR), which recognizes viral RNA (reviewed in ref. [6, 7]).
Although it has been known that DNA can stimulate immune responses since as early as 1908 by Mechnikov [8], and numerous studies have demonstrated that the recognition of double-stranded DNA (dsDNA) by innate immune sensors contributes to the development of systemic lupus erythematosus (SLE), a well-known autoimmune disease [9], the dsDNA sensor within immune cells remained unidentified throughout the entire twentieth century. Before the identification of the dsDNA sensor, several groups made a great contribution to the field in 2008 and 2009 by identifying an ER protein, STING (stimulator of interferon genes), as a key component in DNA-mediated innate immunity [10,11,12,13]. In 2013, Dr. Chen’s group ultimately determined that cGAS is the direct cytosolic DNA sensor and that it activates innate immunity by activating type I IFN expression [14, 15].
Cytosolic DNA triggers the activation of cGAS-cGAMP-STING signaling. This signaling not only plays critical roles in the host defense against microbial infection, but also has been demonstrated to be involved in the antitumor immune response, and numerous studies have suggested that the activation of STING is a novel and promising strategy to treat cancer. In this review, we focus on STING and the cancer immune response and elaborate on the master roles of STING activation in regulating the cancer-immunity cycle.
STING induces the production of type I IFN and activates the innate immune system
Whether caused by leakage from the nucleus or mitochondria or induced by viruses or bacteria, cytoplasmic DNA is a danger signal. Once in the cytoplasm, dsDNA or single-stranded DNA (ssDNA) is sensed by a DNA sensor protein, cGAS, in a sequence-independent but length-dependent manner; cGAS catalyzes the synthesis of 2′3’-cyclic GMP-AMP (2′3’-cGAMP) by using ATP and GTP as substrates [14, 15], and it acts as a second messenger to bind and activate STING.
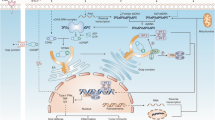
STING is a protein with four putative transmembrane domains and resides in the endoplasmic reticulum (ER) [12, 16], and it is widely expressed in both immune cells (including innate immune cells and adaptive immune cells) and non-immune cells. As a sensor of cyclic dinucleotides (CDNs), including both endogenous 2′3’-cGAMP catalyzed by cGAS in the presence of DNA and exogenous c-di-AMP, c-di-GMP or 3′3’-cGAMP from bacteria, STING binds to these small molecules, is activated, and translocates from the ER to the perinuclear area with the help of iRhom2, wherein STING activates the kinase TANK-binding kinase 1 (TBK1), which phosphorylates STING. Phosphorylated STING recruits interferon regulatory factor 3 (IRF3), which is phosphorylated by TBK1 and forms a homodimer to enter the nucleus and activates the transcription of type I IFNs and inflammatory cytokines and chemokines (Fig. 1) [17]. Notably, since cGAMP could be transferred via gap junction and through viral packaging, thus cGAMP may also activate STING in cells where cytoplasmic dsDNA is not available [18,19,20]. The modification and interaction with the components in this signaling pathway has been reviewed previously [17, 21, 22].

DNA-driven cGAS-cGAMP-STING signaling mediates innate immune response. The left cell exhibits the main components of cGAS-cGAMP-STING signaling pathway and IFN signaling pathway, and the right cell shows that IFN could activate neighbor cells in a paracrine manner and cGAMP could be transferred to neighbor cells through GAP junction
All type I IFNs (including well-documented IFN-α and IFN-β and less well-studied IFN-ε, IFN-κ, IFN-τ and IFN-ω) bind to heterodimer interferon receptors (IFNAR1 and IFNAR2). This results in the recruitment of Janus family kinase1 (Jak1) and tyrosine kinase 2 (Tyk2), and these, in turn phosphorylate and activate IFNAR1 and IFNAR2. The activation of IFNARs causes the recruitment and phosphorylation of effector proteins of the signal transducers and activators of transcription (STAT) family. Phosphorylated STAT1 and STAT2, together with IRF9, transfer to the nucleus, where they enhance the transcription of IFN target genes (reviewed in ref. [21, 23]), leading to the activation of both innate and adaptive immunity.
Numerous studies have shown that the expression levels of Type I IFNs and Type I IFN-induced genes in cancer cells positively correlate with T-cell infiltration in the tumor microenvironment [21]. Most importantly, IFNAR or STAT1 knockout mice fail to reject immunogenic tumors due to the less efficient induction of DC recruitment to tumors and the priming and expansion of CD8+ T cells in vivo [24,25,26]. Consistent with these studies, many previous studies also revealed that type I IFNs contribute to the control of tumors both in vivo and in vitro [27, 28]. These studies suggest that type I IFNs play central roles in the antitumor response. However, recent studies have suggested that type I IFNs may also impair anticancer immunity and even cause unexpected treatment failure for cancer. For example, IFN-β has been shown to induce the production of programmed cell death ligand 1 (PD-L1) and programmed cell death ligand 2 (PD-L2) in tumor cells [29, 30], which contributes to immune escape by cancer cells. Moreover, type I IFNs have been reported to be associated with resistance to radiotherapy and chemotherapy due to type I IFNs inducing DNA damage resistance in multiple cancer types [31, 32]. Additionally, type I IFNs have been revealed to contribute to unexpected autoimmune toxicity during cancer immunotherapy in the clinic [33]. Taken together, even though type I IFNs play central roles in anticancer immunity, immunotherapy directly based on type I IFNs may not be applicable in cancer treatment in the clinic.
It is currently believed that inducing the production of type I IFNs is one of the major mechanisms for STING signaling-mediated anticancer immunity. However, there is some evidence suggesting that STING also regulates anticancer immunity in a type I IFN-independent manner, which implies a broader application of STING (beyond IFNs) in cancer immunotherapy.
Activation of STING is a promising strategy for the cancer immunotherapy
Recent studies have suggested that STING signaling is necessary for the anticancer immune response based on the following observations: on the one hand, STING knockout mice and IRF3 knockout mice show impaired spontaneous T-cell responses against tumors [34, 35]; on the other hand, STING agonists show a favorable effect in promoting the infiltration of T cells into the tumor microenvironment [36, 37]. Moreover, numerous studies using the STING agonists to treat cancers demonstrate that activation of STING is a promising strategy for the cancer immunotherapy.
Actually, before identified the STING signaling, a chemotherapeutic agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA), first synthesized in 2002 as an antivascular agent, shows a promising anticancer effect, although the target molecules of DMXAA is unknown at the time [38]. Further studies show that the anticancer effect of DMXAA is associated with activation and infiltration of CD8+ T cells in murine models of several cancer types [39] and is dependent on type I INF production [40]. In 2012, DMXAA was finally shown to target STING and activate STING dependent type I INF induction [41]. As the first applied STING agonist in cancer immunotherapy, DMXAA showed promising antitumor activity in mice, but unfortunately, it failed in clinical trials because DMXAA does not preferentially bind to human STING [42, 43]. However, these researches strengthened the confidence of scientists to develop STING agonists to treat cancer. Nowadays, it has been demonstrated that STING activation is effective in anticancer in various cancer types, including hematological malignancies (such as acute myeloid leukemia and lymphoma) and solid tumors (such as lung cancer and melanoma). The roles of STING activation in different cancer types are summarized in Table 1.
In addition to DMXAA, there are other types of STING agonists have been developed, and the anticancer effect of those agents has been tested or under evaluated in clinic. CDNs, such as cGAMP and c-di-AMP, synthesized or acquired from microbes, represent the natural agents to bind and activate STING. However, these STING agonists are nonpenetrating [68], thus they must be delivered into cells via vectors, such as liposomes or nanoparticles [69]. Currently, some groups are developing novel CDN derivatives to perform clinical trials [70, 71]. In contrast, a very recent study reported a novel STING agonist, diABZIs, which is a small molecule developed based on amidobenzimidazole (ABZI) symmetry rather than CDNs that showed strong and systemic antitumor activity in a mouse colon cancer model [71]. The clinical studies using the STING agonists in different cancer types are summarized in Table 2.
STING signaling regulates the cancer-immunity cycle
Cancer cell death results in the exposure of cancer antigens; antigen-presenting cells (APCs), typically referred to as dendritic cells (DCs), capture the antigens and present them to T cells, and induce the activation of effector T cells. Next, effector T cells reach the tumor site and infiltrate tumors, where cytotoxic T lymphocytes (CTLs) identify and kill cancer cells. In turn, dead cancer cells release more antigens, which participate in the process above. This cyclic process is defined as the cancer-immune cycle [72]. The cancer-immunity cycle has become a research hotspot in recent years and provides a theoretical basis for tumor immunotherapy. There are a series of stimulatory and inhibitory factors involved in this cyclic process [72]. STING, as a stimulator of type I IFN production, has been demonstrated by an increasing number of studies to act as a master regulator and mediator in each step of the cancer-immunity cycle (Fig. 2).

Activation of STING positively regulates each step of cancer-immunity cycle
STING facilitates the release of cancer cell antigens
Tumor cells are main reason of producing cancer antigens, arise due to genome instability and high exposure to few oncogenes. However, these antigens cannot clearly seen, due to mutation or deletion of the MHC-coding genes in the cancer cells [73], which makes tumor to deceive the immune system. Therefore, APCs has ability to consume the proteins and even mRNAs coding for cancer antigens released by inactive tumor cells, which makes them to appear on the surface of APCs. Thus, this release starts the development of the cancer-immune cycle.
Recent studies have found that the activation of STING can directly trigger cancer cell death. Tang et al. reported that the STING agonist 3′3’-cGAMP is cytotoxic to malignant B cells and induces apoptosis in vitro and in vivo [60]. Mechanistically, they found that 3′3-cGAMP binds to STING and causes the phosphorylation and activation of STING in mouse embryonic fibroblasts. However, this agonist promotes the degradation of STING protein upon binding to it, and this process requires STING to interact with the ER stress sensor IRE-1. Unlike mouse embryonic fibroblasts, the 3′3’-cGAMP-STING interaction causes STING to aggregate in malignant B cells and leads to rapid apoptosis of these cells [60]. In addition to this, researches showed that the infection with human T-cell leukemia virus (HTLV-1) in monocytes, which become a reason of reversing transcription intermediates of HTLV-1, in order to collaborate with STING within the cytoplasm. This causes the production of an IRF3-Bax complex, which results in apoptosis of HTLV-1-infected monocytes [74].
Recently, it has been demonstrated that major histocompatibility complex class II (MHC-II) causes apoptosis of hematopoietic malignant cells [75, 76]. It has been revealed that STING protein is associated with MHC-II and mediates apoptosis of B lymphoma cells. Mechanistically, MHC-II aggregation results in tyrosine phosphorylation of STING, which triggers the activation of the extracellular signal-regulated kinase (ERK) signaling pathway and this process is necessary for MHC-II-mediated cell death signaling in a murine B lymphoma cell line [16]. Although MHC-II molecules have been reported to express in various cancer types [77,78,79], it is still not clear about the roles of the interaction of STING and MHC-II in inducing apoptosis of non-hematopoietic malignant cells currently. These studies suggest that STING activation and/or overexpression may trigger cell apoptosis and cause the release of tumor antigens in certain cancer types.
Activation of STING signaling is necessary for cancer antigen presentation
It has been demonstrated that radiation and chemotherapeutic agents induce antitumor immune responses depending on type I IFN when used to directly attack cells, and that STING is essential for such radiation-induced immune responses [50, 80]. Emerging evidence also indicates that dying cells can release endogenous adjuvant and facilitate activation of APCs [81]. When suffering nonphysiological damage, tumor cells release numerous danger-associated molecular patterns (DAMPs), which can trigger host immune responses [82]. Tumor cell-derived DNA is one of the most important DAMPs. DNA released from dead tumor cells can be found within the cytosol of intratumoral DCs [34]. Tumor-derived DNAs can be recognized by cytoplasmic DNA receptors in dendritic cells, macrophages, and other APCs and activate the cGAS-STING pathway to induce the expression of type I IFN [50].
DCs are the most potent professional APCs, and DC activation and antigen presentation are regulated by multiple factors, and type I IFN plays a particularly crucial role in the regulation of DCs. As early as 1998, T. Luft et al. demonstrated that type I IFN enhances the terminal differentiation of DCs [83]. Since then, R.L. Paquette [84] and L.G. Radvanyi [85] have found that type I IFN also facilitates the maturation of DCs. Recent studies have found that in addition to promoting DC maturation by inducing the expression of type I IFN, cGAMP or other STING agonists can directly activate DCs in vitro, and enhance presentation of tumor-associated antigens to CD8+ T cells [86, 87]. Furthermore, activation of STING signaling in DCs can induce additional protein expression to promote cross-presentation and T-cell activation [88]. Therefore, these studies suggest that, in order to generate adaptive antitumor immunity, STING must be activated by tumor-derived DNA or cGAMP for IFN expression and DC-mediated cross-priming.
STING signaling is responsible for the priming and activation of T cells
The priming and activation of T cells involve multiple signals, including T cell receptor (TCR) recognition and interaction with costimulatory molecules. In addition, cytokines play important roles in T-cell activation. It has been revealed that spontaneous T-cell priming and activation occur in the tumor microenvironment of some solid tumors [89]. Current research suggests that spontaneous tumor antigen-specific T-cell priming appears to be dependent on DC and type I IFN production in host cells [26].
Recently, Seng-Ryong Woo et al. reported that spontaneous T-cell priming was severely debilitated in STING-deficient and IRF3-deficient mice [35], and Olivier Demaria et al. also observed the same phenomenon in STING knockout mice compared with WT mice [36], which suggests that STING signaling may be necessary for the expansion of T cells. In addition, Olivier Demaria et al. [36] and Juan Fu et al. [90] both reported that mice with B16 melanoma treated with cGAMP showed an increase in CD8+ T-cell infiltration in the tumor microenvironment. These results imply that STING activation could facilitate T-cell priming and activation in the tumor microenvironment. However, these studies did not elaborate the detail mechanisms of STING signaling regulating this process. Since DCs and type I IFNs play critical roles in the priming and activation of T cells, and it has been revealed that tumor-derived DNA activates DCs and induces production of type I IFN in the tumor microenvironment [34], thus activation of STING signaling in DCs plays important and even exclusive roles in the spontaneous T cell responses against tumors. When it comes to applying STING agonists to stimulate T cell responses against tumors, T cells could be directly activated by STING agonists [91, 92] and indirectly activated by type I IFN produced by STING activated DCs.
Activation of STING pathway promotes the trafficking and infiltration of T cells to tumors
Before recognizing and killing cancer cells, CTLs must traffic to and infiltrate the tumor tissue. Chemokines play essential roles in regulating the development, priming, functions, homing and retention of T cells (reviewed in ref. [93, 94]). Previous studies demonstrated that the infiltration of CD8+ T cells in the tumor microenvironment is associated with C-X-C motif chemokine ligand 9 (CXCL9), C-C motif chemokine 5 (CCL5) and C-X-C motif chemokine ligand 10 (CXCL10) [95], and the expression of CXCL9 and CXCL10 could be induced in response to type I IFN production by APCs [96], which suggests that APCs play important roles in the trafficking and infiltration of CD8+ T cells. Recently, L Corrales et al. reported that elevated expression of CXCL9 and CXCL10 in DCs is associated with the activation of the STING pathway and contributes to trafficking and infiltration of CD8+ T cells in a xenograft animal model [97]. In addition to DCs, some other immune cells have also been found to be involved in STING-mediated T-cell trafficking. For example, Ohkuri T et al. observed macrophage aggregation after intratumoral injection of cGAMP in mice; however, no aggregation was observed in STING knockout mice. After depletion of mouse macrophages, the antitumor effect induced by cGAMP disappeared, and the mechanistic analysis revealed that STING-induced migrating tumor macrophages express high levels of T-cell-recruiting chemokines, such as CXCL10 and C-X-C motif chemokine ligand 11 (CXCL11), which then contribute to CD8+ T-cell trafficking to the tumor site [98]. In another study, it has been revealed that intratumoral injection of STING agonist (c-di-GMP) activated STING/type I IFN signaling in the CD11b+ brain-infiltrating leukocytes (instead of CD11c+ DCs), in which CXCL10 and CCL5 expression was increased and then contributed to the migration of CD8+ T cells into the glioma [37]. These results show that activation of the STING pathway in APCs and other immune cells can induce the expression of cytokines and thereby promote T-cell trafficking.
Other than immune cells, recent researches showed that the STING activation within endothelial cells causes the infiltration of T cells into solid tumors. Demaria and colleagues found that spontaneous infiltration of CD8+ T cells in an engrafted melanoma is significantly reduced in STING knockout mice compared with WT mice. Furthermore, they demonstrated that intratumoral injection of cGAMP promotes the infiltration of CD8+ T cells into engrafted melanoma [36]. Mechanistically, they revealed that STING-induced IFN-β contributes to the infiltration of CD8+ T cells because the blockage of IFN signaling by anti-IFNAR antibodies or IFNAR ablation completely abolished CD8+ T-cell infiltration [36]. By detecting the expression of intracellular IFN-β within tumor-cell-derived single cells, the authors revealed that IFN-β-producing cells in the tumor express low levels of CD45 (a general marker of hematopoietic cells) but high levels of CD31 and vascular endothelial growth factor receptor 2 (VEGFR-2) (the specific marker of endothelial cells), suggesting that activation of STING pathway by exogenous STING agonists in endothelial cells, instead of DC cells or other immune cells, facilitate the infiltration of CD8+ T cells into the tumor microenvironment [36]. Consistently, another study also found that STING expression in endothelial cells is positively correlated with the infiltration level of CD8+ T cells and prolonged survival in several human cancer types (eg. colon and breast cancer) by using immunohistochemistry staining [59]. However, authors revealed that non-hematopoietic cells play important roles in the infiltration of CD8+ T cells into tumor microenvironment by employing bone morrow chimeric mice models, they did not show the direct evidence to illustrate the accurate roles of STING activation in endothelial cells in the process of T cell infiltration [59]. These results revealed an unexpected role of endothelial cells within the tumor microenvironment in cancer immunity, and suggested that STING activation in endothelial cells is necessary for the infiltration of CTLs.
Adhesion to endothelial cells is a necessary step for the infiltration of T cells into the tumor microenvironment. Current studies have demonstrated that vascular endothelial growth factor (VEGF) and other cytokines secreted by cancer cells inhibit the expression of molecules on endothelial cells that mediate the adhesion of T cells or induce the expression of molecules that trigger cell death of effecter T cells (reviewed in ref. [99, 100]). Moreover, the depletion of CD8+ T cells has been shown to abrogate the therapeutic efficacy of VEGF inhibition by using an anti-VEGFR antibody in a certain cancer model [101]. Thus, inhibition of VEGF signaling promotes the infiltration of T cells into the tumor microenvironment. Consistent with these studies, Hannah and colleagues demonstrated that STING agonists (10 μg of cGAMP or 25 μg ofRR-CDA) treatment combined with VEGFR2 blockade (DC101) enhanced the infiltration of CD8+ T cells in the tumor microenvironment and induces complete tumor regression [59], this exciting result suggests that simultaneously targeting STING and VEGF signaling represents a promising strategy for cancer therapy. However, it must be aware that combined using immunotherapy and anti-angiogenic therapy targeting VEGF or VEGFR may be not effective in certain conditions, because it has been indicated that VEGF inhibition is not beneficial in some human solid tumor types (NSABP-C-08; clinicaltrials.gov: NCT00096278) and even results in progression in certain cancer types (reviewed in [102, 103]). These unexpected phenomenons may be partially explained by that the blockade of VEGF may inhibit the infiltration of T cells by suppressing the proliferation of endothelial cells within tumors in some conditions because appropriate level of VEGF is necessary for maintaining the number of endothelial cells.
Although multiple studies have found that injection of a STING agonist in a tumor-bearing mouse model enhanced the infiltration of T cells into the tumor microenvironment [37, 90], and several types of cells, such as DCs, macrophages and endothelial cells, have been identified to help infiltration of T cells into tumor microenvironment in responding to activation of STING pathway by exogenous STING agonists in different models, the direct effect of STING activation within T cells on their trafficking and infiltration is not evaluated currently. An in vitro study showed that exogenous STING agonist DMXAA activates STING signaling, and then induces type I IFN production and IFN-stimulated gene expression [91, 92], thus the STING activated CTLs by exogenous STING agonists may mirror the response of innate cells and induce more CTLs to migrate and infiltrate into tumor microenvironment. However, further studies are needed to detect this hypothesis.
STING activation is necessary for the recognition and killing of cancer cells by T cells
Antigen binding by MHC followed by recognition and interaction with the TCR is a critical step for T-cell recognition of cancer cells [104]. After recognizing tumor cells, activated CTLs can release cytokines, such as IFN-γ and other factors, to mediate tumor cell death [105].
Numerous studies have reported that STING activation promotes the antitumor effect of CD8+ T cells. It has been reported that antigen-specific CD8+ T-cell responses were diminished in STING-deficient in a murine radiation-mediated antitumor immunity model [50]. Consistently, another study also revealed that the CD8+ T-cell response to tumor-associated antigens was diminished in both STING-deficient and IRF3-deficient mice [35]; these data suggest that host-cell STING and IRF3 are required for spontaneous CD8+ T-cell activity against immunogenic tumors. Furthermore, Ohkuri T et al. found that STING-deficient mice had fewer IFN-γ-producing CD8+ T cells but increased infiltration of immune-suppressing cells, such as CD11b+Gr-1+ immature myeloid suppressor cells and CD25+Foxp3+ regulatory T (Treg) cells, in the tumor microenvironment [37], whereas STING agonist CDN treatment promoted cross-presentation and helped T cells recognize tumor cells [106]. These data suggest significant contributions of STING to T-cell-mediated antitumor immunity via enhancement of type I IFN signaling in the tumor microenvironment.
STING activation negatively regulates cancer immunity
Current studies show that STING activation facilitates the antitumor immune response in most conditions; however, emerging studies also suggest a potential inhibitory effect of STING activation on antitumor immune responses.
Although numerous studies have suggested that STING activation by exogenous cGAMP facilitates the priming and activation of T cells, two recent independent critical studies showed that STING activation in T cells prevents their proliferation and even promotes their death [91, 92]. The proliferation of T lymphocytes with constitutively active STING mutations was found to be impaired; the impairment was dependent on nuclear factor κB (NF-κB) instead of TBK1 and IRF3, due to mitotic errors resulting from STING relocalization to the Golgi apparatus after activation [91]. In another study, it was shown that STING agonist DMXAA activates the cell stress pathways within T cells and finally induces cell death of T cells [92]. These two studies suggest that STING activation in T cells could directly impair the adaptive immune system.
Additionally, it has been suggested that activation of STING signaling also activates immune suppressive cells in certain conditions. We evaluated the relationship between STING expression and the infiltration of 28 types of immune cells in 17 human malignant tumor types based on the TCGA data set and showed that the STING pan-cancer expression level is positively correlated with the infiltration of almost all types of immune cells, including both antitumor immune cells, such as DCs and CTLs, and immune-suppressing cells, such as myeloid-derived suppressor cells (MDSCs) and Tregs [107]. Our unexpected finding is consistent with several previous studies. A study in HPV+ tongue squamous cell carcinoma (TSCC) indicated that activated STING has no impact on cancer cell viability but promotes the induction of immunosuppressive cytokines, such as IL-10, which facilitated the infiltration of Tregs [67], whereas enriched Tregs can express IL-10 to inhibit the proliferation and activity of antigen-specific T cells [104]. In another study, Lemos and colleagues found that STING signaling contributes to the growth of Lewis lung carcinoma (LCC) by promoting the infiltration of MDSCs while decreasing the infiltration of CD8+ T cells in the tumor microenvironment [108]. Mechanistically, they revealed that the indoleamine 2,3-dioxygenase (IDO) activity is elevated significantly in LCC tumor microenvironment from WT mice compared with STING knockout or IFNAR knockout mice. Moreover, the effect of STING promoting tumor growth in LCC model is attenuated either knocking out IDO gene or following treatment with IDO inhibitors [108], suggesting that induction of IDO plays central roles in STING activation mediated tumor growth.
IDO is considered an enzyme, which accelerates the transformation of tryptophan into kynurenine. This is incurred due to innate immune response. It also plays a counter-regulatory role in the inflammation and activation of T cells [109]. IDO is also responsible to vanquish the effector’s T cells by doing metabolic depletion of tryptophan and formation of kynurenine. The depletion tryptophan restricts the escalation of both CD8+ and CD4+ T cells, from the local microenvironment, through blocking the ribosomal translation, incurred due to amino acid withdraw (this process is controlled by molecular stress-response pathways, reviewed in ref. [110,111,112]). On the other hand, kynurenine boost the differentiation of Foxp3+ Tregs, along with negatively regulating the dendritic cell immunogenicity via binding and activating a ligand-activated transcription factor AhR (aryl hydrocarbon receptor) [113, 114].
Enhanced IDO activity is commonly observed in the tumor microenvironment and is believed to be associated with cancer immune evasion (reviewed in ref. [115]). In addition to a recent study directly demonstrated that STING activation contributes to tumor growth [108], there are also many previous studies found that systemic treatment with DNA-containing nanoparticles stimulates IDO activity in many mouse tissues due to STING activation in innate immune cells [116], which activates Tregs and suppresses the T cell responses [117].
Notably, although the impact of STING agonists on the activation of IDO in the immune system or tumor microenvironment has not yet been evaluated, this potential effect on IDO activation must be investigated before applying STING agonists to treat cancer, whereas combined using IDO inhibitors may enhance the immunotherapeutic effect of STING agonists.
In addition to Tregs, programmed cell death1 (PD-1) and other immune checkpoint molecules are also involved in inhibiting Tcell-mediated immunity [118, 119]. Recent studies showed that the activation of STING by c-di-GMP in infiltrated CD8+ T cells results in increased expression of PD-1 pathway components in multiple murine cancer types, including colon, tongue squamous carcinoma, pancreatic carcinoma and head and neck squamous cell carcinoma models [55, 90]. However, combined with the PD-1 pathway blockade, the increased expression of PD-1 is beneficial to the antitumor effect of STING agonisits [55, 90]. Together, these studies suggest that apart from playing positive roles in anticancer immune response, STING may hamper the antitumor immune response after it is inappropriately activated (Fig. 3).

The positive and negative roles of STING activation in antitumor immune response. On the one hand, STING facilitates antitumor immune response through promoting the infiltration of effector cells and eradication of tumor cells. On the other hand, constant STING activation may hamper immune response by inducing the infiltration of immune suppressive cells, such as Treg and MDSC, and upregulating the expression of PD-L1 on tumor cells and PD-1 on T cells. Moreover, STING activation is associated with the enhanced activity of IDO, an enzyme catalyzing the transformation of tryptophan into kynurenine. Diminished tryptophan restricts the proliferation of T cells whereas elevated kynurenine promotes differentiation of Tregs but hampers antigen presenting ability of DCs. Additionally, aberrant STING activation also directly inhibits T cell proliferation and even promotes apoptosis of lymphocytes
Non-immune functions of STING
In addition to regulating anticancer immunity, the non-immune functions of STING are emerging.
Firstly, STING activation results in cell apoptosis. For instance, STING agonists cause apoptosis of certain immune cells, including B cells and even T cells, in vitro and in vivo [60, 91, 92]. In addition to immune cells, activation of STING signaling also induces hepatocyte apoptosis in early alcoholic liver disease. Ethanol causes ER stress and triggers phosphorylation and activation of IRF3 by interacting with STING, activated IRF3 associates with Bax and induces apoptosis of hepatocytes, whereas deficiency of STING prevents hepatocyte apoptosis [120].
Secondly, STING mediates autophagy. For example, by sensing bacterial or viral PAMPs, STING signaling is activated and triggers ER stress; subsequently, STING localizes to autophagosomes from the ER, which provides a homeostatic mechanism to balance immunity and survival after infection [121, 122]. Liu et al. reported that STING directly interacts with LC3 and induces autophagy; however, cGAMP binding to STING activates the immune response, but the complex fails to interact with LC3 and reduces autophagy [123].
A very recent study confirmed that STING translocates to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) upon binding cGAMP, and this STING-containing ERGIC, which is a membrane source for the autophagosome biogenesis, through autophagy-related protein 5 (ATG5) and WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) dependent pathway [124]. No doubt, the STING molecule regulates autophagy process, but the crosstalk between autophagy and immune response upon cGAMP binding STING needs further experimentation to explore.
Thirdly, STING also regulates cell proliferation by regulating the cell cycle. Ranoa and colleagues found that STING knockout in human and murine cancer cells lead to increased proliferation compared with wild-type controls. Mechanistically, they revealed that STING deficiency results in activation of cyclin-dependent kinase 1 (CDK1) and facilitates onset of S and M phase of the cell cycle in P53-activated P21 dependent manner [125]. This study implies that STING not only regulates cell death or survival, but also affects cell proliferation.
Fourthly, STING activation contributes to normalization of the tumor vasculatures. Chemotherapeutic agent DMXAA was firstly designed as an antivascular agent before it was identified to target STING [38], this widely used STING agonist shows rapid and strong antivascular activity in tumors but not in normal tissues, and it is effective to control tumor growth by regulating the vasculatures in various murine cancer models (reviewed in ref. [126]). Consistently, a very recent study also reported that intratumoral injection of other STING agonists (cGAMP or RR-CDA) normalizes the tumor vasculatures in spontaneous or implanted cancers, but this phenomenon is not observed in the STING deficient mice, which implies that STING activation is necessary for the normalization of the tumor vasculatures [59]. Mechanistically, they revealed that endothelial STING activation upregulates vascular stabilizing genes, such as Angpt1, Pdgfrb, and Col4a, in a type I IFN signaling dependent manner [59]. Notably, when combined with VEGFR2 blockade, STING agonists cause the complete regression of immunotherapy-resistant tumors [59]. These studies suggest that STING signaling in the tumor microenvironment regulates angiogenesis.
Finally, several studies found that activation of STING facilities cancer metastasis. It has been reported that the metastatic cancer cells transfer cGAMP to the astrocyte through carcinoma-astrocyte gap junctions, in which cGAMP activates STING pathway and induces production of inflammatory cytokines, these factors activate STAT1 and NF-κB pathway in cancer cells and thereby facilitate the survival and growth of metastatic cancer cells [127]. A similar study was done, which found that chromosomal instability also become a reason of accumulation of micronuclei in the cytoplasm of cancer cells, which results in activation of the STING pathway and downstream the NF-κB signaling, thereby promoting cancer metastasis [128].
Taking these considerations together, it is necessary to evaluate the non-immune functions of STING before using STING agonists to treat cancer in the clinic.
Concluding remarks and perspectives
Since STING plays a critical role in innate immunity, the potential application of STING regulation in infectious diseases, autoimmune diseases and cancer has attracted great interests. In this review, we focused on the roles of STING in cancer immunity by elaborating on its effect at each step of the cancer-immunity cycle. Conclusively, STING is a potent regulator of cancer immunity functioning at each step of the cancer-immunity cycle, and thus activation of STING represents a promising strategy for cancer immunotherapy by developing safe and efficient STING agonists. However, accompanied with the STING-mediated activation of antitumor immune responses, potential immune inhibitory effects of STING are emerging and nonnegligible. In addition to this, it has been observed that STING activation also contributes in cancer initiation and progression, by activating cancer associated inflammation, when it induces type I IFN responses. Thus, it must be thoroughly evaluated, before the STING agonists are used to stimulate the anticancer immune response, and combined using immune checkpoint blockade therapy, such as IDO inhibitor, anti-PD-L1 and anti-PD-1 antibodies, may increase the therapeutic effects of STING agonists in the clinic.
Availability of data and materials
Not applicable.
Abbreviations
- ABZI:
-
Amidobenzimidazole
- AhR:
-
Aryl hydrocarbon receptor
- APC:
-
Antigen-presenting cells
- ATG5:
-
Autophagy-related protein 5
- CCL5:
-
C-C motif chemokine 5
- CDK1:
-
Cyclin-dependent kinase 1
- CDN:
-
Cyclic dinucleotide
- cGAMP:
-
Cyclic GMP-AMP
- CTL:
-
Cytotoxic T lymphocytes
- CXCL10:
-
C-X-C motif chemokine ligand 10
- CXCL11:
-
C-X-C motif chemokine ligand 11
- CXCL9:
-
C-X-C motif chemokine ligand 9
- DAMP:
-
Danger-associated molecular pattern
- DC:
-
Dendritic cell
- DMXAA:
-
5,6-dimethylxanthenone-4-acetic acid
- dsDNA:
-
Double-strand DNA
- EBV:
-
Epstein-Barr virus
- ER:
-
Endoplasmic reticulum
- ERGIC:
-
Endoplasmic reticulum-Golgi intermediate compartment
- ERK:
-
Extracellular signal-regulated kinase
- HPV:
-
Human papilloma virus
- HTLV-1:
-
Human T-cell leukemia virus
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IRF3:
-
Interferon regulatory factor 3
- Jak1:
-
Janus family kinase1
- LCC:
-
Lewis lung carcinoma
- LPS:
-
Lipopolysacchride
- MDSCs:
-
Myeloid-derived suppressor cell
- MHC-II:
-
Major histocompatibility complex class II
- NF-κB:
-
Nuclear factor κB
- NLR:
-
NOD-like receptor
- NSCLC:
-
Non-small cell lung cancer
- PAMP:
-
Pathogen-associated molecular pattern
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand 1
- PD-L2:
-
Programmed cell death ligand 2
- PRR:
-
Pattern recognition receptor
- R, R-CDA:
-
RP, RP dithio c-di-AMP
- R, R-CDG:
-
RP, RP dithio c-di-GMP
- RLR:
-
RIG-like receptor
- SCLC:
-
Small cell lung cancer
- SLE:
-
Systemic lupus erythematosus
- ssDNA:
-
Single-stranded DNA
- STAT:
-
Signal transducers and activators of transcription
- STING:
-
Stimulator of interferon genes
- TBK1:
-
TANK-binding kinase 1
- TCR:
-
T cell receptor
- TLR:
-
Toll-like receptor
- Treg:
-
T regulaory cell
- TSCC:
-
Tongue squamous cell carcinoma
- Tyk2:
-
Tyrosine kinase 2
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- WIPI2:
-
WD repeat domain phosphoinositide-interacting protein 2
References
Kienle GS. Fever in Cancer treatment: Coley’s therapy and epidemiologic observations. Glob Adv Health Med. 2012;1:92–100.
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000;343:338–44.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Wang X, Smith C, Yin H. Targeting toll-like receptors with small molecule agents. Chem Soc Rev. 2013;42:4859–66.
Motta V, Soares F, Sun T, Philpott DJ. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev. 2015;95:149–78.
Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90.
O'Neill LA. DNA makes RNA makes innate immunity. Cell. 2009;138:428–30.
Ablasser A, Hertrich C, Wassermann R, Hornung V. Nucleic acid driven sterile inflammation. Clin Immunol. 2013;147:207–15.
Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674.
Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92.
Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106:8653–8.
Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–50.
Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–91.
Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–30.
Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014–26.
Tao J, Zhou X, Jiang Z. cGAS-cGAMP-STING: the three musketeers of cytosolic DNA sensing and signaling. IUBMB Life. 2016;68:858–70.
Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–4.
Bridgeman A, Maelfait J, Davenne T, Partridge T, Peng Y, Mayer A, Dong T, Kaever V, Borrow P, Rehwinkel J. Viruses transfer the antiviral second messenger cGAMP between cells. Science. 2015;349:1228–32.
Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–6.
Corrales L, Matson V, Flood B, Spranger S, Gajewski TF. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017;27:96–108.
Galluzzi L, Vanpouille-Box C, Bakhoum SF, Demaria S. SnapShot: CGAS-STING Signaling. Cell. 2018;173:276 e1.
Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev. 2012;250:317–34.
Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, Schreiber RD. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722–9.
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–16.
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003.
Gresser I, Bandu MT, Brouty-Boye D. Interferon and cell division. IX. Interferon-resistant L1210 cells: characteristics and origin. J Natl Cancer Inst. 1974;52:553–9.
Gresser I, Bourali C. Antitumor effects of interferon preparations in mice. J Natl Cancer Inst. 1970;45:365–76.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Morimoto Y, Kishida T, Kotani SI, Takayama K, Mazda O. Interferon-beta signal may up-regulate PD-L1 expression through IRF9-dependent and independent pathways in lung cancer cells. Biochem Biophys Res Commun. 2018;507:330–6.
Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490–5.
Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017;31:353–69.
Walsh SR, Bastin D, Chen L, Nguyen A, Storbeck CJ, Lefebvre C, Stojdl D, Bramson JL, Bell JC, Wan Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J Clin Invest. 2019;129:518–30.
Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36:250–6.
Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–42.
Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A. 2015;112:15408–13.
Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, Watkins SC, Sarkar SN, Okada H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2:1199–208.
Ching LM, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-Dimethylxanthenone-4-acetic acid. Br J Cancer. 2002;86:1937–42.
Jassar AS, Suzuki E, Kapoor V, Sun J, Silverberg MB, Cheung L, Burdick MD, Strieter RM, Ching LM, Kaiser LR, Albelda SM. Activation of tumor-associated macrophages by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid induces an effective CD8+ T-cell-mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res. 2005;65:11752–61.
Roberts ZJ, Ching LM, Vogel SN. IFN-beta-dependent inhibition of tumor growth by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA). J Interf Cytokine Res. 2008;28:133–9.
Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 2012;287:39776–88.
Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–25.
Lara PN Jr, Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:2965–71.
Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr, Duttagupta P, Kortylewski M, Kline J. STING pathway activation stimulates potent immunity against acute myeloid leukemia. Cell Rep. 2016;15:2357–66.
Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, Akira S, Matsuda T, Kawai T. DNA-containing Exosomes derived from Cancer cells treated with Topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol. 2017;198:1649–59.
Pantelidou C, Sonzogni O, De Oliveria TM, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via Intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast Cancer. Cancer Discov. 2019;9:722–37.
Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–10.
Gaston J, Cheradame L, Yvonnet V, Deas O, Poupon MF, Judde JG, Cairo S, Goffin V. Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget. 2016;7:77205–24.
Chen J, Markelc B, Kaeppler J, Ogundipe VML, Cao Y, McKenna WG, Muschel RJ. STING-dependent interferon-lambda1 induction in HT29 cells, a human colorectal Cancer cell line, after gamma-radiation. Int J Radiat Oncol Biol Phys. 2018;101:97–106.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–52.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736.
Baird JR, Bell RB, Troesch V, Friedman D, Bambina S, Kramer G, et al. Evaluation of explant responses to STING ligands: personalized Immunosurgical therapy for head and neck squamous cell carcinoma. Cancer Res. 2018;78:6308–19.
Lu S, Concha-Benavente F, Shayan G, Srivastava RM, Gibson SP, Wang L, Gooding WE, Ferris RL. STING activation enhances cetuximab-mediated NK cell activation and DC maturation and correlates with HPV(+) status in head and neck cancer. Oral Oncol. 2018;78:186–93.
Gadkaree SK, Fu J, Sen R, Korrer MJ, Allen C, Kim YJ. Induction of tumor regression by intratumoral STING agonists combined with anti-programmed death-L1 blocking antibody in a preclinical squamous cell carcinoma model. Head Neck. 2017;39:1086–94.
Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, Allen C. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination STING activation and PD-1 pathway blockade. Cancer Immunol Res. 2016;4:1061–71.
Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019;129:1211–28.
Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung Cancer. Cancer Discov. 2019;9:646–61.
Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3’-cGAMP, induces M2 macrophage repolarization. PLoS One. 2014;9:e99988.
Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, Kim S, Kim G, Chon HJ, Kim C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. 2019;130:4350–64.
Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, Hu CC. Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 2016;76:2137–52.
Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49:754–63.e4.
Zhang CX, Ye SB, Ni JJ, Cai TT, Liu YN, Huang DJ, et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019;26(11):2314–28.
Ghaffari A, Peterson N, Khalaj K, Vitkin N, Robinson A, Francis JA, Koti M. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer. 2018;119:440–9.
Jing W, McAllister D, Vonderhaar EP, Palen K, Riese MJ, Gershan J, Johnson BD, Dwinell MB. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer. 2019;7:115.
Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, Cheung FS, Lim WK, Tan PH, Ngeow J, Gasser S. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate Cancer cells. Immunity. 2016;44:1177–89.
Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T-cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5:676–84.
Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, Qin-Gang H, Yan-Hong N, Ya-Yi H. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-Jun/CCL22 signal. Biochim Biophys Acta. 1852;2015:2494–503.
Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ. Liposomal delivery enhances immune activation by STING agonists for Cancer immunotherapy. Adv Biosyst. 2017;1:1600013.
Konate K, Dussot M, Aldrian G, Vaissiere A, Viguier V, Ferreiro Neira I, Couillaud F, Vives E, Boisguerin P, Deshayes S. Peptide-based nanoparticles to rapidly and efficiently “Wrap’n roll” siRNA into cells. Bioconjug Chem. 2019;30:592–603.
Berger G, Lawler SE. Novel non-nucleotidic STING agonists for cancer immunotherapy. Future Med Chem. 2018;10:2767–9.
Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–43.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Tsioulias GJ, Triadafilopoulos G, Goldin E, Papavassiliou ED, Rizos S, Bassioukas P, Rigas B. Expression of HLA class I antigens in sporadic adenomas and histologically Normal mucosa of the Colon. Cancer Res. 1993;53:2374–8.
Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe. 2013;14:422–34.
Setterblad N, Blancheteau V, Delaguillaumie A, Michel F, Becart S, Lombardi G, Acuto O, Charron D, Mooney N. Cognate MHC-TCR interaction leads to apoptosis of antigen-presenting cells. J Leukoc Biol. 2004;75:1036–44.
Nagy ZA, Hubner B, Lohning C, Rauchenberger R, Reiffert S, Thomassen-Wolf E, et al. Fully human, HLA-DR-specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8:801–7.
Pisapia L, Barba P, Cortese A, Cicatiello V, Morelli F, Del Pozzo G. EBP1 protein modulates the expression of human MHC class II molecules in non-hematopoietic cancer cells. Int J Oncol. 2015;47:481–9.
Park IA, Hwang SH, Song IH, Heo SH, Kim YA, Bang WS, Park HS, Lee M, Gong G, Lee HJ. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLoS One. 2017;12:e0182786.
He Y, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, Yu H, Zhou C, Hirsch FR. MHC class II expression in lung cancer. Lung Cancer. 2017;112:75–80.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7.
Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89.
Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804.
Luft T, Pang KC, Thomas E, Hertzog P, Hart DN, Trapani J, Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–53.
Paquette RL, Hsu NC, Kiertscher SM, Park AN, Tran L, Roth MD, Glaspy JA. Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J Leukoc Biol. 1998;64:358–67.
Radvanyi LG, Banerjee A, Weir M, Messner H. Low levels of interferon-alpha induce CD86 (B7.2) expression and accelerates dendritic cell maturation from human peripheral blood mononuclear cells. Scand J Immunol. 1999;50:499–509.
Skrnjug I, Guzman CA, Rueckert C. Cyclic GMP-AMP displays mucosal adjuvant activity in mice. PLoS One. 2014;9:e110150.
Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114:1637–42.
Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–70.
Bui JD, Schreiber RD. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol. 2007;19:203–8.
Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52.
Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med. 2017;214:1769–85.
Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J Immunol. 2017;199:397–402.
Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–16.
Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33:496–504.
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85.
Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol. 2002;71:669–76.
Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–30.
Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother. 2017;66:705–16.
Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73.
Shimizu K, Iyoda T, Okada M, Yamasaki S, Fujii SI. Immune suppression and reversal of the suppressive tumor microenvironment. Int Immunol. 2018;30:445–54.
Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, Hicklin DJ, Jaffee EM, Emens LA. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951–9.
Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–21.
Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15:5020–5.
Murphy KP, Travers P, Walport M, Janeway C. Janeway’s immunobiology. New York: Garland science; 2014.
Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res. 2003;9:84–92.
Lirussi D, Ebensen T, Schulze K, Trittel S, Duran V, Liebich I, Kalinke U, Guzman CA. Type I IFN and not TNF, is Essential for Cyclic Di-nucleotide-elicited CTL by a Cytosolic Cross-presentation Pathway. EBioMedicine. 2017;22:100–11.
An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-Cancer. Mol Ther Nucleic Acids. 2018;14:80–9.
Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, Mellor AL. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–81.
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3.
Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42.
Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–8.
Palm CA, Smukler SM, Sullivan CC, Mutuo PK, Nyadzi GI, Walsh MG. Identifying potential synergies and trade-offs for meeting food security and climate change objectives in sub-Saharan Africa. Proc Natl Acad Sci U S A. 2010;107:19661–6.
Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:19961–6.
Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193–207.
Huang L, Lemos HP, Li L, Li M, Chandler PR, Baban B, McGaha TL, Ravishankar B, Lee JR, Munn DH, Mellor AL. Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. J Immunol. 2012;188:4913–20.
Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol. 2013;191:3509–13.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13.
Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, Szabo G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110:16544–9.
Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell. 2017;171:809–23.e13.
Liu Y, Gordesky-Gold B, Leney-Greene M, Weinbren NL, Tudor M, Cherry S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe. 2018;24:57–68.e3.
Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26:1735–49.
Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, Chen ZJ. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–6.
Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, et al. STING promotes homeostasis via regulation of cell proliferation and chromosomal stability. Cancer Res. 2019;79:1465–79.
Daei Farshchi Adli A, Jahanban-Esfahlan R, Seidi K, Samandari-Rad S, Zarghami N. An overview on Vadimezan (DMXAA): The vascular disrupting agent. Chem Biol Drug Des. 2018;91:996–1006.
Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–8.
Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–72.
Acknowledgements
None.
Funding
This study was supported by the National Natural Scientific Foundation of China (No. 81872435 and No. 81672930 to Tongsen Zheng, No. 81871976 to Xiaobo Li), the national youth talent support program for Tongsen Zheng (W03070060), the Fok Ying Tung Education Foundation (No. 151037), the Academician Yu Weihan Outstanding youth foundation of Harbin Medical University for Tongsen Zheng.
Author information
Authors and Affiliations
Contributions
LXB and ZTS offered main direction and significant guidance of this manuscript. ZYY, AX, ZX and QY drafted the manuscript and illustrated the figures for the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhu, Y., An, X., Zhang, X. et al. STING: a master regulator in the cancer-immunity cycle. Mol Cancer 18, 152 (2019). https://doi.org/10.1186/s12943-019-1087-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1087-y