
Abstract
The response of the abomasal transcriptome to gastrointestinal parasites was evaluated in parasite-susceptible and parasite-resistant Angus cattle using RNA-seq at a depth of 23.7 million sequences per sample. These cattle displayed distinctly separate resistance phenotypes as assessed by fecal egg counts. Approximately 65.3% of the 23 632 bovine genes were expressed in the fundic abomasum. Of these, 13 758 genes were expressed in all samples tested and likely represent core components of the bovine abomasal transcriptome. The gene (BT14427) with the most abundant transcript, accounting for 10.4% of sequences in the transcriptome, is located on chromosome 29 and has unknown functions. Additionally, PIGR (1.6%), Complement C3 (0.7%), and Immunoglobulin J chain (0.5%) were among the most abundant transcripts in the transcriptome. Among the 203 genes impacted, 64 were significantly over-expressed in resistant animals at a stringent cutoff (FDR < 5%). Among the 94 224 splice junctions identified, 133 were uniquely present: 90 were observed only in resistant animals, and 43 were present only in susceptible animals. Gene Ontology (GO) enrichment of the genes under study uncovered an association with lipid metabolism, which was confirmed by an independent pathway analysis. Several pathways, such as FXR/RXR activation, LXR/RXR activation, LPS/IL-1 mediated inhibition of RXR function, and arachidonic acid metabolism, were impacted in resistant animals, which are potentially involved in the development of parasite resistance in cattle. Our results provide insights into the development of host immunity to gastrointestinal nematode infection and will facilitate understanding of mechanism underlying host resistance.
Similar content being viewed by others


Introduction
Gastrointestinal (GI) nematode infections in ruminants remain a major impediment to the efficient production of both meat and dairy products, and therefore, represent a major constraint on global food availability. These GI infections have a significant economic impact on the U.S. cattle industry, with an estimated annual cost of ~$2 billion per year in lost productivity and increased operating expenses. Nematode infections of the GI tract impact numerous production traits. Among the most prominent effect is a reduction in weight gain that may cause decreased bodyweights of up to 14% [1]. Although the impact is particularly evident in young calves, substantial evidence suggests that infection produces long lasting effects on the productivity of adult cattle [2]. In dairy cows, parasitic infections reduce milk yield between 1.2 and 2.2 kg milk/cow per day [3]. Infections also negatively impact carcass quality and reproductive performance, including calving rate and calf mortality [4]. Potential economic loss resulting from GI nematode infections is clearly recognized by producers and veterinarians, as evidenced by the fact that approximately 99% of feedlots and 69% of dairies use a parasiticide in their operations [5].
Among 41 bovine GI nematodes, species from the genera Ostertagia, Cooperia, and Nematodirus are arguably the most important cattle parasites in temperate regions of the world, as assessed by their negative economic impact [6]. Development of protective immunity and resistance to these GI nematodes relies upon the precise control of expression of the host genome. It is evident that the evolution of regulatory programs controlling the transcriptome occurs at a rapid rate comparable to that of other genomic processes. Understanding these regulatory elements is crucial towards unraveling their functional relevance. Comparative transcriptomic analysis has emerged as a promising means for unraveling the molecular basis and regulatory networks underlying complex traits such as host resistance. While recent progress has been made with regard to genes associated with nematode resistance in small ruminants [7–9], an in-depth comparison and characterization of transcriptomic responses of cattle populations that harbor varying degrees of resistance to parasitic nematodes is not yet available. Sequencing steady-state RNA in a biological sample (RNA-seq technology) using next-generation sequencing platforms (e.g. Illumina) overcomes many limitations of previous technologies, such as microarrays and real-time PCR. Most importantly, RNA-seq has been shown to elucidate previously inaccessible complexities in the transcriptome, such as allele-specific expression and involvement of novel promoters and isoforms [10], detection of alternative splicing [11], RNA editing [12], novel transcripts [13], all in conjunction with quantitative evaluation of transcript abundance [14]. In this study, we utilized RNA-seq technology to characterize the bovine transcriptome response of nematode-resistant and nematode-susceptible heifers to identify molecular mechanisms that underlie host resistance to GI nematodes in cattle.
Materials and methods
Animals and parasitology
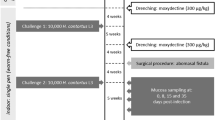
Six 12-month-old Angus heifers that differed with regard to susceptibility to GI nematode infection (3 resistant and 3 susceptible) were used in this experiment. These heifers were from a selective breeding program for parasite resistance that was initiated at our facilities in 1991, using parental stock originating from the Wye Angus herd [15, 16]. Once the initial breeding females were identified, semen from high and low EPG (eggs per gram of feces) bulls was used to produce calves of desired phenotypes. Calves were kept with their dams on pastures with extremely low numbers of parasites prior to weaning. When the median age of the contemporary group was 205 days, calves were weaned and placed on pastures infected with the two most common nematode parasites of cattle, Ostertagia ostertagi and Cooperia oncophora. The calves were monitored weekly for a number of parasitologic and immunologic parameters along with selected measurements of animal growth. The calves were kept pastured for a minimum of 120 days. Replacement animals were selected for secondary challenge experiments, while parasitologic and immunologic parameters were collected from animals chosen for slaughter. This program resulted in resource populations selected for the fecal egg trait (high or low fecal eggs counts or eggs per gram, EPG; high or low parasite resistance, respectively). Based on actual weekly EPG counts and sire expected progeny difference (EPD) values for EPG, a total of six heifers were selected for this study. Three heifers used in this study were classified as susceptible (high EPG EPD value) and the remaining 3 were classified as resistant (low EPG EPD value).
At the end of the grazing season, all heifers were treated with a combination of 10 mg fenbendazole and 0.5 mg moxidectin per kg of body weight to remove existing GI parasites transmitted from the infected pastures. After resting for 30 days on concrete to preclude further parasite exposure, the heifers were orally infected with a single dose of combined O. ostertagi and C. oncophora infective L3 larvae (8.5 × 104 and 1.0 × 105O. ostertagi and C. oncophora L3 larvae, respectively, per animal) and housed on concrete for an additional 20 days, allowing the experimental infection to progress. EPG was monitored during the resting period to ensure that the drug treatment eliminated all pre-existing parasites. The infective L3 larvae were obtained from cultures maintained at the USDA-ARS Beltsville facilities. The heifers were handled according to a protocol approved by the Beltsville Agricultural Research Center Animal Care and Use Committee, following Institutional Animal Care and Use Committees (IACUC) guidelines. The heifers were sacrificed at 20 days post infection (dpi). Worms were counted from the contents of the abomasum and small intestine. The full-thickness of folds from the fundic abomasa were collected, minced into 1-2 cm pieces, and snap frozen in liquid nitrogen prior to storage at -80°C until total RNA was extracted.
RNA extraction and sequencing using RNA-seq technology
Total RNA was extracted using Trizol followed by DNase digestion and Qiagen RNeasy column purification as previously described [17]. The RNA integrity was verified using an Agilent Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA) with a RIN value > 7.0. High-quality RNA was processed using an Illumina RNA-seq sample prep kit following the manufacturer's instruction (Illumina, San Diego, CA, USA). Final RNA-seq libraries were validated and sequenced at 36bp/sequence read using an Illumina GAIIx sequencer at a depth of approximately 23.7 million sequences per sample (mean ± SD = 23 723 620 ± 7 447 499 per sample).
Data analysis and bioinformatics
23 632 bovine genes in the Bovine Official Gene Set version 2 (OGS2.0) [18] were first mapped to the bovine reference genome (Btau4.0) using Genomic Short-read Nucleotide Alignment Program or GSNAP [19]. The best mapping position of each gene (≤ 10 kb intron span, ≥ 95% identity, ≥ 90% coverage, and minimum tail length of 5% of coding sequences) was extracted. Accordingly, 20 809 of the 23 632 bovine genes were uniquely mapped. After removing ambiguously mapped genes, 18 834 genes were used for RNA-seq data analysis.
Raw sequence reads were then checked using several layers of quality control filtering to remove low-quality reads. Raw reads with ≥ 2 ambiguous nucleotides (N) were discarded. Trimming removed approximately 1% of input raw reads and led to the retention of 99% of raw reads. The input reads after cleansing were mapped to the reference genome with gene coordinates using Bowtie (v0.12.7), an ultrafast and memory-efficient short-read aligner using a Burrows-Wheeler index [20]. Approximately 68.5% of trimmed reads mapped to the bovine genome (mean ± SD = 68.54% ± 2.48%). Only reads with one unique best match in the reference genome were used for subsequent analyses. The read depth of each gene was computed based on the coordinates of mapped reads and gene locations in the reference genome and was normalized using a method that corrects for biases introduced by RNA composition and differences in the total numbers of uniquely mapped reads in each sample [21]. Only genes having ≥ 20 uniquely mapped reads (mean of all 6 samples) were further analyzed. The R package edgeR was used to test the null hypothesis that expression of a given gene is not different between the two groups [21]. The normalized read counts were also analyzed using the DEGseq algorithm [22]. The DEGseq built-in function "samWrapper" that is recommended for testing RNA-seq data with biological replication was used to detect differential expression. Candidate genes were first sorted based on P value (P < 0.05) and fold change (2-fold as a cutoff). Genes identified as candidates for differential expression were further filtered with a false discovery rate (FDR) of < 5% to account for multiple testing. Differentially-expressed genes in the transcriptome were further analyzed using Gene Ontology (GO) analysis (GOseq). Over-representation of certain GO terms was determined based on Fisher's exact test. A multiple correction control (permutation to control false discovery rate [23]) was implemented to set up the threshold to obtain the lists of significantly over-represented GO terms. The candidate genes were analyzed using IPA v9.0 for pathways (Ingenuity Systems, Redwood City, CA, USA).
Tophat (v1.2.0) was used to map input reads to the reference genome [11] and identify potential splice junctions or splicing variants. Only reads with one unique match in the reference genome were used for subsequent analyses. The maximum allowed intron size was 5kb (a conservative parameter to avoid a high false discovery rate). At each potential splice junction, spanning reads were counted. Potential splice junctions were compared to annotated splice sites. To identify differential splice junctions between two groups, normalized read counts of splice junctions were required to be eight times different between resistant and susceptible groups. An unpaired t-test was performed on normalized sequence read counts. Splice junctions at P ≤ 0.05 were considered candidates junctions that were differentially regulated between resistant and susceptible groups.
Real-time RT-PCR
Real-time or quantitative RT-PCR (qPCR) was performed as previously described [17]. Briefly, the cDNA synthesis was performed using an iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). Real-time RT-PCR analysis was carried out with an iQ SYBR Green Supermix kit (Biorad) using 200 nM of each amplification primer and the 1st-strand cDNA (100 ng of the input total RNA equivalents) in a 25 μL reaction volume as described. The amplification was carried out on an iCycler iQ™ Real Time PCR Detection System (BioRad) with the following profile: 95°C-60 s; 40 cycles of 94°C-15 s, 60°C-30 s, and 72°C-30 s. A melting curve analysis was performed for each primer pair. The gene encoding for phospholipase A2, group IVA (cytosolic, calcium-dependent) (PLA2G4A), which has a relatively constant expression level across all experiment samples, was used as an endogenous control. Relative gene expression data was calculated using the 2-ΔΔCT method. The fold change was normalized against the susceptible group.
Results
EPG and worm counts
Mean weekly EPG values were 8.8 ± 1.6 (Mean ± SD) and 31.3 ± 10.4 for low-EPG (resistant) and high-EPG (susceptible) heifers, respectively (N = 3) during the 6-month grazing season (Figure 1). In accordance with the experimental design, this difference was statistically significant (P < 0.05). Although a temporal fluctuation in weekly EPG values was evident, resistant heifers shed constantly fewer parasite eggs in feces than did susceptible heifers. Over the grazing period, resistant heifers gained more weight (P < 0.05) than susceptible heifers (159.0 ± 32.8 vs 92.1 ± 20.4 lbs). Similarly, resistant heifers displayed a numerically, but not statistically, greater (P > 0.05) gain in hip height over the experiment period (Table 1). Serum pepsinogen levels between both resistant and susceptible groups were statistically indistinguishable. The mean number of total parasite worms (both O. ostertagi and C. oncophora) recovered from resistant heifers (5 200 ± 3 191) after the experimental challenge were numerically less than those of susceptible heifers (5 923 ± 3 203), but not significantly less (P > 0.05). These worm burden data were not unexpected because the population under study has never been selected for worm burdens as an indicator trait in the applied breeding program. Additionally, the worm counts were obtained from an experimental infection with a high dose of infective larvae that is not typically encountered by calves under nature exposure.

Weekly mean fecal egg counts (eggs per gram, EPG or FEC) of resistant and susceptible Angus heifers grazing on infected pasture between April to October. Y-axis represents mean weekly fecal egg counts. * indicates a significant difference in EPG between resistant and susceptible groups (P < 0.05). ** P < 0.01; *** P < 0.001.
General characteristics of the bovine abomasal transcriptome
20 809 of the 23 633 bovine genes (88%) were uniquely mapped to the bovine genome (Bta4.0). Among these, 18 834 genes were unambiguously mapped and were used for RNA-seq analysis. 14 549 to 15 432 of the 18 834 genes had at least one copy of their transcripts expressed in the bovine abomasal transcriptome (61.6 to 65.3% of all bovine genes). 11 474 to 13 015 genes had ≥ 10 sequence hits in the bovine abomasal tissue. 13 758 genes were expressed in all bovine abomasal samples tested, probably representing the core component of the bovine abomasal transcriptome. The most abundant transcript in all 6 abomasal samples tested was a gene (Gene ID: BT14427) located on Bos taurus autosome (BTA or chromosome) 29 whose function is unknown but represents 10.38% of sequence reads in the transcriptome. The next most abundant transcripts included an unknown gene (BT10810, 2.36%), polymeric immunoglobulin receptor (PIGR, 1.60%), a gene on BTA11 (BT28533, 1.29%), complement C3 (0.73%), growth arrest-specific protein 7 (0.67%), pre-B lymphocyte protein 3 (0.60%), liver fatty acid-binding protein (0.56%), IgG Fc-binding protein (0.53%), and immunoglobulin J chain (0.49%). The 10 most abundant protein-coding genes accounted for 19.21% of all sequence reads in the bovine abomasal transcriptome.
Differentially expressed genes
Normalized sequence counts were analyzed using both edgeR and DEGseq algorithms. A total of 203 genes met 2 criteria: unadjusted P value < 0.05 and 2-fold difference in normalized read counts between resistant and susceptible animals (Additional file 1). These candidate genes were further filtered with a stringent cutoff (FDR < 5%). Sixty four genes were significantly different between resistant and susceptible animals at FDR < 5% (Table 2). These genes had a significantly higher ratio of normalized sequence counts between resistant and susceptible groups. For example, common salivary protein BSP10, form A (BT12506 or SPLUNC2A) was expressed 57.68 fold higher in resistant heifers. Similarly, the sequence counts of intelectin (ITLN2) in the abomasum of resistant heifers were 51.98 times higher than in that of susceptible heifers. Mucin 12 (MUC12) and fatty acid binding protein 6, ileal (FABP6, intestinal bile acid-binding protein or gastrotropin) were significantly over-expressed in resistant animals. Several apolipoproteins (APOA1, A4, B100, and C2) were also over-expressed in resistant heifers. Transcripts for alpha-inducible protein 27 and 27-like 2 (IFI27 and IFI27L2) were more abundant in resistant than in susceptible animals.
Splicing variants
A total of 94 224 potential splice junctions spanned by ≥ 1 sequence read were identified using TopHat. Among them, 139 junctions displayed a significantly different number of sequence reads between resistant and susceptible groups (FDR < 5%). These 139 junctions were distributed on 28 autosomes and the X chromosome. There were no junctions on BTA12 that had significantly different numbers of sequence reads between the two groups. However, the distribution of these 139 junctions did not appear to be random, and the number of these junctions was not proportional to the physical length of the chromosomes. The vast majority of these junctions were unique to one of the two groups. For example, 90 of the 139 junctions were observed in all 3 resistant heifers but absent in 3 susceptible heifers. On the other hand, 43 were only present in susceptible heifers. Some of these unique junctions were observed in 9 of the 64 differentially expressed genes. For instance, 8 sequences (normalized mean counts) spanning an intron-exon junction (Intron position start 33 668 350 and end 33 669 502) on BTA19 were observed only in susceptible heifers and occurred inside a gene named peripheral myelin protein 2 (BT22962). A unique junction in the gene GDPD2 (BT16585) was observed only in resistant animals. The 10 most abundant unique junctions from each group are listed in Table 3.
Gene Ontology (GO) and pathway analyses
Over-representation of GO terms was determined based on Fisher's exact test and filtered further using a multiple correction control at FDR < 5%. As Table 4 shows, the GO enrichment of genes under study was predominantly associated with lipid metabolism.
To gain insights into pathways involved in the development of parasite resistance, we analyzed the differentially expressed genes using Ingenuity pathways analysis software, IPA. Among the 7 regulatory networks identified (data not shown), the primary function of 3 networks was related to lipid metabolism. The primary function of the 3rd regulatory network was involved in antimicrobial response and inflammation. A total of 12 pathways were significantly impacted (P < 0.05) and possibly involved in the development of host resistance to parasitic infection in cattle (Table 5). FXR/RXR activation was the pathway most significantly impacted in resistant heifers (P = 3.66E-07) with at least 8 of the 203 differently expressed genes involved, including APOA1, APOB, APOC2, APOC3, FABP6, and IL18. The other pathways significantly impacted in resistant animals during parasitic infection included LXR/RXR activation (P = 2.78E-04), LPS/IL-1 mediated inhibition of RXR function (P = 8.80E-04), and arachidonic acid metabolism (P = 4.68E-03). In addition, acute phase response signaling was also impacted in resistant heifers (P < 0.05).
Real-time RT- PCR confirmation
The expression of 10 genes at mRNA level in the fundic abomasum was examined using real-time RT-PCR. The mRNA level of cholecystokinin B receptor (CCKBR, NM_174262), a receptor for gastrin, was extremely low in the fundic abomasum. The mRNA levels of gastrin (GAST, NM_173915) and pepsinogen 5, group I (pepsinogen A) (PGA5, NM_001001600) were reliably detected. PGA5 expression level appeared to be higher in resistant animals. However, the difference was not statistically significant due to a large variation while no changes in gastrin mRNA level between susceptible and resistant animals were detected. The expression of MUC12 was barely detectable (≤ 40Ct). The mRNA levels of glucosaminyl (N-acetyl) transferase 3, mucin type (GCNT3, NM_205809), mucin 2 (MUC2, NM_001245997), and galectin 15-like (LGALS13, XM_593263) were moderately abundant but no differences were detected between susceptible and resistant animals, consistent with the RNAseq results.
Expression levels of BPI fold containing family A, member 2A (BPIFA2A or SPUNC2A, NM_174803), bovine putative ISG12(a) protein (IFI27, NM_001038050), lectin, galactoside-binding, soluble, 3 (LGALS3, NM_001102341), and bovine collectin-46 (CL46, NM_001001856), were significantly higher in resistant animals, consistent with RNAseq data (Figure 2).

The expression profiles of SPUNC2A, LGALS3, IFI27, and CL46 in the bovine fundic abomasum of susceptible and resistant heifers. The expression value at the mRNA level was detected using quantitative RT-PCR. The expression value of one of the susceptible animals was set as 1.0. The fold change as calculated using the 2-ΔΔCT method and normalized against the susceptible group (mean ± SD). SPUNC2A = BPI fold containing family A, member 2A (NM_174803); IFI27 = bovine putative ISG12(a) protein (NM_001038050); LGALS3 = lectin, galactoside-binding, soluble, 3 (NM_001102341), and CL46 = bovine collectin-46 (NM_001001856).
Discussion
The parameters of resistance to GI nematode infections in cattle while yet to be precisely defined, generally include decreased worm establishment and reduced parasite fecundity. It has long been known that host genetic factors play a significant role in determining susceptibility and resistance. Among eight factors determining EPG variation, additive genetic variation is predominant and accounts for ~30% of the variation in EPG [24]. Estimates of heritability for parasite indicator traits in ruminants are phenotype-dependent. In small ruminants, the heritability of adult worm length at the end of the first grazing season is very strong at 0.62 [24], whereas the heritability of EPG is moderate, ranging from 0.14 to 0.33 in Creole goats [25]. In cattle, the heritability of EPG released during the 1st grazing season is approximately 0.30 [26]. The ability of calves to recognize parasitic antigens is also under the control of host genetics [27]. Several studies suggest that there exist significant differences in the ability of cattle to resist GI nematode infections, and 3 major responder types can be readily identified in outbreed cattle populations [28, 29]. Worm establishment (worm burden) is predominantly influenced by host responder types. The ability of intermediate and high responders to mount a more effective and rapid immune response compared to low responders is sustained after secondary infection, providing more evidence that genetics may play an important role in regulating host resistance. The finding that the different responder types, based on parasitological variables, also feature a different immune response is very interesting since this also provides the opportunity to study the influence of genetic components of the host immune response [28]. These observations have spurred efforts to develop resource populations and identify genes and QTL that underlie the resistance trait and to develop criteria for selective breeding [30].
A vigorous and effective mucosal immunity is essential for resistance to GI nematode infection in ruminants. The resistant phenotype is often manifested in the host transcriptome. For example, resistant sheep breeds are able to more rapidly up-regulate Th2 cytokines than susceptible breeds [31]. In Angus cattle, our evidence suggests that resistant heifers can better maintain inflammatory responses at the sites of infection, especially during early stages of infection [16]. In the current study, we conducted an in-depth transcriptomic analysis to identify molecular mechanisms that underlie the development of host resistance in cattle, taking advantage of a resource population developed via selective breeding. Our results suggest that among the 94 224 splice junctions identified, 133 were uniquely present in either resistant or susceptible cattle, possibly representing novel splicing variants that have implications in the development of host resistance. We identified 203 candidate genes that displayed significantly different numbers of sequences between resistant and susceptible animals at a combined cutoff value P < 0.05 and 2-fold.
The transcripts from 16 genes, including gastrin-releasing peptide (GRP) and macrophage-stimulating 1 (MST1), had a significantly higher number of sequences in susceptible cattle. GRP has been reported to be down-regulated by parasitic infection in a helminth-mouse system. Our results suggest that parasitic infection in susceptible cattle may have a negative impact on the host enteric nervous system that extends beyond its role in modulating normal functions of host epithelial, immune, and muscle cells [32].
Among the 187 genes with more abundant transcripts in the abomasum of resistant heifers, a notable feature was the up-regulation of various lectins. At least 4 lectins, such as bovine-specific collectin 46 (CL-46), C-type lectin domain family 12 member A (CLEC12A), galectin 3 (LGALS3), and intelectin 2 (ITLN2), had significantly more abundant transcripts in the abomasum of resistant cattle, which confirmed our previous study utilizing high-density DNA oligo arrays [33]. ITLN2 and several C-type lectins, such as collectin 11 (COLEC11), cattle-specific collectin-46, and conglutinin, as well as galectins were strongly up-regulated in the abomasal mucosa of immune cattle developed using multiple rounds of drug-attenuated infections [6, 33]. ITLN2 expression is regulated by Th2 cytokine IL-4 [34]. Its elevated expression is observed in the sheep abomasums in response to Teladorsagia circumcincta infection, Dictyocaulus filaria natural infection [35], and Haemonchus contortus infection [36]. Most pertinent, this gene is naturally deleted in the genome of the susceptible mouse strain, C57BL/10, but is present in the genome of a nematode-resistant mouse strain, BALB/c, suggesting that this gene may serve a protective role in the innate immune response to Trichinella infection [37]. Cattle-specific collectin-46 has been suggested to provide the first line of defense against pathogens without eliciting a general inflammatory reaction [38]. Galectins also play an important role in innate immunity, including serving as receptors for pathogen-associated molecule patterns (PAMP), which is integral in recognizing carbohydrate moieties on the cell surface of parasites, activating various immune cells, participating in cytotoxicity, modulating innate immunity via binding to IgA, and promoting the reconstruction of damaged tissues as receptors for damage-associated molecular patterns (DAMP) [39]. Together, our results suggest that lectins may play an important role in invoking effective host immune responses and in the development of host resistance.
Our evidence also indicates that alterations in lipid metabolism may be necessary to the development of host resistance. The top function of 3 of the 7 regulatory networks identified was associated with lipid metabolism. GO terms associated with genes that were differently expressed between resistant and susceptible animals were also predominantly related to lipid metabolism (Table 4). Lipid metabolism is significantly regulated in the bovine small intestine. In response to C. oncophora infection, lipid balance in the GI tract during parasitic infection may be disrupted [17]. Polyunsaturated fatty acids (PUFA), especially those in omega-3 (n-3) and omega-6 (n-6) families, such as arachidonic acid and linoleic acid, have long been known to have strong immunomodulatory effects [40] and may serve as a potent inhibitor for Th1 response. In cattle, dietary supplementation with fish oil (omega-3 PUFA) results in a 24% reduction in EPG in calves that are infected with O. ostertagi and C. oncophora[41]. The treatment also leads to an increased percentage of immature parasites, indicating that PUFA may enhance protective immunity against parasitic infections. Interestingly, arachidonic acid metabolism was among the pathways most significantly impacted in resistant animals (Table 5). Arachidonic acid (AA) is one of the important PUFA-associated membrane phospholipids. When liberated from the plasma membrane, AA can be oxidized, via a series of enzymatic steps, to a variety of eicosanoids, including prostaglandins, thromboxanes, prostacycline, and leukotrienes. Eicosanoids act as signaling molecules and stimulate a variety of responses in their target cells, such as innate immune responses [42], inflammation, and smooth-muscle contraction. Dietary n-3 PUFA has been used to attenuate tissue AA levels and subsequent eicosanoid formation. Recently, worm killing activities of AA have been demonstrated [43]. In mice, a single oral dose of AA led to a significant reduction of total worm burden of Schistosoma. AA-mediated parasite killing is suggested to be due to excessive activation of parasite neutral sphingomyelinase, leading to sphingomyelin hydrolysis into ceramide and phosphorylcholine [43]. In addition, products of the 5-lipoxygenase pathway, a part of AA metabolism, are important mediators of inflammation. 5-lipoxygenase plays a major role in controlling parasite burden of Trypanosoma cruzi in mice [44]. Detailed link between lipid metabolism and the development of protective immunity and host resistance to parasitic infections in cattle is worthy of further investigation.
The three most significant pathways impacted in resistant animals are associated with retinoid X receptor (RXR). These pathways included FXR/RXR activation, LXR/RXR activation, and LPS/IL-1 mediated inhibition of RXR functions. RXR acts as a master coordinator of numerous signaling pathways [45] via dimerizing with other nuclear receptors, such as liver X receptor (LXR), farnesoid X receptor (FXR), and vitamin D receptor (VDR). This partnership exerts transcriptional control and leads to distinct functions ranging from cell proliferation and differentiation to lipid metabolism. In addition, RXR can bind to a variety of natural and synthetic ligands, including omega-3 unsaturated fatty acids [45], which in turn stimulate transcriptional activation by RXR partners. While retinoic acid receptors (RAR) bind all-trans retinoic acid (RA) and its 9-cis isomer (9-cis RA), which convey most of the activity of RA, only 9-cis RA and docosahexaenoic acid (DHA) are suggested to be endogenous RXR ligands [46]. RA can inhibit cytokine expression including reduction of TNFα, iNOS, IL-6, and IL-1β at the mRNA level [47]. Recently, it has been observed that LPS-specific regulatory networks in which NF-κB plays a critical role in the mouse mucosa overlap with the LPS/IL-1β mediated inhibition of RXR functions [48]. The importance of RXR in cattle during C. oncophora infection has also recently been recognized [17]. Our future work will focus on the mechanistic link between RXR-related signaling pathways and the development of host resistance to GI nematode infection in cattle.
References
Coop RL, Sykes AR, Angus KW: The pathogenicity of daily intakes of Cooperia oncophora larvae in growing calves. Vet Parasitol. 1979, 5: 261-269. 10.1016/0304-4017(79)90015-3.
Sanchez J, Dohoo I: A bulk tank milk survey of Ostertagia ostertagi antibodies in dairy herds in Prince Edward Island and their relationship with herd management factors and milk yield. Can Vet J. 2002, 43: 454-459.
Barger IA, Gibbs HC: Milk production of cows infected experimentally with trichostrongylid parasites. Vet Parasitol. 1981, 9: 69-73. 10.1016/0304-4017(81)90008-X.
Gross SJ, Ryan WG, Ploeger HW: Anthelmintic treatment of dairy cows and its effect on milk production. Vet Rec. 1999, 144: 581-587. 10.1136/vr.144.21.581.
Feedlot 99: Part III. Health management and biosecurity in U.S. 1999, Feedlots, National Animal Health Monitoring System
Li RW, Gasbarre LC: Gene expression in the bovine gastrointestinal tract during nematode infection. Veterinary Parasitology. Edited by: LaMann G. 2010, New York: Nova Biomedical Press, 157-178.
Diez-Tascon C, Keane OM, Wilson T, Zadissa A, Hyndman DL, Baird DB, McEwan JC, Crawford AM: Microarray analysis of selection lines from outbred populations to identify genes involved with nematode parasite resistance in sheep. Physiol Genomics. 2005, 21: 59-69. 10.1152/physiolgenomics.00257.2004.
Knight JS, Baird DB, Hein WR, Pernthaner A: The gastrointestinal nematode Trichostrongylus colubriformis down-regulates immune gene expression in migratory cells in afferent lymph. BMC Immunol. 2010, 11: 51-10.1186/1471-2172-11-51.
Kadarmideen HN, Watson-Haigh NS, Andronicos NM: Systems biology of ovine intestinal parasite resistance: disease gene modules and biomarkers. Mol Biosyst. 2011, 7: 235-246. 10.1039/c0mb00190b.
Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML: A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008, 321: 956-960. 10.1126/science.1160342.
Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009, 25: 1105-1111. 10.1093/bioinformatics/btp120.
Picardi E, Horner DS, Chiara M, Schiavon R, Valle G, Pesole G: Large-scale detection and analysis of RNA editing in grape mtDNA by RNA deep-sequencing. Nucleic Acids Res. 2010, 38: 4755-4767. 10.1093/nar/gkq202.
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu AL, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJ, Hoodless PA, Birol I: De novo assembly and analysis of RNA-seq data. Nat Methods. 2010, 7: 909-912. 10.1038/nmeth.1517.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5: 621-628. 10.1038/nmeth.1226.
Gasbarre LC: Effects of gastrointestinal nematode infection on the ruminant immune system. Vet Parasitol. 1997, 72: 327-337. 10.1016/S0304-4017(97)00104-0.
Li RW, Sonstegard TS, Van Tassell CP, Gasbarre LC: Local inflammation as a possible mechanism of resistance to gastrointestinal nematodes in Angus heifers. Vet Parasitol. 2007, 145: 100-107. 10.1016/j.vetpar.2006.11.015.
Li RW, Gasbarre LC: A temporal shift in regulatory networks and pathways in the bovine small intestine during Cooperia oncophora infection. Int J Parasitol. 2009, 39: 813-824. 10.1016/j.ijpara.2008.11.007.
Childers CP, Reese JT, Sundaram JP, Vile DC, Dickens CM, Childs KL, Salih H, Bennett AK, Hagen DE, Adelson DL, Elsik CG: Bovine Genome Database: integrated tools for genome annotation and discovery. Nucleic Acids Res. 2011, 39: D830-D834. 10.1093/nar/gkq1235.
Wu TD, Nacu S: Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010, 26: 873-881. 10.1093/bioinformatics/btq057.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-10.1186/gb-2009-10-3-r25.
Robinson MD, Oshlack A: A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11: R25-10.1186/gb-2010-11-3-r25.
Wang L, Feng Z, Wang X, Wang X, Zhang X: DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010, 26: 136-138. 10.1093/bioinformatics/btp612.
Zeeberg BR, Qin H, Narasimhan S, Sunshine M, Cao H, Kane DW, Reimers M, Stephens RM, Bryant D, Burt SK, Elnekave E, Hari DM, Wynn TA, Cunningham-Rundles C, Stewart DM, Nelson D, Weinstein JN: High-Throughput GoMiner, an 'industrial-strength' integrative gene ontology tool for interpretation of multiple-microarray experiments, with application to studies of Common Variable Immune Deficiency (CVID). BMC Bioinformatics. 2005, 6: 168-10.1186/1471-2105-6-168.
Stear MJ, Boag B, Cattadori I, Murphy L: Genetic variation in resistance to mixed, predominantly Teladorsagia circumcincta nematode infections of sheep: from heritabilities to gene identification. Parasite Immunol. 2009, 31: 274-282. 10.1111/j.1365-3024.2009.01105.x.
Mandonnet N, Aumont G, Fleury J, Arquet R, Varo H, Gruner L, Bouix J, Khang JV: Assessment of genetic variability of resistance to gastrointestinal nematode parasites in Creole goats in the humid tropics. J Anim Sci. 2001, 79: 1706-1712.
Gasbarre LC, Leighton EA, Sonstegard TS: Role of the bovine immune system and genome in resistance to gastrointestinal nematodes. Vet Parasitol. 2001, 98: 51-64. 10.1016/S0304-4017(01)00423-X.
Gasbarre LC, Leighton EA, Davies CJ: Influence of host genetics upon antibody responses against gastrointestinal nematode infections in cattle. Vet Parasitol. 1993, 46: 81-91. 10.1016/0304-4017(93)90049-S.
Kanobana K, Vervelde L, van d, Eysker M, Ploeger HW: Characterization of host responder types after a single Cooperia oncophora infection: kinetics of the systemic immune response. Parasite Immunol. 2001, 23: 641-653. 10.1046/j.1365-3024.2001.00426.x.
Sonstegard TS, Gasbarre LC: Genomic tools to improve parasite resistance. Vet Parasitol. 2001, 101: 387-403. 10.1016/S0304-4017(01)00563-5.
Coppieters W, Mes TH, Druet T, Farnir F, Tamma N, Schrooten C, Cornelissen AW, Georges M, Ploeger HW: Mapping QTL influencing gastrointestinal nematode burden in Dutch Holstein-Friesian dairy cattle. BMC Genomics. 2009, 10: 96-10.1186/1471-2164-10-96.
Terefe G, Lacroux C, Andreoletti O, Grisez C, Prevot F, Bergeaud JP, Penicaud J, Rouillon V, Gruner L, Brunel JC, Francois D, Bouix J, Dorchies P, Jacquiet P: Immune response to Haemonchus contortus infection in susceptible (INRA 401) and resistant (Barbados Black Belly) breeds of lambs. Parasite Immunol. 2007, 29: 415-424. 10.1111/j.1365-3024.2007.00958.x.
McKay DM, Fairweather I: A role for the enteric nervous system in the response to helminth infections. Parasitol Today. 1997, 13: 63-69. 10.1016/S0169-4758(96)10079-X.
Li RW, Hou Y, Li C, Gasbarre LC: Localized complement activation in the development of protective immunity against Ostertagia ostertagi infections in cattle. Vet Parasitol. 2010, 174: 247-256. 10.1016/j.vetpar.2010.08.037.
French AT, Bethune JA, Knight PA, McNeilly TN, Wattegedera S, Rhind S, Miller HR, Pemberton AD: The expression of intelectin in sheep goblet cells and upregulation by interleukin-4. Vet Immunol Immunopathol. 2007, 120: 41-46. 10.1016/j.vetimm.2007.07.014.
French AT, Knight PA, Smith WD, Pate JA, Miller HR, Pemberton AD: Expression of three intelectins in sheep and response to a Th2 environment. Vet Res. 2009, 40: 53-
Rowe A, Gondro C, Emery D, Sangster N: Sequential microarray to identify timing of molecular responses to Haemonchus contortus infection in sheep. Vet Parasitol. 2009, 161: 76-87. 10.1016/j.vetpar.2008.12.023.
Pemberton AD, Knight PA, Gamble J, Colledge WH, Lee JK, Pierce M, Miller HR: Innate BALB/c enteric epithelial responses to Trichinella spiralis: inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J Immunol. 2004, 173: 1894-1901.
Hansen S, Holm D, Moeller V, Vitved L, Bendixen C, Reid KB, Skjoedt K, Holmskov U: CL-46, a novel collectin highly expressed in bovine thymus and liver. J Immunol. 2002, 169: 5726-5734.
Sato S, St-Pierre C, Bhaumik P, Nieminen J: Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunol Rev. 2009, 230: 172-187. 10.1111/j.1600-065X.2009.00790.x.
Hwang D: Fatty acids and immune responses--a new perspective in searching for clues to mechanism. Annu Rev Nutr. 2000, 20: 431-456. 10.1146/annurev.nutr.20.1.431.
Muturi KN, Scaife JR, Lomax MA, Jackson F, Huntley J, Coop RL: The effect of dietary polyunsaturated fatty acids (PUFA) on infection with the nematodes Ostertagia ostertagi and Cooperia oncophora in calves. Vet Parasitol. 2005, 129: 273-283. 10.1016/j.vetpar.2005.01.009.
Peters-Golden M, Canetti C, Mancuso P, Coffey MJ: Leukotrienes: underappreciated mediators of innate immune responses. J Immunol. 2005, 174: 589-594.
El Ridi R, Aboueldahab M, Tallima H, Salah M, Mahana N, Fawzi S, Mohamed SH, Fahmy OM: vitro and in vivo activities of arachidonic acid against Schistosoma mansoni and Schistosoma haematobium. Antimicrob Agents Chemother. 2010, 54: 3383-3389. 10.1128/AAC.00173-10.
Borges CL, Cecchini R, Tatakihara VL, Malvezi AD, Yamada-Ogatta SF, Rizzo LV, Pinge-Filho P: 5-Lipoxygenase plays a role in the control of parasite burden and contributes to oxidative damage of erythrocytes in murine Chagas' disease. Immunol Lett. 2009, 123: 38-45. 10.1016/j.imlet.2009.02.002.
Lefebvre P, Benomar Y, Staels B: Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab. 2010, 21: 676-683. 10.1016/j.tem.2010.06.009.
de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, Perlmann T: Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science. 2000, 290: 2140-2144. 10.1126/science.290.5499.2140.
Motomura K, Ohata M, Satre M, Tsukamoto H: Destabilization of TNF-alpha mRNA by retinoic acid in hepatic macrophages: implications for alcoholic liver disease. Am J Physiol Endocrinol Metab. 2001, 281: E420-E429.
Saban MR, O'Donnell MA, Hurst RE, Wu XR, Simpson C, Dozmorov I, Davis C, Saban R: Molecular networks discriminating mouse bladder responses to intravesical bacillus Calmette-Guerin (BCG), LPS, and TNF-alpha. BMC Immunol. 2008, 9: 4-10.1186/1471-2172-9-4.
Acknowledgements
The authors would like to thank Drs Louis Gasbarre, Tad Sonstegard and Steve Schroeder for their kind support and assistance. We also want to express our gratitude to Alicia Beavers, Ashley Sperling, Debbie Hebert and Joanne Wilson for their excellent technical assistance. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U. S. Department of Agriculture (USDA). The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RWL conceived the study, conducted the experiment, analyzed the data, and drafted the manuscript. MR and AVC assisted in the experiment and contributed to the interpretation of results. All authors read and approved the final manuscript.
Electronic supplementary material
13567_2011_118_MOESM1_ESM.XLSX
Additional file 1: Genes with significantly different read counts between resistant and susceptible cattle in response to parasitic infection. A total of 203 genes met 2 criteria: unadjusted P value < 0.05 and 2-fold difference in normalized read counts between resistant and susceptible animals. (XLSX 48 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Li, R.W., Rinaldi, M. & Capuco, A.V. Characterization of the abomasal transcriptome for mechanisms of resistance to gastrointestinal nematodes in cattle. Vet Res 42, 114 (2011). https://doi.org/10.1186/1297-9716-42-114
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1297-9716-42-114