
Abstract
Background
Cryptosporidium parvum is an enteric protozoan parasite with zoonotic importance and can cause cryptosporidiosis in humans as well as domestic and wild animals worldwide. The IId subtype family (SF) is one of the most prevalent subtypes of C. parvum. Some clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) protein systems have been developed to detect nucleic acid with high flexibility, sensitivity and specificity.
Methods
By integrating recombinase polymerase amplification and the Cas12a/crRNA trans-cleavage system (termed ReCTC), we established end-point diagnostics by observing fluorescence readouts with the naked eye under blue light and on-site diagnostics using a lateral flow strip (LFS) biosensor.
Results
Our ReCTC-based diagnoses can detect as little as a single copy of a cloned C. parvum 60-kDa glycoprotein (GP60) gene, 10 oocysts per gram (OPG), clinical fecal sample without tedious extraction of genomic DNA and have no cross-reactivity with other SFs of C. parvum or other common enteric parasitic protozoa.
Conclusions
This study provided a new strategy for direct identification of the IId SF of C. parvum free of highly trained operators and expensive special equipment.
Graphical Abstract

Similar content being viewed by others


Background
Cryptosporidium is an enteric protozoan parasite that can infect various vertebrate species, including humans, nonhuman primates and domestic and wild animals worldwide [1]. It has been demonstrated that cryptosporidiosis is the second greatest cause of diarrhea and mortality in children after rotavirus [2,3,4]. Currently, 44 valid Cryptosporidium species have been identified [5, 6], among which Cryptosporidium parvum and Cryptosporidium hominis are the predominant species that mainly infect humans and cause > 90% of the human infections [5, 7]. C. parvum is also one of the most common species detected in ruminants, especially in young-aged animals [8].
Based on the analysis of the DNA sequence encoding the 60-kDa glycoprotein (GP60), a subtyping tool has been established and most commonly used in studies of the transmission of C. parvum in humans and ruminants [7]. A total of 19 subtype families (SFs) (IIa-IIi and IIk-IIt) have been identified for C. parvum [5], among which the IId SF is found in both humans and ruminants and is responsible for zoonotic cryptosporidiosis reported in Europe, Middle East, Asia and Australia [7, 9]. Moreover, in China, all C. parvum isolates characterized thus far belong to IId subtypes [9].
Because of the common existence and high virulence of the C. parvum IId SF, a point-of-care diagnostic method, which can directly distinguish infections caused by the IId SF, is necessary. The diagnosis of Cryptosporidium spp. can be divided into the following categories: optical microscopy, antigens or genetic material detection, and the detection of antibodies against Cryptosporidium spp. [5]. Among these diagnoses, direct identification of IId SF can only be approached by DNA detection such as PCR amplification combined with nucleotide sequencing [10]. However, this method is time-consuming and complicated, requires highly trained operators and expensive special equipment and is not applicable for on-site clinical disease monitoring, especially in resource-limited areas.
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) protein systems have been reported to recognize and cleave specific nucleic acid sequences (namely cis-cleavage) [11, 12]. Some Cas proteins, including Cas12a, Cas12b, Cas13a and Cas14, have exhibited nonspecific trans-cleavage activity by cleaving non-target sequences when they are activated by recognizing a specific target sequence [13,14,15,16]. This collateral effect has been developed into Cas12a/Cas13a-based nucleic acid detection methods termed HOLMES (one-hour low-cost multipurpose highly efficient system), DETECTR (DNA endonuclease-targeted CRISPR trans reporter) and SHERLOCK (specific high-sensitivity enzymatic reporter unlocking) [14, 17, 18]. In DETECTR, CRISPR RNA (crRNA) is designed specifically to target double-strand DNA (dsDNA) located downstream of a short T-rich protospacer-adjacent motif (PAM); therefore, the target DNA works as an activator to trigger both cis- and trans-cleavage of Cas12a. The fluorophore quencher labeled reporter (FQ Reporter) in the system is then cut, and a fluorescence signal is released and measured [14].
This universal molecular diagnostic methodology has been applied in recent studies to detect African swine fever virus (ASFV) [19, 20] and beta-coronavirus severe acute respiratory syndrome (SARS)-CoV-2 [21], mycoplasma [22] and to identify Mycobacterium tuberculosis infection and nontuberculous mycobacteria infection [23]. In the present study, by designing a SF-specific crRNA probe, integrating recombinase polymerase amplification and the Cas12a/crRNA trans-cleavage system (termed ReCTC), we established an end-point diagnostic method by observing fluorescence readouts with the naked eye under blue light and an on-site diagnostic method using a lateral flow strip (LFS) biosensor to detect C. parvum IId SF nucleic acid rapidly and accurately.
Materials and methods
Reagents and oligonucleotides
All the primers, quenched fluorescent DNA reporter (FAM-TTATT-BHQ1) and lateral flow strip test reporter (FAM-TTATT-biotin) were ordered from Sangon Biotech (Shanghai, China). Partial sequences of C. parvum gp60 gene of six SFs (IIa–IIf) were synthesized and cloned into pUC57 vectors and transformed into E. coli DH5α, respectively (also from Sangon Biotech). All the nucleotide sequences are listed in Additional file 1: Table S1. Recombinant Francisella novicida Cas12a (FnCas12a) protein was purchased from Tolo Biotech (Shanghai, China). HiScribe™ T7 High Yield RNA Synthesis Kits were purchased from New England Biolabs (Ipswich, MA, USA). The NucAway™ Spin Columns was purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). The TwistAmp® Basic kit was purchased from TwistDx Ltd. (Hertfordshire, UK). The TIANprep mini plasmid kit was purchased from Tiangen Biotech (Shanghai, China). The lateral flow strip biosensor was purchased from Zoonbio Biotechnology (Nanjing, China). Recombinant DNase I (RNase free) and RNase inhibitor were purchased from TaKaRa Bio Inc. (Dalian, China). Other regents used in this study were purchased from Solarbio Science & Technology Co., Ltd. (Beijing, China).
Cryptosporidium parvum oocyst counting and DNA extraction
Subtype IIdA19G1 oocysts of C. parvum were used in this study. Oocysts were purified and counted according to a previous study [24] and stored in phosphate buffer solution (PBS) at 4 °C. N-lauroylsarcosine sodium salt (LSS) was used in the crude DNA extraction, and a homemade DNA banding column was used to purify DNA from the crude extracts (Additional file 1: Figure S1). One μicroliter 10% LSS solution was added to 100 μl PBS containing C. parvum oocysts. After intensive mixing, the oocyst suspension was incubated in boiling water for 5 min. Then, heating of the water was stopped and the incubation was retained with the afterheat of boiled water for another 10 min. The lysate was then added to the homemade DNA banding column using a 1-ml syringe and was pushed through the column under the syringe pressure; 100 μl 10 mM Tris solution was added to the column using a new syringe. After incubating for 5 min at room temperature, DNA was eluted in Tris solution under the syringe pressure.
Design and preparation of crRNA
The gp60 gene has been used in the identification of SFs of C. parvum; thus, we chose it as the target sequence for the design of crRNA. To determine the highly conserved regions with SF specificity, sequences of different SFs were downloaded from GenBank and aligned using ClustalX 2.1 (http://www.clustal.org/clustal2/), and a 24-nucleotide (nt) sequence closely following a T nucleotide-rich protospacer adjacent motif (PAM) was chosen as the target sequence and synthesized containing T7 promotor (Additional file 1: Table S1). The two synthesized reverse complementary crDNA oligonucleotides were annealed to form dsDNA templates, and the crRNA was transcribed using a HiScribe™ T7 High Yield RNA Synthesis Kit followed by DNase I digestion and NucAway™ Spin Column purification. The concentration of purified crRNA was measured using NanoDrop One (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Recombinase polymerase amplification (RPA) assay
Isothermal recombinase polymerase amplification (RPA) was used to promote the concentration of target dsDNA. Ten pairs of RPA primers were designed (Additional file 1: Table S1), and TwistAmp® Basic kit was used to perform the RPA assay according to the instruction manual. Briefly, the 50-μl total reaction volume was made up of 29.5 μl rehydration buffer, 500 nM of each primer, 5 μl extracted DNA, 2.5 μl 280 nM magnesium acetate (MgOAc) and sterile nuclease-free water up to 50 μl. The reaction was performed at 37 °C for 30 min. The MgOAc was added onto the inner surface of the tube lid while all the other reagents were added to the bottom of the tube that was then carefully capped. A short spin to mix the MgOAc into the solution was applied, which triggered the RPA reactions.
FnCas12a/crRNA trans-cleavage assay
FnCas12a trans-cleavage assays were performed mainly according to previous studies [14, 25] with some modification. In brief, the 20-μl FnCas12a reaction system included 50 μM FnCas12a, 1 μM purified crRNA, 2 μl target DNA (unpurified RPA products), 1.25 μM collateral ssDNA (FAM-TTATT-BHQ1 reporter for the fluorescence assay or FAM-TTATT-biotin for the lateral flow strip assay), 20 U RNase inhibitor and 2 μl 10 × FnCas12a nuclease reaction buffer. The reaction system was incubated at 37 °C for 1 h, and the fluorescence was detected and imaged with a Tanon-5200 Multi Fluorescence Imager (Tanon Science & Technology Co., Ltd., Tianjin, China). A parallel test was also performed, and the real-time fluorescence value was recorded using a qTOWER3G qPCR system (Analytik Jena, Germany) every 5 min.
Construction of the LFS assay
To enable the on-site diagnosis of C. parvum IId SF, a lateral flow readout based on the cleavage of the designed FAM-TTATT-biotin ssDNA reporter was used, which allowed reading the results without sophisticated instruments and skilled technicians. The FAM-TTATT-biotin ssDNA reporter can specifically bind to anti-FITC antibody conjugated with Au nanoparticles and form a complex. When the ssDNA in the reporter is not degraded by FnCas12a, this complex can be captured by the biotin ligand fixed at the control line. On the contrary, when the ssDNA is degraded, biotin is released from the complex, and the complex without biotin cannot be captured by the biotin ligand fixed at the control line but by the IgG antibody at the test line.
In the LFS assay, 20 μl of the ReCTC reaction system was incubated at 37 °C for 1 h followed by mixing with 250 μl of ddH2O. Then, 80 μl of the mixture was loaded onto the LFS pad and incubated at room temperature for 3 min. The strips were then photographed using a smartphone camera.
PCR amplification of Cryptosporidium parvum gp60 gene
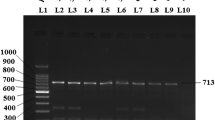
To assess our ReCTC-based detection, a nested PCR amplification based on the SSU rRNA loci of Cryptosporidium was used, mainly according to the previous study [26]. Primers used are listed in the Additional file 1: Table S1, and PCR was performed in 25-μl reaction mixtures consisting of 2 μl of DNA preparation (or first PCR product), 1 × KOD-Plus buffer, 200 μM dNTPs, 1 mM MgSO4, 300 nM of each primer and 1.5 units of KOD-Plus DNA polymerase (Toyobo, Osaka, Japan). The amplification program consisted of an initial denaturation step at 94 °C for 5 min, followed by 35 cycles of 95 °C for 45 s, 55 °C for 45 s and 72 °C for 1 min, with a final extension of 72 °C for 10 min. Then, the PCR products were analyzed electrophoretically on 1% agarose gel stained with DNAGREEN (Tiandz, Beijing, China) and visualized under UV light. To determine the species and subtype of the Cryptosporidium detected, positive secondary PCR products were sequenced bidirectionally by SinoGenoMax Biotechnology Co., Ltd. (Beijing, China), and the generated sequences were aligned in Clustal X software, version 2.1 (http://www.clust al.org/), with reference sequences downloaded from GenBank.
Results
Design and preparation of crRNA
By multiple alignment of the gp60 gene, we identified a fragment that was conserved in IId SF but not in other SFs. A 24-nt sequence of this fragment was chosen as the target sequence, and the crRNA sequence was determined (Additional file 1: Table S1; Figure S2). The synthesized single-strand crDNA oligonucleotides were annealed and transcribed to crRNA. Following DNase I digestion and NucAway™ Spin Column purification, the concentration was measured using NanoDrop One. To ensure the stability of the in vitro transcription assay, three sets of replications were carried out, and concentrations of the final crRNA ranged from 577.1 to 608.3 ng/μl with a mean concentration of 593.2 ng/μl (Fig. 1 and Additional file 1: Figure S3).

Absorbance curves of purified crRNA. The crRNA was transcribed from crDNA annealed from two reverse complementary single-strand oligonucleotides. The transcribed crRNA dealt with DNase I and was purified using the NucAway™ Spin Column
Optimization of the RPA assay
A number of conserved RPA primers were designed based on the gp60 gene of C. parvum IId SF (GenBank accession no. FJ839877). A set of primers named F01333 and R32535 (Additional file 1: Table S1) showed the highest amplification efficiency by using the TwistAmp Basic Kit (TwistDx, UK) (Additional file 1: Figure S4) and was chosen as the best primer pair. According to the manufacturer's instructions, the optimal reaction temperature was 39 °C. To enable our whole ReCTC assay workflow worked well under 37 °C, we also compared the RPA efficiency at 37 and 39 °C, and the result showed that the primers F01333 and R32535 could work well at both temperatures (Additional file 1: Figure S5).
Feasibility verification of ReCTC-based detection
Based on the ReCTC system, we intended to establish the C. parvum IId SF detection methods using both the fluorescence and LFS as read-out reporters. Together with the FnCas12a, the designed crRNA and the target dsDNA sequence with a TTN PAM sequence form a triplex. The recognition of the target dsDNA by the designed crRNA can induce the activation of the FnCas12a, which subsequently cleaves the FAM-TTATT-BHQ1 to emit 520-nm fluorescence under 488-nm light or cleaves the FAM-TTATT-biotin reporter to show visible test lines on the LFS (Fig. 2).

Schematic of the RPA and CRISPR-Cas12a-based detection assay. a Diagram of Cryptosporidium parvum chromosome 6 showing primers, target sequence and crRNA. RPA primers are indicated by black rectangles; the PAM and target sequences are represented by red and blue rectangles, respectively. b Schematic of ReCTC-based diagnosis workflow. The RPA amplicon is used directly as the input of the ReCTC-based detection, and a ternary complex forms if the target DNA exists. F fluorophore, Q quencher, B biotin, F FAM
The fluorescence signal was strong enough to be observed by the naked eye, and distinct differences between positive and negative samples were observed using the CRISPR/Cas12a-based fluorescence detection assay (Fig. 3a, b). To monitor the real-time progress of the reactions and to determine the minimum reaction time in which significant results can be observed, the fluorescence intensity was recorded every 5 min, and the real-time fluorescence curve is shown in Fig. 3c, indicating that 60 min is enough for the observation of the significant difference in fluorescence intensity between positive and negative tests.

Feasibility verification of the ReCTC-based detection. a ReCTC-based fluorescence reaction products showed no signal under visible light. b Obvious fluorescence signal can be observed under UV light by the naked eye. P1 and P2: positive results, N1 and N2: negative results. c Real-time fluorescence intensity curves of the ReCTC-based detection involving FAM-TTATT-BHQ1 reporter
In the LFS assay, to avoid false-positive results, various concentrations (5–200 nM) of the FAM-TTATT-biotin ssDNA reporter were tested on LFS pads without the addition of any target DNA samples. False-positive results were observed when 15 nM or lower concentrations of the FAM-TTATT-biotin ssDNA reporter were used, and the false-positive effects were eliminated when using concentrations of 20-nM FAM-TTATT-biotin ssDNA reporter or higher (Fig. 4). At 15 nM or lower concentrations, there was not enough FAM-TTATT-biotin ssDNA reporter to combine all the Au nanoparticles labeled by the anti-FITC antibody, so a portion of the nanoparticles that did not bind to the FAM-TTATT-biotin ssDNA reporter kept moving forward until they reached the test band to cause the false-positive result. Therefore, 20 nM was chosen as the optimal concentration of reporter for the ReCTC-based LFS detection.

Optimization of the reporter concentration for the ReCTC-based LFS detection. Various concentrations (200, 100, 50, 20, 15, 10, 5 nM) of FAM-TTATT-biotin ssDNA reporter were tested to avoid false-positive and -negative results. The concentrations used were labeled on the LFS pads, and false-positive results wwew eliminated with 20 nM FAM-TTATT-biotin ssDNA reporter or higher concentrations
Sensitivity of the ReCTC-based detection
The limit of detection (LOD) of the ReCTC was evaluated using a cloned C. parvum IId family gp60 gene with a conserved PAM (TTC) region cloned in pUC57 plasmid (Additional file 1: Table S2) and verified with PCR amplification. The cloned plasmid DNA was serially diluted to different concentrations and subsequently applied to the ReCTC. Results of fluorescence detection showed that samples with concentrations of 1.0 × 10–18 M or higher target plasmid DNA showed an obvious fluorescence signal compared to samples with lower concentrations and negative control (Fig. 5a). Similar results were also observed in the ReCTC-based LFS detection. The lowest concentration of samples that showed a test line was 1.0 × 10–18 M, indicating the LOD of the ReCTC-based LFS detection was also 1.0 × 10–18 M target plasmid DNA (Fig. 5b).

Sensitivity of the ReCTC-based detection. Sensitivity test of ReCTC-based fluorescence (a) and LFS (b) assay using cloned recombinant plasmid DNA. The LOD of both the fluorescence and LFS assay was determined as 1.0 × 10–18 M cloned recombinant plasmid DNA. A1–A8: The concentrations of cloned recombinant plasmid DNA were 1.0 × 10–12, 1.0 × 10–15, 1.0 × 10–18, 1.0 × 10–19, 1.0 × 10–20, 1.0 × 10–21, 1.0 × 10–22, 1.0 × 10–23 M, respectively. Sensitivity test of ReCTC-based fluorescence (c) and LFS (d) assay using crude DNA extracted from purified oocysts. The LOD of both the fluorescence and LFS assay was determined as one and ten oocysts per milliliter, respectively. C1–C8: The numbers of oocysts per milliliter were equivalent to 1 × 105, 1 × 104, 1 × 103, 1 × 102, 1 × 101, 1, 0.1 and 0, respectively. The concentrations of cloned recombinant plasmid DNA and the numbers of oocysts per milliliter used in the sensitivity test of LFS assay (b, d) were indicated on the LFS pads
The sensitivity of the ReCTC-based fluorescence detection and LFS detection was also tested using crude DNA extracted from purified oocysts. The concentration of purified oocysts was adjusted to 1 × 106 oocysts per milliliter before the DNA extraction. The concentration of extracted DNA was 9.3 ng/μl and serially diluted to concentrations of 9.3 × 10–7 to 9.3 × 10–1 ng/μl, which were equivalent to 0.1 to 1 × 105 oocysts per milliliter, respectively. In the ReCTC-based fluorescence detection, samples with crude genomic DNA of 9.3 × 10–6 or higher showed observable fluorescence signals, representing an LOD of one oocyst per milliliter (Fig. 5c and Additional file 1: Figure S6). In the ReCTC-based LFS detection assay, the sample with 9.3 × 10–5 ng/μl DNA showed a faint test line and samples with higher input target DNA showed strong test lines, indicating that ten oocysts per milliliter was detected with the ReCTC-based LFS detection (Fig. 5d).
Specificity of the ReCTC-based detection
The specificity of the ReCTC-based detection was verified using the recombinant pUC57 plasmid DNA containing the gp60 gene of six different C. parvum SFs (IIa, IIb, IIc, IId, IIe and IIf) (Additional file 1: Table S2). Genomic DNA extracted from several prevalent Cryptosporidium species (C. andersoni, C. hominis, C. meleagridis, C. muris, C. bovis and C. ryanae) and other intestinal protozoa (Enterocytozoon bieneusi, Giardia duodenalis, Blastocystis hominis and Cyclospora cayetanensis) were also included in the specificity test. As shown in Fig. 6a, only samples of C. parvum IId SF exhibited strong fluorescence signal, which could be observed by the naked eye and verified by the qTOWER3G real-time PCR system (Additional file 1: Figure S7). The LFS detection further confirmed the specificity of the ReCTC, and a clear test line was observed only on the LFS where C. parvum IId SF recombinant pUC57 plasmids DNA was added (Fig. 6b).

Specificity of the ReCTC-based detection. Recombinant pUC57 plasmid DNA containing gp60 gene of IIa, IIb, IIc, IId, IIe and IIf SFs of C. parvum (1–6) and genomic DNA of C. andersoni, C. hominis, C. meleagridis, C. muris, C. bovis, C. ryanae, Enterocytozoon bieneusi, Giardia duodenalis, Blastocystis hominis and Cyclospora cayetan (7–16) were included. a Specificity test of the ReCTC-based fluorescence detection assay. Only the sample of C. parvum IId SF exhibited a strong fluorescence signal. b Specificity test of the ReCTC-based LFS detection assay. A clear test line was observed only on the LFS where C. parvum IId SF recombinant pUC57 plasmids DNA was added
Performance of the ReCTC-based detection of C. parvum IId SF on clinical samples
To assess the clinical utility of the ReCTC-based detection, fresh fecal specimens were collected from 30 preweaned calves. The specimens were filtered through a 7.62-cm-diameter sieve with a pore size of 45 μm and applied to the crude DNA extraction. The extracted crude DNA samples were initially screened using nested PCR based on the gp60 gene of C. parvum, and results of sequencing and alignment revealed that the positive rate of C. parvum IId SF was 23.3% (7/30) (Fig. 7a). All the crude DNA samples were then tested by ReCTC-based detection. Consistent with the PCR results, distinct fluorescence signals were observed in all the PCR-positive samples by the naked eye and no such signal in all the PCR-negative samples, showing an accordance rate of 100% with PCR-based nucleotide sequencing (Fig. 7b). The ReCTC-based LFS detection also was in 100% accordance with the conventional PCR sequencing method (Fig. 7c).

Validation of ReCTC-based detection of C. parvum IId SF in clinical cattle samples. Clinical fecal samples from 30 dairy cattle were tested by a conventional nested PCR sequencing method, b our ReCTC-based fluorescence and c LFS detection. Both the ReCTC-based fluorescence and LFS detection agreed 100% with the conventional nested PCR sequencing method
To validate the ReCTC diagnostic accuracy in human samples, eight clinical DNA samples from diarrhea patients that had been detected as positive for C. parvum IId SF were tested using ReCTC-based detection. Fluorescence signals were observed in all eight samples by the naked eye in the ReCTC-based fluorescence assay (Fig. 8a). The positive results were also confirmed by the fluorescence values recorded by the qTOWER3G qPCR system (Additional file 1: Figure S8). In the ReCTC-based LFS assay, all eight clinical DNA samples showed the test bands on the LFS pads (Fig. 8b).

Validation of ReCTC-based detection of C. parvum IId SF in positive clinical human samples. Clinical human fecal DNA samples collected from inpatients that had been identified as positive for C. parvum IIdA19G1 were subjected to a ReCTC-based fluorescence and b LFS detection. N negative control
Discussion
In this study, we combined isothermal recombinase polymerase amplification with the Cas12a/crRNA trans-cleavage system to establish the ReCTC method and used fluorescence or chromatography readout as signal reporting. We found that ReCTC was able to detect C. parvum IId SF with high sensitivity and specificity. The use of CRISPR/Cas12a to detect infectious antigens has been reported in many other studies on detecting ASFV [20], human papillomavirus (HPV) [14] and the recently emerged coronavirus, SARS-CoV-2 [21]. Here, we first applied this method in detection of the zoonotic parasitic protozoan C. parvum, and the ReCTC-based fluorescence or LFS assay exhibited advantages in point-of-care use without the need for technical expertise, ancillary equipment or power (LFS assay).
In previous Cas12a off-target studies, a mismatch tolerance pattern was observed [27, 28], and PAM mutations or mismatches in the PAM-adjacent region inhibited trans-cleavage when the activator was a dsDNA [14]. This stringent principle gives the Cas12a trans cleavage-based diagnosis good specificity, which was confirmed in our specificity test with several other common intestinal pathogens and other subtypes of C. parvum.
The currently reported CRISPR/Cas12a biosensing systems generally consist of three key parts: signal amplification, signal transducing and signal reporting [29]. In our ReCTC-based diagnosis, the RPA was chosen as the signal amplification method for its mild temperature requirements. Our results indicated that the RPA assay worked at 37 °C as well as 39 °C, which made the ReCTC more convenient as the downstream Cas12a trans-cleavage system was also conducted at 37 °C. A water/metal bath kettle, constant temperature incubator or even body heating can carry out the ReCTC reaction.
Two elements were used in the signal reporting part in this study: a fluorescence readout and LFS biosensor, corresponding to different reporters, FAM-TTATT-BHQ1 and FAM-TTATT-biotin, respectively. When the FAM-TTATT-BHQ1 reporter was used, bright green fluorescence signals could be observed by the naked eye under blue light with a Tanon-5200 Multi Fluorescence Imager. In resource-limited areas, a much cheaper device, Blue Light Gel Imager (Sangon Biotech. Inc., Shanghai, China; 440–485 nm), is recommended [22]. When FAM-TTATT-biotin was used in the signal reporting part, the readout could be observed using a LFS biosensor, which did not rely on technical expertise, ancillary equipment or power. This trait makes ReCTC-based LFS detection a real on-site diagnostic tool for C. parvum IId SF in field conditions.
As a biosensor, the CRISPR/Cas12a/crRNA system acts as a signal converter, converting the existence of target dsDNA to the presence of fluorescence or colorimetric signals by cutting the collateral ssDNA reporter [29]. Once the high endonuclease activity of FnCas12a was activated by the amplified target dsDNA, it could cleave a large number of collateral ssDNA reporters [25]. Together with RPA, the signal amplification part, the efficient cleavage activity of FnCas12a makes ReCTC-based detection highly sensitive.
In conclusion, by integrating recombinase polymerase amplification and Cas12a/crRNA trans-cleavage (termed ReCTC), we established an end point diagnostic method by observing fluorescence readouts with the naked eye under blue light and an on-site diagnostic method using a lateral flow strip (LFS) biosensor. Our ReCTC-based diagnoses could detect the IId subtype family of C. parvum from clinical fecal samples independent of professional technicians, expensive instruments or cumbersome operations. The newly established ReCTC-based detection displayed one and ten copy sensitivity in pure and complex samples, respectively, and the specificity was also confirmed to be robust. The ReCTC-based detection also agreed 100% with the conventional PCR sequencing method, which is the current gold standard commonly used in the diagnosis of C. parvum IId SF. Further optimization of the ReCTC assay as a one-pot reaction should be done in our future research to detect C. parvum IId SF from the clinical samples in the field more rapidly and simply.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, Longxian Zhang: zhanglx8999@henau.edu.cn.
Abbreviations
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas:
-
CRISPR-associated
- SF:
-
Subtype family
- ReCTC:
-
Recombinase polymerase amplification and Cas12a/crRNA trans-cleavage
- LFS:
-
Lateral flow strip
- GP60:
-
60 KDa glycoprotein
- OPG:
-
Oocysts per gram
- PCR:
-
Polymerase chain reaction
- HOLMES:
-
One-hour low-cost multipurpose highly efficient system
- DETECTR:
-
DNA endonuclease-targeted CRISPR trans reporter
- SHERLOCK:
-
Specific high-sensitivity enzymatic reporter unlocking
- crRNA:
-
CRISPR RNA
- dsDNA:
-
Double-strand DNA
- PAM:
-
Protospacer-adjacent motif
- FQ Reporter:
-
Fluorophore quencher labeled reporter
- ASFV:
-
African swine fever virus
- SARS-CoV-2:
-
Beta-coronavirus severe acute respiratory syndrome
- PBS:
-
Phosphate buffer solution
- LSS:
-
N-Lauroylsarcosine sodium salt
- RPA:
-
Recombinase polymerase amplification
- LOD:
-
Limit of detection
- HPV:
-
Human papillomavirus
References
Xiao L, Fayer R, Ryan U, Upton SJ. Cryptosporidium taxonomy: recent advances and implications for public health. Clin Microbiol Rev. 2004;17:72–97.
Ryan U, Hijjawi N. New developments in Cryptosporidium research. Int J Parasitol. 2015;45:367–73.
Striepen B. Parasitic infections: time to tackle cryptosporidiosis. Nature. 2013;503:189–91.
Wang RJ, Li JQ, Chen YC, Zhang LX, Xiao LH. Widespread occurrence of Cryptosporidium infections in patients with HIV/AIDS: Epidemiology, clinical feature, diagnosis, and therapy. Acta Trop. 2018;187:257–63.
Cunha FS, Peralta JM, Peralta RHS. New insights into the detection and molecular characterization of Cryptosporidium with emphasis in Brazilian studies: a review. Rev Inst Med Trop Sao Paulo. 2019;61:e28.
Holubová N, Tůmová L, Sak B, Hejzlarová A, Konečný R, McEvoy J, et al. Description of Cryptosporidium ornithophilus n. sp. (Apicomplexa: Cryptosporidiidae) in farmed ostriches. Parasit Vectors. 2020;13:340.
Xiao L. Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol. 2010;124:80–9.
Wang R, Zhao G, Gong Y, Zhang L. Advances and perspectives on the epidemiology of bovine Cryptosporidium in China in the past 30 years. Front Microbiol. 2017;8:1823.
Wang R, Zhang L, Axen C, Bjorkman C, Jian F, Amer S, et al. Cryptosporidium parvum IId family: clonal population and dispersal from Western Asia to other geographical regions. Sci Rep. 2014;4:4208.
Khan A, Shaik JS, Grigg ME. Genomics and molecular epidemiology of Cryptosporidium species. Acta Trop. 2018;184:1–14.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–4.
Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM, et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–9.
Harrington LB, Burstein D, Chen JS, Paez-Espino D, Ma E, Witte IP, et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science. 2018;362:839–42.
Yang H, Gao P, Rajashankar KR, Patel DJ. PAM-dependent target DNA recognition and cleavage by C2c1 CRISPR-Cas endonuclease. Cell. 2016;167:1814–28.
Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–42.
Li S, Cheng Q, Wang J, Li X, Zhang Z, Gao S, et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018;4:20.
He Q, Yu D, Bao M, Korensky G, Chen J, Shin M, et al. High-throughput and all-solution phase African Swine Fever Virus (ASFV) detection using CRISPR-Cas12a and fluorescence based point-of-care system. Biosens Bioelectron. 2020;154:112068.
Lu S, Li F, Chen Q, Wu J, Duan J, Lei X, et al. Rapid detection of African swine fever virus using Cas12a-based portable paper diagnostics. Cell Discov. 2020;6:18.
Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat Biotechnol. 2020;38:870–4.
Wang B, Wang R, Wang D, Wu J, Wang J, Liu H, et al. Cas12aVDet: A CRISPR/Cas12a-based platform for rapid and visual nucleic acid detection. Anal Chem. 2019;91:12156–61.
Xiao G, Zhang S, Liang Z, Li G, Fang M, Liu Y, et al. Identification of Mycobacterium abscessus species and subspecies using the Cas12a/sgRNA-based nucleic acid detection platform. Eur J Clin Microbiol Infect Dis. 2020;39:551–8.
Cui Z, Song D, Qi M, Zhang S, Wang R, Jian F, et al. Revisiting the infectivity and pathogenicity of Cryptosporidium avium provides new information on parasitic sites within the host. Parasit Vectors. 2018;11:514.
Zhang M, Liu C, Shi Y, Wu J, Wu J, Chen H. Selective endpoint visualized detection of Vibrio parahaemolyticus with CRISPR/Cas12a assisted PCR using thermal cycler for on-site application. Talanta. 2020;214:120818.
Peng MM, Matos O, Gatei W, Das P, Stantic-Pavlinic M, Bern C, et al. A comparison of Cryptosporidium subgenotypes from several geographic regions. J Eukaryot Microbiol. 2001;48:28S-31S.
Kim D, Kim J, Hur JK, Been KW, Yoon SH, Kim JS. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34:863–8.
Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–74.
Li Y, Li S, Wang J, Liu G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019;37:730–43.
Acknowledgements
We thank Yuhang Zhang and Yafei Chang for technical assistance.
Funding
This work was supported, in part, by the National Key Research and Development Program of China (2017YFD0501305, 2019YFC1605700) and the National Natural Science Foundation of China (U1904203). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
Design of the work: FY, XL and LZ. Acquisition and analysis of the data: FY, KZ, YW, DL and JH. Supervision of the project: SZ and LZ. Grant funding: LZ. Writing—original draft: FY. Writing—review and editing: ZC, SZ, XL and LZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All research procedures used in this work were approved by the Institutional Review Board of Henan Agricultural University (approval no. IRB-HENAU-20190820-02). The collection of cattle fecal samples conformed to the Chinese Laboratory Animal Administration Act of 1988. The use of positive human samples in this work complied with Declaration of Helsinki 1975, as revised in 2013.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Figure S1. Absorbance curves of purified crRNA. The crRNA was transcribed from crDNA annealed from two reverse complementary single-strand oligonucleotides. The transcribed crRNA was treated with DNase I and was purified using the NucAway™ Spin Column. Figure S2. Schematic of the RPA and CRISPR-Cas12a-based detection assay. A. Diagram of Cryptosporidium parvum chromosome 6 showing primers, target sequence and crRNA. RPA primers are indicated by black rectangles; the PAM and target sequences are represented by red and blue rectangles, respectively. B. Schematic of ReCTC-based diagnostic workflow. The RPA amplicon is used directly as the input of the ReCTC-based detection, and a ternary complex forms if the target DNA exists. F, fluorophore; Q, quencher; B, biotin; F, FAM. Figure S3. Feasibility verification of the ReCTC-based detection. A. ReCTC-based fluorescence reaction products showed no signal under visible light. B. Obvious fluorescence signal can be observed under UV light by the naked eye. P1 and P2: positive results; N1 and N2: negative results. C. The real-time fluorescence intensity curves of the ReCTC-based detection involving FAM-TTATT-BHQ1 reporter. Figure S4. Optimization of reporter concentration for the ReCTC-based LFS detection. Various concentrations (200, 100, 50, 20, 15, 10, 5 nM) of FAM-TTATT-biotin ssDNA reporter were tested to avoid false-positive and -negative results. The concentrations used were labeled on the LFS pads, and false-positive results were eliminated with 20 nM or higer FAM-TTATT-biotin ssDNA reporter concentrations. Figure S5. Sensitivity of the ReCTC-based detection. A, B. Sensitivity test of ReCTC-based fluorescence (A) and LFS (B) assay using cloned recombinant plasmid DNA. The LOD of both the fluorescence and LFS assay was determined as 1.0 × 10-18 M cloned recombinant plasmid DNA. A1–A8: The concentrations of cloned recombinant plasmid DNA were 1.0 × 10-12, 1.0 × 10-15, 1.0 × 10-18, 1.0 × 10-19, 1.0 × 10-20, 1.0 × 10-21, 1.0 × 10-22, 1.0 × 10-23 M, respectively. C, D. Sensitivity test of ReCTC-based fluorescence (C) and LFS (D) assay using crude DNA extracted from purified oocysts. The LOD of both the fluorescence and LFS assay was determined as one and ten oocysts per milliliter, respectively. C1–C8: The numbers of oocysts per milliliter were equivalent to 1 × 105, 1 × 104, 1 × 103, 1 × 102, 1 × 101, 1, 0.1 and 0, respectively. The concentrations of cloned recombinant plasmid DNA and the numbers of oocysts per milliliter used in the sensitivity test of the LFS assay (B, D) were indicated on the LFS pads. Figure S6. Specificity of the ReCTC-based detection. Recombinant pUC57 plasmid DNA containing the gp60 gene of IIa, IIb, IIc, IId, IIe and IIf SFs of C. parvum (1–6) and genomic DNA of C. andersoni, C. hominis, C. meleagridis, C. muris, C. bovis, C. ryanae, Enterocytozoon bieneusi, Giardia duodenalis, Blastocystis hominis and Cyclospora cayetan (7–16) were included. A. Specificity test of the ReCTC-based fluorescence detection assay. Only the sample of C. parvum IId SF exhibited a strong fluorescence signal. B. Specificity test of the ReCTC-based LFS detection assay. A clear test line was observed only on the LFS where C. parvum IId SF recombinant pUC57 plasmid DNA was added. Figure S7. ReCTC-based detection of C. parvum IId SF on clinical cattle samples. Clinical fecal samples from 30 dairy cattle were tested by a conventional nested PCR sequencing method (A) and our ReCTC-based fluorescence (B) and LFS (C) detection. Both the ReCTC-based fluorescence and LFS detection agreed 100% with the conventional nested PCR sequencing method. Figure S8. ReCTC-based detection of C. parvum IId SF on positive clinical human samples. Clinical human fecal DNA samples collected from inpatients that had been identified as positive for C. parvum IIdA19G1 were subjected to ReCTC-based fluorescence (A) and LFS (B) detection. N: negative control. Table S1. Nucleotide sequences used in this study. Table S2. Partial sequences of C. parvum gp60 gene of six subtype families (IIa–IIf) cloned into the pUC57 vectors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, F., Zhang, K., Wang, Y. et al. CRISPR/Cas12a-based on-site diagnostics of Cryptosporidium parvum IId-subtype-family from human and cattle fecal samples. Parasites Vectors 14, 208 (2021). https://doi.org/10.1186/s13071-021-04709-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04709-2