
Abstract
At present, after extensive studies in the field of cancer, cancer stem cells (CSCs) have been proposed as a major factor in tumor initiation, progression, metastasis, and recurrence. CSCs are a subpopulation of bulk tumors, with stem cell-like properties and tumorigenic capabilities, having the abilities of self-renewal and differentiation, thereby being able to generate heterogeneous lineages of cancer cells and lead to resistance toward anti-tumor treatments. Highly resistant to conventional chemo- and radiotherapy, CSCs have heterogeneity and can migrate to different organs and metastasize. Recent studies have demonstrated that the population of CSCs and the progression of cancer are increased by the deregulation of different epigenetic pathways having effects on gene expression patterns and key pathways connected with cell proliferation and survival. Further, epigenetic modifications (DNA methylation, histone modifications, and RNA methylations) have been revealed to be key drivers in the formation and maintenance of CSCs. Hence, identifying CSCs and targeting epigenetic pathways therein can offer new insights into the treatment of cancer. In the present review, recent studies are addressed in terms of the characteristics of CSCs, the resistance thereof, and the factors influencing the development thereof, with an emphasis on different types of epigenetic changes in genes and main signaling pathways involved therein. Finally, targeted therapy for CSCs by epigenetic drugs is referred to, which is a new approach in overcoming resistance and recurrence of cancer.
Similar content being viewed by others


Introduction
Despite the myriad of studies and advances in cancer treatment, cancer remains one of the main causes of death globally. The rapid detection of cancer is significantly beneficial in the treatment thereof through common therapy such as surgery, chemotherapy and radiotherapy. However, a small subpopulation of cells have been detected in various tumors that are resistant to therapy and remain inside the tumor. Here, these cells grow and cause recurrence, metastasis and spread of new tumors. Being referred to as cancer stem cells (CSCs) and [1], having the ability of self-renewal and differentiation into different cell types. Due to these characteristics, CSCs can form heterogeneous lineages of cancer cells which are also preserved during metastasis. CSCs heterogeneity is one of the main problems of treatment because these cells ensure the survival of cancer cells in difficult conditions and are resistant to treatment [2,3,4]. Therefore heterogeneity of CSCs increases the viability of tumor cells and their invasion into other tissues [5, 6]. CSCs are located in an microenvironment called niche, which help them to maintain their characteristics [7]. Different theories have suggested that CSCs may have been produced from normal stem cells/progenitor by oncogenic mutations or from cancer cells and or differentiated cells by abnormal genetic and epigenetic changes (Fig. 1) [8]. CSCs have specific markers that distinguish them from other tumor cells, several of which are specific to different types of cancer and even different stages of cancer development [5]. CSCs have a high ability to metastasize, which can lead to recurrence of the disease years after primary tumor treatment [9]. A plethora of evidence has demonstrated that genetic and epigenetic changes by inducing self-renewal, differentiation and metastasis in CSCs, render significant resistance to chemo- and radiotherapy [1, 10,11,12,13]. CSCs can spread the cancer in sites farther from the main tumor, survive and multiply in the new organs, and thus cause metastatic tumors, which leads to the return of the disease and is known as one of the main causes of death in patients [14, 15]. Moreover, studies have shown that CSCs can exit the cell cycle and enter a state of quiescence (G0 phase) that increases the survival thereof against treatment. Therefore, part of the resistance of CSCs can also be attributed to the quiescence state and lack of DNA synthesis, while most chemotherapeutic agents affect cells that actively divide and synthesize DNA [10]. In addition, with regard to excision repair cross-complementation group 1 (ERCC1) expression involved in DNA nucleotide excision repair (NER) pathway and O(6)-methylguanine-DNA methyltransferase, observations have been made that a DNA-repairing enzyme is higher in CSCs than non-CSCs [7, 16]. Researchers have also elucidated that, compared with other tumor cells. DNA damage checkpoint responses such as NBS1, Chk1 and Chk2 are more expressed in CSCs, which can help repair damaged DNA caused by drugs. Overexpression of DNA-repairing kinases in CSCs also protects DNA from radiation-induced damage, with the DNA damage repair system in CSCs actually being more efficient [10]. In addition, overexpression and high activity of ROS scavenger enzyme such as catalase, superoxide reductase, superoxide dismutase, glutathione reductase, glutathione peroxidase has been observed in CSCs that reduces the ROS produced during treatment compared to other cancer cells. Reducing the ROS generation reduces the damage caused by chemotherapy and radiotherapy to the DNA, lipids, and cell proteins in CSCs [15, 17, 18]. Overexpression of multi-drug resistance (MDR) proteins such as ATP-binding cassette (ABC) transporters in CSCs is another factor in the resistance mechanism of them. For example, expression of ABCB1, ABCB5, ABCC1, and ABCG2 in CSCs has been reported in association with the resistance of these cells to chemotherapy [15, 19].

Proposed models for cancer stem cells (CSCs) origin in cancer development. In the normal differentiation process, a cell differentiates to form two cells, differentiated and primitive. A finally differentiated cell is formed from precursor progenitor cell and eventually subject to apoptosis. CSC may originate from a normal stem cell, a normal progenitor cell, or a normal differentiated cell by genetic mutation which will activate self-renewal genes in them. Also cancer cells via EMT can change to CSCs
Abnormal epigenetic alternations in DNA methylation, histone modifications, RNA methylations, and noncoding RNAs have been demonstrated to be vital in maintenance and survival of CSCs, tumor initiation and progression [11, 20, 21]. It was observed that hypermethylation of the promoters and deregulation of histone modifications, inhibited the expression of tumor suppressor genes in the CSCs [22, 23]. Key signaling pathways which regulate self-renewal of adult stem cells, including Wnt/β-catenin, Notch, Hedgehog, and TGF-β/BMP, are altered in CSCs through epigenetic changes. Theses deregulation can maintain the stemness, differentiation, and resistance of CSCs to drugs [10, 12].
As widely regarded, CSCs are resistant to conventional treatment and have been identified as one of the important challenges to overcome in cancer therapy [24]. Recently, CSCs have been considered for the design of new antitumor drugs that target epigenetic changes and pathways involved in the formation thereof [6, 25]. The most important epi-drugs are inhibitors of DNA methyltransferase and histone deacetylase, which have exhibited favorable results in the elimination of CSCs and cancer treatment [26].
For the aforementioned reasons, identifying the characteristics of CSCs and the factors affecting their generation and maintenance thereof is important for targeted cancer treatment. In the present review, the existing research is explored on CSC features, epigenetic modifications, and the impact thence on key signaling pathways in CSCs resistances. Finally, it promises new insights into epigenetic targets for CSC resistance eradication are revealed.
Cancer stem cells: biology, origin, and their niche
Cancer stem cells were identified in the late 1990s and have been of considerable interest to scientists throughout the twenty-first century. In acute myeloid leukemia (AML), Bonnet and Dick first detected cells having similar characteristics to normal hematopoietic stem cells with the same markers (CD34+ and CD38−) that were much more prone to developing leukemia, which were referred as leukemic stem cells (LSCs) or CSCs [6]. CSCs were subsequently found in solid tumors such as breast, brain, colon, glioblastoma, pancreas, lung, prostate, and melanoma [25], accounting for about 0.1–10% of tumor cells [27]. CSCs have been identified as tumor initiator cells that also have an integral function in development, metastasis, recurrence, and resistance to treatment [28].
The presence of markers on the surface of CSCs, such as receptors and antigens is one of the best diagnostic methods for these cells in different tumors. Yet, the expression of these markers in CSCs is lower than tumor cell-specific antigens (tumor markers). Furthermore, definition of "marker" is not linked to its overexpression, but their characteristic feature is that they are expressed in a specific cell class similar to stem cells [27]. These markers can be located within the cytoplasm or located on the cell surface and act as surface antigens [5]. By illustration, the high activity of aldehyde dehydrogenase1 (ALDH1) in the cytoplasm is known as a marker of CSCs in a range of cancers, such as breast, endometrial, gastric, leukemia and colon cancer [5, 27, 29]. Numerous studies have discovered a wide range of specific markers related to CSCs. In general, as major markers of CSCs, the expression of CD133, CD24 and CD44 has been identified in solid tumors, while CD34 and CD38 are the most important markers in blood cancers [29]. Notably, the expression level of these markers and their isotypes thereof are diverse in different types of tumors [27]. As an example, ESA+/CD44+/CD24−, and ALDH1+ were detected in breast cancer stem cells (BCSCs); CD133+ in colon, glioblasoma, gastric and lung; EpCAM, CD133/EpCAM, CD90+ ESA+CD133+ CD44+CD24+ in liver; and ESA+/CD44+/CD24+ in pancreatic CSCs (Table 1) [25, 30,31,32].
Regardless of extensive findings, the origin of CSCs has not been properly identified, although various theories have been presented. Up to date, one approach demonstrates that CSCs can arise from somatic stem cells (Fig. 1) [73, 74]. In addition, deregulation of signaling pathways in adult stem cells, containing Wnt, Sonic Hedgehog ligand, Notch and BMI1 causes the formation of CSCs [73, 75]. Another theory is that progenitor cells are the source of the CSCs. The abundance of these cells in the population of adult tumors relative to stem cells is the reason for expressing this hypothesis. However, these cells have partial ability to self-renewal [76]. Another group of researchers suggests that CSCs originated from differentiated and mature cells that have acquired characteristics similar to stem cells. In fact, it is proposed that genetic and epigenetic changes in differentiation genes, tumor suppressor genes, or signaling pathways related to pluripotency have led to ability of non-stem cancer cells to self-renew and stemness (Fig. 1) [73, 76].
Researchers have shown the prevalence of epithelial to mesenchymal transition (EMT) in cancer cells and its significant association with CSCs, which play a vital role in resistance to chemotherapy (Fig. 1) [77]. EMT is a complex reprogramming system in which epithelial cells miss their differentiation and polarity properties and become mesenchymal cells, leading to increased motility, migration, invasion, dealing with immune system responses, and resistance to apoptosis [78]. It has been seen in the context of cancer, EMT induces stem cell-like features in tumor cells and stimulates the formation of CSCs [77]. Therefore, CSCs show the EMT phenotype. It has been observed that the expression level of E-cadherin in CSCs is low. Simultaneously, the overexpression of mesenchymal markers in them increases the ability of invasion and metastasis in CSCs [79]. Extensive studies have shown that the start of EMT is induced by PI3K-Akt, transforming growth factor-β (TGF-β), epithelial growth factor (EGF), fibroblast growth factor (FGF), signaling pathways such as MAPK/ERK, hedgehog, Wnt/ β -catenin, Notch and NF-кB, along with cytokines such as Interleukin-6 (IL-6) [80,81,82]. Upregulation of EMT transcription factors increases the ability to initiate tumors in various cancer cells. For example, overexpression of the transcription factors Snail, Twist, and FoxC2 in human mammary epithelial cells increases EMT initiation in cells, leading to the development of phenotypes similar to CSCs [83]. Furthermore, various studies have proved the epigenetic changes, signals from the tumor microenvironment, and dedifferentiation of non-CSC tumor cells to CSCs promote the EMT and the stemness in the cells through affecting the signaling pathways, [79]. For example, it is said that TGF-β plays a vital role in the induction of EMT in normal and disease conditions [84]. Doherty et al. reported the non-stem cell breast cancer cells exposed to TBF-β-induced mesenchymal/CSC-like characteristics in the cells. Inhibition of TBF-β removed the mesenchymal and CSC phenotype, and the cells returned to epithelial and non-CSC phenotype [85]. Another study showed that Wnt and TGF-β act as autocrine signals in breast epithelial cells, activate the EMT pathway in in primary mammary epithelial cells, and ultimately increase migration and self-renewal [86]. Therefore, compounds that eliminate factors affecting in EMT induction are known to be an effective antitumor strategy in CSC eradication [87].
As it said, CSCs are located in an exceptional area in the tumor microenvironment called the niche. Niche is composed of cell components and secreted factors that preserve the characteristics of the CSCs, maintain their plasticity, protect them from immune responses, induces EMT in them, and increase their ability to invade and metastasize [88, 89]. Fibroblastic cells, immune cells, blood vessels, growth factors, cytokines, and extracellular matrix proteins are present in the niche could affect the CSCs properties (Fig. 2) [89]. As the tumor progresses to more aggressive states, the tumor microenvironment and especially CSC niches determine the fate of CSCs [90]. Also, stromal support cells and other cells in the niche directly and indirectly regulate the number of stem cells, their proliferation, and self-renewal (Fig. 2). In addition to their tumor initiation and progression function, niches protect CSCs from chemotherapy drugs [91]. One of the main components in tumor niche is cancer-associated fibroblasts (CAFs) engaged in the induction of EMT, angiogenesis, metastasis, and drug resistance. CSCs activate the conversion of normal fibroblasts to overproliferative CAFs by the upregulation of Wnt signaling pathway and activating Hedgehog signaling in niche [73]. Another study states that a group of CAFs with CD10 and GPR77 markers produces niches for CSCs that help CSCs survive and promote tumor progression and resistance to chemotherapy. Other studies also have suggested that one of the leading roles of CAFs is to produce niches to preserve CSC. CSCs produce CAFs through signaling pathways, and in contrast, CAFs maintain the spread and survival of CSCs by producing niches [92, 93]. CAFs caused drug resistance in cancer by maintaining stemness in CSCs through secreting NRG1 and activating NF-κB, Wnt, and Notch3 signaling pathways [7].

Schematic representation of the cancer stem cell microenvironment or niche. Progression of tumor needs a cooperative interplay between CSCs and their niche. The CSC niche made of various cells including mesenchymal stem cells (MSCs), endothelial cells, cancer-associated fibroblasts (CAFs) and immune cells, tumor-associated macrophages (TAMs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), T cells and B cells. These cells secrete various growth factors and cytokines which promote tumorigenesis, tumor progression, and immunosuppression
Mesenchymal stem cells (MSCs) are another family of essential cells in that can differentiate into different cell types. They are multipotent and involved in cancer cell proliferation, metastasis, and angiogenesis. MSCs preserve CSCs phenotype by secreting different types of cytokines into the tumor milieu [89]. For example, MSCs increased the population of CSCs by activating the NF-κB signaling pathway through the secretion of IL-6, IL-8, CXCL7, and CXCL12 or BMP2 signaling in various cancers. In addition, mesenchymal stem cells increase the resistance of CSCs to treatment [7, 94]. Tsai et al. reported that MSCs increased the number of colorectal CSCs in mice by activating the IL-6/STAT3 pathway [95]. Another study showed MSCs overexpressed miR-199a in breast cancer that led to abnormal expression of different types of microRNAs and the formation of CSCs [96].
Due to the proliferation of tumor cell leads to hypoxia in the tumor microenvironment, CSCs in niche are mainly associated with microvascular endothelial cells that have a significant role in maintaining CSCs niche in hypoxia conditions. Studies have shown that hypoxia is an essential element of the niche environment that enhance CSC number and maintains stemness [97, 98]. Exposure to hypoxic conditions induces hypoxia-inducible transcription factors (HIFs) in many solid tumors. HIFs have an essential role in regulating the stem cell phenotype through Oct4, SOX2, NANOG markers, MAPK/ERK, Wnt and Notch signaling pathways [99]. Zhang et al. reported HIF-1α-induced stemness phenotype and self-renewal ability in glioblastoma CSCs and inhibited their differentiation, so that its knockdown decreased the tumor’s progression[100]. Another study reported HIF-1α-activated Notch signaling pathways and thereby increased the expression of Notch downstream genes such as Hes1 and Hes2, which play a major role in the preserving leukemia stem cells [101]. Therefore, HIF-1α plays several roles in the tumor, including regulation of angiogenesis in hypoxia, establishing and preserving the CSCs niche, promoting the stem cell phenotype, and increases the ability of self-renewal, proliferation, and tumorigenesis in CSCs. Markers linked with the stem cell phenotype, such as CD44 and CD133, increases under the influence of hypoxia [98]. CSCs under hypoxia can promote angiogenesis through direct or indirect pathways. They can differentiate into vascular endothelium cells, thereby producing their niche or secreting angiogenesis-inducing factors. Angiogenesis-promoting factor VEGF is expressed more in CSCs than non-CSCs [102, 103].
Therefore, CSCs rely on the cells and factors present in a niche to maintain their function and population. They also themselves secrete factors and signaling molecules to maintain niche structure and function. CSCs and niche act together to provide favorable conditions for the maintenance of CSCs and tumor progression [91].
Key molecular signaling pathways, such as Wnt, Hedgehog, Notch, TGF-β, which are precisely regulated in normal stem cells, are deregulated in CSCs. These pathways’ abnormal activity plays a crucial function in maintaining the characteristics of CSCs such as self-renewal, differentiation, survival, and cell proliferation, which are outlined in the next sections [104].
Drug resistance mechanisms of CSCs
As described in the previous sections, CSCs as a subpopulation of cancer cells play an important role in drug resistance and cancer recurrence due to their self-renewal properties and differentiation into cancer heterogeneous cells. They can remain as a small subset of tumor tissue after surgery, radiotherapy, and chemotherapy and promote the cancer [24]. Drug resistance is one of the major problems in cancer therapies, leading to treatment failure [105, 106]. Multidrug resistance (MDR) refers to a general phenomenon that resists the broad spectrum of drugs, not only to a specific one [107, 108]. MDR severely succeeds in the effectiveness of the treatment of the different drugs with a similar structure [106, 109]. It has been widely investigated that CSCs have significantly endogenous resistance mechanisms against radiation and chemotherapy compared to non-CSCs [104,105,106,107,108]. For example, according to the Bao et al. study, patients with an irradiated therapy regimen showed glioblastoma cells resistance in the second irradiated recommended that CSCs are not eliminated and then began to self-renew after therapy [110]. The mechanism of glioblastoma CSC's high-resistance to radiotherapy is well defined by Carruthers et al.'s study [111]. They reported fundamental activation of DNA damage reaction due to the generation of a higher level of replicative stress in those cells is the primary response to this phenomenon. Thus, targeting DNA damage checkpoint pathways in CSCs may reveal a promising strategy.
Consequently, chemotherapy and radiotherapy treatment is more effective on most non-CSCs but not CSCs [112]. It seems that drug resistance is closely related to the similarities between CSCs and normal SCs [10]. As SCs play an essential role in keeping the pool of cells in an organism; preserve these SCs is biologically crucial and necessary. Consequently, CSCs, by the aid of different mechanisms, escape from cell senescence or apoptosis [113].
Drug resistance in CSCs has been identified through various mechanisms like (1) increase in anti-apoptotic proteins, e.g., Bcl-2, Bcl-X and c-FLIP [114, 115], (2) overexpression of ABC transporter proteins and detoxifying enzymes [115,116,117], (3) cell cycle quiescence [118, 119], (4) improved DNA repair capacity [115, 116], (5) enhance the activity of aldehyde dehydrogenase (ALDH) [120], (6) active essential pro-survival signaling molecules like as NOTCH, Wnt/β-catenin, and NF-κB [120,121,122], (7) improvement in activities of phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR), and (8) maternal embryonic leucine zipper kinase (MELK) [114, 123].
CSCs have been well described by the unusual expression of apoptotic and anti-apoptotic proteins, which participate in survival pathways. Upregulation of transcriptional redox-sensing factor Nrf2 increases CSC’s survival rate via promoting Bcl-2 and Bmi-1 gene transcripts. The oncogene BMI-1 is a key component of polycomb repressive complex 1 [114]. Following the durability of CSCs by the high expression of these anti-apoptotic proteins, different efforts have been made in recent years through the direct inhibition of the Bcl-2 family proteins toward therapeutic interventions [124].
ABCC1, ABCB1, and ABCG2 belong to a massive superfamily of ABC transporters recognized as multidrug efflux pumps with ATPase activity. It is demonstrated that upregulation of ABC transporters by CSCs under chemotherapy regime affords protection of CSCs and promote tumor generation [125]. Frank et al. recognized melanoma cancer stem cells by overexpression of ABCB5 (an ABC family member) in these CSCs. It has been shown that ABCB5 can keep melanoma-initiating cells by a pro-inflammatory cytokine signaling pathway [126]. Overexpression of ABCG2 has also been reported in head and neck, lung, brain, osteosarcoma, melanoma, and prostate CSCs [127]. Some studies reported overexpression of ABC transporters also has been identified in side population (SP) of CSCs cells that indicate a stemness phenotype and can be used as candidates for the identification of CSCs [15, 125, 128]. High ABCB1 expression has been observed in SP cells in a number of cancer cell lines compared to other cells [123]. For example, upregulation of ABCB1 and also ABCG2 were found in SP cells in pancreatic cancer cell line [129]. In fact, ABCG2 is known as a marker of CSCs and plays an important role in drug resistance and ABCB1 has also been found in over 50% of resistant tumors [19, 128].
Recently, it is proved that CSCs resistant to chemotherapy could be because they are quiescent in the G0 of the cell cycle. Therefore, chemotherapeutic drugs that affect cell proliferation cannot target them [36, 37]. By understanding the signaling processes and their associated factors that contribute to cell quiescence, appropriate strategies can be used. For example, the dormant phenotype often follows by PI3K-Akt signaling reduction or cell quiescence is remarkably related to mitogenic signals inhibition [130]. Researchers are seeking to find a useful approach to overcome the resistance of CSCs quiescence. Prost et al. showed that agonists of peroxisome proliferator-activated receptor-γ via limiting the expressions of hypoxia-inducible factors 2α (HIF2α) and CITED26 could prevent leukemia CSCs from entering the non-proliferating stage, which is an essential modulator in this process.
Another mechanism that plays a role in radiation resistance is the overexpression of DNA repair proteins that participate in promoting DNA repair in CSCs [116, 131,132,133]. In comparison with the normal tissues or massive tumors, CSCs are resistant to current treatments by the effect on activation of p53 under radiation-induced DNA damage conditions. Consequently, cell cycle arrest and apoptosis are not working accurately. Over different cycles, it causes the accumulation of DNA mutations [134].
Aldehyde dehydrogenases (ALDHs) are a group of NAD(P)+-dependent enzymes with exogenous and endogenous aldehydes detoxification. High ALDH activity could also be considered a CSCs marker in self-renewal, proliferation, differentiation, and drug resistance. Wnt signaling plays a role in transforming dormant CSCs into active CSCs by increasing cell cycle procedure via β-catenin, enhancing the expression of downstream cyclin D1 and MYC. Additionally, MYC increases the expression of the polycomb repressor complex 1 component Bmi-1 and affects the connection of E2F with cyclin E [128]. Based on various investigations, the Notch pathway’s activation is specifically involved in developing cell survival, self-renewal, metastasis and inhibiting apoptosis in CSCs [135, 136]. Deregulation of Notch signaling, specially Notch1 and Notch4, induce self-renewal and metastasis in breast and HCC stem cells. The critical role of the NF-κB pathway in CSCs involves regulating inflammation, self-renewal, or maintenance and metastasis. CD44+ cells manifest the capacity of self-renewal, metastasis, and maintenance of ovarian CSCs with an improved expression of RelA, RelB, and IKKα and mediating nuclear activation of p50/RelA (p50/p65) dimer [137]. Researches have shown that the mTOR signaling pathway is also associated with CSCs metabolism [138]. By an activated mTOR signaling pathway, low folate (LF) stress reprograms metabolic signals show the potential to induce the metastasis and tumorigenicity of lung cancer stem-like cells. As a result, drugs, vaccines, antibodies, and CAR-T cells that target these pathways can be used to develop novel targeted therapy [128]. MELK (a serine/threonine kinase) is expressed in several cancer stem cells populations [139], which implicated in the survival and drug resistance and tumor recurrence in CSCs. According to Kim et al.'s studies, EZH2 is a target of the MELK/FOXM1 Complex and then increases CSC resistance to radiation [140].
Autophagy is another resistance mechanism of CSCs which plays a major role in the maintenance and survival of various CSCs, such as liver, ovarian, breast, pancreatic, osteosarcoma, and gliobastomabrain cells [141]. For example, Pagotto et al. have shown high autophagy activity in ovarian CSCs compared to non-stem counterpart, which plays a fundamental role in maintaining cells and increasing their resistance to chemotherapy. Inhibition of autophagy by reducing CSCs reduced resistance to chemotherapy [142]. Overexpression of autophagy-related proteins such as ATG3, ATG5, ATG7, and ATG12 in colorectal CSCs also increased resistance to photodynamic therapy (PDT) and decreased PDT-induced apoptosis in the cells. Inhibition of autophagy by pharmacological inhibitors increased the sensitivity of colorectal PROM1/CD133+ CSCs to PDT [143]. Self-renewal features give the constant maintenance of the CSC pool through tumor improvement and initiation. Since most of the genes associated with the regulation are oncogenes like Smo, Shh, Gli1, Gli2, and Ptch1, the hedgehog pathway represents an essential part of self-renewal. Active mutation in these genes is associated with active hedgehogs, one of the main reasons for many human cancers. Therefore, targeting self-renewal pathways is a promising strategy for eliminating the CSCs [144].
While there is much information about CSC characteristics that can help us to target CSCs efficiently, there are many difficulties to overcome before fully achieving. In the first step, it is essential to find a way to target the only CSCs to avoid destroying normal tissue stem cells and reduce side effects. Secondly, combinational therapies for effective tumor eradication needs to be considered since CSCs may not eliminate. Thus, CSCs have not been introduced in targeted clinical treatment, but they will be a promising future treatment.
The role of epigenetic modifications in cancer stem cells resistance to chemotherapy and radiotherapy
As described in the previous sections, the presence of CSCs plays an important role in resistance to current treatments [145]. Various studies have shown that epigenetic changes significantly induce the CSC phenotype [146,147,148,149]. Epigenetic modifications involve inherited cell phenotypic changes without changes in nucleotide sequence [3]. Substantial epigenetic alternations in CSCs are described in detail below.
Histone methylation
Histone methylation appears mainly in lysine (K) and arginine (R) residues through the binding of a methyl group to nitrogen atoms in side chains of amino acid or at the N-terminal tails by histone methyltransferases (HMT). Histone methylation is completely conserved during the evolution of animals. Histone lysine methylation can be mono-, di-, or tri-methylation which could activate or inhibit the gene expression [146]. For example, trimethylation at histone H3 lysine 4 (H3K4), H3K36, and H3K79 active transcription and methylation of H3K9, H3K27, and H4K20 repress transcription [147]. Therefore, deregulation of histones methylation because of alternation in gene expression causes various diseases and malignancies. Also, investigations have displayed that histone methylation is different in non-cancer cells and CSCs [148]. Histone methylation has a significant function in regulating the gene expression involved in signaling pathways crucial for maintaining CSCs self-renewal such as Wnt, Notch, and Hg pathways [149]. A study on triple-negative breast cancer stem cells (TNBCSCs) showed that H3K27me3 was abundant in the cells and, conversely, the H3K4me2 level, which has an opposite role H3K27me3, was rarely seen. These abnormal modifications affected key signaling pathways in self-renewal of CSCs, such as Wnt and human gonadotropin-releasing hormone (GNRH), thereby increased survival, tumorigenesis and chemotherapy and radiotherapy resistance in TNBCSCs. Overexpression of DOT1L (the H3K79 methyltransferase), upregulated Rho GTPases and survival proteins in CSCs of HNSCC enhance tumor invasion and chemotherapy resistance. Simultaneously, the inhibition of DOT1L by DOT1L specific small interfering RNAs (siRNAs) reduced the invasion of the tumor and increased chemosensitivity [145]. Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase and member of the Polycomb-group family. Polycomb-group proteins (PcG) regulate gene expression by epigenetic changes. Overexpression of EZH2 by the induction of trimethylation of H3K27 (H3K27me3) and the regulation of gene expression promoted self-renewal and dedifferentiation in oral cancer cells, ultimately leading to tumor progression and invasion [150]. Momparler and Cote reported DNA methylation and polycomb repressive complex 2 (PRC2), a class of PcG proteins, alone or in combination inhibited differentiation in CSCs. As the expression of differentiation programming genes in CSCs was permanently suppressed by histone methylation and/or DNA methylation, these changes were reversible [151]. Alternative research has suggested that EZH2 inhibits the expression of tumor-suppressor genes by inducing H3K27me3. EZH2 absorbed DNMTs in the promoter area of target genes during cancer progression, resulting in DNA and histones’ methylation. These modifications eventually condensed the chromatin structure and silenced the tumor suppressor genes expression. In fact, PcG target genes prefer to be hyper methylated. Due to EZH2 role in promoting the self-renewal of tumor cells, inhibiting them can prevent the formation of CSCs [152]. For example, it has been proven that downregulation of EZH2 in gemcitabine-resistance pancreatic cancer cells increases the sensitivity of cells to gemcitabin through reducing the number of CSCs [153]. Several EZH2 inhibitors have been specifically synthesized, among which EPZ-6438 (E7438) and 3-deazaneplanocin-A (DZNep) have shown significant anti-tumor activity and have been considered for clinical research [151].
Histone acetylation and deacetylation
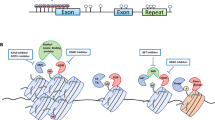
Acetylation and deacetylation of histones are among the principal epigenetic modifications in various diseases such as cancer [154]. Histone acetyltransferase (HAT) binds an acetyl group to the lysine residue in histone and non-histone proteins, and in contrast histone deacetylases (HDAC) removes the acetyl group from them [155]. HATs are divided into two types, type A and B, based on their location. Type A is placed in the nucleus and type B is located in the cytoplasm [156]. Mammalian HDACs are also divided into four classes, each withseveral members (from HDAC1 to HDAC18) [157]. In general, HATs increase the transcription and expression of genes by relaxing chromatin structure and promoting transcription. HDACs cause the silencing of genes by condensation of the chromatin structure and restricting transcription. The imbalance between acetylation and deacetylation of histones by affecting the signaling pathways of differentiation and self-renewal, leading to the formation and preservation of CSCs [154, 155]. For example, Liu et al. reported HDAC3 and HDAC7 were overexpressed in the CSCs that increased the expression of stem cell markers such as SOX2, Oct4 and Nanog. Inhibition of HDAC3 expression by SiRNA inhibited the proliferation and self-renewal of CSCs. It was also observed the amount of acetylated histones H3 (H3Ac) and H4 (H4Ac) in liver CSCs was lower than non-CSCs and increased histone acetylation led to the cell differentiation [158]. Another study found that HDAC1 and HDAC7, generally enhanced in CSCs compared to non-CSCs and their presence is essential for maintaining CSCs phenotype [159]. HATs also have a vital function in regulating the multiple signaling pathways that maintain cell stemness and differentiation such as Wnt/β-catenin, Notch, Hedgehog, TGFβ/BMP, JAK/Stat, FGF/MAPK and Hippo [160]. For example, CBP/p300 (a member of the type A HATs) has been identified as an essential coactivators of the Wnt/β-catenin pathway in CSCs. As shown, the selectively antagonizing the CBP/β-catenin interaction inhibited drug resistance in CSCs by inducing differentiation in them [161]. CBP and p300 are known as coactivators for hundreds of transcription factors. C-Myb is a transcription factor that is overexpressed in AML cells and is needed for self-renewal, survival, and proliferation of CSCs. Reports indicated that P300 is the main coactivator of c-Myb. Therefore, c-Myb and P300 interaction inhibition can be a promising therapeutic agent in the leukemia treatment [162]. Another study showed the amount of hypoacetylation of histone 3 in HNSCC is higher than normal cells. The deregulation of histone aceylation and deacetylation by affecting the transcription process and gene expression disrupted the processes of proliferation, metastasis, DNA repair, and apoptosis. The HDACs suppression and the induction of the acetylation of histones disrupted the processes essential for the formation of CSCs [163]. Researchers also have shown that HDAC11 overexpresses in CSCs of Non-small cell lung cancer (NSCLC). Inhibition of HDAC11 reduced the expression of Sox2 that is an essential transcription factor for the self-renewal of the cells and their viability. In addition, inhibition of the HDAC11 reduced the lung cancer cells growth and their resistance to treatment (Fig. 3) [164].

Schematic illustration of factors related to raising resistance in CSCs. Activation of quiescence, cell survival pathways, enhanced drug efflux, the apoptotic signaling disability, enhanced DNA damage repair, enhanced detoxifying activity, and enhanced scavenging of free radicals are feasible agents lead to the CSCs resistance
DNA methylation
Alternation in DNA methylation plays a vital function in all stages of cancer, such as the formation of CSCs. This modification is performed by DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) which transmit a methyl group from S-adenosylmethionine to the position of 5 cytosine bases [12, 165]. The role of DNMT1 is to keep methylation, and DNMT3A, and DNMT3B are accountable for creating de novo methylation [166]. DNA methylation has been seen to be removed or transmitted to future generations without changing the natural structure of the DNA [165]. Methylation is common in areas rich in CPG dinucleotides, titled as CPG islands. CPG islands are abundant in the gene promoter region; methylation of these areas causes long-term silencing of genes [11]. It has been generally reported that hypomethylation in the oncogenes’ promoter increases their expression and hypermethylation in the tumor suppressor genes promoter causes the silencing of genes in many cancers [167]. Decreased expression of tumor suppressor genes has been shown to lead to increased plasticity in cell phenotype such as EMT, change in cell function, self-renewal and differentiation potential, and finally the formation of CSCs in different cancer cells [168, 169]. Although DNA methylation is normally necessary to maintain normal cell function and physiology, its dysregulation leads to tumorigenesis, increased the formation of CSCs and the ability of self-renewal, increased the cell tolerance to drug toxicity, and resistance to chemotherapy [11, 169]. Mutations in epigenetic regulators such as DNMT3A also promote the formation of CSC in tumors. These, mutations that inactivate epigenetic regulatory genes, activate CSC formation pathways and increase resistance in them [170]. For example, among the AML samples which examined, 44% of the genes associated with DNA methylation changes, with the highest mutation observed on DNMT3A [171]. Mutations can occur in stem cells or adult cells, in both cases causing the cell to lose control of its plasticity and original identity [170]. Investigations have shown that DNA methylation regulate the specific CSC genes expression in solid tumors and leukemia. For example, hypermethylation of WIF1, SFRP2, SFRP5, DKK, WNT5a, APC genes, which act as inhibitors of the Wnt pathway, caused these genes to be silenced. Epigenetic silencing of these genes played a vital role in drug resistance in CSCs, and reactivating them induced apoptosis in the cells [172, 173]. DNA methylation regulate the expression of surface markers of CSCs such as CD44 and CD133, as well as ABC transporters such as ABCG2, which is involved in drug efflux from the CSCs and thus promotes chemotherapy resistance [173]. Research has shown that epigenetic changes, such as DNA methylation, cause CSCs to enter a state of cellular quiescence called a dormant state. In this case, the expression of survival and anti-apoptotic genes is increases, which makes the cells resistant to conventional therapies [23]. For example, upregulation of transcription factors FOXG1 and SOX2 caused CSC phenotype in glioblastoma multiforme (GMB). Hypermethylation in downstream targets of FOXG1 and SOX2 including FOXO3 promoted the cellular dormant process in GMB CSCs [174]. Other researchers have shown that breast CSCs can also be dormant by DNA epigenetic changes, although the exact mechanism by which they enter dormant state has not yet been determined. The amount of DNMT1 also increased in mammary tumors and CSCs and aberrant DNA methylation caused resistance to treatment in CSCs by regulating proteins involved in cell growth [175, 176]. Therefore, targeting DNMTs is an effective strategy in regulating tumor suppressor genes and genes related to differentiation, the formation of CSCs, and creating resistance in them.
RNA methylation
The advancement of technology in recent years, led to the identification the function and regulation of RNA methylation in eukaryotic cells and its importance in biological changes, and the development of related diseases. According to the MODOMICS database, 72 methylation modifications have been identified in RNA, which is about seven times more than those identified in DNA. Most methylation is done on bases in RNA compared to sugar-phosphate backbone [177]. RNA methylation is a post-transcriptional RNA modification that is reversible and affects the RNA stability and translation of mRNA [178]. RNA methylation, especially mRNA and long noncoding RNA, leads to precise regulation of gene expression by impacting the RNA interaction with other cell components [178]. Of all the mRNA modifications, N6-methyladenosine (m6A) is all the more abundant, about 0.1–0.4% of all adenosine. A methyltransferase enzyme complex made up of proteins methyltransferase-like 3 (METTL3), METTL14, and Wilms tumor 1 associated protein (WTAP) which catalyzes this modification [179]. This modification affects all RNA metabolism stages, such as CSC formation, maintenance, and metastasis in various cancers. Deregulation of m6A RNA methylation plays different roles in the cancer initiation, progression and resistance to current treatments [25]. For example, Zhang et al. have shown that hypoxia, which is a definite aspect of the tumor cell environment in breast cancer cells, increase hypoxia-inducible factor (HIF)-1α- and HIF-2α-dependent expression of ALKBH5 as a demethylase that induced m6A demethylation. This led to the demethylation of NANOG mRNA that increased its stability and level. NANOG as a pluripotency factor has an essential function in maintaining the characteristics of the CSCs. Therefore, an increase in NANOG levels led to an increased BCSCphenotype [180]. Cui et al. proposed regulation of m6A RNA modification is significant for the self-renewal of glioblastoma stem cells (GSCs). Downregulation of METTL3 or METTL14 increased self-renewal and tumorigenesis of GSCs. In fact, reducing METTL3 and METTL14 by reducing the m6A RNA methylation levels altered the expression of essential genes in CSCs such as ADAM19. While, the use of the inhibitor of m6A demethylase FTO inhibited self-renewal of GSCs and tumor progression [181]. Therefore, CSCs RNA methylation can be considered as a new therapeutic strategy in glioblastoma [182]. Lin et al. have shown that hypoxia reduces METTL3 expression in human sorafenib-resistant HCC. In fact, deregulation of m6A modification of FOXO3 mRNA by downregulation of METTL3 increased the expression of angiogenesis genes, activated autophagy pathways, and stabilized resistance phenotype in the cells [183]. Another study found increased expression of FTO in cervical squamous cell carcinoma (CSCC). FTO reduced m6A methylation in β-catenin mRNA (an EMT maker) and downregulated its expression, which ultimately led to an increase in chemo-radiotherapy resistance in vitro and in vivo [184]. Researches in colorectal cancer (CRC) have also shown that METTL3 upregulates in the cells and through an m6A-dependent manner and maintains the expression of SRY-box 2 (SOX2) that is a CSC marker. Therefore, METTL3 acted as an oncogene in CRC that promoted the CRC self-renewal, migration, and tumorigenesis in the cells. The emergence of CSCs in CRC has also led to increased resistance to chemotherapy. So that downregulation of METTL3 in SW620 and HCT116 cells increased the sensitivity of cells to oxaliplatin [185]. In general, m6A modification can affect the onset and progression of cancer through the stability of various mRNA oncogenes, increasing the translation of essential genes for cell survival and regulating the immune system [186]. FTO and ALKBH5 act as an m6A demethylase, their inhibitors such as Rhein, meclofenamic acid (MA) and IOX3 have been identified as effective anti-tumor drugs. However, their clinical studies have not yet been confirmed [186]. For example, Cui et al. proposed that the MA2, the ethyl ester form of MA as a FTO inhibitor increased m6A methylation, thereby inhibiting the growth of GSCs [181]. Also MA also enhanced m6A modification in HeLa cells at a concentration-dependent behavior [187].
Noncoding RNA as regulator of epigenetic in CSC drug resistance
Noncoding RNAs (ncRNAs) belong to a class of RNAs that do not translate into protein but regulate the genes expression at the post transcriptional level. Therefore, they have been considered as an important epigenetic regulator in recent years. They are distributed into two main families according to their size: small chain noncoding RNAs contain siRNAs, miRNAs, and piRNAs and long noncoding RNAs (lncRNAs) [188]. NcRNAs have vital function in regulating different signaling pathways related to tumor initiation, progression metastasis and resistance to therapies [20]. They are generally aberrantly expressed in various cancer cells and also CSCs. Investigations have revealed that miRNAs (19–24 nucleotide) have an essential function in the biology of CSCs by regulating the signaling pathways of stemness, differentiation, EMT and carcinogenesis in the cells [189]. The abnormal miRNAs expression can work as a tumor suppressors or an oncogene in various cancer cells [190]. For example, miR-21(an oncogenic miRNA) was upregulated in pancreatic CSCs (PCSCs) and through the impact on the PI3K/AKT pathway affected the process of cell proliferation and chemoresistance [191]. On the other side, miRNA-34a (a tumor suppressor miRNA) was downregulated in pancreatic, glioblastoma, and prostate CSCs contributing to the cells’ self-renewal. The restoration of miR-34a in the cells inhibited tumor regeneration and metastasis [192]. The downregulation of miRNA-34a in gastric cancer also affected CSCs formation’s potential by regulating downstream genes including CD44, Bcl-2, Notch, HMGA2, Nanog, Oct4, SOX-2 and YY1. It is shown that miR-34a overexpression suppresses the self-renewal ability of gastric CSCs and can restore the sensitivity of cisplatin-resistant cells [193]. Tsukasa et al. reported the overexpression of miR-30 family including miR-30a, 30b, or 30c upregulated mesenchymal markers such as CD133+ pancreatic CSCs (PCSCs) and increased migratory and invasive capabilities of the cells. Additionally, LncRNAs (ncRNAs over 200 nucleotides in length) have fundamental function in maintaining CSC populations through regulating the expression of stemness transcription factors, their downstream targets, and pathways related to stem cells. For example, downregulation of LncRNA H19 inhibited the expression of CSCs markers such as CD133, Nonag, Oct4, and Sox2 in glioma stem cells (GSCs) [194]. Another study showed that LincRNA-ROR (a large intergenic noncoding RNAs) expression in glioma tumor cells was lower than normal cells. LincRNA-ROR acted as a tumor suppressor gene in glioma cells; its downregulation elevated the self-renewal capacity of GSCs as well as increased the number of CD133+ GSC by the overexpression of stem cell factor such as KLF4 [195]. LincRNA-p21 was also downregulated in colorectal and glioma CSCs. Its overexpression had an anti-EMT activity in the cells and by inhibiting β-catenin signaling reduced the self-renewal of CSCs [196]. Other studies have shown that Lnc34a overexpresses in colorectal CSCs. It recruited DNMT3A and HDAC1 in MIR34A promoter, led to methylation and deacetylation of the promoter. These epigenetic modifications silenced the MIR34A gene and thus increased CSCs proliferation [197].
Therefore, due to abnormal expression of ncRNA in CSCs and their role in the carcinogenesis and treatment resistance, they can be considered therapeutic targets. Recently, various clinical trials have been done applying nanotechnology to deliver ncRNAs, that the most studies have been conducted on miRNA [198]. For example, miRNA mimics are used to mimic tumor suppressor miRNAs or anti-miRNAs inhibit the oncogenic miRNAs’ function. Nanoparticles are considered efficient delivery systems for transporting RNAs to protect them from nucleases and deliver them to the target cells with minimal toxicity [199,200,201]. For example, scientists synthesized a liposomal nanoparticle that delivers miR-34a mimics to the targeted cancer cells and its clinical trial in humans is in phase 1. Also, many clinical trials based on miRNA therapy have been reported in phase 3 or 4 (https://clinicaltrials.gov) [198, 202].
Epigenetic alterations of signaling pathways in CSC resistance
Several signaling pathways such as Hedgehog, Wnt/B catenin, TGF β/BMP and Notch have a crucial role in maintaining self-renewal, stemness and CSCs differentiation. They are often deregulated in different types of cancer through epigenetic modifications [9]. These abberant epigenetic changes in signaling pathways promote tumor progression, invasion, and resistance by CSCs maintaining [11].
Wnt/β-catenin signaling pathway
The canonical Wnt/β-catenin signaling pathway modulates gene expression by the transcription factor such as β-catenin, which has a key function in cell proliferation, survival, and maintaining CSCs features. In such a way, high Wnt activity is crucial for initiating and maintaining various tumors [170, 203]. The binding of the Wnt ligands to Frizzled and LRP 5/6 coreceptors at the cell surface stabilizes the cytoplasmic accumulation of β-catenin and transports it into the nucleus. β-catenin acts as an activator of transcription of Wnt target genes such as c-myc, c-jun, Axin2, EphB/ephrin-B and Cyclin D1 in the nucleus, which are crucial for the proliferation and differentiation of stem cells (Fig. 4) [204]. The Wnt pathway also affects the expression of CSCs surface markers such as LGR5/GPR49, CD44, CD24, and Epcam in a variety of tissues [205]. Researches have shown that Wnt/beta-catenin signaling induces CD133 expression in hepatoblastoma cells. Increasing CD133 increased resistance to chemotherapy by maintaining CSCs self-renewal, activating the Akt/PKB and Bcl-2 survival pathways [206]. Overexpression of SOX2 promoted the activation of Wnt/β-catenin pathway in CSCs. The Wnt/β-catenin signaling activated EMT program which led to metastasis and treatment resistance [207]. The Wnt/β-catenin pathway is abnormally activated in various cancers through genetic mutation or epigenetic changes [203]. For example, the researchers showed that promoters of genes contributing to Wnt/β-catenin signaling pathways are hypermethylated in breast cancer cells. Moreover, DNA methylation downregulated the expression of Wnt inhibitors including Wnt inhibitory factor 1 (WIF-1), several frizzled-related proteins (SFRP1-5), and Dickkopf-related protein 1 (DKK1) in the tumor cells [208]. In general, it is revealed that the Wnt/β-catenin signaling activity in BCSCs is more than non-CSCs. Therefore, Wnt/β-catenin signaling activity is higher in BCSCs compared to non-CSCs. Inhibition of the Wnt/β-catenin signaling by reducing the genes involved in CSCs reduced tumor formation and metastasis in the cells [209]. DNA methylation inhibited the Wnt pathway’s gene expression, including SFRP, SOX17 (SRY-box 17), and WIF1 in oral cancer [210]. Modification of histones also affects deregulation of Wnt/β-catenin pathway. The H3K27me3 missing in ASCL1 promoter changed activation of Wnt signaling pathway in GCSCs. ASCL1is a strong regulator of the Wnt signaling pathway that is essential for CSC maintenance and tumorigenicity [211]. H3K4me2 and H3K4me3 also reduced in Wnt/β-catenin pathway in CSCs as compared to non-CSCs which contributed to the progression of CSCs phenotype [22]. It has been reported that the EZH2 is essential for maintaining Wnt/β-catenin pathway in CSCs. Overexpression of EZH2 increased downstream expression on β-catenin genes such as vimentin and c-Myc in CSCs. EZH2 inhibition by inactivating the Wnt/β-catenin pathway-induced cell arrest in G1/S-phase [212]. Another study reported p300 and CBP, as HAT, are the main coactivators of the Wnt/β-catenin pathway in CSCs, especially β-catenin mediated transcription [161]. MiRNAs have an essential function in regulating of the expression and function of the Wnt-signaling pathway items [204]. For example, the overexpression of miR-19, miR-501-5p, and miR-744 elevated β-catenin activity, thus, enhancing the expression of CSCs proliferative genes, while overexpression of miR-708-5p and miR-142-3p decreased β-catenin activity in CSCs [213]. Another study reported miR-19b, miR-20a, and miR-92a were overexpressed in GCSCs. They played a notable function in self-renewal and maintenance of CSC by targeting E2F1 and HIPK1 which led to the Wnt/β-catenin signaling pathway activation [214]. MiR-148a is a critical tumor suppressor that is reduced in CSCs and its downregulation increase the cell proliferation, invasion, and chemoresistance by acting on the Wnt/β-catenin signaling pathway. The overexpression of miR-148a in cisplatin-resistance colorectal cancer cells inhibited the expression of WNT10b and activity of β-catenin in the cells. Therefore, the expression of markers of CSCs, cell invasion and migration decreased while cell sensitivity to cisplatin was increased [215].

Graphical representation of CSCs epigenetic regulation. Histone modifications, DNA methylation, RNA methylartion and noncoding RNA molecules (lncRNAs and miRNAs) play important role in CSC biology and plasticity. a lncRNAs interplay between the various layers of epigenetic gene regulation such as histone modifiers or serving as ceRNAs for miRNAs; while miRNAs can act as anti- or pro-CSC regulators. b Histone-modifying enzymes act between CSCs and their non-CSC counterparts, such as HAT, EZH2, and HDAC. c The DNMT1 methyltransferase methylates CpG sites relate to methylation of genes vital for stemness feature, differentiation and quienscent of CSCs. d RNA methylation increase self-renewal of CSCs
Hedgehog signaling pathway
Hedgehog (Hh) is another essential pathway for maintaining self-renewal, the forming CSCs, and chemoresistance. Abnormal Hh signaling pathway activation with different aspects of tumorigenesis causes the development and progression of different types of tumors [216]. Hh signaling also regulates the genes expression of maintaining phenotype and function of CSCs such as Oct4, Sox2 and Bmi1, ALDH1, Wnt2, CCND1, CD44, Twist1, Snail, C-MET, C-MYC and Jagged 1 [128, 217]. Nanog is a key transcription factor for maintaining stemness, self-renewal and differentiation of embryonic stem cell and CSCs, also directly control by Hh signaling pathway [218]. In the presence of Sonic hedgehog ligand (Shh), intermembrane receptor Patched 1 (Ptch1) activates Smoothened (SMO), which in turn causes activation of the Glioma-associated oncogene (Gli) family and their entry into the nucleus. The Gli family controls the expression of Hh signaling target genes as transcription factors (Fig. 5) [217, 219]. Epigenetic modifications can modulate Hh pathway components and lead to the initiation, and progression of various tumors. As an instance, hypomethylation of the Shh promoter region increased the expression of the ligand in breast and gastric cancer cells. Overexpression of Shh activated Hh target genes 101and increased aggressiveness and self-renewal ability of different cancer cells [220, 221]. Histone modifications also modulate Hh signaling pathway. Gli1 and Gli2 proteins are normally acetylated and HDAC1 is essential to activate their transcription. Therefore, overexpression of HDAC1 in cancer cells enhances Hh signaling [219]. Studies showed Gli1 is often active in lung CSCs. The inhibition of Gli1 activity reduced stemness in CSCs and thus decreased tumor growth [222]. Another study reported SMO, Gli and PTCH1 increased tumorigenicity, self-renewal, and migration in CD133+ GSCs. HDAC6 regulated the expression of Gli1, PTCH1, and PTCH2 in these cells [128]. HDAC6 inhibition suppressed Shh/Gli1 signaling pathway in GSCs and increased the sensitivity of cells to radiotherapy [223]. NcRNAs, especially miRNAs and lncRNAs also can regulate Hh signaling activity. For example, Wu et al. reported overexpression of LncHDAC2 increased Hh signaling activity in liver CSCs. LncHDAC2 connected to HDAC2 and reduced the expression of PTCH1 gene, consequently promoted the self-renewal of liver CSCs and tumorigenesis of hepatocellular carcinoma [224]. MiRNAs are known as the main Hh/Gli signaling pathway regulators. Miele et al. reported re-expression of miR-326 suppressed Hh/Gli signaling pathway by downregulation of Nanog, SMO and Gli2 in medulloblastoma CSCs that impaired self-renewal of cells [225]. Another study found that downregulation of miR-324-5p is associated with increased expression of Hh signaling components such as SMO and Gli1 in myeloma stem cells that increased stemness, survival and drug resistance in the cells [226]. MiR-122 also inhibited lung CSCs self-renewal by targeting the Hedgehog, Notch, and Wnt/β-catenin pathways. Overexpression of MiR-122 increased susceptibility of gemcitabine-resistant NSCLC to chemotherapy and radiotherapy [227]. The researchers demonstrated continuous miR‐302–367 cluster expression inhibited the CXCR4 pathway. The CXCR4 repression inhibited the self-renewal and stem cell-like markers in GCSCs by inhibiting the Shh‐Gli‐Nanog network [228].

The main CSCs signaling pathways regulation by epigenetic mechanisms. Epigenetic deregulation of CSC-related signaling pathways enables cancer cells to obtain self-renewal properties and drug resistance characteristics. Hedgehog signaling pathway can be activated by Shh promoter hypomethylation and enhance HDAC1 expression. Wnt/β-catenin signaling can be strengthened by reduced DKK1 inhibitor expression of it via promoter hypermethylation and enhanced H3K27me3 and reduced acetylation at H3K16. Notch signaling focus on genes including Hes1 and Hes5 which can be active by STRAP at their promoter region
Notch signaling pathway
The Notch signaling pathway is an active pathway in cancer cells that has a key function in cell fate, including proliferation, differentiation, metastasis, angiogenesis, and self-renewal. Aberrant function of this pathway has been seen in various cancers [111]. Owing to the Notch signaling role in maintaining CSCs, inhibition of this pathway can be a useful target for treating cancer by eliminating CSCs. Moreover, due to cross-talk of this pathway with other signaling pathways, including Wnt and Hh pathways, its targeting has been considered in clinical trials [229, 230]. Notch is a transmembrane receptor categorized as, Notch-1, 2, 3, and 4. Activation of receptors by binding to Notch ligands (Delta-like [Dll] 1, 3, 4, and Jagged 1, 2) releases Notch intracellular domain (NICD) into the nucleus and activates target genes [231]. Notch Signaling can promote stemness and EMT in cancer cells by affecting transcription factors connected to EMT and stemness including Snail, Slug, SOX2, Nanog and OCT4 [232]. For example, Notch-1 and JAG ligands overexpressed in colon cancer that increased proteins related to EMT and stemness and promoted CSC phenotype in these cells [233]. Notch signaling can work as an oncogene or tumor suppressor based on cell context [234]. Epigenetic changes affect the Notch pathway items and regulate the activity of this pathway. Researchers have shown the high abundance of H3K4me3 in Notch genes activates transcription of Notch related genes in CRC. conversely, the abundance of H3K27me3 in upstream activators of Notch signaling represses gene expression and decreases the expression of stem cell markers in CRC (Fig. 5) [235]. Wang et al. proved that oxaliplatin enhanced the Notch signaling in CRC by reducing the level of H3K27me3 in the Notch 2 transcription initiation region. The Notch pathway increased the expression of stemness-linked genes in cells and by increasing the CSCs formation leads to greater cell resistance. In contrast, the elevation of H3K27me3 level increased the sensitivity of patients to oxaliplatin [236]. Hyperacetylation in the JAGGED2 promoter overexpresses Notch ligand Jagged2 in myeloma cell lines which has a key function in progression of malignant cells [237]. Another study reported Hairy/Enhancer of Split (HES) as a Notch effector and transcriptional inhibitor, suppressed the Notch pathway by hypermethylation of DLL1 gene [232]. Improper Notch-Hes1 activity observed in various CSCs, including glioblastoma, breast, pancreatic, and osteosarcoma, plays a central place in stemness, self-renewal, and maintenance of CSCs [238]. High levels of EZH2 had a direct effect on Notch expression and signaling in BCSCs in an independent behavior with the activity of EZH2 histone methyltransferase that led to the development of CSCs populations, and an increase in tumor initiation [239]. MiRNAs also affect the function of Notch signaling in various cancers. For example, a study showed the expression of miR-26a downregulated in osteosarcoma CSCs. MiR-26a overexpression inhibited Jagged1 function and the Jagged1/Notch signaling, thereby reducing the expression of stem cell markers, tumor progression, and enhanced cells’ sensitivity to chemotherapy [240]. MiR-34a also decreased in BCSCs. MiR-34a expression was inversely related to Notch expression and consequently proliferation, migration and invasion of BCSCs. Overexpression of miR-34a enhanced cells’ sensitivity to chemotherapy by reducing the expression of Notch [229]. Ma et al. showed MiR‐129‐5p decreased self-renewal ability, stemness, proliferation, metastasis and chemoresistance in NSCLC by reducing the Notch signaling receptor delta‐like 1 homolog (DLK1) expression [241]. Downregulation of miR424, miR-222, miR-200b, and let-7c in liver CSCs, increased expression of Notch 3, which is one of the essential genes for the activity of CSCs [242]. Downregulation of miR-200 members also promoted Notch pathway activation in pancreatic adenocarcinomas and aggressive basal type of breast cancer by targeting this pathway component, such as Jagged1 and mastermind-like coactivators MAML2 and MAML3. The activation of Notch signalling maintained the CSCS and increased cells survival, stemness and drug resistance [243].
TGF-β and BMP signaling pathways
Transforming growth factor-beta (TGF-β) is active in different pathways such as cellular homeostasis, cell growth, cell differentiation, and apoptosis in adult and embryonic cells [244]. TGF-β also has an active function in the forming CSCs and resistance chemotherapy and radiotherapy [207]. TGF-β superfamily ligands such as bone morphogenetic proteins (BMPs) bind to type I (that is, signal propagating) and type II (that is, activator) receptors which induce the signaling pathway by activating the SMAD transcription factor in nucleus, open repressive chromatin and regulate target gene expression (Fig. 5) [245]. Scientists have proven that the TGF-β pathway is essential for maintaining the characteristics of CSCs. For example, TGF-β increased the CSC markers’ expression, including CD133 in liver cancer cells, which enhanced the tumor initiation and progression [246]. Kim et al. reported TBF-β-induced EMT in lung cancer cells by downregulation of E-cadherin and overexpression of mesenchymal markers such as vimentin, N-cadherin, slug, fibronectin, and snail. In addition, TBF-β-induced stemness phenotype in the cells by the overexpression of CD87 which could be due to its improper promoter demethylation [82]. Another study reported TGF-β signaling could induce EMT in BCSCs by increasing the population of CSCs to cause treatment resistance. BMP2/7 heterodimer as an TGF-β antagonist decreased TGF-β-driven SMAD signaling and cancer cell progression [7, 247]. TGF-β1 also-induced EMT in hepatocellular carcinoma that increased the density of CSCs [248]. Therefore, deregulation of TGF-β/BMP pathway affects the formation of CSCs [246]. Epigenetic changes can affect the TGF-β ligands or pathway components and TGF-β-related gene expression [245]. Besides, the activated SMAD in the TGF-β/BMP pathway can recruit the epigenetic regulators in nucleus such as HATs, HDACs, DNMTs and lncRNAs which affects the expression of TGF-β target genes [249]. Differentially methylated regions were observed in genes encoding proteins linked to the TGF-β signaling pathway in BCSCs compare to non‐BCSCs. These regions were hypomethylated that led to the overexpression of TGF-β target proteins and affect the regulation of BCSCs differentiation [175]. Another study reported that gene expression associated with the TGF-β signaling pathway increased in chemotherapy-resistant triple-negative breast cancer cells, leading to an increase in CSCs markers. TGF-β type I receptor inhibition and SMAD4 blocked the formation of CSCs and decreased drug resistance in these cells [250]. Scientists have shown that TGFβ1 inhibits DNMT1 and DNMT3β in hepatocellular carcinoma, which led to overexpression of CD133 in the cells by demethylation of CD133 promoter-1. CD133 expression increased the self-renewal, tumor initiation, and resistance to chemotherapy in liver cancer cells [251]. Lee et al. reported that the aberrant methylation of BMP signaling by EZH2 led to the inhibition of the normal differentiation process in glioblastoma stem-like cells and activated their proliferation. Demethylation of the BMP receptor promoter induced the ability of differentiate CSCs and reduced their tumorigenesis [252]. Another study reported miR-495 acted as a tumor suppressor gene in CSCs. Its downregulation induced EMT in the CSCs population in oral squamous cell carcinoma (OSCC) by activating the TGF-β signaling pathway that increased proliferation, migration, and invasion in OSCC [253]. MiR-21 overexpressed about 3–7 times in chemoresistant colon cancer HCT-116 and HT-29 cells rich in CSCs. Mir-21 induced downregulation of TGFβ-receptor-2 (TGFβR2) in the cells. TGF-β demonstrated to affect the Wnt/β-catenin pathway during carcinogenesis. In a way, the TGFbR2 downredulation increased the Wnt/β-catenin signaling and promoted the stemness of colon cancer cells [254]. Pellatt et al. found deregulation of different miRNAs altered the expression of foreign genes in the TGFβ-signaling pathway in colon cancer cells such as TGFBR1, BMP6, BMP2, BMP5, BMP7, BMP7, TGIF1, TGIF2, TFDP1, and TGFβ2 which led to tumor progression [255].
Epigenetic targeting of CSC
Given the extensive role of epigenetic changes in the development of CSCs and the consequent spread of tumors and their resistance to conventional therapies, targeting them can be an promising strategy in eliminating CSCs and cancer treatment [26, 256]. DNMTis and HDACis as two of the most important epigenetic inhibitors in CSCs are described below. The use of these epigenetic drugs alone, in combination with each other, or with chemotherapy and radiotherapy has provided promising results in the treatment of cancer [256].
DNMT inhibitors
Studies have shown that DNMTs are essential for the maintaining CSCs, and inhibiting them can reduce tumor development by removing CSCs [257]. DNMT inhibitors (DNMTis) are the first class of drugs prevent epigenetic changes [11]. For example, the researchers showed that inhibition of DNMT1 reduced the lung CSCs proliferation through reducing the methylation of cell cycle regulators and thus inhibited tumor growth [258]. Desitabin and 5-azacitidine (Aza) are the most common DNMTis, which are in the different process of clinical trials in various cancers. They are cytosine analogs that inhibit DNA methyltransferase’s function by participating in DNA structure and covalently binding to enzymes (Fig. 6) [256, 259]. For example, many studies have shown that the use of Aza and 5-Aza-2′-deoxycytidine (AzadC) has successful results in epigenetic therapies. Wongtrakoongate et al. reported AzadC reduced the population of GSCs by upregulation of microRNA-137, which had an inhibitory effect on CSC proliferation. This treatment also decreased the stemness genes expression in prostate and pancreatic CSCs [260]. Decitabine downregulated genes of OCT4 and NANOG in the prostate CSCs. It upregulated the expression of differentiation-related genes, Nkx3.1, CK5, CK8, and PSA/PSP94; by inducing differentiation in the cells significantly reduced the self-renewal and tumorigenesis in CSCs [148]. Another study showed that the DNMT1 expression in PCSCs was higher than non-CSCs. Inhibition of DNMT1 with zebularine decreased the tumorigenic and self-renewal capacity of CSCs in vivo and chemoresistance by overexpression and activation of silenced miRNAs during DNA hypermethylation, especially the miR-17–92 cluster. Zebularin is a newer inhibitor that is less toxic and can be taken orally [261]. SGI-110 is a new DNMT inhibitor whose low concentrations on ovarian CSCs led to their reprogramming and reduced tumor initiating ability, and increased cell sensitivity to platinum. In such a way, its use after treatment of ovarian CSC with carboplatin, inhibited the growth of ovarian cells by creating profound hypomethalation in cells in vivo [262]. In general, CSCs are resistant to apoptotic drugs due to specific epigenetic changes in them. For example, Capper et al. showed GSCs are resistant to tumor necrosis factor (TNF)-related apoptosis such as TRAIL-based therapies, and also chemotherapy drugs such as temozolomide, carboplatin, paclitaxel and etoposide due to hypermethylation of caspase 8 promoter [19]. Studies have shown decitabin as a DNMT inhibitor can restore caspase 8 in the cells [263]. Li et al. reported DNMTi 5-Azadc blocked stemness and self-renewal in colorectal CSCs by reducing β-catenin activity and downregulation of the Wnt pathway [257]. Another study showed that DNMT1 inhibition reduced tumor formation and metastasis in human and mouse breast cancer cells by lowering the CSC formation in them. Inhibition of DNMT1 enhanced the expression of the tumor suppressor genes Isl1 in mammary tumors and CSCs [176]. Overexpression of all DNMTs in gastric cancer led to the downregulation of tumor suppressor genes and critical genes regulating signaling pathways, proliferation, and apoptosis. Low doses of DNMT inhibitors led to silenced genes’ reactivation in gastric cancer and reduced tumorigenic capacity in gastric CSCs [165].

BMP, TGF-β and the FGF pathways in CSCs. BMP signaling is involved in CSC differentiation. The TGF-β/ Activin/Nodal pathway has different function in CSCs according to cancer type such as CSC self-renewal. TGF-β and the FGF pathways have role in pluripotency of CSCs
HDAC inhibitors
As described, deregulation of histone acetylation and the master epigenetic modification in cancer cells, is due to the overexpression and function of HDACs. Their inhibition can be useful in regulating histone acetylation, controlling the population of CSCs [264]. Different types of HDAC inhibitors (HDACis) are classified as pan-HDACis or isoform-selective HDACis. The pan-HDACis target all HDACs, although; most inhibitors have not targeted class IIa HDACs. The selective HDACis inhibit HDACs in a particular class. The selective inhibitors appear to have better clinical outcomes, although this hypothesis has not yet been proven [265]. Many HDACis have been approved by FDA to treat various cancers or are being trialed [256]. These inhibitors, free or in combination with other drugs, inhibit tumor growth by eradicating CSCs. Studies showed inhibition of class I HDACs (HDAC1-3, 8) is very useful in controlling the population of CSCs. In addition to chromatin remodeling, these inhibitors target the stability and activity of essential proteins for the maintenance of CSCs including transcription factors, including Stat3, HIF-1α, Notch1, β-catenin, c-Jun and NF-κB (Fig. 7) [266]. For example, entinostat is a selective inhibitor of class I HDACs that reduced ALDH-1 activity and CSC markers’ expression in triple-negative breast cancer, such as Bmi-1, Nanog, and Oct-4, and by reactivating the E-cadherin gene, it abolished the EMT phenotype. Therefore, entinostat reduced the CSC population and tumor formation in the primary site and metastasis to the lungs [267]. Another study showed that mocetinostat increased the expression of miR-203 in pancreatic cancer. Overexpression of miR-203 reduced the expression of ZEB1 (an activator of EMT) and CSC markers CD24high/44high and CD133, suppressed stemness traits and increased cell sensitivity to the gemcitabine [268]. AR-42 (OSU-HDAC42), a pan-HDACi, induced apoptosis in leukemic stem cells by blocking NF-κB and Hsp90 activity but does not effect standard hematopoietic stem and progenitor cells [269]. Suberoylanilide hydroxamic acid (SAHA) is another HDAC inhibitor that significantly reduced the tumor sphere formation and proliferation of head and neck cancer cells by reducing Nanog expression in CSCs. Besides, the SAHA removed cells’ resistance to cisplatin and synergistically enhanced the antitumor effect of cisplatin. Consequently, inhibition of HDACs by reducing CSCs increased the cells’ sensitivity to treatment [270]. Vorinostat (also known as SAHA) is an inhibitor of HDAC-1–3 and HDAC 6. This drug’s clinical trials are being performed alone or in combination with other medicines in the different cancer treatments [256]. Quisinostat, an effective inhibitor of class I and II HDACs, synergized the effect of doxorubicin in breast CSCs and non‑CSCs simultaneously [271]. Salvador et al. reported abexinosta, another pan-HDACi, significantly reduced the population of breast CSCs in cells that expressed low levels of long noncoding RNA Xist and induced differentiation in CSC population from low-dose sensitive breast cancer cell lines [272]. MC1742, and MC2625 are newer pan-HDACis that increased acetyl-H3 and acetyl-tubulin and inhibited sarcoma CSC by inducing apoptosis, cellular arrest and differentiation in them [273].

Schematic representation of CSCs epigenome as a target for cancer treatment. Various drugs including Aza-dC (Decitabine), SGI-110 or Zebularine, can reduce DNMT1 protein levels and global methylation. HDAC inhibitors (HDACI), including Mocetinostat, Entinostat and Belinostat, have demonstrated good effect toward inhibitation of HDAC. lncRNAs can be inhibit by GapmeRs or small molecule inhibitors which eventually will be disrupt
Combination of DNMT and HDAC inhibitors
Many studies have reported both DNMTi and HDACi have been approved for the hematologic malignancies treatment by FDA and European Medicines Agency (EMA) [274, 275]. However, in solid tumors, single treatment of these epi-drugs has not yielded favorable results, probably due to their high toxicity, limited bioavailability and low pharmacokinetic effects. However, research has revealed that the combination of HDACs and DNMT inhibitors re-expressed tumor gene suppressor in cancer [275]. High doses of DNMTis cause toxicity, while if combined with HDACis, lower amounts are consumed. Therefore, the combination of these epi-drugs with each other shows potent effects compared to each drug alone and causes synergistic treatment [26]. Aza and decitabine have been approved in the treatment of MDS and AML. They are in Phase II clinical trial in the treatment of ovarian, prostate, and melanoma cancer and combination with HDAC is to treat metastasic melanomas. However, their instability and low bioavailability limited their use [276]. Another study reported the combination of decitabine and SAHA reduced proliferation, self-renewal and EMT processes in PCSCs by reducing expression miR-34a [264]. The phase I/II clinical trials of the combination of Aza and entinostat in patients with refractory metastatic non-small cell lung cancer demonstrated promising treatment results and were utterly tolerable. The general patients’ survival was correlated with these inhibitors’ combined action, which led to the silencing of genes associated with lung cancer [277]. Pathania et al. noted the combination of Aza and butyrate, an HDACi, stopped the growth of CSCs in a mouse mammary tumor model. RNA-seq analysis in CSCs showed that this combination inhibited growth signaling molecules such as RAD51 AP1 and SPC25 by elimination modifications in the chromatin structure [274]. Combination therapy with DNMTi and HDACi also restores the of ER-negative breast cancer cells’ sensitivity to endocrine therapy. These findings support the hypothesis that the combination of DNMT and HDAC inhibitors virtually eliminates CSCs and improves the treatment of refractory and recurrent cancers. However, these inhibitors’ simultaneous use requires a close attention and testing together with clinical barriers that need to be addressed [278] (Table 2).
Conclusion
Cancer resistance and recurrence is one the most important pressing concerns in conventional cancer treatments. Scientists are aware that there is a small population of CSCs in different tumors that create resistance to radiotherapy and chemotherapy, which can be attribute to the abilities to self-renew, differentiate and proliferate indefinitely. These cells can exit the cell cycle for a long time and remain in a quiescent state, rendering treatment difficult and allowing the cells to survive conventional therapies [288]. The dormant phenotype can remain in the CSCs for decades, eventually leading to recurrence of the tumor. In fact, one of the main causes of death in cancer has been reported to be the dormancy state of CSCs, which can cause metastasis and recurrence of the disease after several decades [23, 130, 289]. In addition, due to the ability to change the cell cycle checkpoints and the efficient DNA damage repair system thereof, CSCs have high survival capacity, which makes them resistant to chemotherapy drugs such as cisplatin, oxaliplatin, doxorubicin, daunorubicin and methotrexate [7, 16]. Moreover, CSCs can escape apoptosis by mutating and inactivating apoptotic genes, such as p53, and the overexpression of anti apoptotic proteins, such as AKT and BCL2. High activity of Notch and hedgehog signaling pathways can also activate anti-apoptotic signaling pathways in CSCs [7, 290, 291].
Acting as one of the main agents of MDR in cancers, the overexpression of ABC transporters such as ABCB1, ABCC1, and ABCG2 have been reported in CSCs that protect cells from cytotoxic agents. Abnormal epigenetic changes and signaling pathways directly or indirectly affect their ABC expression in CSCs [117, 290]. Studies have revealed that ABC transpoters expression is increased by promoter hypomethylation of these genes. This can be seen in the promoter methylation of the ABCG2 transporter regulating the expression in CSCs. Further observations have been made that increasing the activity of the DNMTs by melatonin decreased the expression of this gene in brain CSCs through hypermethylating the ABCG2 promoter [292], while ABCC1 overexpression was also observed to be regulated in the CSCs by activating the Notch pathway, which causes cell resistance [288].
Hence, considering the role of CSCs in therapeutic resistance, recognizing the characteristics of CSCs among other tumor cells and the factors affecting the formation will be beneficial in the design of targeted drugs for cancer. In the present review, recent information about the biology and characteristics of CSCs was explored, including CSCs markers, CSCs niche, the relationship between EMT and CSCs, the factors that create resistance and influencing the generation of them. An integral factor in the formation and maintenance of CSCs in the epigenetic modifications that promote tumorgenesis and metastasis by deregulation of gene expression and vital cell signaling pathways. Numerous studies have demonstrated that the deregulation of DNA methylation, histone modifications, RNA methylations, miRNAs and LncRNAs in stem cells or adult cells will downregulate the expression of tumor-suppressor genes, upregulate the expression of oncogenes, activate the expression of CSCs surface markers and ultimately induce CSCs formation pathways and maintain the properties of them which is achieved by altering cell function and increasing plasticity, self-renewal, differentiation, and survival in the cells. These abnormal epigenetic changes also affect the expression and function of ligands or signaling pathway items in connection with the preservation the CSC characteristics such as Wnt/β-catenin, Notch, Hedgehog, and TGF-β/BMP. Aberrant activities of aforementioned signaling pathways effectuated by increasing the genes expression has been demonstrated to contribute to the maintenance of phenotype and function of CSCs, which increases the population thereof and has a critical function in the initiation, progression and invasion of various types of cancer. By inducing EMT and overexpression of metastasis-related genes therein, deregulation of epigenetic alternation can also increase the ability of metastasis and invasion of CSCs to different tissues, causing disease relapse even after therapy. Therefore, these epigenetic changes provide promising prospects for treatment and have been considered by scientists to generally eliminate CSCs and overcome chemotherapy resistance. In particular, recognizing epigenetic signatures in different genes in CSCs, which are currently considered as clinical biomarkers [293,294,295], is integral to the prevention and treatment of cancer. In existing research, epigenetic inhibitors have exhibited favorable results in treatment. In addition, several epi-drugs are at various stages of clinical trials and some have been approved by the FDA, such as decitabine, azacitidine, and vorinostat, with the combination of these inhibitors showing synergistic effects and favorable results in treatment. However, further research is required to determine the more specific features of CSCs in the design of targeted treatment strategies, so that treatment is performed directly on CSCs rather than on stem cells similar thence. Because the niche of CSCs is related to the niche of normal stem cells, this has led to a lack of specific treatment in some cases and also involved healthy tissues [296]. Thus, to prevent recurrence and resistance of various cancers and increase the lifespan of the patient, one of the greatest strategies in cancer treatment is to target CSCs directly and specifically.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable request by permission of institute and department chairman’s.
Abbreviations
- ABC:
-
ATP-binding cassette
- ABCG2:
-
ATP-binding cassette sub-family G member 2
- AML:
-
Acute myeloid leukemia
- ALDH1:
-
Adehyde dehydrogenase1
- APC:
-
Adenomatous polyposis coli
- ASCL1:
-
Achaete-scute family BHLH transcription factor 1
- ATLL:
-
Adult T cell leukemia/lymphoma
- Aza:
-
5-Azacitidine
- AzadC:
-
5-Aza-2′-deoxycytidine
- BCSCs:
-
Breast cancer stem cells
- BMI1:
-
B-Lymphoma Mo-MLV insertion region 1 homolog
- BMPs:
-
Bone morphogenetic proteins
- CAFs:
-
Cancer-associated fibroblasts
- CLL:
-
Chronic lymphocytic leukemia
- CMML:
-
Chronic myelomonocytic leukemia
- CNS:
-
Central nervous system
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cells
- CSCC:
-
Cervical squamous cell carcinoma
- CTCL:
-
Cutaneous T cell lymphoma
- DKK1:
-
Dickkopf-related protein 1
- Dll:
-
Delta-like
- DLK1:
-
Delta‐like 1 homolog
- DNMTs:
-
DNA methyltransferases
- DNMTis:
-
DNMT inhibitors
- DZNep:
-
3-Deazaneplanocin A
- ECM:
-
Extracellular matrix
- EGF:
-
Epithelial growth factor
- EMA:
-
European Medicines Agency
- EMT:
-
Epithelial-to-mesenchymal transition
- EpCAM:
-
Epithelial cell adhesion molecule
- ERCC1:
-
Excision repair cross-complementation group 1
- ESCs:
-
Embryonic stem cells
- EZH2:
-
Enhancer of zeste homolog 2
- FAD:
-
Flavin adenine dinucleotide
- FDA:
-
Food and Drug Administration
- FGF:
-
Fibroblast growth factor
- GBM:
-
Glioblastoma multiforme
- Gli:
-
Glioma-associated oncogene
- H3Ac:
-
Acetylated histones H3
- H3K27:
-
Histone H3 lysine 27
- H3K36:
-
Histone H3 lysine 36
- H3K4:
-
Histone H3 lysine 4
- H3K79:
-
Histone H3 lysine 79
- H3K9:
-
Histone H3 lysine 9
- H3K27me3:
-
Trimethylation of H3K27
- H4K20:
-
Histone H4 lysine 20
- HAT:
-
Histone acetyltransferases
- HCC:
-
Hepatocellular carcinoma
- HDAC:
-
Histone deacetylase
- HDACis:
-
HDAC inhibitors
- HES:
-
Hairy/Enhancer of Split
- HGF:
-
Hepatocyte growth factor
- Hh:
-
Hedgehog
- HIFs:
-
Hypoxia-inducible factors
- HKMT:
-
Histone lysine methyltransferase
- HMT:
-
Histone methyltransferases
- HNSCC:
-
Head and neck squamous cell carcinoma
- IL-6:
-
Interleukin-6
- GSCs:
-
Glioblastoma stem cells
- GNRH:
-
Gonadotropin-releasing hormone
- KDM:
-
Histone lysine demethylase
- lncRNAs:
-
Long noncoding RNAs
- LSCs:
-
Leukemic stem cells
- m6A:
-
N6-methyladenosine
- MDS:
-
Myelodysplastic syndrome
- METTL3:
-
Methyltransferase-like 3
- miRNAs:
-
Micro-RNAs
- MLL:
-
Mixed lineage leukemia
- MLL2:
-
Mixed lineage leukemia protein 2
- MPM:
-
Malignant pleural mesothelioma
- MSCs:
-
Mesenchymal stem cells
- ncRNAs:
-
Non coding RNAs
- NER:
-
Nucleotide excision repair
- NF-κB:
-
Nuclear factor kappa b
- NICD:
-
Notch intracellular domain
- NSCLC:
-
Non-small cell lung cancer
- NSCs:
-
Neural stem cells
- OCT4:
-
Octamer-binding transcription factor 4
- OSCC:
-
Oral squamous cell carcinoma
- PcG:
-
Polycomb-group proteins
- PCSCs:
-
Pancreatic CSCs
- PTCH1:
-
Patched receptor
- RCC:
-
Renal cell carcinoma
- PRC2:
-
Polycomb repressive complex 2
- SAHA:
-
Suberoylanilide hydroxamic acid
- SFRP:
-
Several frizzled-related protein
- Shh:
-
Sonic hedgehog ligand
- SiRNA:
-
Small interfering RNAs
- SCLC:
-
Small cell lung cancer
- SMAD Proteins:
-
Sma and Mad proteins
- SMO:
-
Smoothened
- SNAIL:
-
Snail family zinc finger 1
- SOX2:
-
SRY-box 2
- STRAP:
-
Serine–threonine kinase receptor-associated protein
- TGF-β:
-
Transforming growth factor-β
- TGFbR2:
-
TGFb-receptor-2
- TICs:
-
Tumor initiating cells
- TNBC:
-
Triple-negative breast cancer
- TNF:
-
Tumor necrosis factor
- TWIST1:
-
Twist-related protein 1
- WIF-1:
-
Wnt inhibitory factor 1
- WTAP:
-
Wilms tumor 1 associated protein
- ZEB1:
-
Zinc finger E-box-binding homeobox 1
References
Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25:1–18.
Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017;2017:1–17.
Sun XX, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015;36:1219–27. https://doi.org/10.1038/aps.2015.92.
Seoane J. Division hierarchy leads to cell heterogeneity. Nature. 2017;549:164–6.
Kuşoğlu A, Biray Avcı Ç. Cancer stem cells: a brief review of the current status. Gene. 2019;681:80–5. https://doi.org/10.1016/j.gene.2018.09.052.
Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol Mech Dis. 2016;11:47–76.
Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci. 2017;18:2574.
Thi L, Phi H, Sari IN, Yang Y, Lee S, Jun N, et al. Review article cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:1–6.
Clevers H. The cancer stem cell: premises, promises and challenges (review). Nat Med. 2011;17:313–9. https://doi.org/10.1038/nm.2304.
Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev. 2016;49:25–36. https://doi.org/10.1016/j.ctrv.2016.07.001.
Toh TB, Lim JJ, Chow EKH. Epigenetics in cancer stem cells. Mol Cancer. 2017;16:1–20.
Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6:620–36.
Das T. Deciphering the cancer puzzle: cancer stem cells being the pivotal piece. J Stem Cell Res Transplant. 2017;4:1025.
Zhou Y, Xia L, Wang H, Oyang L, Su M, Liu Q, et al. Cancer stem cells in progression of colorectal cancer. Oncotarget. 2018;9:33403–15.
Zhou H, Zhang J, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther. 2021. https://doi.org/10.1038/s41392-020-00430-1.
Das PK, Islam F. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells. 2020;6:1–21.
Skvortsova I, Debbage P, Kumar V, Skvortsov S. Radiation resistance: cancer stem cells (CSCs) and their enigmatic pro-survival signaling. Semin Cancer Biol. 2015. https://doi.org/10.1016/j.semcancer.2015.09.009.
Wang K, Zhang T, Dong Q, Nice EC, Huang C, Wei Y. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013;4:e537–610. https://doi.org/10.1038/cddis.2013.50.
Kenneth KW, Fu LW. Multidrug resistance transporters—roles in maintaining cancer stem-like cells, Stem Cells in Clinic and Research, Ali Gholamrezanezhad, IntechOpen. 2010. https://doi.org/10.5772/23867. Available from: https://www.intechopen.com/books/stem-cells-in-clinic-and-research/multidrug-resistance-transporters-roles-in-maintaining-cancer-stem-like-cells.
Yang X, Liu M, Li M, Zhang S, Hiju H, Sun J, et al. Epigenetic modulations of noncoding RNA: a novel dimension of Cancer biology. Mol Cancer Mol Cancer. 2020;19:1–12.
Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression review-article. Cell Death Dis. 2018. https://doi.org/10.1038/s41419-017-0129-x.
Li G, Wang D, Ma W, An K, Liu Z, Wang X, et al. Transcriptomic and epigenetic analysis of breast cancer stem cells. Epigenomics. 2018;10:765–83.
Ferrer AI, Trinidad JR, Sandiford O, Etchegaray JP, Rameshwar P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020;39:721–38.
Phi LTH, Sari IN, Yang Y-G, Lee S-H, Jun N, Kim KS, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018;2018:1–16.
Yu Z, et al. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51. https://doi.org/10.1016/j.biocel.2012.08.022.
Ghasemi S. Cancer’s epigenetic drugs: where are they in the cancer medicines? Pharmacogenomics J. 2019. https://doi.org/10.1038/s41397-019-0138-5.
Gopalan V, Islam F, Lam AK. Surface markers for the identification of cancer stem cells. Methods Mol Biol. 2018;1692:17-29. https://doi.org/10.1007/978-1-4939-7401-6_2.
Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–40.
Yadav AK, Desai NS. Cancer stem cells: acquisition, characteristics, therapeutic implications, targeting strategies and future prospects. Stem Cell Rev Rep. 2019;15:331–55.
Yiming L, Yunshan G, Bo M, Yu Z, Tao W, Gengfang L, et al. CD133 overexpression correlates with clinicopathological features of gastric cancer patients and its impact on survival: a systematic review and meta-analysis. Oncotarget. 2015;6:42019–27.
Olsson E, Honeth G, Bendahl PO, Saal LH, Gruvberger-Saal S, Ringnér M, et al. CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer. 2011;11:418.
Yamashita T, Wang XW. Review series Cancer stem cells in the development of liver cancer. J Clin Investig. 2013;123:1911–8.
Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–6.
Carvalho MJ, Laranjo M, Abrantes AM, Torgal I, Botelho MF, Oliveira CF. Clinical translation for endometrial cancer stem cells hypothesis. Cancer Metastasis Rev. 2015;34:401–16.
Reid PA, Wilson P, Li Y, Marcu LG, Bezak E. Current understanding of cancer stem cells: review of their radiobiology and role in head and neck cancers. Head Neck. 2017;39:1920–32.
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci. 2007;104:973–8.
Murillo-Sauca O, Chung MK, Shin JH, Karamboulas C, Kwok S, Jung YH, et al. CD271 is a functional and targetable marker of tumor-initiating cells in head and neck squamous cell carcinoma. Oncotarget. 2014;5:6854–66.
Elkashty OA, Abu Elghanam G, Su X, Liu Y, Chauvin PJ, Tran SD. Cancer stem cells enrichment with surface markers CD271 and CD44 in human head and neck squamous cell carcinomas. Carcinogenesis. 2020;41:458–66.
Allegra E, Trapasso S. Role of CD44 as a marker of cancer stem cells in head and neck cancer. Biol Targets Ther. 2012;6:379.
Faber A. CD44 as a stem cell marker in head and neck squamous cell carcinoma. Oncol Rep. 2011;26:321–6.
Simard EP, Torre LA, Jemal A. International trends in head and neck cancer incidence rates: differences by country, sex and anatomic site. Oral Oncol. 2014;50:387–403.
Panigoro SS, Kurnia D, Kurnia A, Haryono SJ, Albar ZA. ALDH1 Cancer stem cell marker as a prognostic factor in triple-negative breast cancer. Int J Surg Oncol. 2020;2020:1–7.
Kong Y, Lyu N, Wu J, Tang H, Xie X, Yang L, et al. Breast cancer stem cell markers CD44 and ALDH1A1 in serum: distribution and prognostic value in patients with primary breast cancer. J Cancer. 2018;9:3728–35.
Han L, Gao X, Gu X, Guo W, Ma M, Qi X, et al. Prognostic significance of cancer stem cell marker CD133 expression in breast cancer. Int J Clin Exp Med. 2017;10:4829–37.
Borgna S, Armellin M, di Gennaro A, Maestro R, Santarosa M. Mesenchymal traits are selected along with stem features in breast cancer cells grown as mammospheres. Cell Cycle. 2012;11:4242–51.
Wang Z, Wang Q, Wang Q, Wang Y, Chen J. Prognostic Significance of CD24 and CD44 in breast cancer: a meta-analysis. Int J Biol Mark. 2017;32:75–82.
Harris KS, Kerr BA. Prostate cancer stem cell markers drive progression, therapeutic resistance, and bone metastasis. Stem Cells Int. 2017;2017:1–9.
Klemba A, Purzycka-Olewiecka JK, Wcisło G, Czarnecka AM, Lewicki S, Lesyng B, et al. Surface markers of cancer stem-like cells of ovarian cancer and their clinical relevance. Współczesna Onkol. 2018;2018:48–55.
Suster NK, Virant-Klun I. Presence and role of stem cells in ovarian cancer. World J Stem Cells. 2019;11:383–97.
Mokhtari M, Zakerzade Z. EPCAM expression in colon adenocarcinoma and its relationship with TNM staging. Adv Biomed Res. 2017;6:56.
Wang Z, Tang Y, Xie L, Huang A, Xue C, Gu Z, et al. The prognostic and clinical value of CD44 in colorectal cancer: a meta-analysis. Front Oncol. 2019;9:309.
Chen S, Song X, Chen Z, Li X, Li M, Liu H, et al. CD133 expression and the prognosis of colorectal cancer: a systematic review and meta-analysis. PLoS ONE. 2013;8:e56380.
Abotchie PN, Vernon SW, Du XL. Gender differences in colorectal cancer incidence in the United States, 1975–2006. J Women’s Health. 2012;21:393–400.
Fiedorowicz M, Khan MI, Strzemecki D, Orzeł J, Wełniak-Kamińska M, Sobiborowicz A, et al. Renal carcinoma CD105−/CD44− cells display stem-like properties in vitro and form aggressive tumors in vivo. Sci Rep. 2020;10:5379.
Corrò C, Moch H. Biomarker discovery for renal cancer stem cells. J Pathol Clin Res. 2018;4:3–18.
Peired AJ, Sisti A, Romagnani P. Renal cancer stem cells: characterization and targeted therapies. Stem Cells Int. 2016;2016:1–12.
Baeza Pérez G, Calaf G, Montalvo Villalba M, Salgado Prieto K, Caba BF. Frequency of hematologic malignancies in the population of Arica, Chile. Oncol Lett. 2019;18:5637–44.
Jiang Z, Wu D, Lin S, Li P. CD34 and CD38 are prognostic biomarkers for acute B lymphoblastic leukemia. Biomark Res. 2016;4:23.
Fathi E, Farahzadi R, Sheervalilou R, Sanaat Z, Vietor I. A general view of CD33 + leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. BLOOD Res. 2020;55:10–6.
Cap J, Babusikova O, Kaiserova E, Jamarik M. Expression of CD10, CD19 and CD34 markers in bone marrow samples of children with precursor B-cell acute lymphoblastic leukemia in clinical and hematological remission. Neoplasma. 1998;45:231.
Rego EM, Garcia AB, Carneiro JJ, Falcão RP. Immunophenotype of normal and leukemic bone marrow B-precursors in a Brazilian population. A comparative analysis by quantitative fluorescence cytometry. Braz J Med Biol Res. 2001;34:183–94.
Mao X, Zhang X, Xue X, Guo G, Wang P, Zhang W, et al. Brain tumor stem-like cells identified by neural stem cell marker CD15. Transl Oncol. 2009;2:247–57.
He J, Liu Y, Zhu T, Zhu J, DiMeco F, Vescovi AL, et al. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol Cell Proteomics. 2012;11:M111.010744.
Natri HM, Wilson MA, Buetow KH. Distinct molecular etiologies of male and female hepatocellular carcinoma. BMC Cancer. 2019;19:951.
Chen Y, Yu D, Zhang H, He H, Zhang C, Zhao W, et al. CD133 + EpCAM + phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int J Biol Sci. 2012;8:992–1004.
Xiang Y, Yang T, Pang B, Zhu Y, Liu Y. The progress and prospects of putative biomarkers for liver cancer stem cells in hepatocellular carcinoma. Stem Cells Int. 2016;2016:1–14.
Kumar D, Gorain M, Kundu G, Kundu GC. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol Cancer. 2017;16:7.
Civenni G, Walter A, Kobert N, Mihic-Probst D, Zipser M, Belloni B, et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011;71:3098–109.
Kitson SJ, Rosser M, Fischer DP, Marshall KM, Clarke RB, Crosbie EJ. Targeting endometrial cancer stem cell activity with metformin is inhibited by patient-derived adipocyte-secreted factors. Cancers (Basel). 2019;11:653.
Roudi R, Madjd Z, Korourian A, Mehrazma M, Molanae S, Sabet MN, et al. Clinical significance of putative cancer stem cell marker CD44 in different histological subtypes of lung cancer. Cancer Biomark. 2014;14:457–67.
Tachezy M, Zander H, Wolters-Eisfeld G, Müller J, Wicklein D, Gebauer F, et al. Activated leukocyte cell adhesion molecule (CD166): an “inert” cancer stem cell marker for non-small cell lung cancer? Stem Cells. 2014;32:1429–36.
Zakaria N, Satar NA, Abu Halim NH, Ngalim SH, Yusoff NM, Lin J, et al. Targeting lung cancer stem cells: research and clinical impacts. Front Oncol. 2017;7:80.
Das A, Dutta A, Paul S, Dutta A, Bhattacharya A, Banerjee S, et al. Cancer stem cells, their origin and niche: a search for the therapeutic target. J Stem Cell Res Med. 2017;2:1–11.
Rossi F, Noren H, Jove R, Beljanski V, Grinnemo KH. Differences and similarities between cancer and somatic stem cells: therapeutic implications. Stem Cell Res Ther. 2020;11:1–16.
Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50:117–25.
Szafarowski T, Szczepanski MJ. Cancer stem cells in head and neck squamous cell carcinoma. Otolaryngol Pol. 2014;68:105–11.
Loret N, Denys H, Tummers P, Berx G. The role of epithelial-to-mesenchymal plasticity in ovarian cancer progression and therapy resistance. Cancers (Basel). 2019;11:1–22.
Jayachandran A, Dhungel B, Steel JC. Epithelial-to-mesenchymal plasticity of cancer stem cells: therapeutic targets in hepatocellular carcinoma. J Hematol Oncol. 2016;9:1–12. https://doi.org/10.1186/s13045-016-0307-9.
Abell AN, Johnson GL. Implications of mesenchymal cells in cancer stem cell populations: relevance to EMT. Curr Pathobiol Rep. 2014;2:21–6.
Bharti R, Dey G, Mandal M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: a snapshot of IL-6 mediated involvement. Cancer Lett. 2016;375:51–61. https://doi.org/10.1016/j.canlet.2016.02.048.
Tanabe S, Quader S, Cabral H, Ono R. Interplay of EMT and CSC in cancer and the potential therapeutic strategies. Front Pharmacol. 2020;11:1–8.
Kim BN, Ahn DH, Kang N, Yeo CD, Kim YK, Lee KY, et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci Rep. 2020;10:1–11. https://doi.org/10.1038/s41598-020-67325-7.
Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–94. https://doi.org/10.1038/ncb2976.
Mano SS, Uto K, Ebara M. Fluidity of poly(ε-caprolactone)-based material induces epithelial-to-mesenchymal transition. Int J Mol Sci. 2020;21:1757.
Doherty MR, Smigiel JM, Junk DJ, Jackson MW. Cancer stem cell plasticity drives therapeutic resistance. Cancers. 2016;8:1–13.
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah K, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–40. https://doi.org/10.1016/j.cell.2011.04.029.
Zhang J, Tian X, Xing J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J Clin Med. 2016;5:1–18.
Najafi M, Mortezaee K, Ahadi R. Cancer stem cell (a)symmetry and plasticity: tumorigenesis and therapy relevance. Life Sci. 2019;231:116520.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2016;16:225–38.
Fessler E, Dijkgraaf FE, De Sousa E, Melo F, Medema JP. Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett. 2013;341:97–104. https://doi.org/10.1016/j.canlet.2012.10.015.
Noll JE, Vandyke K, Zannettino ACW. The role of the “cancer stem cell niche” in cancer initiation and progression. In: Adult Stem Cell Niches, Sabine Wislet-Gendebien, IntechOpen. 2014. https://doi.org/10.5772/58598.
Liu J, Li P, Wang L, Li M, Ge Z, Noordam L, et al. Cancer-associated fibroblasts provide a stromal niche for liver cancer organoids that confers trophic effects and therapy resistance. Cmgh. 2021;11:407–31. https://doi.org/10.1016/j.jcmgh.2020.09.003.
Valenti G, Quinn HM, Heynen GJJE, Lan L, Holland JD. Cancer stem cells regulate cancer-associated fibroblasts via activation of hedgehog signaling in mammary gland tumors. Cancer Res. 2017;77:2134–48.
Cuiffo BG, Karnoub AE. Mesenchymal stem cells in tumor development. Cell Adhes Migr. 2012;6(220):230.
Tsai K, Yang S, Lei Y, Tsai C, Chen H, Hsu C, et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology. 2011;141:1046–56. https://doi.org/10.1053/j.gastro.2011.05.045.
Cuiffo BG, Campagne A, Bell GW, Lembo A, Orso F, Lien EC, et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. 2014;15:762–74. https://doi.org/10.1016/j.stem.2014.10.001.
Collet G, El Hafny-Rahbi B, Nadim M. Hypoxia-shaped vascular niche for cancer stem cells. Contemp Oncol. 2015;19:A39.
Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102(789):95.
Tong WW, Tong GH, Liu Y. Cancer stem cells and hypoxia-inducible factors (review). Int J Oncol. 2018;53:469–76.
Zhang S, Luo X, Wan F, Lei T. The roles of hypoxia-inducible factors in regulating neural stem cells migration to glioma stem cells and determinating their fates. Neurochem Res. 2012;37:2659–66.
Zhang Q, Han Z, Zhu Y, Chen J, Li W. Role of hypoxia inducible factor-1 in cancer stem cells (review). Mol Med Rep. 2021;23:1–14.
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–30. https://doi.org/10.1038/nature09557.
Matsuda S, Yan T, Mizutani A, Sota T, Hiramoto Y, Prieto-Vila M, et al. Cancer stem cells maintain a hierarchy of differentiation by creating their niche. Int J Cancer. 2014;135:27–36.
Matsui WH. Cancer stem cell signaling pathways. Med (United States). 2016;95:S8-19.
Yamamoto Y, Yoshioka Y, Minoura K, Takahashi R, Takeshita F, Taya T, et al. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Mol Cancer. 2011;10:135.
Schmidt F, Efferth T. Tumor heterogeneity, single-cell sequencing, and drug resistance. Pharmaceuticals. 2016;9:33.
Efferth T, Konkimalla VB, Wang Y-F, Sauerbrey A, Meinhardt S, Zintl F, et al. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin Cancer Res. 2008;14:2405–12.
Longley D, Johnston P. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–92.
Naik PP, Das DN, Panda PK, Mukhopadhyay S, Sinha N, Praharaj PP, et al. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncol. 2016;62:122–35.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60.
Carruthers RD, Ahmed SU, Ramachandran S, Strathdee K, Kurian KM, Hedley A, et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018;78:5060–71.
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–7.
da Silva SD, Hier M, Mlynarek A, Kowalski LP, Alaoui-Jamali MA. Recurrent oral cancer: current and emerging therapeutic approaches. Front Pharmacol. 2012;3:149.
Safa AR. Resistance to cell death and its modulation in cancer stem cells. Crit Rev Oncog. 2016;21:203–19.
Hong M, Tan H, Li S, Cheung F, Wang N, Nagamatsu T, et al. Cancer stem cells: the potential targets of chinese medicines and their active compounds. Int J Mol Sci. 2016;17:893.
Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova I-I. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–67.
Begicevic R-R, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18:2362.
Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–34.
Takeishi S, Nakayama KI. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci. 2016;107:875–81.
Vassalli G. Aldehyde dehydrogenases: not just markers, but functional regulators of stem cells. Stem Cells Int. 2019;2019:1–15.
Wang R, Sun Q, Wang P, Liu M, Xiong S, Luo J, et al. Notch and Wnt/β-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget. 2016;7:5754–68.
Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Emerging targets for glioblastoma stem cell therapy. J Biomed Res. 2016;30(19):31.
Yuan Z, Mo J, Zhao G, Shu G, Fu H, Zhao W. Targeting strategies for renal cell carcinoma: from renal cancer cells to renal cancer stem cells. Front Pharmacol. 2016;7:423.
Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu H-Y, Lin L-T, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78-103.
Moitra K. Overcoming multidrug resistance in cancer stem cells. Biomed Res Int. 2015;2015:1–8.
Wilson BJ, Saab KR, Ma J, Schatton T, Pütz P, Zhan Q, et al. ABCB5 maintains melanoma-initiating cells through a proinflammatory cytokine signaling circuit. Cancer Res. 2014;74:4196–207.
Su C, Zhang J, Yarden Y, Fu L. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct Target Ther. 2021;6:1–15. https://doi.org/10.1038/s41392-021-00499-2.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020. https://doi.org/10.1038/s41392-020-0110-5.
McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor-initiating cells. Expert Opin Drug Metab Toxicol. 2016;12:633–44.
Chen W, Dong J, Haiech J, Kilhoffer M-C, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1–16.
Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L, et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene. 2017;36:6680–90.
Macha MA, Rachagani S, Qazi AK, Jahan R, Gupta S, Patel A, et al. Afatinib radiosensitizes head and neck squamous cell carcinoma cells by targeting cancer stem cells. Oncotarget. 2017;8:20961–73.
Colak S, Medema JP. Cancer stem cells—important players in tumor therapy resistance. FEBS J. 2014;281:4779–91.
Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: the root of tumor recurrence and metastases. Semin Cancer Biol. 2017;44:10–24.
Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006;66:1517–25.
Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18.
Gonzalez-Torres C, Gaytan-Cervantes J, Vazquez-Santillan K, Mandujano-Tinoco EA, Ceballos-Cancino G, Garcia-Venzor A, et al. NF-κB participates in the stem cell phenotype of ovarian cancer cells. Arch Med Res. 2017;48:343–51.
Chen W-J, Huang R-FS. Low-folate stress reprograms cancer stem cell-like potentials and bioenergetics metabolism through activation of mTOR signaling pathway to promote in vitro invasion and in vivo tumorigenicity of lung cancers. J Nutr Biochem. 2018;53:28–38.
Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK—a conserved kinase: functions, signaling, cancer, and controversy. Clin Transl Med. 2015;4:1–8.
Kim S-H, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015;4:226–38.
Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019;26:690–702. https://doi.org/10.1038/s41418-019-0292-y.
Pagotto A, Pilotto G, Mazzoldi EL, Nicoletto MO, Frezzini S, Pastò A, et al. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017;8:e2943. https://doi.org/10.1038/cddis.2017.327.
Wei MF, Chen MW, Chen KC, Lou PJ, Lin SYF, Hung SC, et al. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy. 2014;10:1179–92.
Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997;57:2369–72.
Bourguignon LYW, Wong G, Shiina M. Up-regulation of histone methyltransferase, DOT1L, by matrix hyaluronan promotes MicroRNA-10 expression leading to tumor cell invasion and chemoresistance in cancer stem cells from head and neck squamous cell. J Biol Chem. 2016;291:10571–85.
Jambhekar A, Dhall A, Shi Y. Roles and regulation of histone methylation in animal development. Nat Rev Mol Cell Biol. 2019;20:625–41. https://doi.org/10.1038/s41580-019-0151-1.
Grønbæk K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS. 2007;115:1039–59.
Zagorac S, Garcia-Bermejo L, Sainz B. The epigenetic landscape of pancreatic cancer stem cells. Epigenomes. 2018;2:10.
Ortiz-sánchez E. Overview: epigenetic regulation in cancer stem cells by methylation. 2014;1(1):1017.
Castilho RM, Squarize CH, Almeida LO. Epigenetic modifications and head and neck cancer: implications for tumor progression and resistance to therapy. Int J Mol Sci. 2017;18:1506.
Momparler RL, Côté S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin Investig Drugs. 2015;24:1–13.
Deshmukh A, Binju M, Arfuso F, Newsholme P, Dharmarajan A. Role of epigenetic modulation in cancer stem cell fate. Int J Biochem Cell Biol. 2017. https://doi.org/10.1016/j.biocel.2017.07.003.
Chen Y, Ren B, Yang J. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management H2A. Signal Transduct Target Ther. 2020. https://doi.org/10.1038/s41392-020-00252-1.
Liu N, Li S, Wu N, Cho K. Acetylation and deacetylation in cancer stem-like cells. Oncotarget. 2017;8(89315):89325.
Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenet. 2016;345:1–11. https://doi.org/10.1186/s13148-016-0225-2.
Furdas SD, Kannan S, Sippl W, Jung M. Review article—Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch Pharm. 2012;345:7–21.
Ververis K, Karagiannis TC. Overview of the classical histone deacetylase enzymes and histone deacetylase inhibitors. ISRN Cell Biol. 2012;2012:1–12.
Liu C, Liu L, Shan J, Shen J, Xu Y, Zhang Q, et al. Histone deacetylase 3 participates in self-renewal of liver cancer stem cells through histone modification. Cancer Lett. 2013;339:60–9. https://doi.org/10.1016/j.canlet.2013.07.022.
Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36:1707–20. https://doi.org/10.1038/onc.2016.337.
Trisciuoglio D, Di Martile M, Del Bufalo D. Emerging role of histone acetyltransferase in stem cells and cancer. Stem Cells Int. 2018;2018:1–11.
Kahn M. Wnt signaling in stem cells and cancer stem cells: a tale of two coactivators. Prog Mol Biol Transl Sci. 2018. https://doi.org/10.1016/bs.pmbts.2017.11.007.
Uttarkar S, Piontek T, Dukare S, Schomburg C, Schlenke P, Berdel WE, et al. Small-molecule disruption of the Myb/p300 cooperation targets acute myeloid leukemia cells. Mol Cancer Ther. 2016;15:2905–15.
Le J. Histone modifications: targeting head and neck cancer stem cells. World J Stem Cells. 2014;6:511.
Bora-Singhal N, Mohankumar D, Saha B, Colin CM, Lee JY, Martin MW, et al. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci Rep. 2020;10:1–20.
Norollahi SE, Mansour-Ghanaei F, Joukar F, Ghadarjani S, Mojtahedi K, Gharaei Nejad K, et al. Therapeutic approach of cancer stem cells (CSCs)in gastric adenocarcinoma; DNA methyltransferases enzymes in cancer targeted therapy. Biomed Pharmacother. 2019;115:108958. https://doi.org/10.1016/j.biopha.2019.108958.
Dricu A, Purcaru SO, Buteica AS, Tache DE, Daianu O, Stoleru B, Dobrescu AM, Daianu T, Tataranu LG. DNA Methylation, stem cells and cancer, methylation - from DNA, RNA and histones to diseases and treatment, Anica Dricu, IntechOpen, 2012. https://doi.org/10.5772/53263. Available from: https://www.intechopen.com/books/methylation-from-dna-rnaand-histones-to-diseases-and-treatment/dna-methylation-stem-cells-and-cancer.
Keyvani-Ghamsari S, Khorsandi K, Gul A. Curcumin effect on cancer cells’ multidrug resistance: an update. Phyther Res. 2020;34:ptr.6703.
Peter AJ, Baylin SB. The epigenomic of cancer. Cell. 2007;128:683–92.
Ponnusamy L, Mahalingaiah PKS, Chang Y-W, Singh KP. Role of cellular reprogramming and epigenetic dysregulation in acquired chemoresistance in breast cancer. Cancer Drug Resist. 2019;2:297–312.
Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. 2017;3:372–86. https://doi.org/10.1016/j.trecan.2017.04.004.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson G, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
He B, Lee AY, Dadfarmay S, You L, Xu Z, Reguart N, et al. Secreted frizzled-related protein 4 is silenced by hypermethylation and induces apoptosis in β-catenin—deficient human mesothelioma cells. Cancer Res. 2005;65:743–8.
Crea F, Danesi R, Farrar WL. Cancer stem cell epigenetics and chemoresistance. Epigenomics. 2009;1:63–79.
Bulstrode H, Johnstone E, Marques-torrejon MA, Ferguson KM, Bressan RB, Blin C, et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017;31:757–73.
Contre L, Paoli I, Université AM, El Helou R, Wicinski J, Guille A, et al. A distinct DNA methylation signature defines breast cancer stem cells and predicts cancer. Stem Cells. 2014;32:1–9.
Pathania R, Ramachandran S, Elangovan S, Padia R, Yang P, Cinghu S, et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun. 2015;6:1–11. https://doi.org/10.1038/ncomms7910.
Mongan NP, Emes RD, Archer N. Detection and analysis of RNA methylation. F1000Research. 2019;8:1–12.
Romano G, Veneziano D, Nigita G, Nana-Sinkam SP. RNA methylation in ncRNA: classes, detection, and molecular associations. Front Genet. 2018;9:1–9.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m 6 A RNA methylation. Nat Rev Genet. 2014;15:293–306.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Kalkan R. Glioblastoma stem cells as a new therapeutic target for glioblastoma. Clin Med Insights Oncol. 2015;9:95–103.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m 6 A methylation regulates sorafenib resistance in liver cancer through FOXO 3-mediated autophagy. EMBO J. 2020;39:1–19.
Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613. https://doi.org/10.1016/j.biopha.2019.108613.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:1–15.
Niu Y, Wan A, Lin Z, Lu X, Wan G. N6-Methyladenosine modification: a novel pharmacological target for anti-cancer drug development. Acta Pharm Sin B. 2018;8:833–43. https://doi.org/10.1016/j.apsb.2018.06.001.
Toh JDW, Sun L, Lau LZM, Tan J, Low JJA, Tang CWQ, et al. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N6-methyladenosine demethylase FTO. R Soc Chem. 2015;6:112–22.
Wei JW, Huang K, Yang C, Kang CS. Non-coding RNAs as regulators in epigenetics (review). Oncol Rep. 2017;37:3–9.
Huang T, Alvarez A, Hu B, Cheng SY. Noncoding RNAs in cancer and cancer stem cells. Chin J Cancer. 2013;32:582–93.
Lou W, Liu J, Gao Y, Zhong G, Ding B, Xu L. MicroRNA regulation of liver cancer stem cells. Am J Cancer Res. 2018;8(1126):1141.
Bimonte S, Barbieri A, Leongito M, Palma G, Vecchio V, Falco M, et al. The role of miRNAs in the regulation of pancreatic cancer stem cells. Stem Cells Int. 2016;2016:8352684.
Agostini M, Knight RA. miR-34: from bench to bedside ABSTRACT. Ontarget. 2014;5:872.
Xiong S, Hu M, Li C, Zhou X, Chen H. Role of MIR-34 in gastric cancer: from bench to bedside (review). Oncol Rep. 2019;42:1635–46.
Yan H, Bu P. Non-coding RNAs in cancer stem cells. Cancer Lett. 2018;421:121–6. https://doi.org/10.1016/j.canlet.2018.01.027.
Feng S, Yao J, Chen Y, Geng P, Zhang H, Ma X, et al. Expression and functional role of reprogramming-related long noncoding RNA (lincRNA-ROR) in glioma. J Mol Neurosci. 2015;56:623–30.
Chi HC, Tsai CY, Tsai MM, Yeh CT, Lin KH. Roles of long noncoding rnas in recurrence and metastasis of radiotherapy-resistant cancer stem cells. Int J Mol Sci. 2017;18:1903.
Amirkhah R, Naderi-Meshkin H, Shah JS, Dunne PD, Schmitz U. Alternative splicing and noncoding RNA deregulation in colorectal cancer. Cells. 2019;8:929.
Wang W, Han C, Sun Y, Chen T, Chen Y. Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12:1–15.
Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. MiRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016;6:235–46.
Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622–38. https://doi.org/10.1038/nrd4359.
Awasthi R, Madan JR, Malipeddi H, Dua K, Kulkarni GT. Therapeutic strategies for targeting non-coding RNAs with special emphasis on novel delivery systems. Non-coding RNA Investig. 2019;3:11–11.
Osaki M, Okada F, Ochiya T. MiRNA therapy targeting cancer stem cells: a new paradigm for cancer treatment and prevention of tumor recurrence. Ther Deliv. 2015;6:323–37.
De SF, Vermeulen L. Wnt signaling in cancer stem cell biology. Cancers. 2016;8:60.
Onyido EK, Sweeney E, Nateri AS. Wnt-signalling pathways and microRNAs network in carcinogenesis: experimental and bioinformatics approaches. Mol Cancer. 2016;15:1–17. https://doi.org/10.1186/s12943-016-0541-3.
Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab Investig. 2016;96:116–36.
Mavila N, Thundimadathil J. The emerging roles of cancer stem cells and Wnt/beta-catenin signaling in hepatoblastoma. Cancers (Basel). 2019;11:1406.
Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234:116781.
Klarmann GJ, Decker A, Farrar WL. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics. 2008;3:59–63.
Jang G, Kim J, Cho S, Park K, Jung J. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Nat Publ Gr. 2015;5:1–15. https://doi.org/10.1038/srep12465.
Towle R, Truong D, Hogg K, Robinson WP, Poh CF, Garnis C. Global analysis of DNA methylation changes during progression of oral cancer. Oral Oncol. 2013;49:1033–42. https://doi.org/10.1016/j.oraloncology.2013.08.005.
Rheinbay E, Suvà ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3:1567–79.
Wen Y, Cai J, Hou Y, Huang Z, Wang Z. Role of EZH2 in cancer stem cells: from biological insight to a therapeutic target. Oncotarget. 2017;8:37974–90.
Khan AQ, Ahmed EI, Elareer NR, Junejo K, Steinho M, Uddin S. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells. 2019;8:1–33.
Wu Q, Yang Z, Wang F, Hu S, Yang L, Shi Y, et al. proliferation of gastric cancer stem cells. J Cell Sci. 2013;126:4220–9.
Shi L, Xi J, Xu X, Peng B, Zhang B. MiR-148a suppressed cell invasion and migration via targeting WNT10b and modulating β-catenin signaling in cisplatin-resistant colorectal cancer cells. Biomed Pharmacother. 2019;109:902–9. https://doi.org/10.1016/j.biopha.2018.10.080.
Yang Y, Li X, Wang T, Guo Q, Xi T, Zheng L. Emerging agents that target signaling pathways in cancer stem cells. J Hematol Oncol. 2020;13:1–18.
Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers. 2015;7:1554–85.
Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010;29:2646–58. https://doi.org/10.1038/emboj.2010.131.
Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta BBA Rev Cancer. 2010;1805:181–208. https://doi.org/10.1016/j.bbcan.2010.01.003.
Cui W, Wang L, Wen Y, Song M, Li B, Chen X, et al. Expression and regulation mechanisms of Sonic Hedgehog in breast cancer. Cancer. 2010;101:927–33.
Akbar A, Elham S, Rashidy-Pour A, Mansour-Ghanaei F, Nemati S, Joukar F, et al. Cancer signaling pathways with a therapeutic approach: an overview in epigenetic regulations of cancer stem cells. Biomed Pharmacother. 2018;108:590–9.
Pietrobono S, Gagliardi S, Stecca B. Non-canonical hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond smoothened. Front Genet. 2019;10:1–20.
Yang W, Liu Y, Gao R, Yu H, Sun T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018;415:164–76. https://doi.org/10.1016/j.canlet.2017.12.005.
Wu J, Zhu P, Lu T, Du Y, Wang Y, He L, et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J Hepatol. 2019;70:918–29. https://doi.org/10.1016/j.jhep.2018.12.015.
Miele E, Po A, Begalli F, Antonucci L, Mastronuzzi A, Marras CE, et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer. 2017;17:1–12.
Tang B, Xu A, Xu J, Huang H, Chen L, Su Y, et al. MicroRNA-324-5p regulates stemness, pathogenesis and sensitivity to bortezomib in multiple myeloma cells by targeting hedgehog signaling. Int J Cancer. 2018;142:109–20.
Chandimali N, Huynh DL, Zhang JJ, Lee JC, Yu DY, Jeong DK, et al. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2019;26:292–304. https://doi.org/10.1038/s41417-018-0050-1.
Fareh M, Turchi L, Virolle V, Debruyne D, Almairac F, De-La-Forest Divonne S, et al. The miR 302–367 cluster drastically affects self-renewal and infiltration properties of glioma-initiating cells through CXCR4 repression and consequent disruption of the SHH-GLI-NANOG network. Cell Death Differ. 2012;19:232–44. https://doi.org/10.1038/cdd.2011.89.
Asadzadeh Z, Mansoori B, Mohammadi A, Aghajani M, Haji-Asgarzadeh K, Safarzadeh E, et al. microRNAs in cancer stem cells: biology, pathways, and therapeutic opportunities. J Cell Physiol. 2019;234:10002–17.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64. https://doi.org/10.1038/nrclinonc.2015.61.
Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. 2017;36:1–13.
Mahalingaiah P, Chicago A, Chang Y, Singh K. Role of cellular reprogramming and epigenetic dysregulation in acquired chemoresistance in breast cancer. Cancer Drug Resist. 2019;2:297–312.
Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G. Notch-1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J Cell Biochem. 2015;116:2517–27.
Kuang SQ, Fang Z, Zweidler-McKay PA, Yang H, Wei Y, Gonzalez-Cervantes EA, Boumber Y, Garcia-Manero G. Epigenetic inactivation of Notch–Hes pathway in human B-cell acute lymphoblastic leukemia. PLoS ONE. 2013;8:1–10. https://doi.org/10.1371/journal.pone.0061807.
Jin L, Vu T, Yuan G, Datta PK. STRAP promotes stemness of human colorectal cancer via epigenetic regulation of the NOTCH pathwaya. Cancer Res. 2017;77:5464–78.
Wang Q, Chen X, Jiang Y, Liu S, Liu H, Sun X, et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J Mol Cell Biol. 2020;12:125–37.
Ghoshal P, Nganga AJ, Moran-Giuati J, Szafranek A, Johnson TR, Bigelow AJ, et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009;69:4380–7.
Liu ZH, Dai XM, Du B. Hes1: a key role in stemness, metastasis and multidrug resistance. Cancer Biol Ther. 2015;16:353–9.
Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, et al. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A. 2014;111:3098–103.
Lu J, Song G, Tang Q, Yin J, Zou C, Zhao Z, et al. MiR-26a inhibits stem cell-like phenotype and tumor growth of osteosarcoma by targeting Jagged1. Oncogene. 2016;36:1–11.
Ma Z, Cai H, Zhang Y, Chang L, Cui Y. MiR-129–5p inhibits non-small cell lung cancer cell stemness and chemoresistance through targeting DLK1. Biochem Biophys Res Commun. 2017;490:309–16. https://doi.org/10.1016/j.bbrc.2017.06.041.
Zhao J, Fu Y, Wu J, Li J, Huang G, Qin L. The diverse mechanisms of miRNAs and lncRNAs in the maintenance of liver cancer stem cells. Biomed Res Int. 2018;2018:8686027.
Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770–82.
Nilendu P, Kumar A, Kumar A, Pal JK, Sharma NK. Breast cancer stem cells as last soldiers eluding therapeutic burn: a hard nut to crack. Int J Cancer. 2018;142:7–17.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. https://doi.org/10.1038/nrm3434.
Liu S, Chen S, Zeng J. TGF-β signaling: a complex role in tumorigenesis (review). Mol Med Rep. 2018;17:699–704.
Buijs JT, Van Der Horst G, Van Den Hoogen C, Cheung H, De Rooij B, Kroon J, et al. The BMP2/7 heterodimer inhibits the human breast cancer stem cell subpopulation and bone metastases formation. Oncogene. 2012;31:2164–74. https://doi.org/10.1038/onc.2011.400.
Fan Q, Jing Y, Yu G, Kou X, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial—mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014. https://doi.org/10.1016/j.canlet.2014.05.008.
Bai J, Xi Q. Crosstalk between TGF-β signaling and epigenome. Acta Biochim Biophys Sin (Shanghai). 2018;50:60–7.
Balko JM, DuggerBhola NE, Teresa C, Kuba MG, Sánchez V, Sanders M, et al. TGF-β inhibition enhances chemotherapy.pdf. J Clin Investig. 2013;123:1348–58.
You H, Ding W, Rountree CB. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-β. Hepatology. 2010;51:1635–44.
Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80.
You X, Zhou Z, Chen W, Wei X, Zhou H, Luo W. MicroRNA-495 confers inhibitory effects on cancer stem cells in oral squamous cell carcinoma through the HOXC6-mediated TGF-β signaling pathway. Stem Cell Res Ther. 2020;11:1–14.
Yu Y, Kanwar SS, Patel BB, Oh PS, Nautiyal J, Sarkar FH, et al. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFβr2) in colon cancer cells. Carcinogenesis. 2012;33:68–76.
Pellatt AJ, Mullany LE, Herrick JS, Sakoda LC, Wolff RK, Samowitz WS, et al. The TGFβ-signaling pathway and colorectal cancer: associations between dysregulated genes and miRNAs. J Transl Med. 2018;16:1–22. https://doi.org/10.1186/s12967-018-1566-8.
Dzobo K, Senthebane DA, Ganz C, Thomford NE, Wonkam A, Dandara C. Advances in therapeutic targeting of cancer stem cells within the tumor microenvironment: an updated review. Cells. 2020;9(8):1896. https://doi.org/10.3390/cells9081896.
Li S, Han Z, Zhao N, Zhu B, Zhang Q, Yang X, et al. Inhibition of DNMT suppresses the stemness of colorectal cancer cells through down-regulating Wnt signaling pathway. Cell Signal. 2018;47:79–87. https://doi.org/10.1016/j.cellsig.2018.03.014.
Liu CC, Lin JH, Hsu TW, Su K, Li AFY, Hsu HS, et al. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136:547–59.
Oki Y, Issa J-P. Review: Recent clinical trials in epigenetic therapy. Rev Recent Clin Trials. 2008;1:169–82.
States U. Evolving strategies for therapeutically targeting cancer stem cells. Adv Cancer Res. 2016. https://doi.org/10.1016/bs.acr.2016.04.003.
Zagorac S, Alcala S, Bayon GF, Kheir TB. DNMT1 Inhibition Reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Eur PMC Funders Gr. 2017;76:4546–58.
Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74:4922–36.
Wu Y, Alvarez M, Slamon DJ, Koeffler P, Vadgama JV. Caspase 8 and maspin are downregulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer. 2010;10:1–12.
Bayat S, Shekari M, Choupani J, Reza M. Biomedicine and pharmacotherapy HDACis (class I), cancer stem cell, and phytochemicals: cancer therapy and prevention implications. Biomed Pharmacother. 2018;97:1445–53. https://doi.org/10.1016/j.biopha.2017.11.065.
Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenet. 2012;4:5.
Lin PC, Hsieh HY, Chu PC, Chen CS. Therapeutic opportunities of targeting histone deacetylase isoforms to eradicate cancer stem cells. Int J Mol Sci. 2018;19:1939.
Schech A, Kazi A, Yu S, Shah P, Sabnis G. Histone deacetylase inhibitor entinostat inhibits tumor-initiating cells in triple-negative breast cancer cells. Mol Cancer Therap. 2015;14:1848–58.
Meidhof S, Brabletz S, Lehmann W, Preca B, Mock K, Ruh M, et al. ZEB 1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7:831–47.
Guzman ML, Yang N, Sharma KK, Balys M, Corbett CA, Jordan CT, et al. Selective activity of the histone deacetylase inhibitor AR-42 against leukemia stem cells: a novel potential strategy in acute myelogenous leukemia. Mol Cancer Ther. 2014;13:1979–90.
Kumar B, Yadav A, Lang JC, Teknos TN, Kumar P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer. 2015;6:169–81.
Hii LW, Chung FFL, Soo JSS, Tan BS, Mai CW, Leong CO. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res Treat. 2020;179:615–29. https://doi.org/10.1007/s10549-019-05504-5.
Salvador MA, Wicinski J, Cabaud O, Toiron Y, Finetti P, Birnbaum D, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low xist expression. Clin Cancer Res. 2013;19(6520):6532.
Lattanzi G, Baldini N, Mai A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J Clin Chem. 2015;2625:4063–79.
Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi J-H, et al. Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res. 2016;76:3224–35.
Azad NS, Yin J, Oberg AL, Flynn P, Adkins D, Sharma A, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat: a phase 2 consortium/stand Up 2 cancer study. Oncotarget. 2017;8:35326–38.
Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM, et al. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie. 2012;94:2280–96. https://doi.org/10.1016/j.biochi.2012.07.025.
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non small cell lung cancer. Cancer Discov. 2012;1(598):607.
Huang Y, Davidson NE. Targeting tumorigenicity of breast cancer stem-like cells using combination epigenetic therapy: something old and something new. J Thorac Dis. 2016;8:2971–4.
Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res. 2005;11:3604–8.
Steensma DP. Decitabine treatment of patients with higher-risk myelodysplastic syndromes. Leuk Res. 2009;33:S12–7. https://doi.org/10.1016/S0145-2126(09)70228-0.
Frye R, Myers M, Axelrod KC, Ness EA, Piekarz RL, Bates SE, et al. Romidepsin: a new drug for the treatment of cutaneous T-cell lymphoma. Clin J Oncol Nurs. 2012;16:195–204.
Richardson PG, Laubach JP, Lonial S, Moreau P, Yoon SS, Hungria VT, et al. Panobinostat: a novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert Rev Anticancer Ther. 2015;15:737–48.
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA Approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52.
Zhang L, Tong X, Li J, Huang Y, Hu X, Chen Y, et al. Apoptotic and autophagic pathways with relevant small-molecule compounds, in cancer stem cells. Cell Prolif. 2015;48:385–97.
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:1–39. https://doi.org/10.1038/s41392-019-0095-0.
Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21:2666–70.
Ledford H. Gene-silencing drug approved outrage over changes to EPA chemical assessments. Nature. 2018;560:8–9.
Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta Gen Subj. 2014;1840:3494–502. https://doi.org/10.1016/j.bbagen.2014.09.017.
Bruschini S, Ciliberto G, Mancini R. The emerging role of cancer cell plasticity and cell-cycle quiescence in immune escape. Cell Death Dis. 2020;11:11–3. https://doi.org/10.1038/s41419-020-2669-8.
Cho Y, Kim YK. Cancer stem cells as a potential target to overcome multidrug resistance. Front Oncol. 2020;10:1–10.
Murayama T, Gotoh N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist. 2019;2:457–70.
Zappe K, Cichna-Markl M. Aberrant DNA methylation of ABC transporters in cancer. Cells. 2020;9:2281.
Lathia J, Liu H, Matei D. The clinical impact of cancer stem cells. Oncologist. 2020;25:123–31.
Romano M, De Francesco F, Pirozzi G, Gringeri E, Boetto R, Di Domenico M, et al. Expression of cancer stem cell biomarkers as a tool for a correct therapeutic approach to hepatocellular carcinoma. Oncoscience. 2015;2:443–56.
Hernandez-Vargas H, Sincic N, Ouzounova M, Herceg Z. Epigenetic signatures in stem cells and cancer stem cells. Epigenomics. 2009;1:261–80.
Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015;7:1185–201.
Acknowledgements
The authors gratefully acknowledge all the people who helped us do this project.
Funding
This paper was not funded by any source.
Author information
Authors and Affiliations
Contributions
KK conceived the original idea and designed the article. SK-G contributed to the data collection and drafts the manuscript. KK and AR revised and corrected the final version of manuscript. MKZ designed the figures. The figures were designed in BioRender.com. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Keyvani-Ghamsari, S., Khorsandi, K., Rasul, A. et al. Current understanding of epigenetics mechanism as a novel target in reducing cancer stem cells resistance. Clin Epigenet 13, 120 (2021). https://doi.org/10.1186/s13148-021-01107-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01107-4