
Abstract
Background
According to scientific recommendations, paratransgenesis is one of the solutions for improving the effectiveness of the Global Malaria Eradication Programme. In paratransgenesis, symbiont microorganisms are used for distorting or blocking the parasite life-cycle, affecting the fitness and longevity of vectors or reducing the vectorial competence. It has been revealed recently that bacteria could be used as potent tools for double stranded RNA production and delivery to insects. Moreover, findings showed that RNase III mutant bacteria are more competent for this aim. Asaia spp. have been introduced as potent paratransgenesis candidates for combating malaria and, based on their specific features for this goal, could be considered as effective dsRNA production and delivery tools to Anopheles spp. Therefore, we decided to characterize the rnc gene and its related protein to provide the basic required information for creating an RNase III mutant Asaia bacterium.
Methods
Asaia bacteria were isolated from field-collected Anopheles stephensi mosquitoes. The rnc gene and its surrounding sequences were characterized by rapid amplification of genomic ends. RNase III recombinant protein was expressed in E. coli BL21 and biological activity of the purified recombinant protein was assayed. Furthermore, Asaia RNaseIII amino acid sequence was analyzed by in silico approaches such as homology modeling and docking to determine its structural properties.
Results
In this study, the structure of rnc gene and its related operon from Asaia sp. was determined. In addition, by performing superimposition and docking with specific substrate, the structural features of Asaia RNaseIII protein such as critical residues which are involved and essential for proper folding of active site, binding of magnesium ions and double stranded RNA molecule to protein and cleaving of dsRNA molecules, were determined.
Conclusions
In this study, the basic and essential data for creating an RNase III mutant Asaia sp. strain, which is the first step of developing an efficient RNAi-based paratransgenesis tool, were acquired. Asaia sp. have been found in different medically-important vectors and these data are potentially very helpful for researchers studying paratransgenesis and vector-borne diseases and are interested in applying the RNAi technology in the field.
Similar content being viewed by others

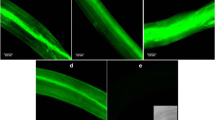

Background
Vector-borne diseases (VBD) have enormous impacts on human life [1,2,3]. Malaria is one of the most important protozoan infectious diseases, caused in humans by five different members of the genus Plasmodium. Based on the World Health Organization (WHO) annual report, there were approximately 219 million clinical malaria cases in 2017 which resulted in almost 0.5 million deaths [4]. The main at-risk groups for malaria disease are children under the age of five years, but it also affects other risk groups such as pregnant women and non-immune adults [5].
After the failure of Global Malaria Eradication program [6], the WHO has been updating the malaria policy instruments beginning with a consultative review process involving the scientific recommendations and findings of scientists in 2007 [7]. Drug, vaccine, vector control, modeling, M&E and surveillance, integration strategies, health systems and operational research and diagnostics and young investigators/basic research consultative groups were considered. Three main subjects were recommended by the vector control consultative group: designing new and more efficient insecticides, developing of transgenic insects and applying paratransgenesis [8].
Paratransgenesis is a kind of “Trojan horse” for controlling and blocking the transmission of vector-borne diseases [9]. This method is described as “the using of different symbiotic microorganisms for delivering molecules with anti-parasitic or anti-vector effects to the wild vector population” [2, 10,11,12,13,14,15].
Recently, different research studies have been performed on symbiont microorganisms such as viruses, fungi, yeasts and bacteria as paratransgenesis agents [14, 16,17,18,19,20,21,22,23,24,25]. Given the fact that bacteria are more available and simpler than other microorganisms, they have been at the focus of research for developing paratransgenesis tools against VBD. Therefore, numerous studies have been performed to reach this goal, and different bacteria have been introduced as suitable candidates for paratransgenesis, especially for combating malaria [2, 9, 10, 12, 13, 16, 20, 24, 26,27,28,29,30,31,32,33,34,35,36,37,38,39]. Among the introduced candidates, bacteria of the genus Asaia have special characteristics that differentiate them from the other introduced bacteria [11, 40, 41]. Asaia spp. belong to the acid acetic alpha proteobacteria and are stably associated with some mosquito species such as Anopheles spp., Culex spp. and Aedes spp. Thus, Asaia spp. have been the target of studies on vector microbiota [42,43,44,45,46]. Asaia spp. have some features that persuade researchers to consider these bacteria as appropriate candidates for paratransgenic control of malaria and other VBD. Cultivation in cell-free media, colonization in different parts of the vector’s body (gut, salivary gland, reproductive organs, etc.), simple transformation capacity, transstadial and transovarial transmission and cryo-preservation capability are the main specific features of the species of this genus that discriminates it from the other symbiont microorganisms [40, 41, 46, 47]. Recently, bacteria have been introduced and used as dsRNA producing agents and delivery vehicles to insects for triggering the RNAi pathway against the targeted genes [48,49,50,51,52,53,54,55,56,57]. Therefore, with regard to the unique features of Asaia spp., unsuitable environmental conditions of malaria endemic regions, drawbacks of the research-based dsRNA administration routes to insects and the costs of in vitro dsRNA production, these bacteria could be considered as valuable and cost-effective candidates for applying the RNAi technology against malaria and other VBD in endemic regions.
Nevertheless, there is an important point that should be considered for increasing the effectiveness of the developed method based on symbiont bacteria and RNAi technology. Studies have shown that the dsRNA-specific endonuclease (RNase III) mutant bacteria are more efficient for producing the dsRNA molecules in quantity in comparison with their wild strains [49, 54, 57,58,59,60].
In fact, dsRNA substrates are subjected to cleavage by a specific superfamily of dsRNA endonucleases that existed in most of prokaryotes and eukaryotes [61,62,63,64,65]. Ribonuclease III (RNase III), an endoribonuclease, is encoded by the rnc gene, is an Mg2+ dependent enzyme that is important for ribosomal RNA (rRNA) processing, cellular defense against the viral infections and post-transcriptional gene expression control [61, 63,64,65,66,67]. Hence, we decided to characterize the rnc locus and its related protein in a field-isolated Asaia sp. by genomics, proteomics and in silico approaches to provide the basic and essential information for developing an rnc mutant Asaia strain. Our final goal was to utilize the information to create a strain which could be used as a pluripotent and robust dsRNA production tool and delivery vehicle against target genes in Anopheles sp. (larvae and adults) to control malaria vectors.
Methods
Primer design
16S rRNA gene primers were used from our previous study [46, 67]. To characterize the rnc gene from the isolated Asaia, full characterized sequences of the rnc gene from different acetic acid bacteria species were aligned using MEGA v.7.0 software [68]. After alignment, nine conserved regions were selected for designing the five forward (F1, F2, F3, F4 and F5) and four reverse primers (R1, R2, R3 and R4) to determine the middle part of the sequence of the rnc gene from the isolated Asaia (Table 1). Thereafter, according to the middle part of the sequence, eight gene-specific primers (forward: F652, F624, F566 and F518; reverse: R32, R161, R182 and R245) were designed to carry out the 3ʹ- and 5ʹ-genome walking (Table 1).
In the next step, rnc-clF and rnc-clR primers were designed to amplify and clone the coding sequence of the rnc gene in pET-28a expression vector (Table 1). All primers in this study were designed using the GeneRunner software (v.4.0.9.68 beta) and their specificity was evaluated by the nucleotide BLAST web-based software (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Specimen collection and rearing
Adult and larval specimens of Anopheles stephensi were collected from Sistan and Baluchistan, Hormozgan and Fars provinces, in the southern regions of Iran. Eggs and larvae from the F1 generation of An. stephensi were first reared in the local insectaries, then sent to the Malaria and Vector Research Group (MVRG) insectary in Pasteur Institute of Iran, Tehran, where they were raised. On arrival, the larvae and adults were identified morphologically to species according to the Iranian Anopheles key [69] and identifications were confirmed with molecular amplification of the internal transcribed spacer 2 (ITS2) and the cytochrome c oxidase subunit 1 (cox1) gene for An. stephensi. Insectary conditions were designed to mimic the local field conditions: 60–80% humidity, a temperature range of 26–28 °C and a 12-h light/dark photocycle in near-axenic conditions during both the developmental and adult stages [70, 71].
Isolation of Asaia sp. and molecular confirmation of the isolated bacteria
To prevent the probable surface contamination, all mosquitoes were initially immersed in 70% ethanol, rinsed by the sterile phosphate-buffered saline (PBS) and dissected separately. The larvae and adults were dissected under a steromicroscope under a biosafety level II biological laminar flow cabinet. The midgut of each sample was dissected, ground and then individually inoculated in Asaia-specific enrichment medium at 30 °C for three days with shaking at 180× rpm [72]. The ingredients of this medium were 2.0% d-sorbitol, 0.5% peptone, 0.3% yeast extract and 100 ppm cycloheximide at pH 3.5 with hydrochloric acid adjustment to provide specific conditions for Asaia sp. growth and isolation [72]. Subsequently, turbid tubes which indicated to the growth of bacteria were selected for plating on CaCO3 agar medium overnight to get the separated and morphologically defined colonies. This medium contained 2.0% d-glucose, 0.5% ethanol, 0.7% yeast extract, 0.8% CaCO3 and 1.2% agar at pH 8.6 [72]. In the next step, separated colonies were cultured in LB broth medium for preservation at – 80 °C and DNA extraction.
DNA was extracted from 1 ml of the overnight culture of bacteria in LB broth medium using the MBST genomic DNA extraction kit (MBST, Tehran, Iran) according to the manufacturer’s instruction. For molecular characterization and confirmation of the isolated colonies, AsaF and AsaR primers were used to amplify a 1200-bp amplicon of the 16S rRNA gene (Table 1). All PCR reactions were performed in a total volume of 20 μl. The reaction mixture contained 400 nM of each primer, 1 unit of Taq DNA polymerase, 0.2 mM (of each) dNTPs, 2 µl of 10× reaction buffer, 1.5 mM MgCl2 and 150 ng of genomic DNA as template. The amplification profile was as follows: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, annealing at 62 °C for 30 s and extension at 72 °C for 1 min; with a final extension step at 72 °C for 10 min.
Amplified fragments were recovered using agarose gel extraction kit (GeneAll Biotechnology, Seoul, South Korea) according to the manufacturer’s instructions and TA-cloned into a pTG19-T cloning vector (Vivantis, Selangor, Malaysia). Plasmids of the positive colonies were purified using a plasmid extraction kit (GeneAll Biotechnology) and subjected to direct sequencing (Microgen, Seoul, South Korea) by universal primers. Sequencing results were analyzed and edited by Chromas v.2.31 and final alignment was performed by nucleotide BLAST web-based software for confirmation (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Molecular characterization of the rnc gene sequence
Five forward and four reverse primers were designed for determining the middle part of the sequence of the rnc gene. Twenty PCR reactions were performed based on the possible combinations of F1, F2, F3, F4 and F5 as forward and R1, R2, R3 and R4 as reverse primers (Table 1). The amplification profile of the performed reactions was as follows: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, annealing at 62 °C for 30 s and extension at 72 °C for 50 s; with a final extension step at 72 °C for 10 min. The amplicon size of each reaction was estimated based on the genetically related species and finally, amplicons within the expected size ranges and higher length in comparison with the others were selected for TA-cloning and sequencing.
To achieve the full sequence of the rnc gene, the rapid amplification of genomic ends method was used. This step was performed based on our previous study [73]. In brief, for determining the 5ʹ-end of the rnc gene, single-stranded DNA (ssDNA) molecules were created by R182 as the outermost gene specific primer (with regard to the 5ʹ-end of the rnc gene middle part sequence) in the first step. Then, Genome Walking primers (GWPs) (A-G) were used for dsDNA production in seven separate reactions. Next, R162-UAP-N1 and R32-UAP-N2 primer pair combinations were used for the nest-1 and nest-2 reactions, respectively (Table 1). All PCR conditions and ingredients were the same as in our previous study [73]. In brief, ssDNA molecules were synthetized using R182 according to the following program: initial denaturation at 94 °C for 5 min; 25 cycles of 94 °C for 30 s, annealing at 65 °C for 30 s and extension at 72 °C for 3 min; with a final extension step at 72 °C for 10 min. Double stranded DNA molecules were synthetized using GWPs according to the following program: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, annealing at 28 °C for 30 s and extension at 72 °C for 3 min; with a final extension step at 72 °C for 10 min. Nest-1 and nest-2 reactions using the R162-UAP-N1 and R32-UAP-N2 primer pairs were performed with the following program: initial denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, annealing at 60 °C for 30 s and extension at 72 °C for 3 min; with a final extension step at 72 °C for 10 min. Finally, the related amplicons of different GWPs within acceptable size range were selected for TA-cloning in pTG19-T (Vivantis) and sequencing.
The 3ʹ-end sequence determination was performed in the same way as the 5’-end sequence determination, except for the use of different specifically designed primers for this region. In brief, ssDNA molecules were synthesized by F652 primer as the outermost gene specific primer. As previously, GWPs (A-G) were used for dsDNA synthesis in seven separate reactions. Next, F624-UAP-N1 and F566-UAP-N2 primer pairs were used to perform two asymmetric nested-PCR reactions (Table 1). Selection of amplicons was performed as for the 5ʹ-end sequence determination and the selected amplicon were TA-cloned and sequenced.
In the next step, three parts of the rnc gene sequences (middle, 3ʹ-end and 5ʹ-end) were assembled using GeneStudio Pro software v.2.2.0.0 and the full-length coding sequence (CDS) of the rnc gene from the isolated Asaia was defined. Finally, rnc-clF and rnc-clR primers were designed to amplify this region for recombinant RNase III production in Escherichia coli (Table 1).
Recombinant RNase III expression and its proteomics analysis
To express the RNAse III, CDS of rnc gene was amplified by the Expand High Fidelity PCR System (Roche, Penzberg, Germany) by rnc-clF and rnc-clR primers and TA-cloned into pTG19-T vector (Vivantis) in the first step. Then, the coding sequence was sub-cloned into the pET-28a by digestion with HindIII and XhoI restriction enzymes (Thermo Fisher Scientific, Massachusetts, USA). Finally, recombinant protein expression was performed into E. coli BL21 (DE3) strain by 0.4 mM IPTG induction for 15 h [70].
Pre-heated samples at 95 °C for 5 min were run at 90 mA of constant current for 90 min on 12% polyacrylamide gel (Bio-Rad, California, USA) and proteins were analyzed by Coomassie Brilliant Blue stain (Sigma-Aldrich, Missouri, USA). Electro-transfer to nitrocellulose sheets (Amersham, Little Chalfont, UK) was performed as described by Towbin et al. [74] with a trans-blot SD semi-dry transfer cell (Bio-Rad). After transferring, blocking was performed using bovine serum albumin (0.3 g BSA in 15 ml PBS at pH 7.5) at 4 °C overnight. The 6x-His tag rabbit polyclonal antibody (1:4000) (Abcam, Cambridge, UK) was used as the first and horseradish peroxidase-conjugated anti-rabbit antibody (1:5000) (Santa Cruz Biotechnology, Dallas, USA) was used as the second antibody to visualize antigen-antibody interactions. Visualization was done by incubating the membrane with SIGMAFast diaminobenzidine (DAB) with metal enhancer (Sigma-Aldrich). Purification of the recombinant protein was performed by Ni-NTA Agarose (Qiagen, Hilden, USA) in hybrid situation according to the Thermo Fisher Scientific protocol. Finally, quality of the purified recombinant protein was evaluated by SDS-PAGE and confirmed by western blotting as previously mentioned.
Constructs for synthesis and production of dsRNA molecules
Considering the importance of carboxypeptidase-1 enzymes of An. gambiae and An. stephensi in the sexual development of Plasmodium falciparum [70, 75], the cpbAs1 gene of An. stephensi was selected as the target gene for designing and producing dsRNA molecules to evaluate their effects on the target gene expression in the future experiments. This gene has three exons and two introns in An. stephensi and 450-bp from the end of the first exon was selected as the target region for the design of dsRNA. A 150-bp linker in length was used to connect two antiparallel sequences and BamHI and XhoI restriction sites were added to the 5ʹ- and 3ʹ-ends of the designed sequence respectively. This construct was synthesized by ShineGene (Shanghai, China) in pUC57 vector and then sub-cloned into the pET-28a expression vector by digestion with BamHI and XhoI restriction enzymes. At the end, dsRNA expression was performed in E. coli BL21 (DE3) strain by 0.4 mM of IPTG induction for 4 h; the produced dsRNA molecules were purified by TRIZOL Lysis Reagent (Sigma-Aldrich, Missouri, Germany) [76,77,78], recovered from the agarose gel and their quality and quantity were evaluated by agarose gel electrophoresis and Colibri Microvolume Spectrophotometer (Titertek Berthold, Bad Wildbad, Germany), respectively.
Bioactivity analysis
In the first step, concentration of the purified RNase III enzyme was determined by Bradford assay [79]. To perform the bioactivity assay, 1 nmol of the purified dsRNA molecules was incubated with ~ 5 ng of the recombinant purified RNase III enzyme. The cleavage buffer, which contained 30 mM Tris (pH 7.5), 150 mM KCl, 5 mM spermidine, 20 mM MgCl2, 0.1 mM DTT and 0.1 mM EDTA, pH 7.5, was mixed with 1 nmol of dsRNA and incubated at 37 °C for 1 min. The reaction was initiated by adding 10 mM of MgCl2 at 37 °C and the degradation of dsRNA molecules was evaluated on 2% agarose gel electrophoresis after blocking the reaction by adding 20 mM EDTA in different time frames (0, 1, 2 and 4 min) [80,81,82]. In addition, the specificity of the recombinant protein was evaluated on the purified total RNA from the induced bacteria for dsRNA production.
Phylogenetic analysis, modeling, refinement and quantitative evaluation of Asaia RNase III enzyme
After translation of the coding sequence of rnc gene with GeneRunner software v.4.0.9.68 beta, protein BLAST was performed for determining the highly similar proteins with our query sequence; proteins with high identity, coverage and score were selected to perform an alignment. Alignment was performed with Clustal Omega server based on the Clustal W [83] multiple sequence alignment method with some bacterial RNase III proteins such as Aquifex aeolicus, Thermotoga maritima, E. coli, Shigella dysenteriae, Gluconobacter oxydans, Parasaccharibacter apium, Commensalibacter intestine, Acetobacter aceti and Komagataeibacter intermedius, to analyze the conservation of the structurally important residues of RNase III enzyme superfamily in our target sequence [83]. Next, these sequences were analyzed with MEGA v.7.0 software based on the neighbor-joining method with 1000 bootstrap replicates [68].
Moreover, comparative homology modeling (HM) was performed to predict the three-dimensional structure of Asaia RNase III enzyme by using the SWISS-MODEL [84], Phyre2 servers [85] and Modeller v.9.19 software [86] and the best structures were selected according to the QMEAN (Qualitative Model Energy Analysis) and GMQE (global model quality estimation) [87,88,89,90]. SWISS-MODEL and Phyre2 are web-based integrated services for predicting and analysing protein structures, whereas Modeller is a computing system for comparative protein structure modeling. Comparative HM consists of four basic steps: template selection, template target alignment, model construction, and model evaluation and refinement [91]. Models were computed by the SWISS-MODEL and Phyre2 servers in an automated-normal mode that only required the amino acid sequence of the target protein as input. In parallel, based on the ranked discrete optimized potential energy (DOPE) score of 10,000 generated models in Modeller, the top ten predicted models were chosen for refinement and validation.
The stereochemical quality of the predicted protein structures from the both approaches was analyzed with several web tools to enhance the accuracy of evaluation. MolProbity [92], PROCHECK [93], VADAR [94], ERRAT [95], VERIFY_3D [96], ProSA [97] and PROSESS web tools [98] were used for structural analysis. The statistics of non-bonded interactions between the atoms in 3D structure in comparison with a database of the highly refined structures were evaluated by ERRAT and structures with higher scores reflected higher quality [95]. The compatibility of the 3D models with amino acid sequences was analyzed by Verify_3D. A 3D structure is considered as well compatible with its sequence when more than 80% of the amino acids are scored ≥ 0.2. Generally, the ProSA Z-score determines the overall model quality. Z-score plot displays whether the z-score of the input structure is within the range of scores that are typically found for native proteins with similar size. The structural similarity between the model and template was calculated by using the TM-align web server. The template modeling score (TM-score) is a measure of similarity between two protein structures with different 3D structures. Commonly, the structures with a score higher than 0.5 are assumed to have the same fold [99].
Furthermore, the best model was selected and considered for structure refinement by using the GalaxyRefine web server [100]. Then, suggested structure with the highest structural similarity was selected for superimposition and calculation of the root-mean-square deviation (RMSD) by using the Swiss-PdbViewer DeepView v.4.1.0 and UCSF Chimera v.1.11.2 [101].
Molecular docking studies
The crystal structure of dsRNA was retrieved from the template crystal structure (PDB ID: 4M30) in the RCSB Protein Data Bank (PDB) and molecular docking experiments of the suggested structure by SWISS-MODEL was carried out with HADDOCK web server to understand the molecular interactions [102, 103].
Promoter region and transcription termination mechanism analysis
BPROM, which is used for predicting the bacterial operon in immediately upstream regions of the ORF start site (prediction of bacterial promoters, Softberry Inc), was used for analyzing the promoter region of rnc gene based on the sigma 70 of RNA polymerase binding site [104]. Furthermore, ARNold and FindTerm (Softberry Inc.) web server software were used to predict the mechanism of transcription termination for rnc gene [105].
Results
Asaia sp. isolation from the field-collected mosquitoes
After inoculation by surface striking, uniformly smooth white colonies with and without dissolution halo were grown on CaCo3 agar medium. Different morphological colonies were selected for molecular analysis and confirmation. Finally, a smooth-white colony with dissolution halo from Hormozgan Province was selected for performing the next steps and further analysis.
Molecular identification and confirmation of Asaia sp. based on the sequence of the 16S rRNA gene
By performing PCR with Asaia 16S rRNA gene-specific primers, a 1200-bp amplicon was observed after agarose gel electrophoresis in all isolated colonies (with and without dissolution halo) which confirmed the species of isolated bacteria (Additional file 1: Figure S1). Sequence analysis by nucleotide BLAST confirmed their high similarity and identity to the reported sequences of 16S rRNA gene of Asaia species in the GenBank. These sequences were submitted to the GenBank database under the accession numbers MK128664, MK616065, MK616094, MK616097, MK645713, MK645714, MK645722, MK645809, MK645810 and MK645851.
Molecular characterization of the rnc gene
For middle part sequence determination, agarose gel electrophoresis revealed that between the 20 possible primer coupling situations, only combinations of F1 with all reverse primers (R1-R4), F4 with R3 and R4, F5 with R3 and R4 resulted in amplification of the expected amplicons. Because F1 and R1 were the outermost primers and their amplicon had more coverage of rnc gene in comparison with the other amplicons, this combination was selected for confirming with internal primers. Primer combinations F4-R3, F4-R4, F5-R3 and F5-R4 confirmed the relationship of the targeted amplicon to the rnc gene. Upon validation, the 685-bp amplicon generated with the F1-R1 primers was TA-cloned and sequenced. Nucleotide BLAST analysis revealed 99% coverage and 95% identity to the Asaia bogorensis genome (GenBank: AP014690.1). Finally, this part of the rnc gene from the isolated bacterium was submitted to GenBank (MG431206) and used to design the gene-specific primers (GSPs) for determining its upstream and downstream sequences by the genome walking method.
After performing the 3ʹ-genome walking, sequence analysis of the products of different DRTs (A-G) revealed the presence of overlapped regions at their 5ʹ-ends with 3ʹ-end of the middle part of the sequence. Therefore, the longest amplicon (925-bp) which was related to DRT-A was selected and submitted to GenBank (MK190424). The 5’-genome walking amplicons were analyzed likewise, and it was revealed that all of them had an overlapped region at their 3ʹ-end with the 5ʹ-end sequence of the middle part sequence of the rnc gene. The longest amplicon, which was related to DRT-C (897-bp), was submitted to GenBank (MK190423). The nucleotide BLAST analysis revealed its high similarity and homology (≥ 95%) with the A. bogorensis genome (AP014690.1).
Finally, the acquired sequences for the 5ʹ-end, middle part and 3ʹ-end of rnc gene were assembled with GeneStudio software and submitted to GenBank (MG431209). Different probable open reading frames (ORFs) of the assembled sequence were predicted by GeneRunner software and ORF related to RNase III enzyme was determined using protein BLAST. It was interesting that there were two ORFs other than rnc which subsequent analysis revealed were related to era and lep genes (MK190425 and MK190426). Nucleotide BLAST analysis of the coding sequence (CDS) of the rnc gene from the isolated bacterium revealed 100% coverage and 95% identity to the Asaia bogorensis genome with accession number AP014690.1.
rnc gene characterization
Protein BLAST analysis of the coded protein by rnc gene showed that subjects with high identity and query coverage were related to the RNase III enzyme family of different bacteria species. Therefore, these were selected for alignment and phylogenetic tree analysis. Evolutionary analysis revealed that our target protein has a close relationship with those of Acidomonas methanolica, Neoasaia chiangmaiensis and Tanticharoenia sakaeratensis, all belonging to the family Acetobacteraceae. Moreover, our target protein has a more distant relationship with its counterparts in E. coli, Mycobacterium tuberculosis and Streptococcus pneumoniae (Fig. 1).

Phylogenetic tree for RNase III protein from the isolated Asaia sp. RNase III enzyme from the isolated Asaia sp. was aligned with the characterized RNase III enzymes from different bacteria and related species. This alignment and clustering was performed by MEGA7.0 software and neighbor-joining method with bootstrapping, respectively, to increase the accuracy of the created tree. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap values (1000 replicates) is shown next to the branches. The scale-bar corresponds to 0.2 changes per residue. Our target protein is indicated with a black square and the branches of the more similar species are indicated by thick lines. Furthermore, subjects which have low similarity to others (Escherichia coli, Mycobacterium tuberculosis and Streptococcus pneumoniae) are marked by black triangles
According to the study by Court et al. [66] on Aquifex aeolicus RNase III structure, alignment of the amino acid sequences of similar RNase III enzymes by Clustal Omega revealed that the structurally important residues are conserved in all aligned sequences (Fig. 2). The structural signature of RNase III enzymes is a divalent metal ion (such as Mg2+, Mn2+, Co2+ and Ni2+) dependent domain which is crucial for their biological activity. This alignment revealed that the glutamic acids at positions 57, 60, 136 and 84, and aspartic acids at positions 64 and 133 are involved in the construction of the catalytic domain of our target enzyme. Moreover, these residues, that are coordinated with divalent metal ions, were conserved between all aligned sequences. By structural comparison with other characterized RNase III enzymes, the secondary structure of our target protein, its signatures and motifs were determined (Fig. 2).

Conserved domain analysis of RNase III proteins with Clustal Omega. The alignment of our target protein with the structurally characterized bacterial RNase III was performed using the Clustal W multiple sequence alignment method. Conserved residues are indicated with gray rectangles; conserved residues between all characterized bacterial RNase III proteins are indicated with black rectangles. The signature motif consists of 9–10 residues that are almost conserved between all aligned sequences. RNA binding motifs (RBM1-4) and linker sequence are indicated by black lines. In addition, the important residues that are involved in active site and Phe61 which has an important role in dimerization of the enzyme are indicated by black squares and a black circle, respectively
Further analysis of the target sequence and two other ORFs revealed that two ORFs other than rnc encode Era and lepB proteins. Therefore, additional analyses were performed for determining the probable relationship between these coding sequences. Our results revealed that era- and lep-coding sequences are located downstream and upstream, respectively, of the rnc-coding sequence and code Era and LepB proteins. We found that the rnc- and era-coding sequences have four nucleotides which overlap with each other. It means that the start codon (ATG) of the era is before the stop codon (TGA) of the rnc gene. Further analysis revealed that there is an inefficient ribosome-binding site (-GGT-) in comparison with the consensus sequence of shine-dalgarno (-GGAG-) before the start codon of the rnc gene which is located at positions − 5 to − 8 from the start codon of the rnc gene. Moreover, our analysis revealed that the lep gene, which encodes the signal peptidase I, is located in the upstream region of the rnc gene.
3D structure prediction and superimposition
Eight 3D structures were presented by SWISS-MODEL and Phyre2 web servers based on homology modeling. According to QMEAN and GMQE, the best predicted structure with the highest score was related to Aquifex aeolicus RNase III enzyme (strain VF5) (PDB: 4M30). Therefore, it was chosen as the reference molecule for performing further and detailed structural analysis.
Validation of the predicted structure for Asaia RNase III enzyme
The stereochemical quality of protein structures of the ten top predicted models of Modeller and eight presented 3D structures by SWISS-MODEL and Phyre2 web servers were analyzed by utilizing several web tools to enhance the accuracy of evaluation. The related results to the final models of Asaia RNase III are presented in Tables 2 and 3. The number of serious clashes which showed unfavorable all-atom steric overlaps by more than 0.4Å per 1000 atoms was considered as the MolProbity clash score [92]. Better models possess a lower number of clash scores and clash scores of the final model of SWISS-MODEL, Phyre2 and Modeller were 1.66, 3.01 and 1.72, respectively, which were acceptable. The Verify_3D score of the suggested model of SWISS-MODEL, Phyre2 and Modeller was 72.01%, 67.07% and 82.13%, respectively (Table 2).
Moreover, the structural results of ERRAT approved high overall quality of the final predicted 3D structures of SWISS-MODEL, Phyre2 and Modeller with the ERRAT scores which were 91.98%, 82.35% and 100%, respectively. The values for free energy of folding which was evaluated using the VADAR web server were − 383.46, − 191.89 and − 391.65 for the SWISS-MODEL, Phyre2 and Modeller predicted structure, respectively. Ramachandran plot results via PROCHECK showed that the stereochemical quality of the final selected models was satisfactory and an acceptable percentage of residues (89.2%, 85.4% and 93.9%, respectively) were located in the most favored region [93]. The overall quality of final models which was measured using the PROSESS web servers, gained scores of 4.5, 3.5 and 6.5 and admitted the overall quality of final models (Table 2). For three final models, ProSA Z-scores were located within the typical range of scores for the experimentally determined structures (Fig. 3).

ProSA Z-score plots for final models of Modeller (a), Phyre2 (b) and Swiss-model (c). X-ray determined structures are colored light blue and NMR determined structures are colored dark blue. Large black dots show the ProSA Z-score of the predicted models by each method
The results of TM-score revealed that the predicted models by using the Swiss-model and Modeller have the same folding close to the template in comparison with the predicted model with the Phyre2 (Table 3). Therefore, the model generated by using the SWISS-MODEL was chosen for the docking study. The structure and sequence of the predicted models by SWISS-MODEL and Modeller were aligned with 4M30 as a template to determine the differences in the sequence that had led to the variation in folding patterns (Additional file 2: Figure S2).
Analysis of the final model by using the UCSF Chimera software revealed that RNase III of the isolated Asaia sp. bacterium is a globular protein with two important domains: (i) RNase III N-terminal catalytic domain (RIIID) and (ii) C-terminal dsRNA-binding domain (dsRBD) (Additional file 3: Figure S3a–c).
Based on the modeling and superimposition with the reference molecule, the catalytic center of our target protein consists of six acidic residues: four glutamic acids (E57, E60, E136 and E84) and two aspartic acids (D64 and D133), of which E57, E60 and D64 were located in the signature motif (Additional file 3: Figure S3d). In addition, it was revealed that there are four RNA-binding motifs (RBM1–4) in the predicted structure of RNase III enzyme, of which the 1st and 2nd were located in dsRBD and 3rd and 4th in RIIID motifs (Additional file 3: Figure S3e). Furthermore, it was found that each smaller domain (dsRBD) of the enzyme consists of 67 residues and are connected together by a short linker (five residues: 168–172) (Additional file 3: Figure S3f). Configuration and flexibly of the enzyme for identifying and biding to dsRNA molecules are dependent on the structure of this linker [66, 106, 107].
Structural properties of the enzyme
Superimposition and comparison of critical residues of the active site revealed that the targeted residues are completely similar to their counterparts in Aquifex aeolicus RNase III and glutamic acid 60, aspartic acid 133, glutamic acid 136 and aspartic acid 64 are the base of catalytic domain that interact with divalent cations. These residues interact with a divalent cation in each monomer and catalytic domain of the enzyme which is constructed upon the dimerization (Additional file 4: Figure S4) [108].
Comparison of their RMSDs with the reference molecule is presented in Table 4. This comparison revealed that the subtraction RMSDs values of the reference and target molecule at specific positions are less than 0.5 Å implying backbone similarity of the reference and target molecule (Fig. 4).

Structural alignment of predicted models and the crystal structure. a Superimposition of the 3D structure of 4M30 (light blue) with the predicted 3D structures of Asaia RNase III using the SWISS-MODEL (purple) and Modeller (green). b The 3D structure of 4M30 (light blue). c The predicted 3D structures of Asaia RNase III using the SWISS-MODEL (purple). d The predicted 3D structures of Asaia RNase III using the Modeller (green)
The values of overall RMSD between the final refined model and different ribonucleases were 2.369 Å for Bacillus halodurans RNase H (PDB: 1ZBF), 3.514 Å for E. coli RNase D (PDB: 1YT3), 2.845 Å for E. coli RNase E (PDB: 2C4R), 2.730 Å for E. coli RNase R (PDB: 1PCA), 1.266 Å for Pseudomonas aeruginosa RNase T (PDB: 2F96) and 3.886 Å for Thermus thermophilus RNase J (PDB: 3BK1).
Molecular docking analysis
Molecular docking studies could be helpful for figuring out the key residues and possible interacting faces in protein-protein interactions. Interestingly, although Asaia RNase III and Aquifex aeolicus RNase III share only 31% identity, patterns of their interactions with dsRNA were noticeably similar. Binding residues in RNase III (Dimer)-dsRNA interactions were investigated by LigPlot+ software [109] and are presented in Fig. 5.

Interaction mode of the predicted model by Swiss-model and dsRNA in comparison with the 4M30. The complexes resulted from molecular docking which was conducted with HADDOCK. a Monomers of the predicted structure of RNase III are colored green and cyan. In close-up view, the residues of model that are involved in interaction with dsRNA are depicted in yellow and stick representation. b Binding model of dsRBD of Asaia RNase III dimer with dsRNA, the crystal structure of dsRBDs from each monomer is presented in blue and pink. In the close-up view, residues of protein that are involved in binding to dsRNA are illustrated in red and stick representation
Prediction of promoter region and transcription termination mechanism
Analysis with BPROM software revealed the presence of a probable promoter sequence with at least 80% accuracy and specificity at position − 128 from the start codon of the rnc gene. Moreover, evaluation of the mechanism of transcription termination at about 1000-bp of the downstream of ORF sequence of rnc gene by ARNold and FindTerm showed that this phenomenon is not rho-dependent.
Cloning, expression, purification and western blotting analysis of RNase III
After overnight expression, a ~ 27.1 kD molecule was seen by SDS-PAGE analysis in comparison to the control that was confirmed by western blotting analysis based on the presence of 6XHis-tag fusion. The Asaia RNase III recombinant protein was purified by Ni-NTA agarose and the purification process was confirmed by SDS-PAGE and western blotting analysis (Fig. 6).

SDS-PAGE and western blot. After cloning in pET28-a and induction, the recombinant protein was analyzed by SDS-PAGE and western blotting assays. SDS-PAGE: Lane 1: clone with RNase III coding sequence, Lane M1: protein marker; Lane 2: clone without RNase III coding sequence. Western blot: Lane 3: clone with RNase III coding sequence; Lane M2: protein marker; Lane 4: clone without RNase III coding sequence
dsRNA production
Agarose gel electrophoresis of the extracted total-RNAs from E. coli Bl21 (DE3) with and without dsRNA coding sequence after RNase A treatment revealed the presence of an amplicon about 500-bp in size in E. coli Bl21 (DE3) with dsRNA coding sequence that confirmed the production of our target dsRNA molecules (Fig. 7a).

Biological activity of recombinant RNase III enzyme and specificity assay. a Purified dsRNA molecules were incubated with the purified RNase III recombinant protein for 1, 2 and 4 min. Lane M: marker; Lanes 1–4: 0, 1, 2 and 4 min after incubation, respectively. b For the specificity assay, purified total RNAs by QIAZOL from the transformed E. coli BL21 with dsRNA coding sequence plasmid were incubated with the purified recombinant RNase III. To perform this test, 1 nmol of the purified dsRNA molecules were incubated for 1 min with ~5 ng of the recombinant purified RNase III enzyme in appropriate buffer for RNase III enzyme activity. Lane M: DNA ladder; Lane 1: non-incubated sample with the recombinant Asaia RNase III protein; Lanes 2 and 3: incubated RNAs with the recombinant Asaia RNase III protein. The incubated samples were loaded on agarose gel three folds higher than the non-incubated sample for better visualization and analyses of dsRNA cleavaging. These analyses revealed that the recombinant Asaia RNase III protein degrades the dsRNA molecules specifically and has no effect on ssRNA molecules. The produced dsRNA is marked in Lane 1 with a white arrow
Enzymatic bioactivity
After stopping the dsRNA degradation assay reactions, the reaction mixtures were analyzed by agarose gel electrophoresis. The analysis showed that the purified recombinant protein degraded dsRNA molecules and its effect on dsRNAs was raised by increasing the incubation time (Fig. 7a). Furthermore, performing specificity analysis on the total purified RNAs revealed that it degraded dsRNA molecules specifically and had no effect on ssRNA molecules (Fig. 7b).
Discussion
The double-stranded RNA endonucleases superfamily is divided to four classes: bacterial RNase IIIs, Rnt1p (in yeasts and fungi), Drosha and Dicer (in mammals) [66, 109,110,111,112,113]. RNase III (RNase III) is an Mg2+ dependent enzyme that is important for ribosomal RNA (rRNA) processing, cellular defense against the viral infections and post-transcriptional gene expression control [61, 63,64,65,66, 107]. For creating an RNase III mutant bacterium, it is necessary to have basic knowledge about the structure of the rnc gene, its surrounding sequences and structural properties of its coded protein for applying to the point mutations which result in the inhibition of its biological activity. Therefore, we characterized the rnc gene and evaluated the bioactivity and structure of its coded protein from an Asaia sp. which had been isolated from field-collected An. stephensi mosquitoes to provide the fundamental information for developing an efficient paratransgenic tool based on RNAi technology.
Coding sequence analysis of our target gene with the nucleotide BLAST server revealed that it is related to endoribonuclease III gene family and its sequence has considerable similarity with RNase III enzyme of different bacteria, especially in the Acetobacter family [66, 114,115,116]. According to the previous studies on other species of bacteria, rnc, era and recO genes are arranged as an operon: rnc is the first member of this operon and era and recO are located in the downstream region of rnc [117,118,119,120]. Our findings revealed that this arrangement is also seen in the isolated Asaia and that the rnc- and era-coding sequences had four nucleotides that overlapped, which is typical and similar to the previous studies on E. coli, P. aeruginosa, Salmonella typhimurium and Rhodobacter capsulatus [117, 121,122,123]. These findings are very critical for selecting an appropriate strategy for inactivating the RNase III without any side effects on the biological activity of other members of this operon. The presence of a weak Shine-Dalgarno sequence before the start codon of rnc gene is similar with the results of the study by March et al. on E. coli [117]. Moreover, this sequence has one of the shortest reported spaces between the ribosome binding site and the start codon in prokaryotes [117, 124]. It is concluded that this difference reflects the low level of cell demand for products of this operon [117, 118, 121].
Bacterial RNase IIIs consists of two distinct domains: RIIID and dsRBD [64, 66, 125]. The RIIID is composed of ~ 150 residues and includes 9–10 residues as the signature motif which are highly conserved in different species [66, 107, 116]. Based on homology modeling, the RIIID of our target protein is composed of 160 residues which include the signature motif and 16 residues with no counterparts in the reference model (4m30). These residues are seen as a protrusion on the surface of our target protein in the predicted model. Further analysis and alignment with the bacterial RNase III enzymes revealed that there is no equivalent for them in other bacterial RNase III enzymes except in the family Acetobacteraceae; it seems that this domain is specific to this family and has a particular function which should be further analyzed in future investigations. Comparison of our results with previous structural findings on RNase III enzymes of other bacteria revealed that the signature motif of our target protein consists of ten residues (57-ERLEFIGDRV-66) which are located in RIIID [66, 107, 114, 125]. In addition, based on the study by Blaszczyk et al. [111], Phe61 has an important role in the dimerization of enzymes, which is completely conserved among most bacterial RNase enzymes (Fig. 2).
Interaction with divalent metal ions is an important structural feature of RNase III enzymes which has been reported in different studies [66, 106, 111]. According to superimposition analysis, E60, D64 and E136 are involved in interaction with divalent metal ion in our target protein. Gan et al. [115] aligned 100 sequences of bacterial RNase III molecules and found that 15 residues (F19, A28, H31, L59, E60, G63, D64, G85, E136, K174, L177, Q178, Y191, F205 and A237) are conserved with high frequency in all of them. Molecular docking results of the predicted model by SWISS-MODEL with dsRNA were consistent with the findings of Gan et al. [115] and these residues were involved in Dimer-dsRNA complex conformation and their interactions. It was noticeable that all of these residues are conserved in the characterized bacterial RNase IIIs and our target protein (Fig. 2). Furthermore, we calculated the RMSD of these residues with the reference molecule and our results revealed that they have very close topology (Table 4).
The structure of RIIID is unique and is the signature of the RNase III family, but the structure of dsRBD might be seen in other dsRNA-binding proteins [66, 114]. dsRBD has an αβββα structure and there is only one copy of this structure in RNase III proteins, but there could be more than one copy in other dsRNA-binding proteins [66]. According to the in silico structural analysis, the dsRBD of our target protein has an αβββα structure similar to previous studies [64, 66, 67, 106, 107, 111, 125, 126].
Conclusions
According to the fact that RNase III mutant bacteria are more efficient for producing the dsRNA molecules in comparison with the wild strains, characterization of the rnc gene and its related protein from an Asaia sp. was performed in this study. An important finding of our study was determination of the structural details of the rnc operon and its relationship with era and recO genes. Moreover, hot spot and structurally important residues in RNase III protein of Asaia sp. were determined by in silico analysis and docking approaches which is useful in paving the road of creating an rnc mutant Asaia strain; to the best of our knowledge, this study is the first report of the molecular characterization of an rnc gene and its related protein from Asaia species. At a glance, the data acquired in this study are informative for investigators that are interested in researching paratransgenesis, Asaia sp., and RNAi technology for combating malaria and other vector-borne diseases.
Availability of data and materials
The sequences obtained and/or analyzed during the present study are deposited in the GenBank database under the following accession numbers: MK128664, MK616065, MK616094, MK616097, MK645713, MK645714, MK645722, MK645809, MK645810 and MK645851 for the 16S rRNA gene; MK190424 for 3ʹ-genome walking; MK190423 for 5’-genome walking; MG431209 for the full length of the rnc gene; and MK190425 and MK190426 for era and lep genes. All other relevant data are included in the article and its additional files.
Abbreviations
- VBD:
-
Vector-borne diseases
- WHO:
-
World Health Organization
- MVRG:
-
Malaria and Vector Research Group
- ITS2:
-
Internal transcribed spacer 2
- cox1:
-
cytochrome c oxidase subunit 1
- PBS:
-
phosphate-buffered saline
- ssDNA:
-
single-stranded DNA
- GWPs:
-
genome walking primers
- CDS:
-
coding sequence
- HM:
-
homology modeling
- QMEAN:
-
qualitative model energy analysis
- GMQE:
-
global model quality estimation
- DOPE:
-
discrete optimized potential energy
- TM-score:
-
template modeling score
- RMSD:
-
root-mean-square deviation
- PDB:
-
Protein Data Bank
- GSPs:
-
gene-specific primers
- ORFs:
-
open reading frames
- dsRBD:
-
dsRNA-binding domain
- RBM:
-
RNA-binding motifs
- PII:
-
Pasteur Institute of Iran
- INSF:
-
Iran National Science Foundation
- BRC:
-
Biotechnology Research Center
References
Hill CA, Kafatos FC, Stansfield SK, Collins FH. Arthropod-borne diseases: vector control in the genomics era. Nat Rev Microbiol. 2005;3:262–8.
Ramalho-Ortigão M, Coutinho-Abreu IV. Transgenesis, paratransgenesis and transmission blocking vaccines to prevent insect-borne diseases. In: Larramendy ML, editor. Integrated pest management and pest control: current and future tactics. Rijeca: InTech; 2012. p. 581–612.
Flores HA, O’Neill SL. Controlling vector-borne diseases by releasing modified mosquitoes. Nat Rev Microbiol. 2018;16:508–18.
WHO. World malaria report 2017. Geneva: World Health Organization; 2018.
Casares S, Brumeanu T-D, Richie TL. The RTS,S malaria vaccine. Vaccine. 2010;28:4880–94.
Modeling mCGo. A research agenda for malaria eradication: modeling. PLoS Med. 2011;8:e1000403.
Alonso PL, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, et al. A research agenda to underpin malaria eradication. PLoS Med. 2011;8:e1000406.
The malERA Consultative Group on Vector Control. A research agenda for malaria eradication: Vector Control. PLoS Med. 2011;8:e1000401.
Hurwitz I, Hillesland H, Fieck A, Das P, Durvasula R. The paratransgenic sand fly: a platform for control of Leishmania transmission. Parasites Vectors. 2011;4:82.
Beard C, Dotson E, Pennington P, Eichler S, Cordon-Rosales C, Durvasula R. Bacterial symbiosis and paratransgenic control of vector-borne Chagas disease. Int J Parasitol. 2001;31:621–7.
Favia G, Ricci I, Damiani C, Raddadi N, Crotti E, Marzorati M, et al. Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proc Natl Acad Sci USA. 2007;104:9047–51.
Aksoy S, Weiss B, Attardo G. Paratransgenesis applied for control of tsetse transmitted sleeping sickness. In: Aksoy S, editor. Transgenesis and the management of vector-borne disease. Berlin: Springer; 2008. p. 35–48.
Riehle MA, Jacobs-Lorena M. Using bacteria to express and display anti-parasite molecules in mosquitoes: current and future strategies. Insect Biochem Mol Biol. 2005;35:699–707.
Coutinho-Abreu IV, Zhu KY, Ramalho-Ortigao M. Transgenesis and paratransgenesis to control insect-borne diseases: current status and future challenges. Parasitol Int. 2010;59:1–8.
Chavshin AR, Oshaghi MA, Vatandoost H, Yakhchali B, Raeisi A, Zarenejad F. Escherichia coli expressing a green fluorescent protein (GFP) in Anopheles stephensi: a preliminary model for paratransgenesis. Symbiosis. 2013;60:17–24.
Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Enayati AA, et al. Identification of bacterial microflora in the midgut of the larvae and adult of wild caught Anopheles stephensi: a step toward finding suitable paratransgenesis candidates. Acta Trop. 2012;121:129–34.
Lindh J, Borg-Karlson A-K, Faye I. Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop. 2008;107:242–50.
Mancini MV, Spaccapelo R, Damiani C, Accoti A, Tallarita M, Petraglia E, et al. Paratransgenesis to control malaria vectors: a semi-field pilot study. Parasites Vectors. 2016;9:140.
Park JM, You Y-H, Back C-G, Kim H-H, Ghim S-Y, Park J-H. Fungal load in Bradysia agrestis, a phytopathogen-transmitting insect vector. Symbiosis. 2018;74:145–58.
Rangberg A, Mathiesen G, Amdam G, Diep D. The paratransgenic potential of Lactobacillus kunkeei in the honey bee Apis mellifera. Benef Microbes. 2015;6:513–23.
Rasgon JL. Using infections to fight infections: paratransgenic fungi can block malaria transmission in mosquitoes. Future Microbiol. 2011;6:851–3.
Ren X, Hoiczyk E, Rasgon JL. Viral paratransgenesis in the malaria vector Anopheles gambiae. PLoS Pathog. 2008;4:e1000135.
Ricci I, Mosca M, Valzano M, Damiani C, Scuppa P, Rossi P, et al. Different mosquito species host Wickerhamomyces anomalus (Pichia anomala): perspectives on vector-borne diseases symbiotic control. Antonie Van Leeuwenhoek. 2011;99:43–50.
Riehle MA, Moreira CK, Lampe D, Lauzon C, Jacobs-Lorena M. Using bacteria to express and display anti-Plasmodium molecules in the mosquito midgut. Int J Parasitol. 2007;37:595–603.
Thomas MB, Read AF. Can fungal biopesticides control malaria? Nat Rev Microbiol. 2007;5:377–83.
Chavshin AR, Oshaghi MA, Vatandoost H, Pourmand MR, Raeisi A, Terenius O. Isolation and identification of culturable bacteria from wild Anopheles culicifacies, a first step in a paratransgenesis approach. Parasites Vectors. 2014;7:419.
Karimian F, Vatandoost H, Rassi Y, Maleki-Ravasan N, Mohebali M, Shirazi MH, et al. Aerobic midgut microbiota of sand fly vectors of zoonotic visceral leishmaniasis from northern Iran, a step toward finding potential paratransgenic candidates. Parasites Vectors. 2019;12:10.
Wang S, Jacobs-Lorena M. Paratransgenesis applications: Fighting malaria with engineered mosquito symbiotic bacteria. In: Wikel S, Aksoy S, Dimopoulos G, editors. Arthropod vector: controller of disease transmission, vol. 1. Amesterdam: Elsevier; 2017. p. 219–34.
Lindh JM, Terenius O, Faye I. 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microbiol. 2005;71:7217–23.
Wilke ABB, Marrelli MT. Paratransgenesis: a promising new strategy for mosquito vector control. Parasites Vectors. 2015;8:342.
Yadav KK, Chandel K, Bora A, Veer V. Isolation and characterization of Dietzia maris from midgut of Aedes albopictus: a suitable candidate for paratransgenesis. Int J Mosq Res. 2015;2:7–12.
Boissière A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012;8:e1002742.
Raharimalala FN, Boukraa S, Bawin T, Boyer S, Francis F. Molecular detection of six (endo-) symbiotic bacteria in Belgian mosquitoes: first step towards the selection of appropriate paratransgenesis candidates. Parasitol Res. 2016;115:1391–9.
Wang S, Dos-Santos AL, Huang W, Liu KC, Oshaghi MA, Wei G, et al. Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science. 2017;357:1399–402.
Wang S, Ghosh AK, Bongio N, Stebbings KA, Lampe DJ, Jacobs-Lorena M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci USA. 2012;109:12734–9.
Wang S, Jacobs-Lorena M. Genetic approaches to interfere with malaria transmission by vector mosquitoes. Trends Biotechnol. 2013;31:185–93.
You Y-H, Park JM, Yi P-H, Back C-G, Park M-J, Han K-S, et al. Microflora of phytopathogen-transferring Bradysia agrestis: a step toward finding ideal candidates for paratransgenesis. Symbiosis. 2017;71:35–46.
Koosha M, Vatandoost H, Karimian F, Choubdar N, Abai MR, Oshaghi MA. Effect of serratia AS1 (Enterobacteriaceae: Enterobacteriales) on the fitness of Culex pipiens (Diptera: Culicidae) for paratransgenic and RNAi approaches. J Med Entomol. 2018;56:553–9.
Gilbert JA, Medlock J, Townsend JP, Aksoy S, Mbah MN, Galvani AP. Determinants of human African trypanosomiasis elimination via paratransgenesis. PLoS Negl Trop Dis. 2016;10:e0004465.
Damiani C, Ricci I, Crotti E, Rossi P, Rizzi A, Scuppa P, et al. Paternal transmission of symbiotic bacteria in malaria vectors. Curr Biol. 2008;18:R1087–8.
Favia G, Ricci I, Marzorati M, Negri I, Alma A, Sacchi L, et al. Bacteria of the genus Asaia: a potential paratransgenic weapon against malaria. In: Cohen I, Lajtha A, Paoletti R, Lambris JD, editors. Transgenesis and the management of vector-borne disease. Berlin: Springer; 2008. p. 49–59.
Chouaia B, Gaiarsa S, Crotti E, Comandatore F, Degli Esposti M, Ricci I, et al. Acetic acid bacteria genomes reveal functional traits for adaptation to life in insect guts. Genome Biol Evol. 2014;6:912–20.
Chouaia B, Rossi P, Montagna M, Ricci I, Crotti E, Damiani C, et al. Molecular evidence for multiple infections as revealed by typing of Asaia bacterial symbionts of four mosquito species. Appl Environ Microbiol. 2010;76:7444–50.
Crotti E, Damiani C, Pajoro M, Gonella E, Rizzi A, Ricci I, et al. Asaia, a versatile acetic acid bacterial symbiont, capable of cross-colonizing insects of phylogenetically distant genera and orders. Environ Microbiol. 2009;11:3252–64.
De Freece C, Damiani C, Valzano M, D’amelio S, Cappelli A, Ricci I, et al. Detection and isolation of the α-proteobacterium Asaia in Culex mosquitoes. Med Vet Entomol. 2014;28:438–42.
Rami A, Raz A, Zakeri S, Djadid ND. Isolation and identification of Asaia sp. in Anopheles spp. mosquitoes collected from Iranian malaria settings: steps toward applying paratransgenic tools against malaria. Parasites Vectors. 2018;11:367.
Gonella E, Crotti E, Rizzi A, Mandrioli M, Favia G, Daffonchio D, et al. Horizontal transmission of the symbiotic bacterium Asaia sp. in the leafhopper Scaphoideus titanus Ball (Hemiptera: Cicadellidae). BMC Microbiol. 2012;12:S4.
Solis CF, Santi-Rocca J, Perdomo D, Weber C, Guillén N. Use of bacterially expressed dsRNA to downregulate Entamoeba histolytica gene expression. PLoS ONE. 2009;4:e8424.
Tian H, Peng H, Yao Q, Chen H, Xie Q, Tang B, et al. Developmental control of a lepidopteran pest Spodoptera exigua by ingestion of bacteria expressing dsRNA of a non-midgut gene. PLoS ONE. 2009;4:e6225.
Yu N, Christiaens O, Liu J, Niu J, Cappelle K, Caccia S, et al. Delivery of dsRNA for RNAi in insects: an overview and future directions. Insect Sci. 2013;20:4–14.
Zhu F, Xu J, Palli R, Ferguson J, Palli SR. Ingested RNA interference for managing the populations of the Colorado potato beetle, Leptinotarsa decemlineata. Pest Manag Sci. 2011;67:175–82.
Gan D, Zhang J, Jiang H, Jiang T, Zhu S, Cheng B. Bacterially expressed dsRNA protects maize against SCMV infection. Plant Cell Rep. 2010;29:1261–8.
Kumar DR, Kumar PS, Gandhi MR, Al-Dhabi NA, Paulraj MG, Ignacimuthu S. Delivery of chitosan/dsRNA nanoparticles for silencing of wing development vestigial (vg) gene in Aedes aegypti mosquitoes. Int J Biol Macromol. 2016;86:89–95.
Li X, Zhang M, Zhang H. RNA interference of four genes in adult Bactrocera dorsalis by feeding their dsRNAs. PLoS ONE. 2011;6:e17788.
Posiri P, Ongvarrasopone C, Panyim S. A simple one-step method for producing dsRNA from E. coli to inhibit shrimp virus replication. J Virol Methods. 2013;188:64–9.
Sarathi M, Simon M, Venkatesan C, Thomas J, Ravi M, Madan N, et al. Efficacy of bacterially expressed dsRNA specific to different structural genes of white spot syndrome virus (WSSV) in protection of shrimp from WSSV infection. J Fish Dis. 2010;33:603–7.
Timmons L, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–12.
Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2000;2:research0002.
Saksmerprome V, Charoonnart P, Gangnonngiw W, Withyachumnarnkul B. A novel and inexpensive application of RNAi technology to protect shrimp from viral disease. J Virol Methods. 2009;162:213–7.
Somchai P, Jitrakorn S, Thitamadee S, Meetam M, Saksmerprome V. Use of microalgae Chlamydomonas reinhardtii for production of double-stranded RNA against shrimp virus. Aquac Rep. 2016;3:178–83.
Court D. RNA processing and degradation by RNase III. New York: Academic Press; 1993. p. 71–116.
Filippov V, Solovyev V, Filippova M, Gill SS. A novel type of RNase III family proteins in eukaryotes. Gene. 2000;245:213–21.
Krainer A. Eukaryotic mRNA processing. Oxford: IRL Press at Oxford University Press; 1997.
Nicholson AW. Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol Rev. 1999;23:371–90.
Robertson HD, Webster RE, Zinder ND. Purification and properties of ribonuclease III from Escherichia coli. J Biol Chem. 1968;243:82–91.
Court DL, Gan J, Liang Y-H, Shaw GX, Tropea JE, Costantino N, et al. RNase III: genetics and function; structure and mechanism. Annu Rev Genet. 2013;47:405–31.
Djadid ND, Jazayeri H, Raz A, Favia G, Ricci I, Zakeri S. Identification of the midgut microbiota of An. stephensi and An. maculipennis for their application as a paratransgenic tool against malaria. PLoS ONE. 2011;6:e28484.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Shahgudian ER. A key to the anophelines of Iran. Acta Med Iran. 1960;3:38–48
Raz A, Djadid ND, Zakeri S. Molecular characterization of the carboxypeptidase B1 of Anopheles stephensi and its evaluation as a target for transmission-blocking vaccines. Infect Immun. 2013;81:2206–16.
Khamsehnejad MI, Djadid ND, Raz A. Identification, molecular characterization, and in silico structural analysis of carboxypeptidase B2 of Anopheles stephensi. J Med Entomol. 2018;56:72–85.
Yamada Y, Katsura K, Kawasaki H, Widyastuti Y, Saono S, Seki T, et al. Asaia bogorensis gen. nov., sp. nov., an unusual acetic acid bacterium in the alpha-Proteobacteria. Int J Syst Evol Microbiol. 2000;50:823–9.
Alipour H, Raz A, Zakeri S, Djadid ND. Molecular characterization of matrix metalloproteinase-1 (MMP-1) in Lucilia sericata larvae for potential therapeutic applications. Electron J Biotechnol. 2017;29:47–56.
Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4.
Lavazec C, Boudin C, Lacroix R, Bonnet S, Diop A, Thiberge S, et al. Carboxypeptidases B of Anopheles gambiae as targets for a Plasmodium falciparum transmission-blocking vaccine. Infect Immun. 2007;75:1635–42.
Baum JA, Bogaert T, Clinton W, Heck GR, Feldmann P, Ilagan O, et al. Control of coleopteran insect pests through RNA interference. Nat Biotechnol. 2007;25:1322.
Coy M, Sanscrainte N, Chalaire K, Inberg A, Maayan I, Glick E, et al. Gene silencing in adult Aedes aegypti mosquitoes through oral delivery of double-stranded RNA. J Appl Entomol. 2012;136:741–8.
Ongvarrasopone C, Roshorm Y, Panyim S. A simple and cost effective method to generate dsRNA for RNAi studies in invertebrates. ScienceAsia. 2007;33:35–9.
Kruger NJ. The Bradford method for protein quantitation. In: Walker JM, editor. The protein protocols handbook. Berlin: Springer; 2002. p. 15–21.
Amarasinghe AK, Calin-Jageman I, Harmouch A, Sun W, Nicholson AW. Escherichia coli ribonuclease III: affinity purification of hexahistidine-tagged enzyme and assays for substrate binding and cleavage. Methods Enzymol. 2001;342:143–58.
Abou Elela S, Ares M Jr. Depletion of yeast RNase III blocks correct U2 3ʹ end formation and results in polyadenylated but functional U2 snRNA. EMBO J. 1998;17:3738–46.
Blomberg P, Wagner E, Nordström K. Control of replication of plasmid R1: the duplex between the antisense RNA, CopA, and its target, CopT, is processed specifically in vivo and in vitro by RNase III. EMBO J. 1990;9:2331–40.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845.
Webb B, Sali A. Protein structure modeling with MODELLER. In: Kihara D, editor. Functional genomics. Berlin: Springer; 2017. p. 39–54.
Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2010;27:343–50.
Bienert S, Waterhouse A, de Beer TA, Tauriello G, Studer G, Bordoli L, et al. The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res. 2016;45:D313–9.
Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2008;4:1–13.
Coll M, Guasch A, Aviles F, Huber R. Three-dimensional structure of porcine procarboxypeptidase B: a structural basis of its inactivity. EMBO J. 1991;10:1–9.
Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Šali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325.
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91.
Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, et al. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–9.
Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–9.
Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–70.
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Suppl. 2):W407–10.
Berjanskii M, Liang Y, Zhou J, Tang P, Stothard P, Zhou Y, et al. PROSESS: a protein structure evaluation suite and server. Nucleic Acids Res. 2010;38(Suppl. 2):W633–40.
Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9.
Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41:W384–8.
Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30:S162–73.
Van Zundert G, Rodrigues J, Trellet M, Schmitz C, Kastritis P, Karaca E, et al. The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–5.
Wassenaar TA, Van Dijk M, Loureiro-Ferreira N, Van Der Schot G, De Vries SJ, Schmitz C, et al. WeNMR: structural biology on the grid. J Grid Comput. 2012;10:743–67.
Salamov VSA, Solovyevand A. Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW, editor. Metagenomics and its applications in agriculture, biomedicine and environmental studies. Hauppauge: Nova Science Publishers; 2011. p. 61–78.
Gautheret D, Lambert A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol. 2001;313:1003–11.
Blaszczyk J, Tropea JE, Bubunenko M, Routzahn KM, Waugh DS, Ji X. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure. 2001;9:1225–36.
Nicholson AW. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscip Rev RNA. 2014;5:31–48.
Pereira PJB, Segura-Martın S, Oliva B, Ferrer-Orta C, Aviles FX, Coll M, et al. Human procarboxypeptidase B: three-dimensional structure and implications for thrombin-activatable fibrinolysis inhibitor (TAFI). J Mol Biol. 2002;321:537–47.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86.
Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363.
Blaszczyk J, Gan J, Tropea JE, Waugh DS, Ji X. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004;12:457–66.
Carthew RW. Gene silencing by double-stranded RNA. Curr Opin Cell Biol. 2001;13:244–8.
MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol. 2007;17:138–45.
Gan J, Shaw G, Tropea JE, Waugh DS, Court DL, Ji X. A stepwise model for double-stranded RNA processing by ribonuclease III. Mol Microbiol. 2008;67:143–54.
Gan J, Tropea JE, Austin BP, Waugh DS, Ji X. Intermediate states of ribonuclease III in complex with double-stranded RNA. Structure. 2005;13:1435–42.
Wu C-X, Xu X-J, Zheng K, Liu F, Yang X-D, Chen C-F, et al. Characterization of ribonuclease III from Brucella. Gene. 2016;579:183–92.
March PE, Ahnn J, Inouye M. The DNA sequence of the gene (rnc) encoding ribonuclease III of Escherichia coli. Nucleic Acids Res. 1985;13:4677–85.
Takiff HE, Chen S-M. Genetic analysis of the rnc operon of Escherichia coli. J Bacteriol. 1989;171:2581–90.
Wang W, Bechhofer DH. Bacillus subtilis RNase III gene: cloning, function of the gene in Escherichia coli, and construction of Bacillus subtilis strains with altered rnc loci. J Bacteriol. 1997;179:7379–85.
Chen S-M, Takiff H, Barber A, Dubois G, Bardwell J. Expression and characterization of RNase III and Era proteins. Products of the rnc operon of Escherichia coli. J Biol Chem. 1990;265:2888–95.
Anderson P, Matsunaga J, Simons E, Simons R. Structure and regulation of the Salmonella typhimurium rnc-era-recO operon. Biochimie. 1996;78:1025–34.
Rauhut R, Conrad C, Klug G. Identification and analysis of the rnc gene for RNase III in Rhodobacter capsulatus. Nucleic Acids Res. 1996;24:1246–51.
Powell B, Peters HK, Nakamura Y. Cloning and analysis of the rnc-era-recO operon from Pseudomonas aeruginosa. J Bacteriol. 1999;181:5111–3.
Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol Rev. 1983;47:1–45.
Gan J, Tropea JE, Austin BP, Waugh DS, Ji X. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006;124:355–66.
Masliah G, Barraud P, Allain FH-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013;70:1875–95.
Acknowledgements
We thank our colleagues in the Malaria Control program, Center for Diseases Management and Control and our colleagues in Malaria and Vector Research Group (MVRG) and National Insectary of Pasteur Institute of Iran (PII) for their kind assistance during the performing of this project.
Funding
This project was funded by the educational office of PII to MA and Iran National Science Foundation (INSF) Grant 94810113 to AR.
Author information
Authors and Affiliations
Contributions
AR and NDD conceived the study. AR, NDD, MA and MI participated in the design of the study. MA performed the assays and wrote the first draft of the manuscript. ER performed the in silico analysis. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Molecular confirmation of the isolated Asaia sp.. Molecular confirmation was performed based on 16S rRNA gene amplification by the Asaia-specific primers. Lane M: DNA ladder; Lane 1: isolated bacterium; Lane 2: positive control; Lane 3: non-template control. The amplification of a 1200-bp amplicon confirmed that the isolated bacterium is Asaia sp.
Additional file 2: Figure S2.
Sequence alignment of the predicted models and template by Clustal Omega. The sequence alignment of the predicted models by SWISS-MODEL, Modeller and 4M30 (as template) has been depicted to reveal the differences in the sequences that have an effect on the folding pattern. The conserved amino acids are shown in blue. Consensus residues are indicated by black rectangles.
Additional file 3: Figure S3.
Three-dimensional structure of RNase III protein from the isolated Asaia sp. Three-dimensional structure prediction of our target protein was performed using SWISS MODEL and Phyre2 servers based on homology modeling. Our result shows that the RNase III from the isolated Asaia sp. is a globular protein with two distinct subunits which are connected together by a linker. a, d Front view of our target protein. b, e The complex of RNase III and dsRNA molecule. c, f Monomeric lateral view. N-terminal catalytic domains (RIIID), C-terminal dsRNA binding domain (dsRBD), dsRNA molecule, signature motif and liker are illustrated by purple, green, gray, yellow and red, respectively.
Additional file 4: Figure S4.
The active site structure of Asaia sp. RNase III enzyme and its interaction with divalent cations. Six acidic residues are important and involved in catalytic domain construction and interaction with divalent cations. The specific residues have been determined and numbered and divalent ions are indicated with black circles. Their interactions are indicated by black lines.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Asgari, M., Ilbeigikhamsehnejad, M., Rismani, E. et al. Molecular characterization of RNase III protein of Asaia sp. for developing a robust RNAi-based paratransgensis tool to affect the sexual life-cycle of Plasmodium or Anopheles fitness. Parasites Vectors 13, 42 (2020). https://doi.org/10.1186/s13071-020-3889-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-020-3889-6