
Abstract
Bcl-B is a poorly understood protein of the Bcl-2 family that is highly expressed in many healthy tissues and tumor types. Bcl-B is considered an antiapoptotic protein, but many reports have revealed its contradictory roles in different cancer types. In this mini-review, we elucidate the functions of Bcl-B in normal conditions and various pathologies, its regulation of programmed cell death, its oncogene/oncosuppressor activity in tumorigenesis, its impact on drug-acquired resistance, and possible approaches to inhibit Bcl-B.
Similar content being viewed by others


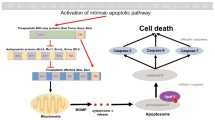
Introduction
The members of the Bcl-2 family of proteins are essential regulators of cell death which control activation of the intrinsic pathway of apoptosis. They are divided into two groups: pro- and antiapoptotic proteins. The former is also separated into two subsets: multidomain effector proteins (Bak and Bax) and regulatory proteins containing only one Bcl-2 homology (BH) region, namely BH3-only proteins (Bim, Bid, Bad, Bmf, Bik, Noxa, Puma, and Hrk). The induction of the intrinsic pathway in response to various stimuli leads to displacement of proapoptotic proteins from their antiapoptotic partners, mitochondrial outer membrane permeabilization, and activation of the caspase cascade that ultimately results in apoptotic cell death. The increased level of prosurvival proteins is responsible for evasion of cell death and promotes carcinogenesis [1,2,3].
The antiapoptotic subfamily includes at least six members (Bcl-2, Bcl-xL, Mcl-1, Bcl-w, Bfl-1, and Bcl-B) [3]. The first three proteins have been studied intensively by many authors [4,5,6]. Recently, Bcl-w and Bfl-1 have also been discussed in detail [7, 8]. However, despite its apoptotic and non-apoptotic functions, Bcl-B remains poorly characterized. In this mini-review, we try to close this gap and summarize the current knowledge concerning the functions of this “unknown” protein (Fig. 1).
Discovery, structure, and protein–protein interactions
Bcl-B (B-cell lymphoma 2 family protein resembling Boo)/Bcl-2L10/Nrh (Bcl-2 like protein 10), is encoded by BCLB gene. This human protein was discovered independently by three groups in 2001 [9,10,11]. Bcl-B is homologous to the murine Bcl-2 protein Boo/Diva, a fact that is reflected in its name [9]. Despite the structural resemblance (the amino acid sequence identity and similarity between Bcl-B and Boo is about 45.5–47% and 60.7%, respectively), there are some differences between these proteins [9, 12]. First, Bcl-B contains 204 amino acids, while Boo is only 191 amino acids long protein [13]. Second, human Bcl-B is widely expressed in many healthy tissues and tumors in adults, while Boo has been found mainly in mouse ovary and testis [9, 14].
Surprisingly, more than twenty years after its discovery, many aspects of Bcl-B remain unclear. The first “open question” is the structural organization of Bcl-B. On the one hand, numerous researchers have reported that Bcl-B has a structure that is typical for various antiapoptotic proteins of the Bcl-2 family [15], with four BH domains (BH1, BH2, BH3-like, and BH4) and a transmembrane (TM) domain [9, 16,17,18,19,20]. On the other hand, some researchers have presented evidence that human Bcl-B/Bcl-2L10 and murine Boo/Diva are characterized by the absence of a BH3 domain [10, 12, 13, 21]. Moreover, Boo/Diva contains an altered BH1 domain that prevents possible interactions with proapoptotic proteins [13].
The second “open question” relates to possible interaction partners of Bcl-B. This protein is able to interact with fewer Bcl-2 family proteins compared with the other antiapoptotic Bcl-2 family proteins. Bcl-B/Bcl-2L10 selectively binds Bax and neutralizes its proapoptotic function [9, 17, 18, 22]. Mutant forms of any of these proteins abrogate the formation of the corresponding complex [18]. However, Bcl-B cannot interact with Bak, Bad, and Bid [18]. Surprisingly, Boo/Diva can bind Bak, but not Bax [21]. Another partner of Bcl-B is the BH3-only protein Bim. Interestingly, Bim and Bax form complexes with the whole subset of the antiapoptotic Bcl-2 family proteins [12]. The main reason for specificity of Bcl-B binding to only these two proteins is still unclear.
Finally, it should be noted that Boo/Diva knockout mice show no abnormalities. The mice are fertile, and their life expectancy is similar to that of wild type mice [23]. The lack of abnormalities suggests several things. First, the functional activity of Bcl-B during embryogenesis could be compensated by other proteins. Second, Bcl-B targeting in humans may result in lower negative effects in normal tissues compared with other proteins of the Bcl-2 family. Notably, Bcl-xL or Mcl-1 knockout in mice leads to embryonic lethality [24, 25]. Moreover, selective inhibitors of Bcl-xL and Mcl-1 have not succeeded in clinical trials due to excessive toxicity [2, 26]. Bcl-2 knockout in mice results in an altered phenotype. Venetoclax, a selective Bcl-2 inhibitor, was approved by the Food and Drug Administration (FDA) several years ago for treatment of various cancers [2].
The role of Bcl-B in normal conditions
Pro- and antiapoptotic properties
Another unclear area is the functional activity of Bcl-B. Is it an apoptotic or a prosurvival protein? Again, the results are controversial. Murine Boo/Diva has demonstrated both proapoptotic [20, 27] and antiapoptotic [21, 28, 29] activities in different in vitro cell models. For example, nucleoside diphosphate kinase NM23-H2 suppresses Bcl-B- or Boo-mediated apoptosis in vitro [16]. According to various reports, human Bcl-B/Bcl-2L10 exerts predominantly antiapoptotic properties [9, 10, 17, 18]. However, Nur77/TR3, an orphan nuclear receptor, can bind to Bcl-B and transform its antiapoptotic phenotype into a proapoptotic one via conformational changes in its structure that expose its BH3 domain and subsequent exertion of proapoptotic activity in plasma and myeloma cells [30, 31]. Of note, Nur77/TR3-dependent transformation has been proposed for Bcl-2 [32]. Importantly, Nur77/TR3 does not contain BH domains. This fact suggests that Bcl-B could interact with other proteins not just via the BH-mediated interface. It is likely that the pro- or antiapoptotic role of Bcl-B and Diva is determined by the cellular context, but this topic requires further investigation.
Autophagy
A possible explanation for the contradictory apoptotic functions of Bcl-B could be that this protein regulates other types of programmed cell death (PCD), in particular, autophagy, which promotes degradation of damaged proteins and organelles. This process might also act as an essential adaptive mechanism for the maintenance of cell survival by preventing apoptosis [33, 34]. Bcl-B binds to the BH3 domain of Beclin 1 and can block this important activator of autophagy. In contrast, Bcl-B suppression induces both apoptosis and autophagy [19, 35]. A similar mechanism of autophagy regulation is known for Bcl-2 and Bcl-xL [36, 37].
Mitophagy is a subtype of autophagy and represents selective elimination of aged and damaged mitochondria in lysosomes. Mitophagy activation usually inhibits apoptosis, but it is also able to promote apoptosis in several situations. Bcl-B/Bcl-2L10 can control the activity of Parkin, an E3 ubiquitin ligase and a key participant of mitophagy. The formation of a Bcl-B/phospho-Parkin complex blocks mitophagy and thus inhibits apoptosis in hepatic stellate cells [38]. Taken together, the disturbed balance between apoptosis and autophagy regulation can underlie the “apoptotic dualism” of Bcl-B (Fig. 1).

The roles of Bcl-B in normal and pathological conditions
The functional activity of all proteins is linked to their subcellular localization. Like other antiapoptotic proteins of the Bcl-2 family, Bcl-B contains a C-terminal TM domain that is responsible for its anchoring in the intracellular membranes. Bcl-B is commonly located in the outer mitochondrial membrane (OMM) [15]. However, prosurvival proteins can also be localized in the endoplasmic reticulum (ER) to regulate intracellular Ca2+ levels and activation of apoptosis [39]. Bcl-B can bind to the inositol 1,4,5-trisphosphate receptor (IP3R) through its BH4-domain and block Ca2+ release from the ER, thereby preventing apoptosis. Bcl-B-mediated regulation of Ca2+ is controlled by IP3R-binding protein (IRBIT): Phospho-IRBIT enhances the action of Bcl-B, but dephosphorylation of IRBIT has the opposite effect [40]. Taken together, these data contribute “contradictions” regarding the apoptotic properties of Bcl-B.
Oogenesis and embryogenesis
Bcl-B and its homologs are highly expressed in mice, buffalo, zebrafish, and human oocytes; ovarian tissue; and early-stage embryos. This protein plays an important role in the development and maintenance of oocytes and embryos [41,42,43,44,45,46]. Boo/Diva and BCL2L10 suppression inhibits oocyte maturation in cultured murine and buffalo oocytes [42, 45], a phenomenon accompanied by alterations in their spindles and chromosome organization [42]. Moreover, Bcl-B is essential for correct microtubule organization in mouse and human oocytes [43, 47]. Interestingly, Bcl-B is mainly found in the cytosol of human oocytes and embryos, whereas in adult tissues it is localized in mitochondria and the ER. Meanwhile, Bcl-B is detected in the nucleus of abnormal embryos and might be a potential biomarker of “embryo quality” [43, 44]. The zebrafish protein Nrz (a homolog of murine Boo) located in the OMM and ER and regulates apoptosis and Ca2+ signaling, thereby controlling cytoskeletal dynamics. It is essential for processes of gastrulation and somitogenesis in zebrafish [39, 48]. Additionally, blastocysts of patients with polycystic ovaries have lower expression of BCL2L10 compared with healthy controls [49]. Taken together, these observations indicate that Bcl-B is crucial for the maintenance of oogenesis and embryogenesis in humans and animals, underlining the importance of apoptosis regulation in physiological processes.
The regulation of Bcl-B and its role in pathology
Transcriptional/translational level
Like all proteins, Bcl-B is regulated at the transcriptional, translational, and posttranslational levels (Fig. 2).

The regulation of Bcl-B and its participation in programmed cell death. P – phosphorylated form of the Parkin protein; Ub – ubiquitin. The figure was prepared by using the elements from Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License
Unfortunately, transcriptional and translational regulation of Bcl-B is understudied. STAT3 is a positive regulator of Bcl-B transcription in melanoma [50]. Additionally, the long noncoding RNA CERNA1 increases the Bcl-2L10 transcription rate via epigenetic regulation in vascular endothelial cells and ovarian cancer [51, 52]. At the translational level, several microRNAs (miRNAs) have been reported to regulate Bcl-B/Bcl-2L10 expression. First, miRNA-1229 negatively affects the Bcl-2L10 level in colorectal cancer [53]. miRNA-18a also downregulates Bcl-2L10, and this change might mediate cell invasion, migration, and proliferation in hepatocellular carcinoma (HCC) [54]. Moreover, miRNA-dependent regulation of Bcl-B might contribute to the development of non-cancer diseases. For example, toxic epidermal necrolysis (a type of severe drug-induced skin reaction) whose precise pathogenesis remains unknown could be associated with excessive keratinocyte apoptosis. Moreover, miR-18a-5p-mediated Bcl-B suppression activates apoptotic death of keratinocytes in patients with toxic epidermal necrolysis [55, 56].
Posttranslational level
The process of proteasomal degradation is an important mechanism by which Bcl-B is regulated [57]. Ubiquilin-1 is a selective Bcl-B regulator that does not interact with other antiapoptotic Bcl-2 family proteins. Ubiquilin-1-dependent monoubiquitinylation of Bcl-B leads to its stabilization and removal from mitochondria to the cytosol [58]. Furthermore, another member of the ubiquilin family, ubiquilin-4, stabilizes Bcl-B and prevents apoptosis in mesothelioma cells, which have high Bcl-B expression [59]. Unfortunately, the mechanisms underlying the stabilization and relocation of Bcl-B after interactions with ubiquilins remain uncertain and require further clarification.
It should be noted that stability of antiapoptotic proteins correlates with their prosurvival activity. Of the six antiapoptotic Bcl-2 family proteins, Bcl-B, Bfl-1, and Mcl-1 are more prone to basal or drug-mediated proteasomal turnover in cancer cells; this turnover could limit their functional activity. Nevertheless, disturbances in the proteasomal degradation machinery of Bcl-B, Bfl-1, and Mcl-1 could facilitate drug resistance or tumor development [60]. Notably, the indirect inhibitor (PaTrin-2) of the specific deubiquitinase of Mcl-1 has been studied [1]. Furthermore, the combination of azacytidine (a chemotherapeutic agent) and erlotinib (an inhibitor of the epidermal growth factor receptor) has a synergetic effect in acute myeloid leukemia (AML) cells and primary AML, and myelodysplastic syndrome (MDS) cells by inducing proteasomal degradation of Mcl-1 and Bcl-B; these data indicate the indirect inhibition of Bcl-B [61]. These facts suggest that the cellular strategies used to control the levels of antiapoptotic Bcl-2 family proteins can be applied for therapy. Accordingly, targeting deubiquitinases or activating ubiquitin ligases of Bcl-B appears to be a potential strategy to eliminate Bcl-B-dependent cancer cells (Fig. 2).
“Dualism” of Bcl-B in carcinogenesis: Oncogene/oncosuppressor activity
As mentioned earlier, Bcl-B is highly expressed in normal and tumor tissues. Its expression has been detected in many types of solid and blood malignancies [14]. However, its role in tumor development and progression is variable. Bcl-B acts as an oncogene in some tumors [50, 62,63,64] but prevents tumorigenesis in others [65,66,67]. Hence, the following question arises: what are the reasons underlying the “ambiguity” of Bcl-B in carcinogenesis?
It is well known that evasion of cell death is one of the hallmarks of cancer that can be achieved by increased expression of the antiapoptotic Bcl-2 family proteins [1, 68]. Indeed, upregulated Bcl-B levels are responsible for tumor promotion in cases of breast cancer [62], melanoma [50, 63], and multiple melanoma (MM) [69]. At the same time, the Bcl-B level correlates with a positive prognosis in patients with HCC and gastric cancer [70, 71]. Moreover, this protein blocks cell migration, angiogenesis, and metastasis, thereby, serving as a oncosuppressor in HCC [70]. How might this be possible?
First, it could be associated with the “apoptotic dualism” of Bcl-B (Fig. 1). Besides its prosurvival activity, Bcl-B is also involved in the regulation of autophagy and Ca2+ signaling, as discussed above. Indeed, Bcl-B can stimulate autophagy in HCC [66], an action that could explain its tumor suppressor activity in this cancer type. Second, the potential mutations in the protein structure of Bcl-B or BCL2L10 polymorphisms could abate its antiapoptotic activity. For example, a BCL2L10 single nucleotide polymorphism (rs2231292, Leu11Arg), which is predicted to be a biomarker of a favorable outcome, has been observed in patients with breast and rectal cancer. It leads to disturbance of the interactions between BCL2L10 and IP3R that, in turn, facilitates Ca2+ release from the ER and activation of Ca2+-dependent cell death [72, 73]. Additionally, patients with this BCL2L10 polymorphism have a diminished risk of the development de novo MDS [74]. Third, epigenetic regulation of Bcl-B has great significance in tumorigenesis: Methylation of the gene could lead to silenced or reduced expression of this protein in HCC [66] and gastric cancer [65, 75, 76]. Finally, a decrease in level of one antiapoptotic protein can be compensated for by an increase in the level of another prosurvival partner, a phenomenon that has been proved in various in vitro and in vivo models. Therefore, mutant cells might contain “low” levels of Bcl-B and “high” levels of Bcl-2, Bcl-xL, Mcl-1, etc. Indeed, the survival of various cancer cells is “dependent” on distinct antiapoptotic proteins [1, 2, 77, 78].
Conclusions
Bcl-B/Bcl-2L10 is a multifaceted protein that exerts both pro- and antiapoptotic activities, allowing it to act as an oncogene as well as an oncosuppressor in different cancer types. These “dual” activities are possibly associated with its negative epigenetic regulation, altered structure due to gene polymorphism, and regulation of other types of PCD. Some data suggest that Bcl-B is involved in autophagy [19, 35] and Ca2+-mediated apoptosis [39, 40]. This protein can probably control other types of PCD, an eventuality that should be elucidated in the near future. Bcl-B participates in oogenesis and embryogenesis, but its knockout in mice does not lead to any negative effects [23], a finding that could be promising in the context of Bcl-B targeting. At present, there is little data about Bcl-B inhibitors. This protein is prone to proteasomal degradation and could be suppressed indirectly [60, 61]. Several compounds have been reported to directly suppress Bcl-B by disrupting complexes with proapoptotic Bcl-2 family proteins in silico and in vitro [79,80,81,82]. Gambogic acid and gossypol, non-selective inhibitors of antiapoptotic Bcl-2 family proteins, can bind to Bcl-B [83, 84]. Importantly, high Bcl-B levels contribute to the development of acquired resistance to various chemotherapeutics, including camptotecin (a topoisomerase inhibitor) [85], busulfan (an alkylating agent) [86], ABT-737 (a non-selective BH3 mimetic) [50, 87, 88], azacytidine (a hypomethylating agent) [64, 89], and cisplatin and dacarbazine (alkylating agents) [50]. Therefore, Bcl-B could be considered an important prognostic marker in cancer. Moreover, potential blockade of Bcl-B in combination with chemotherapeutics or targeted therapy could be a promising anticancer strategy that prevents the appearance of acquired drug resistance and diminishes the possible toxic effects. Additionally, increased Bcl-B gene expression is related to some non-cancer diseases such as toxic epidermal necrolysis [55, 56], affective psychosis [90], and cardiac disorders [91]. To conclude, there is no doubt that Bcl-B plays a role in normal physiological conditions and pathologies. Further investigation will be able to resolve the current contradictions and make Bcl-B a more “understandable” protein.
Data Availability
Not applicable.
References
Pervushin NV, Senichkin VV, Zhivotovsky B, Kopeina GS. Mcl-1 as a barrier in cancer treatment: can we target it now? Int Rev Cell Mol Biol. 2020;351:23–55.
Senichkin VV, Pervushin NV, Zuev AP, Zhivotovsky B, Kopeina GS. Targeting Bcl-2 Family Proteins: What, Where, When? Biochemistry (Mosc). 2020;85:1210–26.
Hatok J, Racay P. Bcl-2 family proteins: master regulators of cell survival. Biomol Concepts. 2016;7:259–70.
Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109.
Li M, Wang D, He J, Chen L, Li H. Bcl-XL: a multifunctional anti-apoptotic protein. Pharmacol Res. 2020;151:104547.
Senichkin VV, Streletskaia AY, Zhivotovsky B, Kopeina GS. Molecular comprehension of Mcl-1: from gene structure to Cancer Therapy. Trends Cell Biol. 2019;29:549–62.
Hartman ML, Czyz M. BCL-w: apoptotic and non-apoptotic role in health and Disease. Cell Death Dis. 2020;11:260.
Wang G, Diepstraten ST, Herold MJ. Last but not least: BFL-1 as an emerging target for anti-cancer therapies. Biochem Soc Trans. 2022;50:1119–28.
Ke N, Godzik A, Reed JC. Bcl-B, a novel Bcl-2 family member that differentially binds and regulates Bax and Bak. J Biol Chem. 2001;276:12481–4.
Zhang H, Holzgreve W, De Geyter C. Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum Mol Genet. 2001;10:2329–39.
Aouacheria A, Arnaud E, Venet S, Lalle P, Gouy M, Rigal D, et al. Nrh, a human homologue of Nr-13 associates with Bcl-Xs and is an inhibitor of apoptosis. Oncogene. 2001;20:5846–55.
Rautureau GJP, Yabal M, Yang H, Huang DCS, Kvansakul M, Hinds MG. The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 2012;3:e443.
Rautureau GJP, Day CL, Hinds MG. The structure of Boo/Diva reveals a divergent Bcl-2 protein. Proteins. 2010;78:2181–6.
Krajewska M, Kitada S, Winter JN, Variakojis D, Lichtenstein A, Zhai D, et al. Bcl-B expression in human epithelial and nonepithelial malignancies. Clin Cancer Res. 2008;14:3011–21.
Cory S, Roberts AW, Colman PM, Adams JM. Targeting BCL-2-like proteins to kill Cancer cells. Trends Cancer. 2016;2:443–60.
Kang Y, Lee D-C, Han J, Yoon S, Won M, Yeom J-H, et al. NM23-H2 involves in negative regulation of diva and Bcl2L10 in apoptosis signaling. Biochem Biophys Res Commun. 2007;359:76–82.
Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of Bax and bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem. 2008;283:9580–6.
Zhai D, Ke N, Zhang H, Ladror U, Joseph M, Eichinger A, et al. Characterization of the anti-apoptotic mechanism of Bcl-B. Biochem J. 2003;376:229–36.
Robert G, Gastaldi C, Puissant A, Hamouda A, Jacquel A, Dufies M, et al. The anti-apoptotic Bcl-B protein inhibits BECN1-dependent autophagic cell death. Autophagy. 2012;8:637–49.
Inohara N, Gourley TS, Carrio R, Muñiz M, Merino J, Garcia I, et al. Diva, a Bcl-2 homologue that binds directly to Apaf-1 and induces BH3-independent cell death. J Biol Chem. 1998;273:32479–86.
Song Q, Kuang Y, Dixit VM, Vincenz C. Boo, a novel negative regulator of cell death, interacts with Apaf-1. EMBO J. 1999;18:167–78.
Bhargavi K, Kalyan Chaitanya P, Ramasree D, Vasavi M, Murthy DK, Uma V. Homology modeling and docking studies of human Bcl-2L10 protein. J Biomol Struct Dyn. 2010;28:379–91.
Russell HR, Lee Y, Miller HL, Zhao J, McKinnon PJ. Murine ovarian development is not affected by inactivation of the bcl-2 family member diva. Mol Cell Biol. 2002;22:6866–70.
Boumela I, Assou S, Aouacheria A, Haouzi D, Dechaud H, De Vos J, et al. Involvement of BCL2 family members in the regulation of human oocyte and early embryo survival and death: gene expression and beyond. Reproduction. 2011;141:549–61.
Sochalska M, Tuzlak S, Egle A, Villunger A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: implications for targeted therapy. FEBS J. 2015;282:834–49.
Roberts AW, Wei AH, Huang DCS. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood. 2021;138:1120–36.
Lee R, Chen J, Matthews CP, McDougall JK, Neiman PE. Characterization of NR13-related human cell death regulator, Boo/Diva, in normal and cancer tissues. Biochim Biophys Acta. 2001;1520:187–94.
Naumann U, Weit S, Wischhusen J, Weller M. Diva/Boo is a negative regulator of cell death in human glioma cells. FEBS Lett. 2001;505:23–6.
Liu NS, Du X, Lu J, He BP. Diva reduces cell death in response to oxidative stress and cytotoxicity. PLoS ONE. 2012;7:e43180.
Luciano F, Krajewska M, Ortiz-Rubio P, Krajewski S, Zhai D, Faustin B, et al. Nur77 converts phenotype of Bcl-B, an antiapoptotic protein expressed in plasma cells and Myeloma. Blood. 2007;109:3849–55.
Banta KL, Wang X, Das P, Winoto A. B cell Lymphoma 2 (Bcl-2) residues essential for Bcl-2’s apoptosis-inducing interaction with Nur77/Nor-1 orphan steroid receptors. J Biol Chem. 2018;293:4724–34.
Lin B, Kolluri SK, Lin F, Liu W, Han Y-H, Cao X, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–40.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52.
Pervushin NV, Senichkin VV, Kapusta AA, Gorbunova AS, Kaminskyy VO, Zhivotovsky B, et al. Nutrient deprivation promotes MCL-1 degradation in an autophagy-independent manner. Biochem (Mosc). 2020;85:1235–44.
He J, Deng L, Liu H, Chen T, Chen S, Xia S, et al. BCL2L10/BECN1 modulates hepatoma cells autophagy by regulating PI3K/AKT signaling pathway. Aging. 2019;11:350–70.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell. 2005;122:927–39.
Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39.
Ding Q, Xie X-L, Wang M-M, Yin J, Tian J-M, Jiang X-Y, et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp Mol Med. 2019;51:1–13.
Popgeorgiev N, Jabbour L, Gillet G. Subcellular localization and Dynamics of the Bcl-2 family of proteins. Front Cell Dev Biol. 2018;6:13.
Bonneau B, Ando H, Kawaai K, Hirose M, Takahashi-Iwanaga H, Mikoshiba K. IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. Elife. 2016;5:e19896.
Kratz E, Eimon PM, Mukhyala K, Stern H, Zha J, Strasser A, et al. Functional characterization of the Bcl-2 gene family in the zebrafish. Cell Death Differ. 2006;13:1631–40.
Yoon S-J, Kim E-Y, Kim YS, Lee H-S, Kim K-H, Bae J, et al. Role of Bcl2-like 10 (Bcl2l10) in regulating mouse oocyte maturation. Biol Reprod. 2009;81:497–506.
Guillemin Y, Lalle P, Gillet G, Guerin J-F, Hamamah S, Aouacheria A. Oocytes and early embryos selectively express the survival factor BCL2L10. J Mol Med (Berl). 2009;87:923–40.
Guérin J-F, Cornut-Thibaut A, Giscard-Destaing S, Pouvreau S, Guillemin Y, Aouacheria A. Subcellular dynamics of the maternal cell death regulator BCL2L10 in human preimplantation embryos. Hum Reprod. 2013;28:729–39.
Huang Y-L, Wang H-J, Chen F-M, Zhao X-L, Fu Q, Zhang P-F, et al. Role of BCL2L10 in regulating buffalo (Bubalus bubalis) oocyte maturation. Theriogenology. 2018;110:1–7.
Liu Y, Xin J, Zhang S, Li Q, Wang W, Chen J, et al. Expression patterns and biological function of BCL2L10 during mouse preimplantation development. Gene Expr Patterns. 2022;46:119285.
Lee S-Y, Kim E-Y, Kim K-H, Lee K-A. Bcl2l10, a new Tpx2 binding partner, is a master regulator of Aurora kinase A in mouse oocytes. Cell Cycle. 2016;15:3296–305.
Bonneau B, Popgeorgiev N, Prudent J, Gillet G. Cytoskeleton dynamics in early zebrafish development: A matter of phosphorylation? Bioarchitecture. 2011;1:216–20.
McCallie BR, Parks JC, Griffin DK, Schoolcraft WB, Katz-Jaffe MG. Infertility diagnosis has a significant impact on the transcriptome of developing blastocysts. Mol Hum Reprod. 2017;23:549–56.
Quezada MJ, Picco ME, Villanueva MB, Castro MV, Barbero G, Fernández NB, et al. BCL2L10 is overexpressed in Melanoma downstream of STAT3 and promotes cisplatin and ABT-737 resistance. Cancers (Basel). 2020;13:78.
Lu W, He X, Su L, Zhao B, Miao J. Effects of annexin A7 inhibitor-ABO on the expression and distribution of long noncoding RNA-CERNA1 in vascular endothelial cells apoptosis. Apoptosis. 2019;24:552–61.
Xu R, Peng H, Yang N, Liu Z, Lu W. Nuclear lncRNA CERNA1 enhances the cisplatin-induced cell apoptosis and overcomes chemoresistance via epigenetic activation of BCL2L10 in Ovarian cancer. Genes Dis. 2023;10:10–3.
Hu L, Fang L, Zhang Z, Yan Z. circTADA2A retards the progression of Colorectal Cancer via regulating miR-1229/BCL2L10 Signal Axis. Cancer Manag Res. 2021;13:6811–21.
Wang X, Lu J, Cao J, Ma B, Gao C, Qi F. MicroRNA-18a promotes hepatocellular carcinoma proliferation, migration, and invasion by targeting Bcl2L10. Onco Targets Ther. 2018;11:7919–34.
Ichihara A, Wang Z, Jinnin M, Izuno Y, Shimozono N, Yamane K, et al. Upregulation of miR-18a-5p contributes to epidermal necrolysis in severe drug eruptions. J Allergy Clin Immunol. 2014;133:1065–74.
Lv P, Huang J, Yang Q, Yang T, Cao X, Liu O, et al. Analysis of circRNA profiles and clinical value in Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol. 2023. https://doi.org/10.1111/exd.14939.
van de Kooij B, Rooswinkel RW, Kok F, Herrebout M, de Vries E, Paauwe M, et al. Polyubiquitination and proteasomal turnover controls the anti-apoptotic activity of Bcl-B. Oncogene. 2013;32:5439–48.
Beverly LJ, Lockwood WW, Shah PP, Erdjument-Bromage H, Varmus H. Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc Natl Acad Sci U S A. 2012;109:E119–126.
Liu F, Pan R, Ding H, Gu L, Yang Y, Li C, et al. UBQLN4 is an ATM substrate that stabilizes the anti-apoptotic proteins BCL2A1 and BCL2L10 in Mesothelioma. Mol Oncol. 2021;15:3738–52.
Rooswinkel RW, van de Kooij B, de Vries E, Paauwe M, Braster R, Verheij M, et al. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood. 2014;123:2806–15.
Lainey E, Wolfromm A, Marie N, Enot D, Scoazec M, Bouteloup C, et al. Azacytidine and erlotinib exert synergistic effects against acute Myeloid Leukemia. Oncogene. 2013;32:4331–42.
Nougarede A, Popgeorgiev N, Kassem L, Omarjee S, Borel S, Mikaelian I, et al. Breast Cancer Targeting through Inhibition of the endoplasmic reticulum-based apoptosis Regulator Nrh/BCL2L10. Cancer Res. 2018;78:1404–17.
Del Bufalo D, Di Martile M, Valentini E, Manni I, Masi I, D’Amore A, et al. Bcl-2-like protein-10 increases aggressive features of Melanoma cells. Explor Target Antitumor Ther. 2022;3:11–26.
Cluzeau T, Robert G, Mounier N, Karsenti JM, Dufies M, Puissant A, et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget. 2012;3:490–501.
Xu JD, Cao XX, Long ZW, Liu XP, Furuya T, Xu JW, et al. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J Pathol. 2011;223:400–9.
Liu X, Hu X, Kuang Y, Yan P, Li L, Li C, et al. BCLB, methylated in hepatocellular carcinoma, is a Starvation stress sensor that induces apoptosis and autophagy through the AMPK-mTOR signaling cascade. Cancer Lett. 2017;395:63–71.
Lee S-Y, Kwon J, Woo JH, Kim K-H, Lee K-A. Bcl2l10 mediates the proliferation, invasion and migration of Ovarian cancer cells. Int J Oncol. 2020;56:618–29.
Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70.
Hamouda M-A, Jacquel A, Robert G, Puissant A, Richez V, Cassel R, et al. BCL-B (BCL2L10) is overexpressed in patients suffering from Multiple Myeloma (MM) and drives an MM-like Disease in transgenic mice. J Exp Med. 2016;213:1705–22.
Bai Y, Wang J, Han J, Xie X-L, Ji C-G, Yin J, et al. BCL2L10 inhibits growth and Metastasis of hepatocellular carcinoma both in vitro and in vivo. Mol Carcinog. 2017;56:1137–49.
Xu JD, Furuya T, Cao XX, Liu XL, Li QQ, Wang WJ, et al. Loss of BCL2L10 protein expression as prognostic predictor for poor clinical outcome in gastric carcinoma. Histopathology. 2010;57:814–24.
Duong MQ, Gadet R, Treilleux I, Borel S, Nougarède A, Marcillat O, et al. Nrh L11R single nucleotide polymorphism, a new prediction biomarker in Breast cancer, impacts endoplasmic reticulum-dependent Ca2 + traffic and response to neoadjuvant chemotherapy. Cell Death Dis. 2023;14:392.
Lee IH, Kang K, Kang BW, Lee SJ, Bae WK, Hwang JE, et al. Genetic variations using whole-exome sequencing might predict response for neoadjuvant chemoradiotherapy in locally advanced rectal cancer. Med Oncol. 2018;35:145.
Fabiani E, Fianchi L, Falconi G, Boncompagni R, Criscuolo M, Guidi F, et al. The BCL2L10 Leu21Arg variant and risk of therapy-related myeloid Neoplasms and de novo myelodysplastic syndromes. Leuk Lymphoma. 2014;55:1538–43.
Mikata R, Fukai K, Imazeki F, Arai M, Fujiwara K, Yonemitsu Y, et al. BCL2L10 is frequently silenced by promoter hypermethylation in gastric cancer. Oncol Rep. 2010;23:1701–8.
Mikata R, Yokosuka O, Fukai K, Imazeki F, Arai M, Tada M, et al. Analysis of genes upregulated by the demethylating agent 5-aza-2’-deoxycytidine in gastric cancer cell lines. Int J Cancer. 2006;119:1616–22.
Tantawy SI, Timofeeva N, Sarkar A, Gandhi V. Targeting MCL-1 protein to treat cancer: opportunities and challenges. Front Oncol. 2023;13:1226289.
Carrington EM, Zhan Y, Brady JL, Zhang J-G, Sutherland RM, Anstee NS, et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017;24:878–88.
Yip KW, Godoi PHC, Zhai D, Garcia X, Cellitti JF, Cuddy M, et al. A TR3/Nur77 peptide-based high-throughput fluorescence polarization screen for small molecule Bcl-B inhibitors. J Biomol Screen. 2008;13:665–73.
Zou J, Ardecky R, Pinkerton AB, Sergienko E, Su Y, Stonich D et al. Selective Bcl-2 inhibitor probes. Probe reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010.
Curpan RF, Simons PC, Zhai D, Young SM, Carter MB, Bologa CG, et al. High-throughput screen for the chemical inhibitors of antiapoptotic bcl-2 family proteins by multiplex flow cytometry. Assay Drug Dev Technol. 2011;9:465–74.
Parthiban A, Sachithanandam V, Lalitha P, Muthukumaran J, Misra R, Jain M, et al. Isolation, characterisation, anticancer and anti-oxidant activities of 2-methoxy mucic acid from Rhizophora apiculata: an in vitro and in silico studies. J Biomol Struct Dyn. 2023;41:1424–36.
Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic bcl-2-family proteins. Cell Death Differ. 2006;13:1419–21.
Zhai D, Jin C, Shiau C-W, Kitada S, Satterthwait AC, Reed JC. Gambogic acid is an antagonist of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2008;7:1639–46.
Yasui K, Mihara S, Zhao C, Okamoto H, Saito-Ohara F, Tomida A, et al. Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res. 2004;64:1403–10.
Valdez BC, Murray D, Ramdas L, de Lima M, Jones R, Kornblau S, et al. Altered gene expression in busulfan-resistant human Myeloid Leukemia. Leuk Res. 2008;32:1684–97.
Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 2012;3:e366.
Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, et al. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic Leukemia to ABT-737. Blood. 2011;118:3579–90.
Vidal V, Robert G, Goursaud L, Durand L, Ginet C, Karsenti JM, et al. BCL2L10 positive cells in bone marrow are an Independent prognostic factor of azacitidine outcome in Myelodysplastic Syndrome and acute Myeloid Leukemia. Oncotarget. 2017;8:47103–9.
Bouwkamp CG, Kievit AJA, Olgiati S, Breedveld GJ, Coesmans M, Bonifati V, et al. A balanced translocation disrupting BCL2L10 and PNLDC1 segregates with affective psychosis. Am J Med Genet B Neuropsychiatr Genet. 2017;174:214–9.
Sciacca FL, Ciaccio C, Fontana F, Strano C, Gilardoni F, Pantaleoni C, et al. Severe phenotype in a patient with homozygous 15q21.2 Microdeletion Involving BCL2L10, GNB5, and MYO5C genes, resembling Infantile Developmental Disorder with Cardiac Arrhythmias (IDDCA). Front Genet. 2020;11:399.
Funding
Open access funding provided by Karolinska Institute. The work was supported by a grant from the non-commercial organization “The Russian Science Foundation” (23-74-30006). The work in the authors’ laboratories is supported (to BZ) by grants from the Swedish (22 2013) and Stockholm (181301) Cancer Societies.
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
NVP performed a major part of literature search, structured the information, and wrote the main part of the text. GSK and BZ conceptualized the review, structured the information, edited the main text, supervised the manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pervushin, N.V., Kopeina, G.S. & Zhivotovsky, B. Bcl-B: an “unknown” protein of the Bcl-2 family. Biol Direct 18, 69 (2023). https://doi.org/10.1186/s13062-023-00431-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13062-023-00431-4