
Abstract
Background
The integration of molecular data from hosts, parasites, and microbiota can enhance our understanding of the complex biological interactions underlying the resistance of hosts to parasites. Haemonchus contortus, the predominant sheep gastrointestinal parasite species in the tropics, causes significant production and economic losses, which are further compounded by the diminishing efficiency of chemical control owing to anthelmintic resistance. Knowledge of how the host responds to infection and how the parasite, in combination with microbiota, modulates host immunity can guide selection decisions to breed animals with improved parasite resistance. This understanding will help refine management practices and advance the development of new therapeutics for long-term helminth control.
Methods
Eggs per gram (EPG) of feces were obtained from Morada Nova sheep subjected to two artificial infections with H. contortus and used as a proxy to select animals with high resistance or susceptibility for transcriptome sequencing (RNA-seq) of the abomasum and 50 K single-nucleotide genotyping. Additionally, RNA-seq data for H. contortus were generated, and amplicon sequence variants (ASV) were obtained using polymerase chain reaction amplification and sequencing of bacterial and archaeal 16S ribosomal RNA genes from sheep feces and rumen content.
Results
The heritability estimate for EPG was 0.12. GAST, GNLY, IL13, MGRN1, FGF14, and RORC genes and transcripts were differentially expressed between resistant and susceptible animals. A genome-wide association study identified regions on chromosomes 2 and 11 that harbor candidate genes for resistance, immune response, body weight, and adaptation. Trans-expression quantitative trait loci were found between significant variants and differentially expressed transcripts. Functional co-expression modules based on sheep genes and ASVs correlated with resistance to H. contortus, showing enrichment in pathways of response to bacteria, immune and inflammatory responses, and hub features of the Christensenellaceae, Bacteroides, and Methanobrevibacter genera; Prevotellaceae family; and Verrucomicrobiota phylum. In H. contortus, some mitochondrial, collagen-, and cuticle-related genes were expressed only in parasites isolated from susceptible sheep.
Conclusions
The present study identified chromosome regions, genes, transcripts, and pathways involved in the elaborate interactions between the sheep host, its gastrointestinal microbiota, and the H. contortus parasite. These findings will assist in the development of animal selection strategies for parasite resistance and interdisciplinary approaches to control H. contortus infection in sheep.
Graphical Abstract

Similar content being viewed by others


Background
Helminths cause significant economic and production losses in small ruminant livestock systems [1]. Haemonchus contortus, the barber’s pole worm, is the most prevalent pathogenic nematode affecting small ruminants in tropical regions [2]. In contrast, chemical treatments, which are the main approaches currently used for its control, are becoming less effective owing to the development of anthelmintic resistance [3].
Therefore, there is a need to develop new alternative control strategies to reduce the parasite burden on production systems. One interesting animal-centric control strategy is to use sheep breeds that are naturally more resistant to gastrointestinal nematodes or, alternatively, to select more resistant individuals within breeds [4]. Some indigenous breeds have adapted to survive with low maintenance requirements and high parasitic challenges [5]. An example of such a breed is the Morada Nova, a native Brazilian hair sheep breed that has high gastrointestinal nematode resistance compared to other tropical-adapted and high-productive sheep breeds [6]. Better molecular characterization of Morada Nova can support the use of this indigenous breed in production systems as a long-term helminth control strategy [7] and advance our understanding of the mechanisms of immune responses and parasite resistance in other sheep breeds.
Knowledge of pathology of haemonchosis is essential for establishing new therapeutics and control strategies. This can be assisted by omics technologies, which can be used to increase our understanding of helminth and host responses [8]. Multi-omics approaches can aggregate various layers of information to elucidate complex host-parasite interactions [9], particularly those that are regulated by several genes with minor effects and are influenced by diverse environmental conditions. Genomics can reveal the genetic mechanisms underlying parasite resistance in hosts, whereas transcriptomics can identify the mechanisms through which hosts respond to infections, or those used by parasites to establish infection and modulate the host immune system. Integrating both host and parasite data quantified through genomic and transcriptomic layers, such as those used in expression quantitative trait loci (eQTL) studies [10], can provide a more in-depth knowledge of interactions.
Another factor in the parasite-host molecular scenario is the host microbiota, which has received increasing attention in recent years owing to the effect of the microbiome on resistance to gastrointestinal nematodes [11], H. contortus burden [12], and Teladorsagia circumcincta female fertility [13] in sheep. Symbiosis between microorganisms and their hosts, particularly ruminants, is crucial for providing nutrients, carbohydrates, and proteins [14]. However, gastrointestinal nematode infections alter the typical host microbiota, resulting in microenvironmental dysbiosis [15, 16]. H. contortus infections change the bacterial genera in the abomasum and rumen [15], and T. circumcincta increases the bacterial population involved in pro-inflammatory processes, which may contribute to the immunopathology of helminth disease [16]. Furthermore, the microbiota may influence parasite and host gene expression, as they can interact with host/parasite cells and affect gene regulation, switching certain cellular processes on or off.
The aim of this study was to improve our understanding of the parasite-host-microbiota interactions that lead to increased resistance to H. contortus in Morada Nova sheep using an integrated multi-omics approach with transcriptome sequencing (RNA-seq) data from sheep abomasum and H. contortus, 50 K sheep genotypes, and 16S ribosomal RNA (rRNA) gene sequencing of the microbiota from sheep feces and rumen content.
Methods
Sample collection and phenotypes
Sheep
The Morada Nova sheep and the phenotypes used in this study have been previously described [17]. Briefly, 274 lambs (136 males and 138 females), the progeny of four sires and 156 dams from the Embrapa Pecuária Sudeste flock, were born in two breeding seasons (March to May 2017 and 2018). All lambs were raised under the same environmental conditions.
Parasitological examination
For egg per gram (EPG) counts, 2 g of feces collected from the rectum were mixed with 28 mL saturated sodium chloride solution and evaluated in a McMaster chamber, then the total number of counted gastrointestinal strongyles eggs was multiplied by 50 [18].
Fecal cultures [19] performed at the end of each artificial infection revealed 100% Haemonchus.
Artificial infection and phenotyping
Lambs (3 months old) were treated with monepantel (2.5 mg/kg; Zolvix®) to eliminate natural infection (96.4% Haemonchus, 2.1% Cooperia, and 1.5% Trichostrongylus) with gastrointestinal nematodes. After 2 weeks, the animals were experimentally infected with 4000 third-stage larvae (L3) of the Echevarria H. contortus isolate [20]. This isolate, retrieved in Brazil in 1991, exhibited anthelmintic susceptibility to levamisole (100%), ivermectin (100%), and albendazole (98.9%) [20], and was obtained before the launch of monepantel on the market.
Phenotyping of individuals was based on EPG obtained on day 0 (day of infection) and in four collections at 7-day intervals after H. contortus prepatent period [21], from days 21–42 (days 21, 28, 35, and 42 after infection). On day 42 of the first artificial infection, the lambs were treated again with monepantel and, after 15 days, were subjected to a new parasitic infection with the same H. contortus isolate, following the same sampling protocol. On day 42 of the second parasitic challenge, the EPG mean was calculated from the ten collection dates, and the animals were ordered based on the mean EPG and ranked as resistant (EPG mean ranging 10–1765) or susceptible (EPG mean ranging 3923–24,112) to H. contortus. Notably, the natural infection, comprising H. contortus with historical multidrug resistance [22], was not totally eliminated after the first and second monepantel treatments, resulting in EPG counts on day 0 in both artificial challenges (Table 1). Therefore, the day of infection (day 0) was included in the calculation of the EPG mean.
All four sires and 79 of the 156 dams were phenotypically evaluated in two parasite challenges, following the same protocol used for the lambs, between September 2018 and January 2019.
Statistical analyses for heritability estimation
Considering the Morada Nova lamb-sire-dam trios, EPG phenotypic information for four sires, 79 dams, and 147 lambs, totaling 230 individuals, were available. These animals were used to build the additive genetic relationship matrix (A matrix) using the AGHmatrix [23] and pedigreemm [24] R (version 4.2.1 [25]) packages, and heritability (h2) was estimated using the NAM package [26].
Selection of lambs for the genome-wide association study (GWAS)
A total of 44 lambs from the top- and bottom-ranked animals (Table 1) were split into two groups with high and low EPG means, with a 21.5 × variation in EPG means between the groups. Lambs were also selected to balance the sex distribution between the groups and, as much as possible, the number of offspring per sire. Blood samples from each of the four sires and 44 lambs were collected through the jugular vein and subjected to DNA extraction [27]. All 48 DNA samples were genotyped using an Illumina 50 K single-nucleotide polymorphism (SNP) array.
Selection of lambs for transcriptome and metabarcoding sequencing
Five resistant and five susceptible animals were selected for transcriptome sequencing (RNA-seq) and metabarcoding (16S rRNA) sequencing. As expected, an unequal distribution of genders occurred [28] with a larger number of females in the resistant group. To better characterize the local host response in the early phase of infection [29], these ten lambs were dewormed with monepantel, allocated to cemented stalls, subjected to a third challenge with 4000 L3 from the same H. contortus isolate, and euthanized 7 days later. The abomasum was retrieved, and fragments of the fundic region were collected, immediately frozen in liquid nitrogen without any additional processing, and stored at – 80 ℃ for RNA extraction. Furthermore, 10 g of feces from the rectal ampulla was collected 2 weeks before slaughter, and 50 mL of rumen content of the slaughtered sheep was stored in liquid nitrogen for microbiota investigation.
RNA-seq
Total RNA was extracted from fragments of the fundic region of the abomasum using QIAzol Lysis Reagent (Qiagen, Germany) and TissueRuptor (Qiagen, Germany), and purified in a silica column with an RNeasy Mini kit (Qiagen, Germany). RNA concentration (ng/µL) and purity (A260/A280 ratio) were estimated in Qubit using an HS RNA kit (Thermo Fisher Scientific, USA) and in NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), respectively. RNA samples were submitted to RNA-seq after quality confirmation through RNA Integrity Number ≥ 7.5 in Agilent 2100 Bioanalyzer (Agilent Technologies, USA). RNA-seq libraries were prepared using the TruSeq Stranded mRNA kit (Illumina, USA) and sequenced using an Illumina HiSeq2500 system. For each sample, approximately 40 million reads in a 100 bp paired-end (PE) protocol were sequenced.
After sequencing, FASTQ files were visualized using FastQC (version 0.11.7 [30]) to assess RNA-seq quality. Trimmomatic (version 0.39 [31]) was used to remove adapters, the first ten initial bases and 2–3 final bases, reads of < 50 bases, and reads with a phred score of < 15 in a 4-bp window.
Initially, three reference sheep genomes were compared: Oar_rambouillet_v1.0 (GCA_002742125.1), Oar_v3.1 Texel (GCA_000298735.1), and ARS-UI_Ramb_v2.0 (GCF_016772045.1) (data not shown). As ARS-UI_Ramb_v2.0 (version 104) showed higher alignment rates and number of transcripts compared to the others, and this genome assembly was selected as the reference for subsequent analysis. HISAT2 (version 2.1.0 [32]) was used to index the sheep reference genome and align the trimmed paired reads. The resultant SAM files were converted to BAM files using SAMtools (version 1.11 [33]), and statistics were generated using MultiQC (version 1.7 [34]). The aligned and sorted BAM files were assembled using StringTie (version 2.1.3 [35]). Gene and transcript read counts were extracted using the Python (version 3.6.6 [36]) prepDE.py3 script in the StringTie pipeline.
For differential gene expression analysis between susceptible and resistant animals, an initial filtering step in R software was used to remove lowly expressed genes with a count sum of < 50, and overexpressed genes with a mean count of > 100,000. The gene count matrix was analyzed using DESeq2 [37], with a Wald test adjusted p-value (q-value) of < 0.1, and using edgeR [38], with a false discovery rate (FDR) of < 0.05. Differential transcript expression was analyzed in ballgown [39] (q-value < 0.1) and DESeq2 (q-value < 0.05). In all analyses, both | fold change (FC) |> 1.5 and adjusted p-values were regarded as thresholds. Regarding p-values, in the absence of results at < 0.05, an adjusted p-value of < 0.1 was used. The differential expression of the overexpressed genes was investigated separately using the t-test, DESeq2, and edgeR. The biological roles of the differentially expressed and overexpressed genes were investigated using GeneCards (version 5.15 [40]).
Variant calling and annotation of RNA-seq data
For variant calling from the abomasum RNA-seq data [41], trimmed PE reads were mapped to the indexed sheep reference genome using the 2-pass mode of STAR (version 2.7.3a [42]). The STAR aligner was used because of its higher tolerance for accepting mismatches and soft clipping compared with the HISAT2 [43]. SAMtools was used to index the BAM files, and Picard (version 2.25.0 [44]) was used to add read groups, mark duplicates, and create a sequence dictionary. GATK (version 4.2.0.0 [45]) with SplitNCigar, BaseRecalibrator including known sheep variants [46], ApplyBQSR, and HaplotypeCaller, was used to call the variants. GATK GenomicsDBImport was used to combine files from different animals, and GenotypeGVCFs was used for joint genotyping. GATK SelectVariants was used to select the SNPs, which were then filtered with VariantFiltration with –cluster-window-size 35, –cluster-size 3, QD < 2.0, FS > 30.0, SOR > 3.0, MQ < 40.0, MQRankSum < − 12.5, and ReadPosRankSum < − 8.0. BCFtools (version 1.9.64 [33]) was used to retrieve hard-filtered variants and remove poor-quality calls per variant (GQ < 20, depth < 3 reads, no calls, MAF < 0.05, missing genotypes > 10%, and multiallelic sites). A total of 51,434 SNPs were identified.
Subsequently, a database for the ARS-UI_Ramb_v2.0 sheep reference genome was created and the variants were annotated using SnpEff (version 5.1 [47]). SnpSift [48] was used to detect high-impact variants, and the caseControl function was used to select homozygous variants between resistant and susceptible sheep (termed RNA-seq genotypes).
Genome-wide association study
DNA from the four sires and 44 extreme phenotype lambs, including the ten lambs used in the RNA-seq study, were extracted through saline precipitation and stored at – 20 ℃. DNA integrity in 1% agarose gel electrophoresis, concentration (ng/µL) and purity (260/280 absorbance ratio ranging 1.8–2.0) in NanoDrop were assessed. The DNA samples were genotyped using an Illumina OvineSNP50v3 chip. Genotyping data were subjected to quality control with snpQC [49], and SNPs with GCScore < 0.5, MAF < 0.05, HWE < 10e-16, call rate < 0.90, and samples with call rate < 0.75 were assigned as missing values. No samples with > 10% missing values were detected. A total of 45,070 SNPs and all 48 samples were included in the following steps. The genotype frequency of SNPs with missing data was imputed using the mean frequency of the other samples. Paternity was verified using a matrix of opposing homozygotes constructed using R [50].
These data were previously used for a case–control GWAS [51] based on the Oar_v3.1 Texel reference genome. In the present study, data from the 44 lambs were reanalyzed using the same reference genome as that used in the RNA-seq analysis. A custom script was used to lift the 50 K genotypes to the ARS-UI_Ramb_v2.0 assembly by mapping the probe sequence to the reference and then re-aligning the nucleotides to the reference strand. The EPG mean normalized by cube-root transformation was used as a phenotype in an snpBLUP GWAS to identify significant SNPs at FDR < 0.05, and h2 for EPG was estimated from the genomic data with the NAM package in R. Only the genotypes from the arrays were used for the GWAS, as no significant results (data not shown) were obtained using the RNA-seq genotypes from the ten lambs.
Genes located 1 Mbp upstream and downstream of each significant SNP from the GWAS analysis were retrieved from the National Center for Biotechnology Information (NCBI) Genome Data Viewer [52], and gene function was investigated using GeneCards and a literature review to identify candidate genes for resistance to H. contortus.
eQTL analysis
The R MatrixEQTL package [53] was used for eQTL analysis using all SNP data (45,070 from the array and 51,434 from RNA-seq genotypes) and the RNA-seq of the ten most extreme animals. Gene and transcript expressions were RNA-seq read counts normalized by DESeq2 variance-stabilizing transformation. Sex and sire were included as covariates. Cis-eQTL were defined as SNPs located up to 1 Mb from the regulated gene, whereas trans-eQTL considered distances of > 1 Mb. From all significant cis- and trans-eQTL (FDR < 0.05) retrieved, only those comprising significant SNPs from GWAS or RNA-seq genotypes and differentially expressed data are reported here.
Analysis of unmapped RNA-seq reads
The RNA-seq study was originally designed to assess gene expression in sheep hosts, but as H. contortus L3 penetrates the mucosal glands of the abomasum, primarily in the fundic region, to molt into L4 [54], the presence of genetic material from the H. contortus parasite in the RNA-seq data was also investigated using reads unmapped to the sheep reference genome. The trimmed PE reads were aligned to the H. contortus reference genome (Haemonchus_contortus.PRJEB506.WBPS17.genomic.fa) and assembled with an annotation file (Haemonchus_contortus.PRJEB506.WBPS17.annotations.gff3) retrieved from the WormBase ParaSite database (version 17 [55]).
Because of the small amount of data and the resultant low read counts, no additional filtering was performed, and differential expression analysis was implemented through DESeq2, edgeR, ballgown, and t-tests using the same parameters previously described for sheep.
Microbiota from sheep feces and rumen content
DNA from the feces and rumen content of the ten lambs selected for RNA-seq was extracted using the Quick-DNA™ Fecal/Soil Microbe Miniprep Kit (Zymo, USA) following the manufacturer’s instructions. Polymerase chain reaction (PCR) amplification of the bacterial and archaeal 16S rRNA genes was performed using a previously described primer set [56]. Libraries were sequenced on an Illumina MiSeq (2 × 250 bp) using an Illumina V3 sequencing kit (Illumina, USA). Raw reads were quality checked, filtered for quality (> Q25), and trimmed at positions 220 (forward) and 175 (reverse), based on aggregated quality plots generated using QIIME 2 [57]. The remaining data were submitted to DADA2 [58] to resolve the sequences into amplicon sequence variants (ASV) and exclude chimeric sequences. Bacterial and archaeal sequences were classified using the SILVA database (version 132 [59]). The conditional quantile regression (ConQuR) approach [60] was used to remove the batch effects caused by multiple sequencing runs and sex. Only the ASVs present in at least five lambs were considered. A total of 526 and 607 bacterial ASVs and 22 and 28 archaeal ASVs from the feces and rumen, respectively, were considered for downstream analyses.
Multi-omics integration
The final step of the analysis was the integration of fecal and ruminal microbiota abundance identified in the 16S rRNA data with the expression identified using RNA-seq. Co-expression network analysis of ASVs, host genes/transcripts, and H. contortus gene/transcript expression was performed using the R CEMiTool package [61]. CEMiTool uses an unsupervised filtering method based on the inverse gamma distribution for each gene selection used in the analyses and applies a soft-thresholding power β to determine a similarity criterion between pairs of features [61]. The CEMiTool parameters were set to apply_vst = TRUE, cor_method = “spearman”, network_type = “signed”, and tom_type = “signed” to construct co-expression networks. The minimum module size was set to 50 and 100 for gene and transcript expression levels, respectively. Genes within significantly associated modules were then subjected to Metascape [62] for biological pathway and gene ontology enrichment analyses, with the input species set to Homo sapiens.
Results and discussion
RNA-seq
The sheep abomasum RNA-seq reads ranged 31.1–41.3 M per sample, with 97.9–98.5% of the reads mapped to the sheep ARS-UI_Ramb_v2.0 reference genome (64.5–70% of proper pairs). For the differential gene expression analysis, 9099 genes with zero counts and 5891 genes with count sums < 50 were removed from the total of 33,800 genes.
Fourteen overexpressed genes (Table 2) were removed and analyzed separately; and no significant differential expression was detected between resistant and susceptible animals. Among these overexpressed genes, PIGR, COX1, and LGALS15 have been reported in the sheep abomasum in a transcriptome study comparing several tissues [63], and PIGR was the most abundant transcript found in the cattle abomasum [64]. These overexpressed genes are involved in gastrointestinal biological processes and infection responses. Pepsin is the principal acid protease of the stomach [65]; lysozymes are involved in bacterial cell wall cleavage [66]; COX1 produces cytoprotective prostaglandins for the stomach [67]; LGALS15 (also known as ovgal11), associated with immune/inflammatory responses and protection against infection [68], and intelectin-2, associated with the gastrointestinal mucus [69], are present in the abomasum of sheep infected with H. contortus; immunoglobulin lambda expression is detected in gastric cancer [70]; and ND4 mutations are considered biomarkers of preneoplastic lesions of the gastrointestinal tract [71].
Of the remaining 18,796 filtered genes (55.6%), 11 differentially expressed genes (DEG) (Table 3) were detected between H. contortus-susceptible and H. contortus-resistant animals using edgeR (Additional file 1: Table S1 and Additional file 3:Fig. S1). Four genes (CCDC85B, LOC121819799, GAST, and LOC101121371) were upregulated in H. contortus-resistant sheep, whereas seven genes (LOC114116426, LOC101116991, LOC101104728, LOC101107420, GNLY, GRP, and IL13) were upregulated in susceptible sheep. Using DESeq2 (Additional file 1: Table S2), only two DEGs were upregulated in susceptible sheep (LOC101116991 and LOC114116426; both were DEGs shown in the edgeR results) (Table 3). The role of IL13 in the resistance to H. contortus is through smooth muscle hypercontraction and increased mucus production, resulting in the detachment of parasites from the gut wall [72]. IL13 upregulation in the susceptible Morada Nova is in accordance with a previous study using infected young sheep of a native breed [73]. However, IL13 has also been shown to be upregulated in adult resistant sheep [72]. In addition to animal age, infection duration should be considered, as genes related to the inflammatory response, T lymphocyte attraction, and leukocyte binding are upregulated in the abomasal lymph nodes of resistant animals at the beginning of T. circumcincta infection but downregulated at later stages, indicating a delayed Th2 response in susceptible animals [29]. Associations between the GAST gene and the response to H. contortus[74] and between the GNLY gene and the response to Trichostrongylus colubriformis [75] have been previously reported.
A subset of ten genes (expressed in at least two samples and count sum of > 50 reads) was observed to be exclusively expressed in susceptible or resistant animals. Of these, six genes (CCDC85B, LOC121819799, GAST, ABCC11, LOC101106468, and LOC121818066) were expressed only in resistant sheep and four genes (LOC101104728, GNGT1, LOC114116426, and GRP) were expressed only in susceptible sheep (Table 4). Six of these genes (CCDC85B, LOC121819799, GAST, LOC101104728, LOC114116426, and GRP) were previously identified as DEGs (Table 3). In addition to the previously discussed genes associated with resistance to gastrointestinal nematodes (such as GAST and IL13), genes involved in adaptation (such as ABCC11 [76] and GNGT1 [77]) were detected.
From a total of 76,900 transcripts, 478 differentially expressed transcripts (DET) from 435 genes (including LOC101116991, LOC114116426, LOC101107420, and IL13 DEGs) were detected using DESeq2 (Additional file 1: Table S3), whereas ballgown (Additional file 1: Table S4 and Additional file 4: Fig. S2) detected only three transcripts (TECPR1, LOC114111361, and LOC121818463 genes).
Variant calling and annotation of RNA-seq data
A total of 884,918 variants were detected in sheep abomasum RNA-seq data. After filtering, 51,434 autosomal SNPs remained and were associated with 264,604 effects, of which 0.04% had high impact and 24.8% had missense effects. Case–control selection of RNA-seq genotypes between resistant and susceptible sheep revealed 16 variants (Additional file 1: Table S5) in three genes: TRAPPC6B (Chr18), MGRN1 (Chr24), and SPCS3 (Chr26). Among these, MGRN1 has been associated with resistance to gastrointestinal nematodes in a previous study [78].
Genome-wide association study
After quality control filtering of the genotype data, 45,070 SNPs from 44 lambs were used in the snpBLUP GWAS. The observed SNP average heterozygosity was 0.38, and the diagonal mean value of the genomic relationship matrix (GRM) was 0.95, indicating no inbreeding in the population. This was also confirmed by the inbreeding coefficient estimation at 0 using the A matrix of the pedigree data. The animals used in this study were raised in the state of São Paulo, which holds 10.4% of the Morada Nova population registered in Brazil [79]. However, high levels of inbreeding have been reported in animals from northwestern Brazil [80]. Production conditions under extensive or semi-intensive systems and animal selection practices vary significantly between Brazilian states; consequently, the genetic background of the breed differs as well [81]. Nonetheless, Morada Nova animals exhibit sufficient genetic variation to be used in breeding programs [80].
The opposing homozygous matrix identified two discordant sire-offspring pairs that needed to be reassigned. In the final data of the 44 lambs, 11 were offspring of sire 1 (with a 28.1 × variation in EPG means between resistant and susceptible half-siblings), seven of sire 2 (7.5 × variation in EPG means), 13 of sire 3 (19.9 × variation in EPG means), and 13 of sire 4 (30.8 × variation in EPG means).
The h2 of EPG was estimated at 0.12 using the GRM and at 0.02 using the pedigree, noting the limited number of animals used to estimate h2, which also impaired the confidence interval calculation. The reported heritability estimates for EPG vary significantly in the range 0.01–0.65, as it is a complex trait that is easily affected by breed, age, parasite, climate, natural versus artificial parasite infection, and other experimental conditions [4]. Further studies with larger numbers of individuals are required to address the reliability of selecting Morada Nova individuals that are resistant to H. contortus based solely on EPG. However, even if not used for animal selection, the genomic estimated breeding value (GEBV) for EPG is considered a good criterion for identifying animals for target-selective anthelmintic treatment [82], which can contribute to the control of gastrointestinal nematodes in flocks.
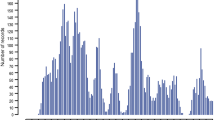
The snpBLUP GWAS (Fig. 1) detected three significantly associated SNPs (FDR < 0.05): rs419988472 (intron variant in the DHRS9 gene) at position Chr2:140323047, OAR11_27473453.1 (rs161041632—exon variant in the MED11 gene) and s08310.1 (rs427555933 – intergenic variant) at positions Chr11:26501006 and Chr11:26595120, respectively, which overlapped in Fig. 1.

snpBLUP genome-wide association study for eggs per gram (EPG) in Morada Nova sheep. Dashed line: FDR < 0.05
Within the ± 1 Mbp window of the significant SNPs, 171 annotated genes were detected (Additional file 1: Table S6). The candidates included genes with reported association with resistance to gastrointestinal nematodes, such as NLGN2 in goats [83] and CXCL16 and CD68 in cattle [84]; specific response and resistance to H. contortus, such as ALOX15 [73] and LRP2 [85], to T. circumcincta, such as RABEP1 [86], to the cestode Echinococcus granulosus, such as TNFSF13 [87], and to the protozoan Toxoplasma gondii in mice, such as NLRP1 [88]. In addition, candidate genes are related to macrophage regulation during helminth infection, such as KDM6B [89], response to Ostertagia ostertagi vaccination in cattle, such as DHRS9 [90], and lipoxygenases (ALOXE3, ALOX12, ALOX12B, and ALOX15) associated with the inflammatory response in mice [91]. There are also genes related to body size and feed efficiency in sheep, such as ALOX12 and TP53 [92], and in cattle, such as TP53 [93], WSCD1 [94], and TRNAC-GCA [95], to adaptability, such as SLC2A4 for hypoxia [96] and CHD3 for disease resistance and climate adaptation in sheep [97], and DVL2 [98] for thermal adaptation in cattle. Among the candidate genes involved in microbiota regulation, ABCB11 (also known as BSEP) plays a role in the transport of bile salts, whose direct microbial activity shapes the gut microbiota [99], and NLGN2 gene expression in horses is affected by anthelmintic treatment, likely through an effect on the microbiota [100].
eQTL analysis
For gene expression, 48,682 significant (FDR < 0.05) eQTL consisting of 426 cis-eQTL (Additional file 2: Table S7) and 48,256 trans-eQTL (Additional file 2: Table S8) were detected, whereas transcript expression resulted in 9,999,256 eQTL: 2540 cis-eQTL (Additional file 2: Table S9) and 9,986,716 trans-eQTL (Additional file 2: Table S10). Among the significant eQTL, no DEG was regulated by significant variants. The lack of cis-eQTL for the significant variants suggests the absence of regulation among genes closely located on the same chromosome; however, the small sample size of this study has limited the power to identify smaller effects; a larger sample size generally results in gains for cis-eQTL mapping [101].
Trans-eQTL were detected for DETs (Table 5), including transcripts of functional candidate genes, such as FGF14 associated with response to H. contortus in sheep [102] and RORC in goats [103]; LRRC8B associated with growth, body conformation, and weight [104]; DTX3 [105] and ZNF789 [97] associated with adaptation; and RAPGEF2 [106] associated with microbiota.
Unmapped read analysis (RNA-seq from H. contortus)
Between 0.4% and 1.0% of the total abomasum RNA-seq data reads were mapped to H. contortus, resulting in 0.1–0.4 M. Most of the 19,776 annotated genes had zero counts, and only 40 genes (0.2%) were retained for further analyses.
The most highly expressed gene detected in all the samples was HCON_00026760, a non-annotated pseudogene. No DEG using DESeq2 (Additional file 1: Table S11), edgeR (Additional file 1: Table S12), or t-test, and no DET using DESeq2 (Additional file 1: Table S13) or ballgown (Additional file 1: Table S14) were detected between the reads of H. contortus retrieved from resistant and susceptible Morada Nova sheep.
Of the 40 genes, nine were exclusively expressed (Additional file 1: Table S15) in H. contortus collected from resistant Morada Nova sheep; however, only in one sample each, whereas 26 genes were expressed exclusively in H. contortus recovered from susceptible sheep (Additional file 1: Table S15), but only four in at least two samples: HCON_00174250 (Epsin-1), HCON_00667215 (Cytochrome c oxidase subunit 1), HCON_00667245 (NADH:ubiquinone reductase [H( +)-translocating]), and HCON_00667255 (NADH dehydrogenase subunit 6). Epsin-1 is an ENTH-domain protein involved in endocytosis and lysosomal protein trafficking, and its silencing in Heterodera avenae, a cereal cyst nematode, results in a 71% reduction in females and eggs [107]. Cytochrome c oxidase, NADH:ubiquinone reductase, and NADH dehydrogenase are mitochondrial genes, which are considered potential targets for anthelmintic treatments because of the unique energy-transducing and anaerobic systems developed by nematode parasites in their adaptation to the low oxygen concentration of the mammalian host gastrointestinal tract [108, 109]. Despite their expression in only one sample each, three genes related to collagen and cuticle composition were expressed exclusively in H. contortus recovered from susceptible sheep (Additional file 1: Table S15). The nematode cuticle interacts with the host immune system and its major protein is collagen [110]. Genes associated with collagen and cuticle development are upregulated in the transition from L3 to L4 in H. contortus [111].
The potential roles of epsin-1, mitochondrial, collagen-, and cuticle-related genes in the establishment and maintenance of infection and parasite fitness in sheep hosts should be further investigated in future studies and considered as potential targets for the development of new therapeutics against H. contortus.
Gene co-expression network analysis
Early establishment of the gut microbiome is crucial for immune system development and maintenance of a healthy gut, including barrier function and mucosal immunity [112]. Moreover, the nematodes modulate their microbiome to provide an adequate environment for their survival [113]. The results obtained here from the microbiome may reflect the identification of taxa consistently associated with infection and resistance to H. contortus and must be interpreted carefully, as significant variations in bacterial populations were detected between trials in 2 years, consecutively, after immunization against and infection by T. circumcincta [114].
Co-expression network analysis detected four functional modules based on the sheep RNA-seq gene data and ASVs (Fig. 2). Among these, three modules (Fig. 2 and Table 6) exhibited a significant association with resistance to H. contortus, whereas one module was not associated with the trait. Notably, the most active module, referred to as M1, encompassed 302 correlated features (77 ASVs) and displayed a positive normalized enrichment score (NES) of 4.87 in the resistant group. In contrast, M2, consisting of 284 features (117 ASVs), exhibited a negative NES of − 3.75 in the resistant group, and M3, containing 240 features (36 ASVs), also exhibited a negative NES of − 2.32 in the resistant group.

Co-expression network analysis of sheep RNA-seq genes and amplicon sequence variants (ASV). The figure displays the normalized enrichment score (NES) of modules (red represents higher activity and blue represents lower activity), with circle size and color intensity proportional to the NES values
Hub genes/genera representative of each of the three modules associated with H. contortus resistance were also identified (Table 6 and Additional file 1: Tables S16 and S17). In the case of M1, the hub features were genes associated with the immune system, such as CALHM6, CD8A, CD8B, and TRGC1. CD8A and CD8B encode subunits of the CD8 protein that occurs on the surface of cytotoxic T cells. A DNA vaccine conferring partial protection against H. contortus infection in goats resulted in increased CD8 + T lymphocyte production and reduced EPG and worm burden in the abomasum [115]. TRGC1 encodes the gamma-constant region of the T cell receptor surface, which recognizes and binds to specific antigens and plays a multifaceted role in tissue homeostasis, autoimmunity, pro- and antitumor activity, and innate and adaptive immune responses [116, 117]. However, no relationship between TRGC1 and nematode infections has been previously described.
The M2 module had three ASVs as hub features: ASV_Bac_1273_R, classified as belonging to the Christensenellaceae R-7 group genus; ASV_Bac_396_R, classified as the Kiritimatiellae class (both found in the rumen); and ASV_Bac_422_S, classified as the Bacteroides genus, from the fecal samples. In addition, the HKDC1 hub gene from M2, which exhibited reduced activity in resistant animals, is associated with glucose use and homeostasis [118].
SLC26A7 and KCNJ16 are hub genes in the M3 module that exhibited lower activity in resistant animals. These genes are related to homeostasis and pH balance [119] and, in addition to HRH2, they impair gastric acid secretion [120, 121]. Previous studies have hypothesized that gastric acid protects against nematode infections, as shown by reduced gastric acid secretion and predisposition to infection by a variety of nematodes, including Ostertagia spp. and T. colubriformis in sheep [122, 123]. H. contortus infection causes a significant increase in the abomasal pH during the early and late stages of infection in goats [124]. Our findings present a correlation between gastric acid secretion and susceptibility to H. contortus. Therefore, compared to resistant animals, susceptible animals are less able to respond to H. contortus infection through gastric acid secretion, which, in turn, results in higher infection rates.
The annotated genes were subjected to pathway analysis to capture biological information using enriched terms (Fig. 3 and Additional file 1: Table S18). For genes in M1, the top pathways and processes included responses to bacteria and positive regulation of immune and inflammatory responses, which are traits intrinsically related to parasite resistance. Nematodes have evolved immunomodulatory mechanisms to suppress host immune responses and promote infection [125]. Knowledge of the mechanisms and target molecules involved in the inflammatory response may provide an effective means of nematode parasite control [126]. For genes in M2, the enriched terms were related to the regulation of insulin-like growth factor transport and uptake by insulin-like growth factor binding proteins, post-translational protein phosphorylation, and degradation of the extracellular matrix. In addition, for genes in M3, gastric acid secretion was highlighted.

Metascape enrichment analysis of statistically enriched ontology terms of hub genes/genera from three functional co-expression modules of sheep RNA-seq genes and amplicon sequence variants (ASV). Heatmap of enriched terms across input gene lists, colored by p-values
Enrichment of the genera classified as Prevotella, Treponema, Christensenellaceae R-7 group, and Methanobrevibacter was observed in the M1 and M2 modules. Four ASVs were observed in both the rumen and feces in M1: ASV_98 and ASV_23, classified as the p-251-o5 and [Eubacterium] coprostanoligenes group bacterial families, and ASV_13 (Methanobrevibacter genus) and ASV_6 (Candidatus Methanomethylophilus), which are two archaea. For M2, five archaeal ASVs, i.e., ASV_3, ASV_4, ASV_8, ASV_31, and ASV_57, which are classified as Methanobrevibacter, were enriched. Among the ASVs, the p-251-o5 family identified in M1 has been described in the fecal microbiota of pigs [127] and in the rumen of ewes and cows [128, 129]. In addition, the [Eubacterium] coprostanoligenes group, an anaerobic bacterium potentially impacting host lipid metabolism [130], was less abundant in Tibetan sheep infected with gastrointestinal nematodes [131], supporting our results and suggesting that it may be a promising target for H. contortus resistance. Christensenellaceae R-7, a hub feature in M2, is associated with amino acid and lipid metabolism and human metabolic health in different disease contexts, including obesity and inflammatory bowel disease [132]. The relative abundance of Christensenellaceae decreases in goats infected with H. contortus [124]. The Bacteroides genus, an ASV from feces in the M2 module, is considered beneficial to hosts as it prevents potential pathogens from colonizing the gut and processes complex molecules into simpler molecules [133].
The hub archaeal ASV, classified as Methanobrevibacter genus in the M2 module, plays a notable role in the gut microbial ecosystem [134] by contributing to the efficient digestion of polysaccharides by consuming the end products of bacterial fermentation [135]. It is considered a potential therapeutic target for managing gastrointestinal disorders [136]. Anthelmintic treatment of adult ewes significantly affects the archaeal community, resulting in increased relative abundance of different Methanobacteria [137]. An increase in Methanobrevibacter has been observed during chronic Trichuris trichiura infections in humans [138]. It was hypothesized that H. contortus infection could increase mucus secretion [139], which would provide energy for adapted microorganisms, including the mucus colonizer Methanobrevibacter [138]. Thus, the potential role of Methanobacteria in controlling H. contortus infections is of interest.
Transcript co-expression network analysis
Co-expression network analysis of RNA-seq transcript data identified 26 functional co-expression modules (Additional file 5: Figure S3 and Additional file 1: Table S19) and hub features (Additional file 1: Table S20). The top modules and hub features (Table 7) were M2 (294 features with 55 ASVs, NES = − 3.85), M25 (123 features with seven ASVs, NES = − 3.29), M6 (263 features with 31 ASVs, NES = 3.66), and M10 (231 features with eight ASVs, NES = 3.4).
The M2 and M25 modules of the transcript networks were less active in resistant animals. Regarding M2, several ASVs were identified as hubs, including the same ASVs found in the gene level M2 module. ASV_Bac_250_R, classified as Verrucomicrobiota and identified as a hub for M2, is associated with mucin degradation, glucose homeostasis, and immunity regulation [140]. Previous studies have described a symbiotic association between Verrucomicrobia and soil nematodes [141], and the abundance of the Verrucomicrobiota phylum was decreased in the abomasum of lambs infected with H. contortus [142], suggesting potential mechanisms for therapeutic interventions.
For M6, five ASVs were identified as hubs: ASV_Bac_1254_R (Prevotella), ASV_Bac_433_R (Ruminococcus gauvreauii group genus), ASV_Bac_115_R (Prevotellaceae NK3B31 group genus), ASV_Bac_269_R (Prevotella), and ASV_Bac_218_S (Lachnospiraceae). In addition, previous studies have shown an increase in Prevotella abundance in the ruminal, fecal, and abomasal microbiota of sheep and goats infected with H. contortus [15, 143]. In the present study, a decrease in Prevotella abundance associated with H. contortus susceptibility was observed, which is consistent with more recent findings after H. contortus infection [124, 142] and other nematode infections in humans [144] and mice [145]. In addition, as the Prevotella genus specializes in complex polysaccharide degradation, such as starch and cellulose, and contributes to the metabolism of dietary fiber [146], its reduction may affect feed digestion, resulting in low weight gain in infected animals [145] and, consequently, lower resilience to parasites.
Regarding pathway-enriched terms (Fig. 4 and Additional file 1: Table S21), the genes associated with each transcript were enriched in the MAPK, Wnt, and EGF/EGFR signaling pathways, morphogenesis, and cellular processes, including the regulation of cell morphogenesis, chromatin organization, and actin cytoskeleton organization. Additionally, transcripts representing the same genes (i.e., ABO, ASAP2, ADGRF5, ARHGAP12, CAPN15, and FN1) were identified in various modules.

A Metascape enrichment analysis of statistically enriched ontology terms. Heatmap of enriched terms across input gene lists, colored by p-values. B Overlap among gene lists: identical genes are linked with purple curves, genes that hit multiple lists are colored with dark orange, and genes unique to a list are colored with light orange
None of the H. contortus genes or transcripts was significantly correlated with the identified modules.
Conclusions
Gene and transcript expression data and genomic markers from Morada Nova sheep were investigated and associated using eQTL to assess their roles in the phenotype of H. contortus resistance. Some candidate genes were related to immune response and parasite resistance (ALOX12, ALOX12B, ALOX15, CD68, CXCL16, DHRS9, DNAH2, FGF14, GAST, GNLY, GP1BA, IL13, KDM6B, LRP2, MGRN1, NLRP1, RABEP1, RORC, and TNFSF13), growth and weight (ENO3, TP53, LRRC8B, TRNAC-GCA, and WSCD1), environmental adaptation (CHD3, DTX3, DVL2, GNGT1, SLC2A4, TRNAG-CCC, and ZNF789), and microbiota regulation (ABCB11, NLGN2, and RAPGEF2). Through eQTL mapping, SNP variants were predicted to regulate the differential expression of transcripts, including candidate genes (DTX3, FGF14, LRRC8B, RORC, and SNF789). These genes represent potential molecular markers for the detection and selection of H. contortus-resistant animals. Transcriptomic and genomic data from hosts, expression data from H. contortus, and ASVs from the microbiota of feces and rumen were integrated to detect the biological functions and pathways resulting in resistance. The genes (mitochondrial, collagen-, and cuticle-related genes) expressed in H. contortus may modulate evasion from the host immune system, facilitating the establishment of infection. Furthermore, the sheep genes (CD8A, CD8B, HKDC1, KCNJ16, SLC26A7, and TRGC1) and biological pathways (positive regulation of the immune system, response to bacteria, inflammatory response, gastric acid secretion, and mucus secretion) identified using this multi-omics approach are potential modulators of host immunity. Additionally, enriched microbiota (Bacteroides, Christensenellaceae R-7, [Eubacterium] coprostanoligenes group, Methanobrevibacter, Prevotellaceae, and Verrucomicrobiota) may regulate host metabolism (homeostasis, glucose use, amino acid and lipid metabolism, and dietary fiber degradation), resulting in resistance to parasite infection. The parasitic genes, biological pathways, and microbiomes identified in this study are potential targets for the development of new therapeutics aimed at increasing host resistance and parasitic control in sheep flocks.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the European Nucleotide Archive repository, under the project PRJEB67593, https://www.ebi.ac.uk/ena/browser/view/PRJEB67593.
Abbreviations
- A matrix:
-
Additive genetic relationship matrix
- ASV:
-
Amplicon sequence variant
- DEG:
-
Differentially expressed gene
- DET:
-
Differentially expressed transcript
- EPG:
-
Eggs per gram of feces
- eQTL:
-
Expression quantitative trait loci
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GEBV:
-
Genomic estimated breeding value
- GRM:
-
Genomic relationship matrix
- GWAS:
-
Genome-wide association study
- h2 :
-
Heritability
- L3 :
-
Third-stage larvae
- L4 :
-
Fourth-stage larvae
- NES:
-
Normalized enrichment score
- PCR:
-
Polymerase chain reaction
- PE:
-
Paired-end
- RNA-seq:
-
Transcriptome sequencing
- SNP:
-
Single nucleotide polymorphism
References
Chagas ACS, Tupy O, Santos IB, Esteves SN. Economic impact of gastrointestinal nematodes in Morada Nova sheep in Brazil. Rev Bras Parasitol Vet. 2022;31:e008722.
Amarante AFT, Bricarello PA, Rocha RA, Gennari SM. Resistance of Santa Ines, Suffolk and Ile de France sheep to naturally acquired gastrointestinal nematode infections. Vet Parasitol. 2004;120:91–106.
Van Wyk JA, Reynecke DP. Blueprint for an automated specific decision support system for countering anthelmintic resistance in Haemonchus spp. at farm level. Vet Parasitol. 2011;177:212–23.
Zvinorova PI, Halimani TE, Muchadeyi FC, Matika O, Riggio V, Dzama K. Breeding for resistance to gastrointestinal nematodes—the potential in low-input/output small ruminant production systems. Vet Parasitol. 2016;225:19–28.
Piedrafita D, Raadsma HW, Gonzalez J, Meeusen E. Increased production through parasite control: can ancient breeds of sheep teach us new lessons? Trends Parasitol. 2010;26:568–73.
McManus C, Louvandini H, Paiva SR, de Oliveira AA, Azevedo HC, de Melo CB. Genetic factors of sheep affecting gastrointestinal parasite infections in the Distrito Federal. Brazil Vet Parasitol. 2009;166:308–13.
Nunes SF, Ferreira J, Paiva SR, de Faria DA, de Sousa JER, Soares CEA, et al. Fine genetic structure of Brazilian white Morada Nova hair sheep breed from semi-arid region. Small Rumin Res. 2022;211:106694.
Britton C, Laing R, McNeilly TN, Perez MG, Otto TD, Hildersley KA, et al. New technologies to study helminth development and host-parasite interactions. Int J Parasitol. 2023;53:393–403.
Swann J, Jamshidi N, Lewis NE, Winzeler EA. Systems analysis of host-parasite interactions. Wiley Interdiscip Rev Syst Biol Med. 2015;7:381–400.
Kristensen VN, Lingjærde OC, Russnes HG, Vollan HKM, Frigessi A, Børresen-Dale AL. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014;14:299–313.
Paz EA, Chua EG, Hassan SU, Greeff JC, Palmer DG, Liu S, et al. Bacterial communities in the gastrointestinal tract segments of helminth-resistant and helminth-susceptible sheep. Anim Microbiome. 2022;4:23.
Al MMA, Sandeman M, Rayment P, Brook-Carter P, Scholes E, Kasinadhuni N, et al. Variation in gut bacterial composition is associated with Haemonchus contortus parasite infection of sheep. Anim Microbiome. 2020;2:3.
Agüero VCG, Esteban-Blanco C, Argüello H, Valderas-García E, Andrés S, Balaña-Fouce R, et al. Microbial community in resistant and susceptible Churra sheep infected by Teladorsagia circumcincta. Sci Rep. 2022;12:17620.
Peng S, Yin J, Liu X, Jia B, Chang Z, Lu H, et al. First insights into the microbial diversity in the omasum and reticulum of bovine using Illumina sequencing. J Appl Genet. 2015;56:393–401.
El-Ashram S, Al Nasr I, Abouhajer F, El-Kemary M, Huang G, Dinçel G, et al. Microbial community and ovine host response varies with early and late stages of Haemonchus contortus infection. Vet Res Commun. 2017;41:263–77.
Cortés A, Wills J, Su X, Hewitt RE, Robertson J, Scotti R, et al. Infection with the sheep gastrointestinal nematode Teladorsagia circumcincta increases luminal pathobionts. Microbiome. 2020;8:60.
Toscano JHB, dos Santos IB, von Haehling MB, Giraldelo LA, Lopes LG, da Silva MH, et al. Morada Nova sheep breed: Resistant or resilient to Haemonchus contortus infection? Vet Parasitol. 2019;276S:1000019.
Ueno H, Gonçalves PC. Manual para diagnóstico das helmintoses de ruminantes. 4th ed. Tokyo: Japan International Cooperation Agency; 1998. p. 14–45.
Roberts F, O’Sullivan P. Methods for egg counts and larval cultures for strongyles infesting the gastro-intestinal tract of cattle. Aust J Agric Res. 1950;1:99.
Echevarria FAM, Armour JL, Duncan JL. Efficacy of some anthelmintics on an ivermectin-resistant strain of Haemonchus contortus in sheep. Vet Parasitol. 1991;39:279–84.
Herath HMPD, Taki AC, Sleebs BE, Hofmann A, Nguyen N, Preston S, et al. Chapter four—advances in the discovery and development of anthelmintics by harnessing natural product scaffolds. Adv Parasitol. 2021;111:203–51.
Chagas ACS, Katiki LM, Silva IC, Giglioti R, Esteves SN, Oliveira MCS, et al. Haemonchus contortus: a multiple-resistant Brazilian isolate and the costs for its characterization and maintenance for research use. Parasitol Int. 2013;62:1–6.
Amadeu RR, Cellon C, Olmstead JW, Garcia AAF, Resende MFR, Muñoz PR. AGHmatrix: R Package to construct relationship matrices for autotetraploid and diploid species: a blueberry example. Plant Genome. 2016;9:1–10.
Bates D, Vazquez AI. pedigreemm: pedigree-based mixed-effects models. R package version 0.3–3. 2014. https://CRAN.R-project.org/package=pedigreemm. Accessed 10 Oct 2023.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2022.
Xavier A, Xu S, Muir WM, Rainey KMNAM. Association studies in multiple populations. Bioinformatics. 2015;31:3862–4.
Niciura SCM, Cruvinel GG, Moraes CV, Bressani FA, Malagó Junior W, Benavides MV, et al. PCR-based genotyping of SNP markers in sheep. Mol Biol Rep. 2018;45:651–6.
Gauly M, Schackert M, Hoffmann B, Erhardt G. Influence of sex on the resistance of sheep lambs to an experimental Haemonchus contortus infection. Dtsch Tierarztl Wochenschr. 2006;113:178–81.
McRae KM, Good B, Hanrahan JP, McCabe MS, Cormican P, Sweeney T, et al. Transcriptional profiling of the ovine abomasal lymph node reveals a role for timing of the immune response in gastrointestinal nematode resistance. Vet Parasitol. 2016;224:96–108.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 10 Oct 2023.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008.
Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Van Rossum G, Drake FL. Python 3 reference manual. Scotts Valley: CreateSpace; 2009. p. 242.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40.
Fu J, Frazee AC, Collado-Torres L, Jaffe AE, Leek JT. ballgown: flexible, isoform-level differential expression analysis. R package version 2.28.0. 2022. https://doi.org/10.18129/B9.bioc.ballgown.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:1.30.1-33.
Brouard JS, Bissonnette N. Variant calling from RNA-seq data using the GATK joint genotyping workflow. Var Calling Methods Mol Biol. 2022;2493:205–33.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Sahraeian SME, Mohiyuddin M, Sebra R, Tilgner H, Afshar PT, Au KF, et al. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat Commun. 2017;8:59.
Broad Institute. Picard toolkit. 2018. http://broadinstitute.github.io/picard. Accessed 10 Oct 2023.
Van Der Auwera GA, O’Connor BD. Genomics in the cloud using Docker GATK and WDL in Terra. 1st ed. Sebastopol: O’Reilly Media; 2020.
Gao Y, Jiang G, Yang W, Jin W, Gong J, Xu X, et al. Animal-SNPAtlas: a comprehensive SNP database for multiple animals. Nucleic Acids Res. 2023;51:D816–26.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program. SnpSift Front Genet. 2012;3:35.
Gondro C, Porto-Neto LR, Lee SH. SNPQC—An R pipeline for quality control of Illumina SNP genotyping array data. Anim Genet. 2014;45:758–61.
Ferdosi MH, Kinghorn BP, van der Werf JHJ, Lee SH, Gondro C. hsphase: An R package for pedigree reconstruction, detection of recombination events, phasing and imputation of half-sib family groups. BMC Bioinformatics. 2014;15:172.
Niciura SCM, Benavides MV, Okino CH, Ibelli AMG, Minho AP, Esteves SN, et al. Genome-wide association study for Haemonchus contortus resistance in Morada Nova sheep. Pathogens. 2022;11:939.
Genome Data Viewer. Bethesda: National Library of Medicine, National Center for Biotechnology Information; 2004. https://www.ncbi.nlm.nih.gov/genome/gdv/browser/genome/?id=GCF_016772045.1. Accessed 10 Oct 2023.
Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–8.
Dansou CC, Kuiseu J, Olounlade PA, Azando EVB, Lagnika L, Aboh AB, et al. Haemonchosis: a review on dreaded strongylosis that affects the zootechnical performance of sheep. J Parasitol Vector Biol. 2021;13:58–70.
Doyle SR, Tracey A, Laing R, Holroyd N, Bartley D, Bazant W, et al. Genomic and transcriptomic variation defines the chromosome-scale assembly of Haemonchus contortus, a model gastrointestinal worm. Commun Biol. 2020;3:656.
Andrade BGN, Bressani FA, Cuadrat RRC, Tizioto PC, De Oliveira PSN, Mourão GB, et al. The structure of microbial populations in Nelore GIT reveals inter-dependency of methanogens in feces and rumen. J Anim Sci Biotechnol. 2020;11:6.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
Ling W, Lu J, Zhao N, Lulla A, Plantinga AM, Fu W, et al. Batch effects removal for microbiome data via conditional quantile regression. Nat Commun. 2022;13:5418.
Russo PST, Ferreira GR, Cardozo LE, Bürger MC, Arias-Carrasco R, Maruyama SR, et al. CEMiTool: a Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinformatics. 2018;19:56.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523.
Xiang R, Oddy VH, Archibald AL, Vercoe PE, Dalrymple BP. Epithelial, metabolic and innate immunity transcriptomic signatures differentiating the rumen from other sheep and mammalian gastrointestinal tract tissues. PeerJ. 2016;2016:e1762.
Li RW, Rinaldi M, Capuco AV. Characterization of the abomasal transcriptome for mechanisms of resistance to gastrointestinal nematodes in cattle. Vet Res. 2011;42:114.
Tang J. 3—Pepsin A. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of proteolytic enzymes. 2nd ed. Cambridge: Academic Press; 2004. p. 19–28.
Wohlkönig A, Huet J, Looze Y, Wintjens R. Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS ONE. 2010;5:e15388.
Roy HK. Cox 3? Am J Gastroenterol. 1999;94:2343.
Dunphy JL, Balic A, Barcham GJ, Horvath AJ, Nash AD, Meeusen ENT. Isolation and characterization of a novel inducible mammalian galectin. J Biol Chem. 2000;275:32106–13.
Rowe A, Gondro C, Emery D, Sangster N. Sequential microarray to identify timing of molecular responses to Haemonchus contortus infection in sheep. Vet Parasitol. 2009;161:76–87.
D’Errico M, de Rinaldis E, Blasi MF, Viti V, Falchetti M, Calcagnile A, et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur J Cancer. 2009;45:461–9.
Sui G, Zhou S, Wang J, Canto M, Lee EE, Eshleman JR, et al. Mitochondrial DNA mutations in preneoplastic lesions of the gastrointestinal tract: a biomarker for the early detection of cancer. Mol Cancer. 2006;5:73.
Patra G, Borthakur SK, Das S, Lalliankimi H, Lalrinkima H. Quantification and expression studies of IL-13 gene in response to experimental infection with Haemonchus contortus in Garole and Sahabadi sheep. Vet Parasitol Reg Stud Rep. 2017;8:99–103.
Guo Z, González JF, Hernandez JN, McNeilly TN, Corripio-Miyar Y, Frew D, et al. Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Sci Rep. 2016;6:26200.
Liu J, Tan M, Xu X, Shen T, Zhou Z, Hunt PW, et al. From innate to adaptive immunity: Abomasal transcriptomic responses of merino sheep to Haemonchus contortus infection. Mol Biochem Parasitol. 2021;246:111424.
Knight JS, Baird DB, Hein WR, Pernthaner A. The gastrointestinal nematode Trichostrongylus colubriformis down-regulates immune gene expression in migratory cells in afferent lymph. BMC Immunol. 2010;11:51.
Zhang CL, Liu C, Zhang J, Zheng L, Chang Q, Cui Z, et al. Analysis on the desert adaptability of indigenous sheep in the southern edge of Taklimakan Desert. Sci Rep. 2022;12:12264.
Wiener P, Robert C, Ahbara A, Salavati M, Abebe A, Kebede A, et al. Whole-genome sequence data suggest environmental adaptation of Ethiopian sheep populations. Genome Biol Evol. 2021;13:evab014.
Carracelas B, Navajas EA, Vera B, Ciappesoni G. Genome-wide association study of parasite resistance to gastrointestinal nematodes in Corriedale sheep. Genes. 2022;13:1548.
McManus C, Facó O, Shiotsuki L, de Paula Rolo JLJ, Peripolli V. Pedigree analysis of Brazilian Morada Nova hair sheep. Small Rumin Res. 2019;170:37–42.
Paim TP, Paiva SR, de Toledo NM, Yamaghishi MB, Carneiro PLS, Facó O, et al. Origin and population structure of Brazilian hair sheep breeds. Anim Genet. 2021;52:492–504.
Lacerda TSA, Caetano AR, Facó O, de Faria DA, McManus CM, Lôbo RN, et al. Single marker assisted selection in Brazilian Morada Nova hair sheep community-based breeding program. Small Rumin Res. 2016;139:15–9.
Laurenson YCSM, Kyriazakis I, Bishop SC. Can we use genetic and genomic approaches to identify candidate animals for targeted selective treatment. Vet Parasitol. 2013;197:379–83.
Bhuiyan AA, Li J, Wu Z, Ni P, Adetula AA, Wang H, et al. Exploring the genetic resistance to gastrointestinal nematodes infection in goat using RNA-sequencing. Int J Mol Sci. 2017;18:751.
Araujo RN, Padilha T, Zarlenga D, Sonstegard T, Connor EE, Van Tassel C, et al. Use of a candidate gene array to delineate gene expression patterns in cattle selected for resistance or susceptibility to intestinal nematodes. Vet Parasitol. 2009;162:106–15.
Liu J, Zhou J, Zhao S, Xu X, Li CJ, Li L, et al. Differential responses of abomasal transcriptome to Haemonchus contortus infection between Haemonchus-selected and Trichostrongylus-selected merino sheep. Parasitol Int. 2022;87:102539.
Pemberton JM, Beraldi D, Craig BH, Hopkins J. Digital gene expression analysis of gastrointestinal helminth resistance in Scottish blackface lambs. Mol Ecol. 2011;20:910–9.
Li X, Jiang S, Wang X, Jia B. Intestinal transcriptomes in Kazakh sheep with different haplotypes after experimental Echinococcus granulosus infection. Parasite. 2021;28:14.
Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. MBio. 2014;5:e01117-e1213.
Lechner A, Bohnacker S, Esser-von BJ. Macrophage regulation & function in helminth infection. Semin Immunol. 2021;53:101526.
Van Meulder F, Van Coppernolle S, Borloo J, Rinaldi M, Li RW, Chiers K, et al. Granule exocytosis of granulysin and granzyme B as a potential key mechanism in vaccine-induced immunity in cattle against the nematode Ostertagia ostertagi. Infect Immun. 2013;81:1798–809.
Terra JK, France B, Cote CK, Jenkins A, Bozue JA, Welkos SL, et al. Allelic variation on murine chromosome 11 modifies host inflammatory responses and resistance to Bacillus anthracis. PLoS Pathog. 2011;7:e1002469.
Signer-Hasler H, Burren A, Ammann P, Drögemüller C, Flury C. Runs of homozygosity and signatures of selection: a comparison among eight local Swiss sheep breeds. Anim Genet. 2019;50:512–25.
Widmann P, Reverter A, Weikard R, Suhre K, Hammon HM, Albrecht E, et al. Systems biology analysis merging phenotype, metabolomic and genomic data identifies non-SMC condensin I complex, subunit G (NCAPG) and cellular maintenance processes as major contributors to genetic variability in bovine feed efficiency. PLoS ONE. 2015;10:e0124574.
An B, Xu L, Xia J, Wang X, Miao J, Chang T, et al. Multiple association analysis of loci and candidate genes that regulate body size at three growth stages in Simmental beef cattle. BMC Genet. 2020;21:32.
King FJM, Visser C, Banga C. Genetic characterization of Mozambican Nguni cattle and their relationship with indigenous populations of South Africa. Livest Sci. 2022;264:105044.
Yang J, Li WR, Lv FH, He SG, Tian SL, Peng WF, et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol Biol Evol. 2016;33:2576–92.
Eydivandi S, Roudbar MA, Karimi MO, Sahana G. Genomic scans for selective sweeps through haplotype homozygosity and allelic fixation in 14 indigenous sheep breeds from Middle East and South Asia. Sci Rep. 2021;11:2834.
Mei C, Gui L, Hong J, Raza SHA, Aorigele C, Tian W, et al. Insights into adaption and growth evolution: a comparative genomics study on two distinct cattle breeds from Northern and Southern China. Mol Ther Nucleic Acids. 2021;23:959–67.
Schubert K, Olde Damink SWM, von Bergen M, Schaap FG. Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol Rev. 2017;279:23–35.
Boisseau M, Dhorne-Pollet S, Bars-Cortina D, Courtot É, Serreau D, Annonay G, et al. Species interactions, stability, and resilience of the gut microbiota - Helminth assemblage in horses. iScience. 2023;26:106044.
Strober BJ, Wen X, Wucher V, Kwong A, Lappalainen T, Li X, et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;18:1318–30.
Estrada-Reyes ZM, Ogunade IM, Pech-Cervantes AA, Terrill TH. Copy number variant-based genome wide association study reveals immune-related genes associated with parasite resistance in a heritage sheep breed from the United States. Parasite Immunol. 2022;44:e12943.
Aboshady HM, Mandonnet N, Félicité Y, Hira J, Fourcot A, Barbier C, et al. Dynamic transcriptomic changes of goat abomasal mucosa in response to Haemonchus contortus infection. Vet Res. 2020;51:44.
Zhang J, Toremurat Z, Liang Y, Cheng J, Sun Z, Huang Y, et al. Study on the association between LRRC8B gene InDel and sheep body conformation traits. Genes. 2023;14:356.
Mengistie D, Edea Z, Tesema TS, Dejene G, Dessie T, Jemal J, et al. Genome-wide signature of positive selection in Ethiopian indigenous and European beef cattle breeds. Anim Gene. 2023;29:200151.
McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, Andreux PA, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE. 2012;7:e39191.
Gantasala NP, Kumar M, Banakar P, Thakur PK, Rao U. Functional validation of genes in cereal cyst nematode, Heterodera avenae, using siRNA gene silencing. In: Dababat AA, Muminjanov H, Smiley RW, editors. Nematodes Small Grain Cereals. Ankara: Food And Agriculture Organization of The United Nations; 2015. p. 353–6.
Kita K, Nihei C, Tomitsuka E. Parasite mitochondria as drug target: diversity and dynamic changes during the life cycle. Curr Med Chem. 2005;10:2535–48.
Murai M, Miyoshi H. Current topics on inhibitors of respiratory complex I. Biochim Biophys Acta—Bioenerg. 2016;1857:884–91.
Nikolaou S, Gasser RB. Prospects for exploring molecular developmental processes in Haemonchus contortus. Int J Parasitol. 2006;36:859–68.
Laing R, Kikuchi T, Martinelli A, Tsai IJ, Beech RN, Redman E, et al. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 2013;14:R88.
Nakandalage R, Guan LL, Malmuthuge N. Microbial interventions to improve neonatal gut health. Microorganisms. 2023;11:1328.
Hodžić A, Dheilly NM, Cabezas-Cruz A, Berry D. The helminth holobiont: a multidimensional host–parasite–microbiota interaction. Trends Parasitol. 2023;39:91–100.
Rooney J, Cortés A, Scotti R, Price DRG, Bartley Y, Clarke KF, et al. Vaccination against the brown stomach worm, Teladorsagia circumcincta, followed by parasite challenge, induces inconsistent modifications in gut microbiota composition of lambs. Parasit Vectors. 2021;14:189.
Zhao GW, Yan RF, Muleke CI, Sun YM, Xu LX, Li XR. Vaccination of goats with DNA vaccines encoding H11 and IL-2 induces partial protection against Haemonchus contortus infection. Vet J. 2012;191:94–100.
Antonacci R, Massari S, Linguiti G, Jambrenghi AC, Giannico F, Lefranc MP, et al. Evolution of the T-cell receptor (TR) loci in the adaptive immune response: the tale of the TRG locus in mammals. Genes. 2020;11:624.
Fichtner AS, Ravens S, Prinz I. Human γδ TCR repertoires in health and disease. Cells. 2020;9:800.
Ludvik AE, Pusec CM, Priyadarshini M, Angueira AR, Guo C, Lo A, et al. HKDC1 Is a novel hexokinase involved in whole-body glucose use. Endocrinology. 2016;157:3452–61.
Calvete O, Reyes J, Martin P, Marazuela M, Barroso A, Escalada J, et al. Alterations in SLC4A2, SLC26A7 and SLC26A9 drive acid—base imbalance in gastric neuroendocrine tumors and uncover a novel mechanism for a co-occurring polyautoimmune scenario. Cells. 2021;10:3500.
Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, et al. Deletion of the chloride transporter Slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem. 2009;284:29470–9.
Alonso N, Zappia CD, Cabrera M, Davio CA, Shayo C, Monczor F, et al. Physiological implications of biased signaling at histamine H2 receptors. Front Pharmacol. 2015;6:45.
Howden CW, Hunt RH. Relationship between gastric secretion and infection. Gut. 1987;28:96–107.
Barker IK, Titchen DA. Gastric dysfunction in sheep infected with Trichostrongylus colubriformis, a nematode inhabiting the small intestine. Int J Parasitol. 1982;12:345–56.
Aboshady HM, Choury A, Montout L, Félicité Y, Godard X, Bambou JC. Metagenome reveals caprine abomasal microbiota diversity at early and late stages of Haemonchus contortus infection. Sci Rep. 2023;13:2450.
Hewitson JP, Grainger JR, Maizels RM. Helminth immunoregulation: the role of parasite secreted proteins in modulating host immunity. Mol Biochem Parasitol. 2009;167:1–11.
Cooper D, Eleftherianos I. Parasitic nematode immunomodulatory strategies: recent advances and perspectives. Pathogens. 2016;5:58.
Azziz G, Giménez M, Carballo C, Espino N, Barlocco N, Batista S. Characterization of the fecal microbiota of Pampa Rocha pigs, a genetic resource endemic to eastern Uruguay. Heliyon. 2023;9:e16643.
Boggio GM, Meynadier A, Daunis-I-Estadella P, Marie-Etancelin C. Compositional analysis of ruminal bacteria from ewes selected for somatic cell score and milk persistency. PLoS ONE. 2021;16:e0254874.
Sbardellati DL, Fischer A, Cox MS, Li W, Kalscheur KF, Suen G. The bovine epimural microbiota displays compositional and structural heterogeneity across different ruminal locations. J Dairy Sci. 2020;103:3636–47.
Wang H, Xia P, Lu Z, Su Y, Zhu W. Metabolome-microbiome responses of growing pigs induced by time-restricted feeding. Front Vet Sci. 2021;8:681202.
Wang X, Zhang Z, Li B, Hao W, Yin W, Ai S, et al. Depicting fecal microbiota characteristic in yak, cattle, yak-cattle hybrid and Tibetan sheep in different eco-regions of Qinghai-Tibetan plateau. Microbiol Spectr. 2022;10:e0002122.
Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019;17:83.
Cheng J, Hu J, Geng F, Nie S. Bacteroides utilization for dietary polysaccharides and their beneficial effects on gut health. Food Sci Hum Wellness. 2022;11:1101–10.
Low A, Lee JKY, Gounot JS, Ravikrishnan A, Ding Y, Saw WY, et al. Mutual exclusion of Methanobrevibacter species in the human gut microbiota facilitates directed cultivation of a Candidatus methanobrevibacter intestini representative. Microbiol Spectr. 2022;10:e00849-e922.
Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci U S A. 2006;103:10011–6.
Ohkusa T, Nishikawa Y, Sato N. Gastrointestinal disorders and intestinal bacteria: advances in research and applications in therapy. Front Med. 2023;9:935676.
Moon CD, Carvalho L, Kirk MR, McCulloch AF, Kittelmann S, Young W, et al. Effects of long-acting, broad spectra anthelmintic treatments on the rumen microbial community compositions of grazing sheep. Sci Rep. 2021;11:3836.
Chen H, Mozzicafreddo M, Pierella E, Carletti V, Piersanti A, Ali SM, et al. Dissection of the gut microbiota in mothers and children with chronic Trichuris trichiura infection in Pemba Island Tanzania. Parasites Vectors. 2021;14:62.
Abosse JS, Terefe G, Teshale BM. Comparative study on pathological changes in sheep and goats experimentally infected with Haemonchus contortus. Surg Exp Pathol. 2022;5:14.
Gryaznova M, Dvoretskaya Y, Burakova I, Syromyatnikov M, Popov E, Kokina A, et al. Dynamics of changes in the gut microbiota of healthy mice fed with lactic acid bacteria and bifidobacteria. Microorganisms. 2022;10:1020.
Karpinska A, Ryan D, Germaine K, Dowling D, Forrestal P, Kakouli-Duarte T. Soil microbial and nematode community response to the field application of recycled bio-based fertilisers in Irish grassland. Sustain. 2021;13:12342.
Xiang H, Fang Y, Tan Z, Zhong R. Haemonchus contortus infection alters gastrointestinal microbial community composition, protein digestion and amino acid allocations in lambs. Front Microbiol. 2022;12:797746.
Li RW, Li W, Sun J, Yu P, Baldwin RL, Urban JF. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci Rep. 2016;6:20606.
Lee SC, Tang MS, Lim YAL, Choy SH, Kurtz ZD, Cox LM, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. 2014;8:e2880.
Houlden A, Hayes KS, Bancroft AJ, Worthington JJ, Wang P, Grencis RK, et al. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS ONE. 2015;10:e0125945.
Gálvez EJC, Iljazovic A, Amend L, Lesker TR, Renault T, Thiemann S, et al. Distinct polysaccharide utilization determines interspecies competition between intestinal Prevotella spp. Cell Host Microbe. 2020;28:838–52.
Acknowledgements
We thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil—Finance Code 001 for the free access to the journals used in the literature review.
Funding
FAPESP (processes no. 2017/01626-1, 2019/02929-3, 2021/11842-9, 2021/02535-5, and 2022/06281-0). AMGI is recipient of a productivity fellowship from CNPq. BGNA was funded by Science Foundation Ireland and co-funded by the European Regional Development Fund through the ADAPT Centre for Digital Content Technology Resource Centre grant number 13/RC/2106_P2.
Author information
Authors and Affiliations
Contributions
SCMN generated, analyzed and interpreted genomic and transcriptomic (GWAS, variant calling, eQTL, and RNA-seq) data. TFC analyzed and interpreted microbiota data and performed multi-omics integration. AMGI and CRO generated and interpreted transcriptomic data. BGA and LCAR generated and interpreted microbiota data. MVB, ACSC, SNE and APM obtained phenotypic data and collected samples. CG analyzed and interpreted genomic and transcriptomic data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols were approved by the Embrapa Pecuária Sudeste Ethics and Animal Experimentation Committee under protocol numbers 04/2017 and 01/2020.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
edgeR analysis of differential gene expression in the abomasum between H. contortus-susceptible and H. contortus-resistant Morada Nova sheep. Table S2. DESeq2 analysis of differential gene expression in the abomasum between H. contortus-susceptible and H. contortus-resistant Morada Nova sheep. Table S3. DESeq2 analysis of differential transcript expression in the abomasum between H. contortus-susceptible and H. contortus-resistant Morada Nova sheep. Table S4. Ballgown analysis of differential transcript expression in the abomasum between H. contortus-susceptible and H. contortus-resistant Morada Nova sheep. Table S5. Annotation and impact of homozygous SNPs between resistant and susceptible Morada Nova sheep detected after case-control variant calling of RNA-seq data. Table S6. Genes located in the +/- 1 Mbp window of each significant SNP by GWAS for EPG in Morada Nova sheep. Table S11. DESeq2 analysis of differential gene expression in H. contortus recovered from resistant and susceptible Morada Nova sheep. Table S12. edgeR analysis of differential gene expression in H. contortus recovered from resistant and susceptible Morada Nova sheep. Table S13. DESeq2 analysis of differential transcript expression in H. contortus recovered from resistant and susceptible Morada Nova sheep. Table S14. Ballgown analysis of differential transcript expression in H. contortus recovered from resistant and susceptible Morada Nova sheep. Table S15. Genes expressed in H. contortus recovered from resistant and susceptible Morada Nova sheep. Table S16. Gene-level features and ASV modules associated with resistance to H. contortus. Table S17. ASV taxonomy of gene co-expression modules from sheep feces and rumen content associated with resistance to H. contortus. Table S18. Gene-level relevant pathways and ASV modules in H. contortus-resistant and H. contortus-susceptible Morada Nova sheep. Table S19. Transcript-level features and ASV modules associated with resistance to H. contortus. Table S20. Transcript functional co-expression modules and ASVs associated with resistance to H. contortus. Table S21. Transcript-level relevant pathways and ASV modules in H. contortus-resistant and H. contortus-susceptible Morada Nova sheep.
Additional file 2: Table S7.
Cis-eQTL for differentially expressed genes between resistant and susceptible Morada Nova sheep. Table S8. Trans-eQTL for differentially expressed genes between resistant and susceptible Morada Nova sheep. Table S9. Cis-eQTL for differentially expressed transcripts between resistant and susceptible Morada Nova sheep. Table S10. Trans-eQTL for differentially expressed transcripts between resistant and susceptible Morada Nova sheep.
Additional file 3: Figure S1.
Differential gene expression and count in abomasum of H. contortus-resistant and H. contortus-susceptible Morada Nova sheep for 11 genes detected using edgeR.
Additional file 4: Figure S2.
Differential transcript expression and count in abomasum of H. contortus-resistant and H. contortus-susceptible Morada Nova sheep for three transcripts detected using ballgown.
Additional file 5: Figure S3.
Co-expression analysis of sheep RNA-seq transcripts and amplicon sequence variants (ASV). The figure displays the normalized enrichment score (NES) (red represents higher activity and blue represents lower activity), with the circle size and color intensity proportional to the NES values.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Niciura, S.C.M., Cardoso, T.F., Ibelli, A.M.G. et al. Multi-omics data elucidate parasite-host-microbiota interactions and resistance to Haemonchus contortus in sheep. Parasites Vectors 17, 102 (2024). https://doi.org/10.1186/s13071-024-06205-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06205-9