
Abstract
Background
S-Adenosylmethionine (SAM) is a critical cofactor involved in many biochemical reactions. However, the low fermentation titer of SAM in methionine-free medium hampers commercial-scale production. The SAM synthesis pathway is specially related to the tricarboxylic acid (TCA) cycle in Bacillus amyloliquefaciens. Therefore, the SAM synthesis pathway was engineered and coupled with the TCA cycle in B. amyloliquefaciens to improve SAM production in methionine-free medium.
Results
Four genes were found to significantly affect SAM production, including SAM2 from Saccharomyces cerevisiae, metA and metB from Escherichia coli, and native mccA. These four genes were combined to engineer the SAM pathway, resulting in a 1.42-fold increase in SAM titer using recombinant strain HSAM1. The engineered SAM pathway was subsequently coupled with the TCA cycle through deletion of succinyl-CoA synthetase gene sucC, and the resulted HSAM2 mutant produced a maximum SAM titer of 107.47 mg/L, representing a 0.59-fold increase over HSAM1. Expression of SAM2 in this strain via a recombinant plasmid resulted in strain HSAM3 that produced 648.99 mg/L SAM following semi-continuous flask batch fermentation, a much higher yield than previously reported for methionine-free medium.
Conclusions
This study reports an efficient strategy for improving SAM production that can also be applied for generation of SAM cofactors supporting group transfer reactions, which could benefit metabolic engineering, chemical biology and synthetic biology.
Similar content being viewed by others


Background
S-Adenosylmethionine (SAM) is one of the most widely used cofactors for group transfer reactions involved in various metabolic processes, and it serves as the main methyl donor for methylation of DNA, proteins and secondary metabolites, as well as 5-deoxyadenosyl radical, 1-aminopropyl, and 2-aminobutyryl donors [1, 2]. SAM-dependent group transfer reactions can be broadly applied in the fields of chemical biology, synthetic biology and metabolic engineering [3,4,5]. In particular, SAM-dependent methylation is the critical step for production of pharmaceuticals and fine chemicals [3, 4]. Moreover, SAM has multiple beneficial effects for human health and has been used as a functional nutriment or drug for the prevention and treatment of liver disease, osteoarthritis and depression [6, 7]. SAM also has the potential for extending lifespan and treating colon cancer [8, 9]. Given the critical functions and market demand, SAM is attracting much interest.
SAM can be synthesized by SAM synthetase from methionine and ATP [7, 10]. Low availability of the precursor methionine is considered as the limiting factor for SAM production [11,12,13]. Consequently, methionine is usually added directly into the medium as a fermentation substrate to produce SAM and various previous studies have aimed to improve SAM production from methionine, including fermentation optimization, conventional mutation breeding, and genetic engineering [7, 14]. The conversion rate from expensive methionine to SAM ranges from 15 to 42% [15,16,17], resulting in high cost. Therefore, development of efficient SAM production methods using methionine-free medium is much needed.
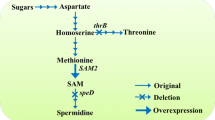
Researchers have attempted to manipulate the SAM biosynthesis pathway to produce SAM using low-cost carbohydrates or aspartate as substrates [13, 15, 18]. In this pathway, aspartate is synthesized from glucose via the glycolytic pathway and the tricarboxylic acid (TCA) cycle and then converted to SAM through the SAM synthesis pathway (Fig. 1), which includes aspartokinase (encoded by lysC), bifunctional aspartokinase/homoserine dehydrogenase (thrA or metL), aspartate-semialdehyde dehydrogenase (asd), homoserine dehydrogenase (hom), homoserine O-succinyltransferase (metA), cystathionine-γ-synthase (metI, metB or YML082W), cystathionine-β-lyase (metC), cystathionine-β-synthase (mccA), methionine synthase (metE) and SAM synthetase (metK or SAM2) [15, 18]. In Escherichia coli, the SAM titer was improved in methionine-free medium via regulation of NADPH and ATP, but remained below 10 mg/L [13, 18]. In Corynebacterium glutamicum, four genes (mcbR, thrB, metB and Ncgl2640) were deleted and two genes (vgb and metK) were overexpressed, resulting in a maximum SAM titer of 196.7 mg/L in methionine-free medium [15]. Although the SAM titer was improved in methionine-free medium, it is still not high enough from a commercial production perspective. Therefore, effort is needed to fully explore the production potential using low-cost substrates.

Metabolic engineering strategies for coupling the engineered SAM synthesis pathway and the TCA cycle in Bacillus amyloliquefaciens. Genes are as follows: aspartokinase (encoded by lysC), bifunctional aspartokinase/homoserine dehydrogenase (thrA or metL), aspartate-semialdehyde dehydrogenase (asd), homoserine dehydrogenase (hom), homoserine O-succinyltransferase (metA), cystathionine-γ-synthase (metI, metB or YML082W), cystathionine-β-lyase (metC), cystathionine-β-synthase (mccA), methionine synthase (metE), SAM synthetase (metK or SAM2), succinyl-CoA synthetase (sucCD)
Coupling to the TCA cycle can be effective for improving the production of lysine and 7-aminodeacetoxycephalosporanic acid [19, 20]. In the SAM synthesis pathway of some bacteria (Fig. 1), succinyl-CoA serves as the cosubstrate with homoserine to synthesize O-succinylhomoserine, which is then converted to cystathionine and succinate. On the other hand, succinyl-CoA can be converted to succinate by succinyl-CoA synthetase in the TCA cycle [19, 20]. Thus, the TCA cycle competes for succinyl-CoA with the SAM pathway. If the native TCA cycle is blocked by disrupting succinyl-CoA synthetase, more succinyl-CoA may be driven to flux into the SAM pathway to promote SAM production. If the SAM pathway is strong enough, it could generate abundant succinate to compensate for the blocked TCA cycle, supporting recovery of cell growth. The SAM pathway and the TCA cycle could, therefore, be coupled to enhance SAM production collaboratively, but this has not yet been demonstrated in practice.
Bacillus species, especially Bacillus subtilis, Bacillus amyloliquefaciens and Bacillus licheniformis, have been widely applied in the fields of medicine, food, cosmetics and agriculture [21, 22]. Due to the rapid growth, high robustness and rich genetic tools, Bacillus species have been engineered as industrial workhorses for production of various products [23], such as engineering B. subtilis to synthesize riboflavin, pyridoxine, N-acetylglucosamine and hyaluronan [24,25,26,27], and engineering B. amyloliquefaciens to produce poly-γ-glutamic acid, levan and menaquinone-7 [28,29,30]. B. amyloliquefaciens is a promising chassis cell and related metabolic engineering tools have been developed. Moreover, B. amyloliquefaciens deficient in tetrahydropicolinate succinylase cannot mediate succinyl-CoA into the lysine pathway, while it may just drive succinyl-CoA into the SAM synthesis pathway by homoserine O-succinyltransferase. Therefore, B. amyloliquefaciens have the potential to couple the SAM synthesis pathway with the TCA cycle to enhance SAM production. The present study aimed to enhance SAM production using B. amyloliquefaciens HZ-12 in methionine-free medium by coupling a strong SAM synthesis pathway with the TCA cycle. Associated genes were engineered systematically to strengthen the SAM pathway and succinyl-CoA synthetase was disrupted to force succinyl-CoA flux through the engineered SAM pathway to enter the TCA cycle, thereby coupling the engineered SAM pathway and the TCA cycle (Fig. 1).
Results and discussion
Effects of key genes on SAM production
SAM synthetase is a critical enzyme that catalyzes SAM synthesis from methionine and ATP, and overexpression of the genes involved can enhance SAM production, especially the SAM2 gene from S. cerevisiae [14, 31]. However, whether B. amyloliquefaciens SAM synthetase gene metK affects SAM production has not been investigated. Therefore, metK from B. amyloliquefaciens and SAM2 from S. cerevisiae were expressed in HZ-12, resulting in recombinant strains HZ-12 (PBmetK) and HZ-12 (PSAM2), respectively. These recombinant strains were fermented for 42 h in initial fermented medium to compare SAM titers. As shown in Fig. 2a, expression of metK and SAM2 significantly improved SAM titers, and the maximum SAM titer (66.22 mg/L) was achieved by HZ-12 (PSAM2). These results indicate that SAM2 is the preferred target gene for enhancement of SAM production, similar to the results of other studies [7, 31].

Effects of key genes on SAM production. a HZ-12 (PBmetK) expressing metK from B. amyloliquefaciens and HZ-12 (PSAM2) expressing SAM2 from S. cerevisiae. b HZ-12 (PBmetA) expressing metA from B. amyloliquefaciens and HZ-12 (PEmetA) expressing metA from E. coli. c HZ-12 (PBmetI) expressing metI from B. amyloliquefaciens, HZ-12 (PEmetB) expressing metB from E. coli, and HZ-12 (PYML) expressing YML082 W from S. cerevisiae. d HZ-12ΔmccA carrying deleted mccA. All data were obtained under the initial fermentation medium. Data are expressed as mean ± standard deviation (SD) from triplicate measurements. Asterisks indicate a significant difference (p < 0.05) compared with the control strain (HZ-12)
Homoserine O-succinyltransferase (encoded by metA) combines homoserine and succinyl-CoA to synthesize O-succinylhomoserine [32]. This step is critical for driving succinyl-CoA into the SAM pathway, hence strengthening the expression of metA is likely to increase competition for succinyl-CoA for SAM synthesis. In E. coli, metA can efficiently enhance the production of methionine, the precursor of SAM [33, 34]. In the present work, metA genes from E. coli and B. amyloliquefaciens were expressed and compared (Fig. 2b). In HZ12 (PEmetA) harboring metA from E. coli, the SAM titer reached 48.28 mg/L after 42 h in initial fermentation medium, higher than that of wild-type strain HZ-12, whereas overexpression of native metA in HZ-12 yielded no significant enhancement. These results confirmed that metA from E. coli is crucial for enhancing SAM production by B. amyloliquefaciens.
Cystathionine-γ-synthase catalyzes the reaction between O-succinylhomoserine and cysteine to produce cystathionine and succinate, a key step in the generation of succinate from the SAM pathway. Overexpression of the cystathionine-γ-synthase gene can also significantly improve methionine production in E. coli [33]. In the present work, cystathionine-γ-synthase genes from HZ-12 (metI), E. coli (metB) and S. cerevisiae (YML082W) were expressed, resulting in recombinant strains HZ-12 (PBmetI), HZ-12 (PEmetB) and HZ-12 (PYML), respectively. As shown in Fig. 2c, expression of metB and YML082 W markedly improved SAM titers in initial fermentation medium, while no significant change was observed for the metI-expressing strain. Since strain HZ-12 (PEmetB) produced the maximum SAM titer of 31.06 mg/L, metB from E. coli was chosen for further investigation.
Cystathionine-β-synthase catalyzes the conversion of homocysteine to cystathionine, a reverse reaction in the SAM synthesis pathway, and knockout of the cystathionine-β-synthase gene can improve SAM production in Pichia pastoris [35]. Therefore, we deleted the mccA gene encoding cystathionine-β-synthase in HZ-12, and the SAM titer reached 42.56 mg/L in HZ-12ΔmccA in initial fermentation medium, much higher than that of HZ-12 (Fig. 2d). In addition, we also manipulated other genes in the SAM synthesis pathway, including expression of bifunctional aspartokinase/homoserine dehydrogenase genes (thrA and metL) [36], and deletion of the anti-antiterminator of the metE gene in HZ-12 [37]. However, expression of thrA and metL significantly reduced SAM production (Additional file 1: Fig. S1), and the SAM titer was only slightly improved after deletion of the anti-antiterminator of the metE gene (Additional file 1: Fig. S2). Various genes influencing SAM synthesis were identified, including SAM2 from S. cerevisiae, metA and metB from E. coli, as well as the native mccA, among which SAM2 had the strongest effect on SAM synthesis.
Combined effects of beneficial SAM synthesis pathway genes
All the aforementioned beneficial genetic manipulations were combined in a single strain. Specifically, SAM2 from S. cerevisiae and metA and metB from E. coli were integrated into the genome of HZ-12, and the mccA gene was deleted, resulting in strain HSAM1. Fermentation profiles of strains HZ-12 and HSAM1 were compared in initial fermentation medium. As shown in Fig. 3a, the SAM titer of HSAM1 increased with the increasing fermentation time, and the maximum titer reached 67.41 mg/L at 48 h, 1.42-fold higher than that of HZ-12 (27.91 mg/mL). The cell growth trend of HSAM1 was similar to that of HZ-12 (Fig. 3a). The above results showed that the SAM titer of strain HSAM1 was not significantly improved over that of strain HZ-12 harboring plasmid PSAM2. This result was probably due to the low gene copy number during integration expression. Each HSAM1 cell carries only a single copy of the integrated SAM2, metA and metB genes, whereas there are 5–20 copies of the SAM2 gene in each HZ-12 (PSAM2) cell due to the high copy number of the pHY300PLK-based plasmid [38].

Effects of combining beneficial genes on SAM synthesis. a SAM titer and cell growth. b Relative transcription levels of SAM synthesis pathway-related genes. c Fold change of key metabolites. All data were obtained under the initial fermentation medium. Data are expressed as mean ± SD from triplicate measurements. Asterisks indicate a significant difference (p < 0.05) between HZ-12 and HSAM1
Transcription levels of native SAM synthesis pathway genes between HSAM1 and HZ-12 were also compared. As shown in Fig. 3b, transcription levels of most native SAM synthesis genes in strain HSAM1 were higher than those in HZ-12, with ~ twofold improvement observed for metA, metC and metE. These results indicate that combining genetic manipulations enhances the overall SAM synthesis pathway. The transcription level of native metK gene showed no significant difference, probably because the integrated SAM2 gene was strong enough to mediate the SAM synthesis. Changes in the key intracellular metabolites of the SAM synthesis pathway were also measured. In HSAM1, the abundance of aspartate and homocysteine, located in the upstream pathway of modified genes, was 22% and 61% lower, respectively, than in HZ-12 (Fig. 3c), indicating that combined downstream genetic manipulations affected upstream substrate consumption. As expected for the intermediate metabolite of the modified gene products, methionine was also decreased significantly (Fig. 3c), probably because the SAM2 gene is more efficient than metA, metB and mccA. The greater substrate consumption further confirmed that the combined genetic manipulations drove metabolic flux for improved SAM synthesis.
Coupling the engineered SAM synthesis pathway with the TCA cycle
The biosynthesis of SAM requires the cosubstrate succinyl-CoA, which is also a key intermediate of the TCA cycle [19, 20]. The TCA cycle competes with the SAM synthesis pathway for succinyl-CoA. If the succinyl-CoA synthetase gene is deleted to block succinyl-CoA consumption by the TCA cycle, the SAM pathway would likely receive more succinyl-CoA, thereby enhancing SAM synthesis. Moreover, the improved SAM pathway would be expected to release more succinate to compensate for the TCA cycle and cell growth. Thus, the SAM pathway would presumably couple with the TCA cycle to coordinate SAM production and cell growth. To verify this hypothesis, succinyl-CoA synthetase was inactivated by deleting the sucC gene from HSAM1, resulting in strain HSAM2. As shown in Fig. 4a, the SAM titer of HSAM2 reached 107.47 mg/L in the initial fermentation medium, representing a 0.59-fold increase over that of HSAM1.

Effects of pathway coupling on SAM synthesis. a SAM titer and cell growth. b Relative transcription levels of SAM synthesis pathway-related genes. c Fold change of key metabolites. All data were obtained under the initial fermentation medium. Data are expressed as mean ± SD from triplicate measurements. Asterisks indicate a significant difference (p < 0.05) between HSAM1 and HSAM2
Transcription analysis was also performed to investigate the effect of sucC deletion on the SAM synthesis pathway. Compared with the control strain, transcription levels of most SAM pathway-associated genes were increased (Fig. 4b), especially the integrated SAM2 gene, and metA and metB from E. coli (designated as EmetA and EmetA), all elevated ~ twofold, which were much higher than that of corresponding native genes (metA, metI and metK) of B. amyloliquefaciens. According to Fig. 1, genes of EmetA, EmetB and SAM2 were more effective than corresponding native genes in B. amyloliquefaciens to improve SAM production, suggesting that those three integrated genes were more efficient to affect the SAM synthesis pathway. Bacteria preferably utilized efficient genes and enzymes to cope with stress perturbations. Therefore, transcriptional levels of these three integrated genes were improved more obviously than that of wild-type genes after deletion of sucC. These results confirmed that deletion of sucC forced more succinyl-CoA through the SAM synthesis pathway. GC–MS analysis showed that the medium substrate aspartate was reduced by 57% after knockout of sucC, and the downstream metabolite methionine was increased by 41% (Fig. 4c), further confirming that the SAM synthesis pathway was enhanced. These results indicate that our pathway coupling strategy could also be applied to enhance methionine production.
Conversely, no significant concentration change was observed for intracellular succinate or fumarate intermediates of the TCA cycle (Fig. 4c), and cell growth did not decline after deletion of sucC (Fig. 4a), suggesting that succinate generated from the engineered SAM pathway likely compensates for the TCA cycle and cell growth. In C. glutamicum, succinyl-CoA synthetase was mutated to couple the TCA cycle and the lysine synthesis pathway, leading to a 0.60-fold increase in lysine yield, and succinate generated from the lysine pathway and the glyoxylate shunt was believed to compensate for the TCA cycle and cell growth in C. glutamicum [19]. Bacillus species usually utilize acetyl-CoA as cosubstrate for lysine synthesis instead of succinyl-CoA [39]. KEGG database analysis showed that, unlike C. glutamicum, B. amyloliquefaciens lacks tetrahydrodipicolinate succinylase, the enzyme responsible for feeding succinyl-CoA into the lysine pathway. According to the KEGG database, a glyoxylate shunt is not present in B. amyloliquefaciens, and the metabolite glyoxylate was not detected in our GC–MS analysis. Therefore, succinate generated from the engineered SAM pathway was believed to compensate for the TCA cycle and cell growth after deletion of sucC from HSAM1. We also compared cell growth between the wild-type strain (HZ-12), the sucC-deficient strain (HZ-12ΔsucC) and the sucC-deficient strain harboring the engineered SAM pathway (HSAM2). As shown in Additional file 1: Fig. S3, cell growth of HZ-12ΔsucC was much lower than HZ-12, indicating that the native SAM pathway could not recover cell growth completely after deletion of sucC. Interestingly, HSAM2 showed no significant difference from HZ-12, confirming that the engineered SAM pathway could compensate for the TCA cycle to maintain normal cell growth in the sucC-deficient strain.
SAM synthesis from HSAM3
To improve SAM production, plasmid PSAM2 carrying the most efficient SAM2 gene was transformed into HSAM2 to generate HSAM3. Then, SAM titers of four key strains (HZ-12, HSAM1, HSAM2 and HSAM3) were compared under initial fermentation medium. Therein, the strain HSAM3 produced the maximum SAM titer of 226.61 mg/L, much higher than other strains (Fig. 5a). To reduce fermentation cost of HSAM3, the medium was optimized by adjustment of glucose and aspartate. Removing the glucose showed no significant effect on SAM titer, while further eliminating the aspartate significantly improved the SAM titer (Additional file 1: Fig. S4). Using optimized fermentation medium without glucose and aspartate, the maximum SAM titer reached 290.35 mg/L in 250 mL flasks containing 25 mL medium (Fig. 5b) and it was further improved to 648.99 mg/L by adding 2.5 mL of 10× optimized fermentation medium at 24 h and 48 h (semi-continuous batch fermentation), which was partly due to the increased cell growth (Fig. 5b). The batch and semi-continuous batch fermentation were also performed in a 3 L bioreactor, while the maximum SAM titer reached 242.01 mg/L and 424.21 mg/L, respectively (Additional file 1: Fig. S5). B. amyloliquefaciens can produce multiple surfactants, which led to the generation of excess foam in the 3 L bioreactor. Therefore, defoamer was added and the agitation speed was maintained at 400 rpm to avoid foam escaping from the bioreactor, which probably hindered SAM production.

SAM synthesis of the strain HSAM3 in flasks. a SAM titer of HSAM3 compared with different strains in initial fermentation medium. b SAM titer and cell growth of HSAM3 under batch and semi-continuous batch fermentation based on the optimized fermentation medium. Data are expressed as mean ± SD from triplicate measurements. Asterisks indicate a significant difference at p < 0.05
This study aimed to improve SAM production using methionine-free medium. Therefore, previously reported SAM production results using methionine-free medium were compared at the flask scale (Table 1). The titer (290.35 mg/L), yield (30.28 mg/g DCW) and productivity (4.84 mg/L h) of HSAM3 in flask batch fermentation were much higher than previously reported [13, 15, 18]. Furthermore, the titer, yield and productivity were further improved by semi-continuous batch fermentation, demonstrating potential industrial applicability.
Conclusions
This study presents an efficient strategy for enhancing SAM production by coupling an engineered SAM synthesis pathway and the TCA cycle. Recombinant plasmid-based expression of SAM2 from S. cerevisiae and metA and metB from E. coli, in combination with deletion of mccA, resulted in significantly enhanced SAM production. Combining these genetic manipulations achieved a maximum SAM titer of 67.41 mg/L using the resulted strain HSAM1, a 1.42-fold increase compared with strain HZ-12. Transcription and metabolite analyses confirmed that the SAM synthesis pathway was strengthened. Unlike C. glutamicum using tetrahydropicolinate succinylase to drive succinyl-CoA into the lysine pathway, B. amyloliquefaciens just harbored homoserine O-succinyltransferase to mediate succinyl-CoA into the SAM pathway. Therefore, deleting the sucC gene forced the succinyl-CoA into the engineered SAM pathway, which was consequently coupled with the TCA cycle to generate a 0.59-fold increase of the SAM titer. Transcription and metabolite analyses also confirmed that pathway coupling diverted the metabolic flux toward the SAM pathway. Addition of plasmid-based expression of SAM2 further increased the maximum SAM titer to 648.99 mg/L in methionine-free medium, much higher than previous reports. Thus, we developed an efficient strategy for enhanced SAM production, and provide a potential SAM production workhorse in the form of engineered B. amyloliquefaciens.
Methods
Strains and plasmids
All strains and plasmids used in this study are listed in Table 2. All resulted B. amyloliquefaciens strains were derived from the wild-type strain B. amyloliquefaciens HZ-12. E. coli DH5α was used to construct plasmids and amplify metA, metB, thrA and metL, and Saccharomyces cerevisiae CICC 31001 was used for amplification of SAM2 and YML082 W genes. Primers used in this study are included in Additional file 1: Table S1.
Medium and culture conditions
Cells were inoculated into LB liquid medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl), and cultured for 12 h at 37 °C with shaking at 200 rpm to generate seed cultures. Seed cultures (inoculum size 3%) were then transferred into the fermentation medium. The 250 mL flask experiment was performed with 25 mL medium at 37 °C and 200 rpm. The 3 L bioreactor experiment was carried out in 1.2 L medium at 37 °C with an agitation speed of 400 rpm and an aeration ratio of 1.6 vvm. The initial fermentation medium comprised 10 g/L glucose, 40 g/L sucrose, 3 g/L aspartate, 10 g/L peptone, 5 g/L beef extract, 2 g/L urea, 6.3 g/L (NH4)2SO4, 2.5 g/L NaCl, 3 g/L KH2PO4 and 4.2 g/L MgSO4·7H2O (pH 6.5), which was used to investigate the effect of different genetic manipulation on SAM titer. For the final strain HSAM3, the initial fermentation was further optimized, and the optimized fermentation medium consisted of 40 g/L sucrose, 10 g/L peptone, 5 g/L beef extract, 2 g/L urea, 6.3 g/L (NH4)2SO4, 2.5 g/L NaCl, 3 g/L KH2PO4 and 4.2 g/L MgSO4·7H2O. Semi-continuous batch fermentation of HSAM3 was performed in optimized fermentation medium by repeated feeding 1/10 volume of 10× optimized fermentation medium at 24 h and 48 h.
Recombinant plasmid expression
Recombinant plasmid expression was carried out based on the procedures reported previously [40, 41]. The metK gene was amplified from B. amyloliquefaciens HZ-12 using primers metK-F and metK-R, and the gene fragment was fused with the P43 promoter amplified from B. subtilis 168 and the TamyL terminator from B. licheniformis WX-02 by splicing with overlapping extension PCR (SOE-PCR) to generate the gene expression module. This was subsequently inserted into the pHY-300PLK plasmid between the BamHI and XbaI restriction sites, resulting in recombinant expression plasmid PBmetK, which was electrotransformed into B. amyloliquefaciens competent cells to generate recombination strain HZ-12 (PBmetK). Other genes were expressed using the pHY-300PLK plasmid following the same procedure.
Homologous recombination
Gene deletion and integration expression were performed by T2(2)-ori-mediated homologous recombination [40, 41]. To delete the mccA gene, primers ΔmccA-A-F/ΔmccA-A-R and ΔmccA-B-F/ΔmccA-B-R were designed to amplify two homologous arms (A and B) surrounding the mccA gene (~ 500 bp), which were further fused by SOE-PCR using primers ΔmccA-A-F/ΔmccA-B-R. The fused fragment was inserted into T2(2)-ori between the XbaI and BamHI sites, resulting in knockout plasmid T2ΔmccA, which was electrotransformed into competent B. amyloliquefaciens cells. Cells were spread onto kanamycin-containing LB plates (20 μg/mL). After PCR verification, positive clones were inoculated into LB liquid medium containing kanamycin (20 μg/mL) and cultured for 8 h at 45 °C with shaking at 180 rpm. After further kanamycin resistance screening and PCR verification, single-crossover strains were picked, transferred into LB liquid medium, and sub-cultured at 37 °C (8 h) several times. Finally, subcultures were spread onto LB plates, and each single colony was streaked onto LB and kanamycin-containing LB plates, respectively, to select kanamycin-sensitive colonies. After further PCR verification, the double-crossover strain HZ-12ΔmccA was obtained. Deletion of sucC was carried out by the same method. For integrant expression, the SAM2 gene expression module was fused with two homologous arms amplified from genomic prophage regions by SOE-PCR, and the fragment was inserted into the T2(2)-ori to generate the integrant expression vector T2-SAM2. The subsequent protocol was performed as described above for gene deletion.
Detection of cell growth
The cell growth was monitored by measuring the OD600. At preset time, aliquots of culture broth were sampled and centrifuged at 12,000×g for 10 min to collect cell pellets. Then, the cell pellets were resuspended with distilled water and separated by centrifugation, which was repeated for three times to remove the residual medium. At last, the cells were resuspended to measure the OD600.
Determination of SAM concentration
After fermentation, a 500 μL broth sample containing cells and medium was mixed with 1.5 mL perchloric acid solution (0.4 M), agitated by vortexing for 10 s every 15 min to extract intracellular and extracellular SAM for 1 h, and mixtures were centrifuged at 12,000×g for 10 min to collect the supernatant. Next, 800 μL supernatant, 95 μL NaOH (2 M) and 15 μL saturated NaHCO3 were mixed and centrifuged at 2500×g for 5 min. The supernatant was further filtered through a 0.22 μm membrane and the filtrate was subjected to SAM analysis by high-performance liquid chromatography (HPLC) [31]. Analysis was performed using a Zorbax Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 μm) at 30 °C on an Agilent 1260 HPLC system (Agilent, USA). Methanol and 40 mM NH4H2PO2 solution containing 2 mM sodium heptane sulfonate served as mobile phases A and B at a ratio of 18:82. The flow rate was 0.8 mL/min and the detection wavelength was 254 nm.
Transcription level analysis
Transcription levels were measured based on the method reported previously [42]. Total RNA from B. amyloliquefaciens cells was extracted with TRIzol Reagent (Invitrogen, USA), and residual DNA was removed by DNase I enzyme (TaKaRa, Japan). The cDNA was amplified with a RevertAid First-Strand cDNA Synthesis Kit (Thermo, USA). The target gene was amplified using corresponding primers in a 20 μL reaction system containing 2 μL cDNA, 1 μL primers, 10 μL SYBR Select Master Mix, and 7 μL DEPC water, and real-time PCR was performed at 95 °C for 5 min followed by 40 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. Fragment specificity was verified through melting curve analysis at 95 °C for 1 min and 60 °C for 1 min, and transcription data were normalized using the 16S rDNA gene of B. amyloliquefaciens as an internal reference.
Metabolite analysis
Intracellular metabolites were measured by gas chromatography–mass spectrometry (GC–MS) [43]. The cell culture was cooled to 9 ± 2 °C using liquid nitrogen and cells were collected by centrifugation at 6080×g for 5 min at 4 °C. Cell pellets were washed twice with 2 mL cold 0.85% (wt/vol) NaCl solution, resuspended in 2 mL cold 75% (v/v) ethanol solution containing 10 μg/L phenethyl acetate as an internal standard, and then frozen in liquid nitrogen for 3 min. Frozen cells were thawed and vortexed for 5 min to extract intracellular metabolites, which were collected by centrifugation and lyophilized. The lyophilized extract was dissolved in 100 μL methoxyamine hydrochloride pyridine solution (20 mg/mL), oximated for 1 h at 37 °C, and derived with 100 μL N-methyl-N-(trimethylsilyl)-trifluoroacetamide for 3 h at 60 °C. Derivatives were determined using a TSQ8000 Evo gas chromatograph/mass spectrometer with a DB-5 MS capillary column (30 m × 0.25 μm × 250 μm) (Thermo Fisher, USA). The initial column temperature (50 °C) was ramped to 110 °C at 10 °C/min and held for 3 min, then raised to 165 °C at 2 °C/min, and finally to 220 °C at 3 °C/min and held for 10 min. The temperature of the MS transfer line and ion source was 280 °C and 300 °C, respectively. The scanned range was 50–650 m/z, and the helium carrier gas low rate was 1.2 mL/min. Aspartate, succinate, fumarate, homocysteine and methionine in samples were identified by comparing the m/z value, retention time, and fragmentation pattern with corresponding standards. Peak retention time alignment and peak area integration were carried out by Thermo TraceFinder 4.1 (Thermo Fisher, USA), and the peak area was normalized using an internal standard. The fold change of each metabolite was calculated according to their peak areas.
Statistical analysis
All data were collected at least in triplicate to calculate the mean value and standard deviation, and significance t tests were used to determine differences at the 95% confidence level using Data Processing System (DPS) 7.05.
Availability of data and materials
Not applicable.
References
Walsh CT, Tu BP, Tang Y. Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem Rev. 2018;118:1460–94.
North JA, Miller AR, Wildenthal JA, Young SJ, Tabita FR. Microbial pathway for anaerobic 5′-methylthioadenosine metabolism coupled to ethylene formation. Proc Natl Acad Sci. 2017;114:E10455–64.
Struck AW, Thompson ML, Wong LS, Micklefield J. S-Adenosyl-methionine-Dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem. 2012;13:2642–55.
Bennett MR, Shepherd SA, Cronin VA, Micklefield J. Recent advances in methyltransferase biocatalysis. Curr Opin Chem Biol. 2017;37:97–106.
Kim SK, Westpheling J. Engineering a spermidine biosynthetic pathway in Clostridium thermocellum results in increased resistance to furans and increased ethanol production. Metab Eng. 2018;49:267–74.
Lu SC, Mato JM. S-Adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–42.
Kanai M, Mizunuma M, Fujii T, Iefuji H. A genetic method to enhance the accumulation of S-adenosylmethionine in yeast. Appl Microbiol Biot. 2017;101:1351–7.
Ogawa T, Tsubakiyama R, Kanai M, Koyama T, Fujii T, Iefuji H, Soga T, Kume K, Miyakawa T, Hirata D, Mizunuma M. Stimulating S-adenosyl-l-methionine synthesis extends lifespan via activation of AMPK. Proc Natl Acad Sci. 2016;113:11913–8.
Guruswamy S, Swamy MV, Choi C-I, Steele VE, Rao CV. S-Adenosyl l-methionine inhibits azoxymethane-induced colonic aberrant crypt foci in F344 rats and suppresses human colon cancer Caco-2 cell growth in 3D culture. Int J Cancer. 2008;122:25–30.
Grillo MA, Colombatto S. S-Adenosylmethionine and its products. Amino Acids. 2008;34:187–93.
Walther T, Topham CM, Irague R, Auriol C, Baylac A, Cordier H, Dressaire C, Lozano-Huguet L, Tarrat N, Martineau N, Stodel M, Malbert Y, Maestracci M, Huet R, André I, Remaud-Siméon M, François JM. Construction of a synthetic metabolic pathway for biosynthesis of the non-natural methionine precursor 2,4-dihydroxybutyric acid. Nat Commun. 2017;8:15828.
Walther T, Calvayrac F, Malbert Y, Alkim C, Dressaire C, Cordier H, François JM. Construction of a synthetic metabolic pathway for the production of 2,4-dihydroxybutyric acid from homoserine. Metab Eng. 2018;45:237–45.
Chen Y, Xu D, Fan L, Zhang X, Tan T. Manipulating multi-system of NADPH regulation in Escherichia coli for enhanced S-adenosylmethionine production. RSC Adv. 2015;5:41103–11.
Chu J, Qian JC, Zhuang YP, Zhang SL, Li YR. Progress in the research of S-adenosyl-l-methionine production. Appl Microbiol Biot. 2013;97:41–9.
Han G, Hu X, Qin T, Li Y, Wang X. Metabolic engineering of Corynebacterium glutamicum ATCC13032 to produce S-adenosyl-l-methionine. Enzyme Microb Technol. 2016;83:14–21.
Chen H, Yang Y, Wang Z, Dou J, Wang H, Zhou C. Elevated intracellular acetyl-CoA availability by acs2 overexpression and mls1 deletion combined with metK1 introduction enhanced SAM accumulation in Saccharomyces cerevisiae. Biochem Eng J. 2016;107:26–34.
Ren WQ, Cai D, Hu S, Xia SS, Wang Z, Tan TW, Zhang QH. S-Adenosyl-l-methionine production by Saccharomyces cerevisiae SAM 0801 using dl-methionine mixture: from laboratory to pilot scale. Process Biochem. 2017;62:48–52.
Chen Y, Lou S, Fan L, Zhang X, Tan T. Control of ATP concentration in Escherichia coli using synthetic small regulatory RNAs for enhanced S-adenosylmethionine production. Fems Microbiol Lett. 2015;362:fnv115.
Kind S, Becker J, Wittmann C. Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway—metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum. Metab Eng. 2013;15:184–95.
Lin B, Fan K, Zhao J, Ji J, Wu L, Yang K, Tao Y. Reconstitution of TCA cycle with DAOCS to engineer Escherichia coli into an efficient whole cell catalyst of penicillin G. Proc Natl Acad Sci. 2015;112:9855–9.
Rey MW, Ramaiya P, Nelson BA, Brody-Karpin SD, Zaretsky EJ, Tang M, de Leon AL, Xiang H, Gusti V, Clausen IG, Olsen PB, Rasmussen MD, Andersen JT, Jørgensen PL, Larsen TS, Sorokin A, Bolotin A, Lapidus A, Galleron N, Ehrlich SD, Berka RM. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillusspecies. Genome Biol. 2004;5:r77.
Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, Junge H, Voigt B, Jungblut PR, Vater J, Süssmuth R, Liesegang H, Strittmatter A, Gottschalk G, Borriss R. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol. 2007;25:1007.
Liu Y, Li J, Du G, Chen J, Liu L. Metabolic engineering of Bacillus subtilis fueled by systems biology: recent advances and future directions. Biotechnol Adv. 2017;35:20–30.
Commichau FM, Alzinger A, Sande R, Bretzel W, Meyer FM, Chevreux B, Wyss M, Hohmann H-P, Prágai Z. Overexpression of a non-native deoxyxylulose-dependent vitamin B6 pathway in Bacillus subtilis for the production of pyridoxine. Metab Eng. 2014;25:38–49.
Shi S, Chen T, Zhang Z, Chen X, Zhao X. Transcriptome analysis guided metabolic engineering of Bacillus subtilis for riboflavin production. Metab Eng. 2009;11:243–52.
Liu Y, Zhu Y, Li J, Shin H-D, Chen RR, Du G, Liu L, Chen J. Modular pathway engineering of Bacillus subtilis for improved N-acetylglucosamine production. Metab Eng. 2014;23:42–52.
Jin P, Kang Z, Yuan P, Du G, Chen J. Production of specific-molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab Eng. 2016;35:21–30.
Feng J, Gu Y, Quan Y, Cao M, Gao W, Zhang W, Wang S, Yang C, Song C. Improved poly-γ-glutamic acid production in Bacillus amyloliquefaciens by modular pathway engineering. Metab Eng. 2015;32:106–15.
Feng J, Gu Y, Quan Y, Zhang W, Cao M, Gao W, Song C, Yang C, Wang S. Recruiting a new strategy to improve levan production in Bacillus amyloliquefaciens. Scientific Rep. 2015;5:13814.
Xu J-Z, Yan W-L, Zhang W-G. Enhancing menaquinone-7 production in recombinant Bacillus amyloliquefaciens by metabolic pathway engineering. RSC Adv. 2017;7:28527–34.
Zhao W, Hang B, Zhu X, Wang R, Shen M, Huang L, Xu Z. Improving the productivity of S-adenosyl-l-methionine by metabolic engineering in an industrial Saccharomyces cerevisiae strain. J Biotechnol. 2016;236:64–70.
Hong K-K, Kim JH, Yoon JH, Park H-M, Choi SJ, Song GH, Lee JC, Yang Y-L, Shin HK, Kim JN, Cho KH, Lee JH. O-Succinyl-l-homoserine-based C4-chemical production: succinic acid, homoserine lactone, γ-butyrolactone, γ-butyrolactone derivatives, and 1,4-butanediol. J Ind Microbiol Biot. 2014;41:1517–24.
Li H, Wang BS, Li YR, Zhang L, Ding ZY, Gu ZH, Shi GY. Metabolic engineering of Escherichia coli W3110 for the production of l-methionine. J Ind Microbiol Biot. 2017;44:75–88.
Huang JF, Liu ZQ, Jin LQ, Tang XL, Shen ZY, Yin HH, Zheng YG. Metabolic engineering of Escherichia coli for microbial production of l-methionine. Biotechnol Bioeng. 2017;114:843–51.
He J, Deng J, Zheng Y, Gu J. A synergistic effect on the production of S-adenosyl-l-methionine in Pichia pastoris by knocking in of S-adenosyl-l-methionine synthase and knocking out of cystathionine-β synthase. J Biotechnol. 2006;126:519–27.
Li H, Wang B, Zhu L, Cheng S, Li Y, Zhang L, Ding ZY, Gu ZH, Shi GY. Metabolic engineering of Escherichia coli W3110 for l-homoserine production. Process Biochem. 2016;51:1973–83.
Epshtein V, Mironov AS, Nudler E. The riboswitch-mediated control of sulfur metabolism in bacteria. Proc Natl Acad Sci. 2003;100:5052.
Uchida I, Makino S, Sekizaki T, Terakado N. Cross-talk to the genes for Bacillus anthracis capsule synthesis by atxA, the gene encoding the frans-activator of anthrax toxin synthesis. Mol Microbiol. 1997;23:1229–40.
Nærdal I, Netzer R, Irla M, Krog A, Heggeset TMB, Wendisch VF, Brautaset T. l-lysine production by Bacillus methanolicus: genome-based mutational analysis and l-lysine secretion engineering. J Biotechnol. 2017;244:25–33.
Wei X, Zhou Y, Chen J, Cai D, Wang D, Qi G, Chen S. Efficient expression of nattokinase in Bacillus licheniformis: host strain construction and signal peptide optimization. J Ind Microbiol Biotechnol. 2015;42:287–95.
Qiu Y, Zhang J, Li L, Wen Z, Nomura CT, Wu S, Chen S. Engineering Bacillus licheniformis for the production of meso-2,3-butanediol. Biotechnol Biofuels. 2016;9:117.
Wei X, Tian G, Ji Z, Chen S. A new strategy for enhancement of poly-γ-glutamic acid production by multiple physicochemical stresses in Bacillus licheniformis. J Chem Technol Biotechnol. 2015;90:709–13.
Meyer H, Weidmann H, Lalk M. Methodological approaches to help unravel the intracellular metabolome of Bacillus subtilis. Microb Cell Fact. 2013;12:69.
Acknowledgements
Not applicable.
Funding
This study was funded by the Project Supported by Da Bei Nong Group Promoted Project for Young Scholar of HZAU (No. 2017DBN011), and the National Natural Science Foundation of China (No. 21877053).
Author information
Authors and Affiliations
Contributions
XW and YD conceived and designed the study. LR, LL, DZ and CJ carried out experiments. All authors performed data analysis and prepared the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Additional tables and figures.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ruan, L., Li, L., Zou, D. et al. Metabolic engineering of Bacillus amyloliquefaciens for enhanced production of S-adenosylmethionine by coupling of an engineered S-adenosylmethionine pathway and the tricarboxylic acid cycle. Biotechnol Biofuels 12, 211 (2019). https://doi.org/10.1186/s13068-019-1554-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-019-1554-0