
Abstract
Acquired resistance to cell death is a hallmark of cancer. The BCL-2 protein family members play important roles in controlling apoptotic cell death. Abnormal over-expression of pro-survival BCL-2 family members or abnormal reduction of pro-apoptotic BCL-2 family proteins, both resulting in the inhibition of apoptosis, are frequently detected in diverse malignancies. The critical role of the pro-survival and pro-apoptotic BCL-2 family proteins in the regulation of apoptosis makes them attractive targets for the development of agents for the treatment of cancer. This review describes the roles of the various pro-survival and pro-apoptotic members of the BCL-2 protein family in normal development and organismal function and how defects in the control of apoptosis promote the development and therapy resistance of cancer. Finally, we discuss the development of inhibitors of pro-survival BCL-2 proteins, termed BH3-mimetic drugs, as novel agents for cancer therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
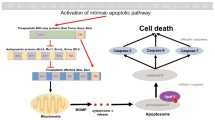
Apoptosis is an important cellular phenomenon critical for the development, survival, and functioning of multi-cellular organisms [1]. Consequently, deregulation of apoptosis is commonly associated with a broad range of diseases, ranging from cancer to degenerative disorders [2]. There are two well-defined pathways to apoptosis - the mitochondrial, also known as the intrinsic, stress-induced or BCL-2 regulated pathway, and the death receptor-induced, also known as the extrinsic pathway [3]. Several proteins participate in the process of programmed cell death, including caspases (cysteinyl aspartate-specific proteases), their adaptors/activators and the BCL-2 (B-cell lymphoma/leukemia-2 gene) protein family members which constitute the critical regulators of apoptosis. The BCL-2 protein family members can be classified into three sub-groups based on their functions and amino acid sequence similarity. This includes the pro-apoptotic BH3-only proteins (BIM, BID, PUMA, BMF, NOXA, BIK, BAD, HRK), the pro-survival proteins (BCL-2, BCL-XL, BCL-W, MCL-1, A1/BFL-1) and the effectors of apoptosis (BAX, BAK, BOK) [4,5,6]. The interaction between the members of the BCL-2 protein subgroups determines whether a cell will undergo apoptosis or survive. In healthy cells the pro-survival BCL-2 proteins restrain the effectors of apoptosis, BAX and BAK, to safeguard their survival. In response to a broad range of stresses, such as nutrient or growth factor deprivation, oxidative stress, γ-irradiation and treatment with diverse cytotoxic drugs, the levels of the pro-apoptotic BH3-only proteins are increased through diverse transcriptional and/or post-transcriptional processes [6,7,8]. The BH3-only proteins can bind with high affinity to the pro-survival BCL-2 proteins, and this unleashes the effectors of apoptosis, BAX and BAK, from their restraint. Upon such activation, BAX and BAK oligomerise and form pores in the outer mitochondrial membrane, thereby causing mitochondrial outer membrane permeabilisation (MOMP) resulting in the release of apoptogenic factors from inside the mitochondria, including cytochrome c and SMAC/DIABLO [9,10,11](Fig. 1). Some BH3-only proteins, including PUMA, BIM and the activated form of BID, called tBID, have also been reported to be able to bind and thereby directly activate BAX and BAK [12], but whether this process is critical for apoptosis initiation has been challenged [13]. MOMP unleashes the cascade of caspases, that cleave hundreds of cellular proteins and thereby drive the ordered demolition of the dying cells [14]. The expression of the various BCL-2 protein family members is stringently controlled at the transcriptional, post-transcriptional, and post-translational levels [6, 15, 16].
The impact of abnormal over-expression of pro-survival BCL-2 proteins as well as abnormally reduced expression of pro-apoptotic BCL-2 family members on tumour development and the resistance of malignant cells to anti-cancer agents are well established [17,18,19]. Therefore, BCL-2 family members and their regulators are attractive targets for the development of anti-cancer therapeutics [20, 21]. This review describes the roles of the different BCL-2 family members in the normal development and functioning of multi-cellular organisms and the impact of their dysregulation in cancer. We also discuss therapeutic strategies to target these regulators of apoptosis for cancer therapy, for example using BH3-mimetic drugs which inhibit selective pro-survival BCL-2 proteins.

The intrinsic pathway of apoptotic cell death is controlled by the BCL-2 protein family. This pathway is activated in response to various stress stimuli, such as oncogene activation or DNA damage. This causes an increase in the levels of the BH3-only proteins (e.g., PUMA, NOXA, BIM, BID, BAD) through diverse transcriptional as well as post-transcriptional processes. For example, the genes for PUMA and NOXA are directly transcriptionally activated by the tumour suppressor TP53/TRP53 (indicated in the dashed red box). The BH3-only proteins bind to the pro-survival BCL-2 proteins (e.g., BCL-2, BCL-XL, MCL-1) with high affinity. This unleashes the pro-apoptotic effector proteins BAK and BAX from their restraint by the pro-survival BCL-2 family members. The effectors of apoptosis, BAX and BAK, are also reported to be activated directly by certain BH3-only proteins, such as PUMA, BIM, and t-BID (the caspase activated form of BID). The activation of BAX and BAK allows these proteins to oligomerise and form pores in the outer mitochondrial membrane. This results in outer mitochondrial membrane permeabilisation (MOMP) causing release of cytochrome c from the space between the inner and the outer mitochondrial membranes into the cytoplasm. Upon release into the cytosol, cytochrome c drives the formation of a heptameric complex of the apoptotic protease activating factor 1 (APAF-1), called the apoptosome, which triggers the caspase cascade that causes the ordered demolition of the cells undergoing apoptosis
The role of pro-survival BCL-2 proteins in organismal development and function
The different pro-survival members of the BCL-2 protein family exert distinct critical roles during organismal development and function. The differences between them are due in part to differences in their expression patterns [22]. BCL-2 is expressed in a broad range of haematopoietic cell subsets, melanocyte progenitors, certain epithelial cell populations in the embryonic kidney and in certain neuronal cell populations. The absence of BCL-2 in mice causes fatal polycystic kidney disease within ~ 30 days post-birth, premature greying of the coat and an abnormal reduction in mature B and T lymphocytes [23, 24]. These defects can all be prevented by the concomitant absence of the pro-apoptotic BH3-only protein BIM [25]. The relatively high levels of BCL-2 during early neurulation in mice (E4.5–8) suggest its role in preventing apoptosis at that stage. BCL-2 expression wanes after the neural tube is formed in the central nervous system (CNS), whereas high levels are maintained in the peripheral nervous system [26]. Notably, however, BCL-2-deficient mice do not have marked defects in the CNS [23, 24], indicating that its role in the survival of these cells can be effectively backed up by other pro-survival BCL-2 family members.
BCL-XL is expressed broadly during embryonic development and its levels are particularly high throughout neuronal ontogeny including in differentiating cells [27]. The absence of BCL-XL in mice leads to embryonic death around E13.5 as a consequence of defects in the survival of certain neuronal cell populations and erythroid progenitors [28]. The loss of BCL-XL causes aberrant apoptosis in post-mitotic immature neurons of the developing brain, spinal cord, and dorsal root ganglion, demonstrating its essential role in the survival of these cell populations [28]. Conditional gene deletion studies have shown that in adult mice BCL-XL is critical for erythropoiesis and the survival of certain cell populations in the kidney [29].
BCL-W is expressed in several tissues, such as the testes, colon, brain and certain myeloid and lymphoid cell populations [30]. BCL-W is essential for spermatogenesis. BCL-W deficient male mice display progressive testicular degeneration with apoptosis of Sertoli cells occurring soon after weaning [31, 32]. Furthermore, abnormal death of Leydig cells is seen in BCL-W deficient males starting at 3 months of age. This causes disruption of the architecture of the testes and sterility. BCL-W knockout mice are otherwise normal in all other cell types examined [32].
MCL-1 is expressed in a broad range of cell types both during embryogenesis and in mice after birth [33]. MCL-1 has a critical role during early embryonic development. Genetic studies revealed the dependency of several cell types on MCL-1 for survival [22]. Mcl-1 gene knock-out mice die prior to implantation around embryonic day 3.5 [34]. Conditional gene knockout studies have shown that MCL-1 plays an essential role in hepatocytes, cardiomyocytes, neuronal cells, intestinal epithelial cells, mammary epithelial cells and several haematopoietic cell subsets [35,36,37,38,39]. Specifically, MCL-1 is required for the survival of haematopoietic stem and progenitor cells (HSPCs), the development of B as well as T cells and NK cells, the formation and maintenance of germinal-centre B cells, the development and survival of plasma cells (PCs) and immature erythroid cells [40,41,42,43,44,45,46].
A1/ BFL-1 is mainly expressed in haematopoietic cells, such as mitogen activated B and T cells, but its absence has only minimal impact on mice [47, 48]. A1-knockdown studies using in vivo expression of shRNAs in the haematopoietic system suggested a role for A1 in mast cell maturation [49], mature B cell survival [50] and early T cell development [51], but this was not replicated in studies of complete A1 gene knockout mice [47].
Regulation of the different members of the BCL-2 protein family
The balance between the various pro-survival and pro-apoptotic members of the BCL-2 protein family is crucial for normal embryonic development and tissue homeostasis after birth. The levels and activity of BCL-2 family members can be controlled through a broad range of transcriptional (transcriptional induction vs. repression), post-transcriptional (mRNA stability, effects of micro-RNAs or lncRNAs) and post-translational (e.g. phosphorylation, proteolytic processing and subcellular localisation) processes [6, 52].
The expression of pro-survival members of the BCL-2 protein family can be transcriptionally regulated by several transcription factors, such as E2F-1, the nuclear factor kappa B (NF-kB) family, and Janus kinase (JAK)-signal transducers and activators of transcription (STAT). Many of these transcription factors are activated by signalling from receptors for diverse cytokines (e.g. IL-2, IL-3, IL-4, IL -6, IL-7) [53] or the stimulation of antigen receptors on B as well as T cells [54].
The expression of BCL-2 protein can be transcriptionally regulated by the NF-κB transcription factor family [55] and by STATs [56]. The expression of BCL-2 is negatively regulated by the miRNAs, miR-15a, and miR-16-1, whereas the RNA binding protein nucleolin has been shown to increase BCL-2 expression by binding to the 3′-UTR, thereby enhancing BCL-2 mRNA stability [57]. BCL-2 protein is a long-lived protein with a half-life of about 20 h [46], and it was reported that this can be impacted by phosphorylation at residue Ser-70 [58].
Like the BCL-2 protein, BCL-XL is also relatively stable with a half-life of ~ 20 h [59]. The expression of BCL-XL can be increased in response to a variety of stimuli, such as IL-2, IL-3, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), colony-stimulating factor-1 (CSF-l), leukaemia inhibitory factor (LIF), erythropoietin (EPO) as well as the stimulation of antigen receptors, which can all promote the survival and/or proliferation of several haematopoietic cell subsets [60]. The transcription factors ETS (erythroblastosis virus E26 oncogene homolog), REL/NF-KB, STAT and AP-1 have all been reported to transcriptionally upregulate expression of the gene encoding BCL-XL [60]. Activated RAS/mitogen-activated protein kinase (RAS/MAP kinase), integrin, vitronectin and hepatocyte growth factor signalling cascades have also been shown to cause an increase in the expression of BCL-XL [61]. The microRNAs miR-5-5p, miR-125b, miR140-5p, miR133a-3p, miR4300, miR-377 and hsa-let-7b-5p are all reported to modulate the expression of BCL-XL [62,63,64].
BCL-W can be transcriptionally regulated by several transcription factors, including NF-κB, MEF2 (myocyte enhancer factor 2), ETS-1 and ETS-2, and C/EBP (CCAAT/enhancer-binding protein) [65]. BCL-W expression is positively regulated by the TCF4 (β-catenin/transcription factor 4) complex and transgenic expression of either dominant-negative TCF4 (TCF4ΔN) or wild-type β-catenin resulted in downregulated or upregulated activity of the promoter for BCL2L2 that encodes BCL-W, respectively [66]. Several miRNAs, including miR-29 and miR-122 [67], were shown to negatively regulate the expression of BCL-W by binding to the 3’-untranslated region (3’-UTR) of the BCL-W transcript[68]. A long non-coding RNA (lncRNA) RP11-436H11.5 functions as a competitive endogenous RNA, and was reported to sequester miR-335-5p which then causes an increase in the levels of BCL-W [68].
The expression of MCL-1 is regulated at the transcriptional, post-transcriptional and post-translational levels [69]. MCL-1 expression can be increased by many cytokines and growth factors, involving a range of signalling pathways [69]. Vascular endothelial growth factor (VEGF) and IL-6 regulate the expression of MCL-1 via autocrine signalling loops [70]. Activation of the MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase) signalling pathway reduces MCL-1 protein degradation and thereby increases its levels [71]. Activation of the NOTCH-1 signalling pathway induces the production of IL-6, thereby increasing the expression of MCL-1 [72]. MCL-1 protein levels are regulated by IL-15 and IL-22 through the JAK/STA3 [73] and phosphatidylinositol-3-kinase (PI3K) signalling pathways [74]. A broad range of miRNAs have been shown to downregulate the expression of MCL-1, including miR-26a [75], miR-15a, miR- 101 and miR-197 [76]. MCL-1 is a short-lived protein with a half-life of approximately 30 min [77]. Several E3 ubiquitin ligases, including MULE [78], SCFFbw7 [79], APC/CCdc20 [80] and SCFB-TrCP [81], regulate the stability of the MCL-1 protein. These ubiquitin ligases prime MCL-1 for proteasomal degradation. Conversely, the de-ubiquitinases USP9X [82] and USP13 [83] have been reported to stabilise the expression of the MCL-1 protein. The PEST domain of MCL-1 contains many phosphorylation sites, including Thr-92, Thr-163, Ser-64, Ser-155, and Ser-159. Phosphorylation of residues in the PEST domain of MCL-1 (region rich in amino acids Proline (P), glutamic acid (E), serine (S) and threonine (T)) by protein kinases, such as CDK1/2 (cyclin- dependent kinase 1/2), GSK3 (glycogen synthase kinase-3), JNK (c-Jun N-terminal kinase) and ERK, has been reported to impact access to different E3 ligases and thereby affect the ubiquitination and stability of MCL-1 [84, 85].
The gene encoding BFL-1/A1 is a direct target of NF-kB transcription factors [86]. The PI3K and JAK/STAT signalling pathways have also been reported to regulate the expression of BFL-1/A1. BFL-1/A1 has a short half-life of ~ 15 min and this is at least in part due to its ubiquitination followed by proteasomal degradation [87, 88].
The expression of the pro-apoptotic BCL-2 family proteins is also highly regulated at the transcriptional, post-transcriptional and post-translational levels [16]. For example, the genes that encode the BH3-only proteins PUMA [89, 90] and NOXA [91] are directly transcriptionally upregulated by the tumour suppressor TP53, and consequently their expression is increased in response to cytotoxic stimuli that cause DNA damage and thereby activate TP53 [89, 91]. PUMA is also important in the response of cells to certain TP53-independent apoptotic stimuli, such as treatment with glucocorticoids or phorbol ester [92]. Of note, it is not well understood how these agents control PUMA expression. The PUMA gene contains binding sites for several transcription factors in its promoter region, exon 1 and intron 1. These transcription factors include TP53, as mentioned, its close relatives TP63 and TP73 which bind to the same response element [93, 94] and also c-MYC, FOXO3a (Forkhead box O3a) which can be activated by growth factor deprivation, C/EBP homologous protein CHOP, and E2F1, the latter two activated by ER stress [95, 96]. Puma transcription can be down-regulated as part of a negative feedback process during TP53 activation. In response to DNA damage, TP53 induces the expression of the transcriptional repressor SLUG, which inhibits TP53-mediated transcription of Puma [97]. Post-translational modifications can also regulate the expression of PUMA. For example, it has been reported that the levels of PUMA can be reduced through phosphorylation at certain sites, including Ser10, which promotes its proteasomal degradation [98].
The gene that encodes NOXA, PMAIP1, can be transcriptionally regulated by various transcription factors, including by the tumour suppressor TP53 [91] and also by HIF-1α, E2F-1, MYC, TP63 and TP73 [99]. HIF-1α causes an increase in the levels of NOXA protein under hypoxic conditions, thereby mediating cell death in a TP53-independent manner [100]. NOXA can also be transcriptionally induced in response to post-translational modifications of IRF-1, IRF-3 and CREB [101]. The NOXA protein can be degraded by the 26S proteasome after priming by K11-linked poly-ubiquitination [102] or through a ubiquitin-independent pathway [103].
Transcription of the BCL2L11 gene, encoding BIM, has been reported to be regulated by FOXO3a [104], c-MYC [105], NF-Y[106], SMAD1/3[107], RUNX1-3[108], c-Jun [109] and RELA [110] although the importance of BIM regulation by the FOXO transcription factors has been questioned [111]. It has also been reported that the promoter for the gene encoding BIM can be epigenetically regulated through methylation of CpG dinucleotides [112]. BIM expression is also regulated by several miRNAs, such as the miR-106b~25 and miR-106a~363 clusters, and most prominently by the mir-17-92 cluster [113,114,115,116]. The activity and stability of the BIM protein is reported to be controlled by the phosphorylation of several residues. Phosphorylation can occur through a JNK-dependent mechanism, which promotes BIM dissociation from dynein light chain 1 (DLC1) [16, 117,118,119] and allows it to move to the mitochondria and induce BAK/BAX activation and apoptosis, or by the MAPK/ERK pathway that promotes BIM degradation and thereby increases cell survival [110]. The importance of the latter process has been questioned and there is evidence that ERK inhibits BIM mediated apoptosis not via a post-translational process but through direct transcriptional repression or induction of miRNAs that target the gene for BIM [120].
BMF expression can be transcriptionally regulated through the MAP kinase and AKT signalling pathways, for example in apoptosis that occurs during mammary epithelial morphogenesis [121]. The expression of BMF can also be epigenetically modulated at the promoter for its gene via CpG islands. Accordingly, treatment with histone deacetylase (HDAC) inhibitors causes a marked increase in the levels of BMF [122].
Binding patterns of the BCL-2 family proteins
Apoptosis signalling is controlled by complex interactions between the pro-survival BCL-2 family members, the pro-apoptotic BH3-only proteins and the effectors of apoptosis, BAX and BAK. The pro-survival BCL-2 proteins can either directly inhibit BAX and BAK by binding to them or by binding to the BH3-only proteins, thereby preventing them from activating the effectors of apoptosis. BIM, PUMA and tBID (the caspase activated form of BID) can bind to all pro-survival BCL-2 proteins with very high affinity [123, 124]. BAD selectively binds to BCL-2, BCL-XL and BCL-W, whereas NOXA selectively binds to MCL-1 and A1 [123, 124]. BAX and BAK also differ in their ability to bind to the pro-survival BCL-2 proteins. BAX can interact with all pro-survival BCL-2 proteins, whereas BAK associates only with MCL-1 and BCL-XL [125]. In contrast, the effector protein BOK is not regulated by the pro-survival BCL-2 proteins or the BH3-only proteins; instead the activity of BOK appears to be regulated mostly by its levels of synthesis and degradation [126, 127].
The role of BCL-2-family proteins in tumorigenesis
Evasion of apoptosis is one of the hallmarks of cancer [128]. Defects in the control of apoptosis that contribute to the development, expansion and therapy resistance of cancer can be caused by abnormally increased expression of pro-survival proteins or abnormally decreased expression of pro-apoptotic proteins.
Aberrantly increased levels of pro-survival BCL-2 proteins
Abnormally high expression of pro-survival BCL-2 proteins is correlated with the development and poor prognosis of various cancers [4, 129, 130]. BCL-2, MCL-1, and BCL-XL are frequently over-expressed in lymphomas and leukaemias [20, 21, 131, 132]. The genomic regions containing the genes encoding MCL-1 and BCL-XL are somatically amplified in ~ 15% of diverse tumour types [133]. The abnormal over-expression of pro-survival BCL-2 family proteins can also be caused by chromosomal translocations or increased gene transcription [134]. However, it is also known that cellular dependence on distinct pro-survival BCL-2 family proteins does not always correlate with expression patterns. Studies using conditional gene targeting or inducible CRISPR platforms revealed that different malignant cells rely on the expression of distinct pro-survival BCL-2 proteins for their sustained survival, such as MCL-1 for MYC-driven lymphomas [135], even though their genes are not over-expressed owing to somatic copy number amplification or chromosomal translocation. This may be because their normal cellular counterparts rely on these same pro-survival BCL-2 proteins for their survival or because stresses present in malignant cells have imposed these dependencies.
The t(14;18) chromosomal translocation causes deregulated over-expression of BCL-2 in human follicular lymphoma (FL) [136, 137]. High levels of BCL-2 were also detected in several other haematological malignancies, including chronic lymphocytic leukemia (CLL), diffuse large B cell lymphoma (DLBCL) and mantle cell lymphoma [138,139,140] and in certain solid tumours, including subsets of brain, breast and lung cancer [141, 142]. Over-expression of BCL-2 greatly accelerates c-MYC driven lymphoma development in mice [143]. Moreover, over-expression of BCL-2 (or other pro-survival BCL-2 proteins) renders both malignant as well as non-transformed cells markedly resistant to diverse anti-cancer agents that kill cells in either a TP53-dependent [144] or TP53-independent manner [145].
Approximately 3% of human cancers of diverse origin carry somatically acquired amplification of the region that harbours the gene for BCL-XL [133]. It has been reported that BCL-XL plays a critical role in the progression of glioma [146] and breast cancer [147]. Human multiple myeloma (MM) cells as well as melanoma cells express high levels of BCL-XL [148, 149] and, accordingly, some of these malignant cells can be killed by treatment with inhibitors of BCL-XL, either on their own or more potently in combination with inhibitors of oncogenic kinases [150,151,152]. High levels of BCL-XL have also been observed in certain lymphomas, such as B cell non-Hodgkin lymphomas, FL, and DLBCL as well as T cell non-Hodgkin lymphomas [153]. Notably, EBV-associated T/NK cell lymphoma cells are dependent on BCL-XL for continued growth and survival [154]. High levels of BCL-XL can be detected in many colorectal cancers and, accordingly, inhibition of BCL-XL impairs adenoma outgrowth in vivo and enhances the efficacy of chemotherapy in colorectal cancer [155].
In gastric cancer, high levels of BCL-W have been reported to promote the survival, migration, and invasion of malignant cells [156]. Furthermore, BCL-W was observed in colorectal adenocarcinomas, with relatively higher levels detected in advanced-stage cancers as compared to localised tumours with better prognosis [157]. It has also been reported that certain lymphoma cells rely on BCL-W for sustained survival [158], but another study was not able to reproduce this finding [159].
MCL-1 is expressed at relatively high levels in many haematological malignancies, including MM and acute myeloid leukaemia (AML), as well as in cancers of the breast, pancreas, prostate, lung, and ovary [160,161,162,163,164]. Approximately 12% of human cancers of diverse origin carry somatically acquired amplifications of the region that harbours the MCL-1 gene [133]. Transgenic mice over-expressing MCL-1 in haematopoietic cells develop B lymphoid [165] or myeloid malignancy, albeit with low incidence and long latency [165, 166]. Moreover, MCL-1 over-expression greatly accelerates the development of c-MYC driven lymphoma [166]. Studies using inducible gene deletion revealed that a broad range of cancer cells, including AML [167], MYC driven B cell lymphomas [135], T cell lymphomas and lung cancer caused by loss of TP53 or mutations in Notch [168,169,170] require MCL-1 for sustained survival and growth. These findings indicate that MCL-1 could be an attractive target for cancer therapy [164] .
Aberrantly decreased levels of pro-apoptotic BCL-2 family proteins are observed in diverse human cancers
The reduction of pro-apoptotic members of the BCL-2 family has also been implicated in the development and therapy resistance of cancer. The levels of the BH3-only proteins BIM and/or PUMA are abnormally low in several cancers [174, 175]. For example, ~ 40% of human Burkitt lymphomas express very low levels of the mRNAs for BIM and/or PUMA, and this was ascribed to epigenetic silencing of their genes [174, 176]. Reduced expression of PUMA has also been reported in subcutaneous melanoma, and this correlated with poor prognosis [177]. The downregulation of BIM due to deletion or hyper-methylation of the gene was reported in mantle cell lymphoma [178] and DLBCLs [176, 179]. Finally, abnormally low levels of NOXA and/or BIM were observed in colon cancer and small-cell lung cancer [180].
There are reports of loss of BAX expression in human cancers, including endometrial and colon cancers [181, 182]. However, the combined loss of BAX and BAK, which would be required to render cells resistant to apoptosis because of the extensive functional overlap of these effectors of apoptosis [127, 183], is only rarely seen in human cancer (e.g. some AML cells) [167], probably because four alleles would need to be mutated to achieve this.
The deletion of pro-apoptotic members of the BCL-2 family can accelerate tumour development in mice
Several studies using gene-targeted mice demonstrated that the absence of pro-apoptotic BCL-2 family members promotes tumour development and renders malignant cells resistant to a broad range of anti-cancer agents. Mice lacking either PUMA [92, 184] or BIM [185], two of the BH3-only proteins that can inhibit all pro-survival BCL-2 proteins, do not spontaneously develop tumours, but mice lacking both of these critical initiators of apoptosis develop plasma cell-like tumours with advanced age [186]. In cells genetically engineered to express oncogenes, the impact of loss of pro-apoptotic BCL-2 family members is even more pronounced. The individual loss of the genes encoding PUMA or BIM (even loss of one allele) substantially accelerates MYC-driven pre-B/B cell lymphoma in mice carrying an Eµ-MYC transgene [187, 188]. The loss of PUMA was also shown to cooperate with the oncogenes H-Ras or E1A in the neoplastic transformation of fibroblasts in culture [189]. Finally, loss of BMF or NOXA has been shown to accelerate γ-irradiation induced thymic T cell lymphoma development in mice [122, 190].
Since the genes that encode the pro-apoptotic proteins PUMA and NOXA are both direct transcriptional targets of TRP53, it was hypothesised that mice lacking PUMA and NOXA (and therefore lacking the capability to undergo TRP53-mediated apoptosis) would develop tumours at the same rate as TRP53-deficient mice. However, in contrast to TRP53-deficient mice which all develop tumours prior to 300 days of age (in the absence of an engineered oncogenic driver), the PUMA-deficient as well as PUMA/NOXA double-deficient mice do not develop cancer spontaneously on a C57BL/6 genetic background [188, 191]. This demonstrates that loss of the pro-apoptotic function of TRP53 alone is not sufficient to cause tumour development, i.e. other cellular processes activated by TRP53, such as the coordination of DNA damage repair, may be even more critical for its ability to suppress tumorigenesis [192].
Defects in the intrinsic apoptotic pathway render malignant cells resistant to a broad range of anti-cancer therapeutics
Resistance to anti-cancer therapy contributes to poor clinical outcomes. Defects in the intrinsic apoptotic pathway, owing to over-expression of pro-survival BCL-2 proteins or the abnormal reduction of pro-apoptotic BCL-2 family members, render both malignant as well as non-transformed cells profoundly resistant to a broad range of anti-cancer therapeutics. This was first demonstrated when lymphoid cells from BCL-2 transgenic mice were found to be resistant to several DNA damage-inducing anti-cancer agents and glucocorticoids [144]. Accordingly, increased levels of BCL-2 expression have been correlated with resistance to several anti-cancer drugs, including 5-fluorouracil, adriamycin and mitomycin, in gastric cancer [193], cisplatin in ovarian cancer [194] and doxorubicin in osteosarcoma and chondrosarcoma [195, 196].
The over-expression of BCL-XL has also been reported to protect tumour cells from a broad range of chemotherapeutic drugs [197, 198]. High levels of BCL-XL driven by STAT5 have been implicated in the resistance of BCR/ABL+ chronic myelogenous leukaemia (CML) to apoptosis [199]. Moreover, in a cisplatin-resistant patient cohort of ovarian cancer, 61.5% of samples displayed over-expression of BCL-XL [200]. A study using a nude mouse tumour xenograft model showed that BCL-XL over-expression rendered ovarian cancer cells resistant to cisplatin, paclitaxel, topotecan and gemcitabine [200].
High levels of MCL-1 have also been associated with the resistance of a broad range of malignant cells to chemotherapeutic agents [164]. Overexpression of MCL-1 causes resistance to various conventional chemotherapeutic agents, such as cisplatin in ovarian cancer cells [201], lapatinib in a human colon cancer cell line [202], rituximab in B-cell malignancies [203] and prednisone in MLL-rearranged infant acute lymphoblastic leukemia [204]. Of note, the downregulation of MCL-1 levels restores the effectiveness of these anti-cancer drugs in several cancer cell lines [205, 206].
Experiments using mice lacking different BH3-only proteins identified which of these proteins are critical for cell killing by different anti-cancer agents. PUMA is required for the killing of cells by DNA damage inducing agents that activate TRP53 [92, 184]. Of note, the combined loss of PUMA, NOXA, and BIM renders MYC-driven lymphoma cells almost completely resistant to killing by DNA damage inducing anti-cancer agents, such as etoposide or cyclophosphamide [207]. BIM and PUMA both contribute to the killing of lymphoid cells by glucocorticoids [92, 185] and BIM is also needed for the killing of malignant cells by inhibitors of oncogenic kinases, such as the BCR-ABL inhibitor Gleevec in chronic myeloid leukaemia (CML) [150, 151, 208]. Malignant as well as non-transformed cells lacking both BAX and BAK are profoundly resistant to all anti-cancer agents tested [183], demonstrating that these effectors of apoptosis are essential for such cell killing and that the killing of cells by these drugs is mediated to a large extent by the induction of apoptosis.
Therapeutic interventions that directly target the apoptotic machinery

Potential strategies targeting the intrinsic apoptotic pathway either by directly targeting the pro-apoptotic or the pro-survival BCL-2 family members or by targeting metabolic pathways and signal transducers to induce apoptosis by causing an increase in pro-apoptotic BH3-only proteins and/or a decrease in the pro-survival BCL-2 family members
There are several different approaches to induce apoptosis of cancer cells for therapy (Fig. 2). The discovery that BH3-only proteins are critical for the initiation of apoptosis triggered by diverse anti-cancer agents, and that genetic deletion of distinct pro-survival BCL-2 proteins can effectively kill certain types of malignant cells, gave rise to the concept that pharmacological inhibitors of pro-survival BCL-2 proteins that mimic the function of BH3-only proteins could be effective in cancer therapy [20, 21]. This led to programs by pharma and biotechnology companies to generate small molecules that mimic the function of pro-apoptotic BH3-only proteins, known as the BH3-mimetic drugs [21]. Since non-transformed cells in healthy tissues also depend on pro-survival BCL-2 proteins for their survival (see above), a notable issue for the clinical use of BH3-mimetic drugs is their on-target toxicity [20, 21].
The first BH3-mimetic compounds ABT- 737 and its orally available derivative ABT-263 (navitoclax) inhibit BCL-2, BCL-XL, and BCL-W. They can induce apoptosis in a broad range of cancer-derived cell lines in vitro and delay the growth of certain tumours in vivo in tumour transplant models [152]. ABT-263/navitoclax was the first BH3-mimetic drug to be tested in patients [209]. ABT-263/navitoclax proved effective in CLL patients in clinical trials but dose-limiting thrombocytopenia, due to the dependence of platelets on BCL-XL for survival [210], hampered the progression of this agent in the clinic [211]. ABT-199 (venetoclax) was therefore developed as a BH3-mimetic drug that is a highly selective BCL-2 inhibitor that potently induces apoptosis in BCL-2 dependent malignant cells [212]. Venetoclax is highly effective in patients with relapsed or refractory CLL [52, 213]. Remarkably, the combination of venetoclax with rituximab (antibody against CD20) led to complete remissions in 51% of CLL patients, with disease-free survival persisting for up to 2 years after completion of therapy [214]. In high-risk relapsed/refractory AML patients, administration of venetoclax in phase 2 clinical trials resulted in complete response/complete response with incomplete blood recovery (CR/CRi) in 19% of patients [215]. Combination therapies for AML including venetoclax are proving even more effective. Considering the clinical benefits of venetoclax as a monotherapy, as well as in combination with standard-of-care anti-cancer drugs, it has now been approved by the FDA and several other regulatory authorities worldwide for the treatment of patients with CLL or AML [20, 21, 216, 217].
Many cancer cells have been shown to depend on BCL-XL for their sustained survival and proliferation, prompting the development of BCL-XL-specific BH3-mimetic drugs. WEHI-539 was the first compound to specifically target BCL-XL [218]. Additional structure-guided design led to the development of A-1155463 and A-1331852, which are both also selective for BCL-XL. A-1155463 exhibited anti-tumour activity in a xenograft model of small cell lung carcinoma (SCLC) in immune-deficient mice [219]. A-1331852 was shown to potently kill several cancer-derived cell lines on its own and cooperates with a broad range of anti-cancer agents in vitro [220]. However, at present, no BCL-XL specific inhibitors have been approved for clinical use, and clinical trials are progressing slowly because of the predicted on-target toxicity to platelets.
Abnormally increased expression of MCL-1 can drive tumorigenesis and often confers a poor prognosis. Therefore, several MCL-1 inhibitors have been developed and assessed in pre-clinical studies, these include S63845 [171], A-1210477 [221], AMG176 [172] and AZD5991 [173]. The in vitro and in vivo potential of the tool compound S63845 was explored in pre-clinical studies in haematological malignancies, such as MM, AML, CML, and c-MYC-driven Burkitt lymphoma [171]. S63845, either alone or in combination with inhibitors of oncogenic kinases, was found to be moderately effective in certain solid tumours, such as breast cancer and prostate cancer [171, 222] and SCLC derived cell lines that are express high levels of MCL-1 but low levels of BCL-XL [223]. There have been limited details on the potency of MIK665/S64315, the related compound that has entered clinical trials. The MCL-1 inhibitor AMG176 has been found to be effective in diverse haematological malignancies [172] and in certain solid tumour derived cell lines, such as breast cancer and non-small cell lung cancer [224]. AZD599, another MCL-1 specific BH3-mimetic drug, was also shown to be effective for the treatment of MM in mouse models, and its effect can be enhanced by co-treatment with the BCL-2 inhibitor venetoclax or the proteasome inhibitor bortezomib [173]. Until now, six MCL-1 inhibitors have entered clinical trials, but some of these trials have been halted due to on-target cardiac toxicity [225, 226]. This toxicity was predicted from genetic studies that had shown that MCL-1 is critical for the survival of cardiomyocytes [37, 227].
The survival of many cancer cells is safeguarded not by a single pro-survival BCL-2 protein but rather by two or even more of these proteins. Hence, effective killing of such cancer cells will require two or more BH3-mimetic drugs, or combined treatment with one BH3-mimetic drug plus one or several standard-of-care anti-cancer agents that cause an increase in BH3-only proteins which then inhibit the pro-survival BCL-2 proteins that are not targeted by the BH3-mimetic drug used [20]. The tolerability of such therapies will need to be carefully determined. For example, it is unlikely that inhibitors of MCL-1 and BCL-XL can be combined safely in patients, given that mice lacking only single alleles of the genes encoding MCL-1 and BCL-XL die on the day of birth because of severe craniofacial and several other defects [228]. Of note, the combination of an MCL-1 inhibitor with a BCL-2 inhibitor was shown to be tolerable in mice, providing a synergistic response that was able to overcome drug resistance in models of DLBCL [229]. AML derived cell lines were more potently killed by the combination of an MCL-1 inhibitor (S63845) and a BCL-2 inhibitor (venetoclax) than by treatment with either agent alone [229]. Other than the BH3-mimetic drugs, antisense oligonucleotides [230] and HDAC inhibitors that can lead to repression of expression of the BCL-2 pro-survival proteins [231] might be a promising approach in targeting the pro-survival members of the BCL-2 protein family.
Since the pro-apoptotic BH3-only proteins are the critical initiators of apoptosis, there should also be a focus on developing novel therapeutic strategies that increase the expression of these pro-apoptotic proteins. Such therapeutic interventions could be used alongside BH3-mimetic drugs to increase their effectiveness. This could be achieved by either using drugs that can boost expression of the BH3-only proteins directly or by developing drugs that can inhibit negative regulators of their expression. It is known that many conventional chemotherapeutic drugs do induce expression of the BH3-only proteins, particularly those drugs that can induce DNA damage. Colon cancer cells exposed to 5-fluorouracil-based anti-cancer therapy display elevated expression of PUMA and BIM and high induction of these proteins is correlated with a better prognosis of the patients [180]. Decreased levels of BIM have been correlated with poor response to diverse inhibitors of oncogenic kinases in several cancers [232] and therefore strategies to boost BIM expression would be anticipated to increase sensitivity both to inhibitors of oncogenic kinases and to BH3-mimetic drugs. Towards targeting negative regulators of BH3-only protein expression, efforts are underway to find TRP53 independent regulators of these proteins. Since the expression of PUMA and BIM can be suppressed by epigenetic modifications, drugs that target epigenetic regulators, such as HDAC inhibitors, might be beneficial for upregulating these initiators of apoptosis, thereby increasing the effectiveness of anti-cancer therapy [233, 234].
There is considerable evidence that cellular metabolism can impact the levels of certain pro-survival as well as pro-apoptotic BCL-2 family members. Of note, alterations in tumour cell metabolism were shown to potentiate the ability of malignant cells to evade apoptosis. The PI3K/AKT pathway provides a link between cell proliferation and cell metabolism. In cancer, the PI3K/AKT pathway is frequently aberrantly activated, for example due to the expression of oncogenic kinases, loss of PTEN as well as mutation or amplification of the gene for PI3K, promoting glucose metabolism [235,236,237]. The oncogenic kinase BCR-ABL regulates expression of the glucose transporter 1 (GLUT 1) via the PI3K/AKT pathway [235]. Thus, several inhibitors of oncogenic tyrosine kinases have been developed that act at least in part by inhibiting the PI3K/AKT pathway to suppresses glucose metabolism [238,239,240]. Moreover, directly targeting the glycolysis pathway with 2-deoxyglucose (2-DOG) was shown to enhance cisplatin induced killing of ovarian cancer cells [241]. Deprivation of nutrients, such as glucose or amino acids, causes a substantial decrease in MCL-1 levels because this causes a reduction in protein translation via activation of AMPK-activated protein kinase (AMPK), leading to the inhibition of mTOR [242]. Reduced glucose metabolism as a consequence of cytokine deprivation causes an increase in the levels of pro-apoptotic PUMA and BIM [243, 244] leading to BAX/BAK mediated apoptosis [245]. These approaches therefore tip the balance between pro-apoptotic and anti-apoptotic proteins towards the induction of apoptosis and the elimination of cancer cells.
NF-κB transcription factors are critical regulators of both the adaptive and innate immune systems [246]. They can promote cell survival by modulating the expression of certain pro-survival BCL-2 family members, most notably BCL-XL and A1/BFL-1 [54, 247]. These findings suggest that inhibitors of NF-kB signalling, such as inhibitors of IKK, an upstream activator of NF-kB, may be used to enhance BH3-mimetic drug or chemotherapeutic drug induced killing of cancer cells by reducing the levels of pro-survival BCL-2 proteins.
MYC is a helix-loop-helix-leucine zipper protein that, as a heterodimer with MAX, binds to a palindromic E-box element CACGTG in DNA and thereby upregulates the expression of specific target genes. In non-transformed cells, the expression of MYC relies on mitogenic signals and MYC is critical for cell volume growth and cell proliferation [248]. Approximately 70% of cancers express high levels of MYC, owing to chromosomal translocations (e.g., in Burkitt lymphoma), genomic amplifications or oncogenic signals. Studies using mice with a regulatable Myc transgene showed that MYC-driven tumour cells die when MYC is removed [249]. Deregulated MYC expression can also enhance the predisposition of cells to undergo apoptosis in response to stress, such as growth factor deprivation [250]. This involves transcriptional processes relating to the MYC relative MNT that causes an increase in BIM [251]. Accordingly, genetic loss of BIM or PUMA reduces MYC-driven apoptosis [187, 252]. Once the mechanisms by which MYC causes an increase in BIM and PUMA are understood, it may become possible to manipulate this process to increase the levels of these pro-apoptotic BH3-only proteins in malignant cells for therapeutic benefit, either alone or in combination with anti-cancer agents, such as BH3-mimetic drugs.
Since BAX and BAK are the critical effectors of apoptosis, it is possible that plasma membrane permeable agents that can activate these proteins could be effective in cancer therapy. However, the safety of such approaches would need to be considered carefully since most, if not all, non-transformed cells express either BAX and/or BAK and would therefore also be targets of such agents. Possibly, activators of BAX and BAK can only be administered safely to patients when conjugated to antibodies or ligands that will direct them preferentially to malignant cells. Such conjugate approaches are already being explored to increase the safety and utility of BH3-mimetic drugs that have considerable on-target toxicities to non-transformed cells, particularly those targeting MCL-1 or BCL-XL [20]. Finally, since the intrinsic and the death receptor activated apoptotic pathways are distinct, albeit converging on the activation of effector caspases [3], it is expected that BH3-mimetic drugs (activating the intrinsic apoptotic pathway) and activators of death receptors, such as the TRAIL receptors, would cooperate in killing malignant cells [253]. Again, the tolerability of such approaches will need to be tested rigorously.
Concluding remarks
The BCL-2 protein family members constitute the crucial regulators of apoptosis. Abnormalities in the expression of pro-survival or pro-apoptotic members of the BCL-2 protein family can promote tumour development and render malignant cells resistant to anti-cancer therapy. The field has developed a detailed understanding of the control of apoptosis and how the different subgroups of the BCL-2 family proteins interact with each other. This understanding has enabled the development of novel anti-cancer drugs, called BH3-mimetics, that can directly activate the apoptosis machinery by inhibiting pro-survival BCL-2 proteins. These compounds have shown efficacy in pre-clinical studies, and some have entered clinical trials for cancer therapy, with the BCL-2 specific inhibitor venetoclax FDA approved for the treatment of patients with CLL or AML. Current efforts are aimed at developing effective and tolerable treatment schedules for the BH3-mimetic drugs that inhibit MCL-1 or BCL-XL and to discover which other anti-cancer agents can be combined with these drugs to achieve effective and safe cancer therapy. We believe that gaining a clearer understanding of how the expression of the pro-apoptotic BH3-only proteins is regulated may lead to insights that can be harnessed to develop novel therapeutics that enhance the expression of these initiators of cell killing. Such agents would be expected to cooperate with BH3-mimetic drugs and standard chemotherapeutics in killing malignant cells.
Data Availability
As this is a review article, no new data are presented, and all published data are referenced.
References
Voss AK, Strasser A (2020) The essentials of developmental apoptosis F1000Research 9. https://doi.org/10.12688/f1000research.21571.1
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462. doi:https://doi.org/10.1126/science.7878464
Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S (1995) Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J 14:6136–6147. doi:0.1002/j.1460-2075.1995.tb00304.x
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326. doi:https://doi.org/10.1126/science.281.5381.1322
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59. doi:https://doi.org/10.1038/nrm2308
Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15:49–63. doi:https://doi.org/10.1038/nrm3722
Strasser A, O’Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69:217–245. doi:https://doi.org/10.1146/annurev.biochem.69.1.217
Lopez J, Tait SWG (2015) Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer 112:957–962. doi:https://doi.org/10.1038/bjc.2015.85
Cosentino K, Garcia-Saez AJ (2017) Bax and bak pores: are we closing the circle? Trends Cell Biol 27:266–275. doi:https://doi.org/10.1016/j.tcb.201611.004
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139. doi:https://doi.org/10.1083/jcb.139.5.1281
Uren RT, Iyer S, Kluck RM (2017) Pore formation by dimeric Bak and Bax: an unusual pore? Philos Trans R Soc Lond B Biol Sci 372:20160218. doi:https://doi.org/10.1098/rstb.2016.0218
Brunelle JK, Letai A (2009) Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci 122:437–441. doi:https://doi.org/10.1242/jcs.031682
O’Neill KL, Huang K, Zhang J, Chen Y, Luo X (2016) Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev 30:973–988. doi:https://doi.org/10.1101/gad.276725.115
Strasser A, Cory S, Adams JM (2011) Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J 30:3667–3683. doi:https://doi.org/10.1038/emboj.2011.307
Huang DCS, Strasser A (2000) BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103:839–842. doi:https://doi.org/10.1016/S0092-8674(00)00187-2
Puthalakath H, Strasser A (2002) Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 9:505–512. doi:https://doi.org/10.1038/sj.cdd.4400998
Yip K, Reed J (2008) Bcl-2 family proteins and cancer. Oncogene 27:6398–6406. doi:https://doi.org/10.1038/onc.2008.307
Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K et al (1996) Immunohistochemical analysis of Bcl-2, Bax, Bcl-X and Mcl-1 expression in prostate cancers. Am J Pathol 148:1567–1576
Khanna KK, Wie T, Burrows SR, Moss DJ, Krajewski S, Reed JC et al (1996) Expression of p53, Bcl-2, Bax, Bcl-X2 and c-myc in radiation-induced apoptosis in Burkitt’s lymphoma cells. Cell Death Differ 3:315–322
Diepstraten ST, Anderson MA, Czabotar PE et al (2022) The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer 22:45–64. doi:https://doi.org/10.1038/s41568-021-00407-4
Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW (2018) BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 34:879–891. doi:https://doi.org/10.1016/j.ccell.2018.11.00
Brinkmann K, Ng AP, de Graaf CA et al (2022) What can we learn from mice lacking pro-survival BCL-2 proteins to advance BH3 mimetic drugs for cancer therapy? Cell Death Differ 29:1079–1093. doi:https://doi.org/10.1038/s41418-022-00987-0
Nakayama K, Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY (1994) Targeted disruption of bcl-2ab in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA 91:3700–3704. doi:https://doi.org/10.1073/pnas.91.9.3700
Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 75:229–240. doi:https://doi.org/10.1016/0092-8674(93)80065-m
Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM (2001) Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell 1:645–653. doi:https://doi.org/10.1016/s1534-5807(01)00083-1
Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ (1994) Bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development 120:301–311. doi:https://doi.org/10.1242/dev.120.2.301
Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB (1993) Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74:597–608. doi:https://doi.org/10.1016/0092-8674(93)90508-n
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I et al (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x- deficient mice. Science 267:1506–1510
Brinkmann K, Waring P, Glasser SP, Wimmer V, Cottle DL et al (2020) BCL-XL exerts a protective role against anemia caused by radiation-induced kidney damage. EMBO J 39. doi:https://doi.org/10.15252/embj.2020105561
O’Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DC, Strasser A (2001) Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ 8:486–494. doi:https://doi.org/10.1038/sj.cdd.4400835
Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD et al (1998) Testicular degeneration in Bclw-deficient mice. Nat Genet 18:251–256
Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N et al (1998) Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA 95:12424–12431. doi:https://doi.org/10.1073/pnas.95.21.12424
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW (1993) MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A 90:3516–3520. doi:https://doi.org/10.1073/pnas.90.8.3516
Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ (2000) Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 14:23–27
Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S et al (2013) Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 27:1351–1364. doi: https://doi.org/10.1101/gad.215855.113
Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T et al (2009) Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology 50:217–1226. doi:https://doi.org/10.1002/hep.23126
Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM et al (2013) Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 27:1365–1377. doi:https://doi.org/10.1101/gad.215871.113
Healy ME, Boege Y, Hodder MC et al (2020) MCL1 Is Required for Maintenance of Intestinal Homeostasis and Prevention of Carcinogenesis in Mice. Gastroenterology 159:183–199. doi:https://doi.org/10.1053/j.gastro.2020.03.017
Fu NY, Rios AC, Pal B, Soetanto R, Lun AT, Liu K, Beck T, Best SA, Vaillant F, Bouillet P, Strasser A, Preiss T, Smyth GK, Lindeman GJ, Visvader JE (2015) EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival. Nat Cell Biol 17:365–375. doi:https://doi.org/10.1038/ncb3117
Vikstrom I, Carotta S, Lüthje K, Peperzak V, Jost PJ, Glaser S, Busslinger M, Bouillet P, Strasser A, Nutt SL, Tarlinton DM (2010) Mcl-1 is essential for germinal center formation and B cell memory. Science 330:1095–1099. doi:https://doi.org/10.1126/science.1191793
Peperzak V, Vikström I, Walker J, Glaser SP, LePage M, Coquery CM, Erickson LD, Fairfax K et al (2013) Mcl-1 is essential for the survival of plasma cells. Nat Immunol 14:290–297. doi:https://doi.org/10.1038/ni.2527
Sathe P, Delconte R, Souza-Fonseca-Guimaraes F et al (2014) Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun 5. doi:https://doi.org/10.1038/ncomms5539
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ (2003) Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426:671–676. doi:https://doi.org/10.1038/nature02067
Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S-I, Akashi K, Korsmeyer SJ (2005) Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307:1101–1104. doi:https://doi.org/10.1126/science.1106114
Turnis ME, Kaminska E, Smith KH, Kartchner BJ, Vogel P et al (2021) Requirement for antiapoptotic MCL-1 during early erythropoiesis. Blood 137:1945–1958. doi:https://doi.org/10.1182/blood.2020006916
Carrington E, Zhan Y, Brady J et al (2017) Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ 24:878–888. doi:https://doi.org/10.1038/cdd.2017.30
Schenk R, Tuzlak S, Carrington E et al (2017) Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ 24:534–545. doi:https://doi.org/10.1038/cdd.2016.156
Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D et al (2017) The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ 24:523–533
Ottina E, Lyberg K, Sochalska M, Villunger A, Nilsson GP (2015) Knockdown of the antiapoptotic Bcl-2 family member A1/Bfl-1 protects mice from anaphylaxis. J Immunol 194:1316–1322. doi:https://doi.org/10.4049/jimmunol.1400637
Sochalska M, Ottina E, Tuzlak S, Herzog S, Herold M, Villunger A (2016) Conditional knockdown of BCL2A1 reveals rate-limiting roles in BCR-dependent B cell survival. Cell Death Differ 23:628–639
Ottina E, Grespi F, Tischner D, Soratroi C, Geley S, Ploner A et al (2012) Targeting antiapoptotic A1/Bfl-1 by in vivo RNAi reveals multiple roles in leukocyte development in mice. Blood 119:6032–6042. doi:https://doi.org/10.1182/blood-2011-12-399089
Delbridge AR, Grabow S, Strasser A, Vaux DL (2016) Thirty years of BCL2: Translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 16:99–109. doi:https://doi.org/10.1038/nrc.2015.17
Jourdan M, De Vos J, Mechti N, Klein B (2000) Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ 7:1244–1252. doi:https://doi.org/10.1038/sj.cdd.4400758
Grossmann M, O’Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S (2000) The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J 19:6351–6360. doi:https://doi.org/10.1093/emboj/19.23.6351
Catz S, Johnson J (2001) Transcriptional regulation of bcl-2 by nuclear factor κB and its significance in prostate cancer. Oncogene 20:7342–7351. doi:https://doi.org/10.1038/sj.onc.1204926
Bhattacharya S, Ray RM, Johnson LR (2005) STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J 392:335–344. doi:https://doi.org/10.1042/BJ20050465
Willimott S, Wagner SD (2010) Post-transcriptional and post-translational regulation of Bcl2. Biochem Soc Trans 38:1571–1575. doi:https://doi.org/10.1042/BST0381571
Ruvolo P, Deng X, May W (2001) Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 15:515–522. doi:https://doi.org/10.1038/sj.leu.2402090
Rooswinkel RW, de Kooji BV, de Vries E et al (2014) Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood 123:2806–2815. doi:https://doi.org/10.1182/blood-2013-08-519470
Sevilla L, Zaldumbide A, Pognonec P, Boulukos KE (2001) Transcriptional regulation of the bcl-x gene encoding the anti-apoptotic Bcl-xL protein by Ets, Rel/NFkappaB, STAT and AP1 transcription factor families. Histol Histopathol 16:595–601. doi:https://doi.org/10.14670/HH-16.595
Grad JM, Zeng XR, Boise LH (2000) Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncology 12:543–549. doi:https://doi.org/10.1097/00001622-200011000-00006
Morales-MartÌnez M, Vega MI (2022) Roles and Regulation of BCL-xL in Hematological Malignancies. Int J Mol Sci 23. doi:https://doi.org/10.3390/ijms23042193
Hernandez-Luna M, Rocha-Zavaleta L, Vega MI, Huerta-Yepez S (2012) Hypoxia inducible factor-1? induces chemoresistance phenotype in non-Hodgkin lymphoma cell line via up-regulation of Bcl-xL. Leuk Lymphoma 54:1048–1055. doi:https://doi.org/10.3109/10428194.2012.733874
Zhou Y, Chen L, Barlogie B, Stephens O, Wu X, Williams DR, Cartron M-A, van Rhee F, Nair B, Waheed S et al (2010) High-risk myeloma is associated with global elevation of miRNAs and overexpression ofEIF2C2/AGO2. Proc Natl Acad Sci USA 107:7904–7909. doi:https://doi.org/10.1073/pnas.0908441107
Uittenbogaard M, Baxter KK, Chiaramello A (2009) Cloning and characterization of the 5’UTR of the rat anti-apoptotic Bcl-w gene. Biochem Biophys Res Commun 389:657–662. doi:https://doi.org/10.1016/j.bbrc.2009.09.049
Lapham A, Adams JE, Paterson A et al (2009) The Bcl-w promoter is activated by beta-catenin/TCF4 in human colorectal carcinoma cells. Genes Cancer 432:112–117. doi:https://doi.org/10.1016/j.gene.2008.12.002
Lin CJ, Gong HY, Tseng HC, Wang WL, Wu JL (2008) miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem Biophys Res Commun 375:315–320. doi:https://doi.org/10.1016/j.bbrc.2008.07.154
Wang K, Jin W, Song Y, Fei X (2017) LncRNA RP11-436H11.5, functioning as a competitive endogenous RNA, upregulates BCL-W expression by sponging miR-335-5p and promotes proliferation and invasion in renal cell carcinoma. Mol Cancer 16:166. doi:https://doi.org/10.1186/s12943-017-0735-3
Opferman JT, Kothari A (2018) Anti-apoptotic BCL-2 family members in development. Cell Death Differ 25:37–45. doi:https://doi.org/10.1038/cdd.2017.170
Véronèse L, Tournilhac O, Verrelle P, Davi F, Dighiero G, Chautard E et al (2009) Strong correlation between VEGF and MCL-1 mRNA expression levels in B-cell chronic lymphocytic leukemia. Leuk Res 33:1623–1626. doi:https://doi.org/10.1016/j.leukres.2009.05.003
Cherla R, Zhang Y, Ledbetter L et al (2018) Coxiella burnetii inhibits neutrophil apoptosis by exploiting survival pathways and antiapoptotic protein Mcl-1. Infect Immun 86:e00504–e00517. doi:https://doi.org/10.1128/IAI.00504-17
Choi B, Chun E, Kim SY, Kim M, Lee KY, Kim SJ (2012) Notch-induced hIL-6 production facilitates the maintenance of self-renewal of hCD34 + cord blood cells through the activation of Jak-PI3K-STAT3 pathway. Am J Pathol 180:351–364. doi:https://doi.org/10.1016/j.ajpath.2011.09.030
Pan B, Wang D, Li L, Shang L, Xia F, Zhang F et al (2019) IL-22 Accelerates thy- mus regeneration via Stat3/Mcl-1 and decreases chronic graft-versus- host disease in mice after allotransplants. Biol Blood Marrow Transplant 25:1911–1919. doi:https://doi.org/10.1016/j.bbmt.2019.06.002
Shenoy AR, Kirschnek S, Häcker G (2014) IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J Immunol 44:2500–2507. doi:https://doi.org/10.1002/eji.201344238
Gao J, Li L, Wu M, Liu M, Xie X, Guo J et al (2013) MiR-26a inhibits proliferation and migration of breast cancer through repression of MCL-1. PLoS ONE 8:e65138. doi:https://doi.org/10.1371/journal.pone.0065138
Shirjang S, Mansoori B, Asghari S, Duijf PHG, Mohammadi A, Gjerstorff M et al (2019) MicroRNAs in cancer cell death pathways: apoptosis and necroptosis. Free Radic Biol Med 139:1–15. doi:https://doi.org/10.1016/j.freeradbiomed.2019.05.017
Adams KW, Cooper GM (2007) Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem 282:6192–6200. doi:https://doi.org/10.1074/jbc.M610643200
Zhong Q, Gao W, Du F, Wang X (2005) Mule/A RF-BP1, a BH3-only E3 ubiquitin ligase,catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121:1085–1095. doi:https://doi.org/10.1016/j.cell.2005.06.009
Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW et al (2011) SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109
Harley ME, Allan LA, Sanderson HS, Clarke PR (2010) Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20 dependent destruction during mitotic arrest. EMBO J 29:2407–2420. doi:https://doi.org/10.1038/emboj.2010.112
Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC et al (2007) Degradation of Mcl-1 by beta-TrCP me diates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 27:4006–4017. doi:https://doi.org/10.1128/MCB.00620-06
Schwickart M, Huang X, Lill JR, Liu J, Dixit VM (2009) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463:103–107
Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z et al (2018) Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun 9:215
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21:749–760. doi:https://doi.org/10.1016/j.molcel.2006.02.009
Thomas LW, Lam C, Edwards SW (2010) Mcl-1; the molecular regulation of protein function. FEBS Lett 584:2981–2989. doi:https://doi.org/10.1016/j.febslet.2010.05.061
Wang CY, Guttridge DC, Mayo MW, Baldwin AS Jr (1999) NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol 19:5923–5929. doi:https://doi.org/10.1128/MCB.19.9.5923
Kucharczak JF, Simmons MJ, Duckett CS, Gelinas C (2005) Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor. Cell Death Differ 12:1225–1239
Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G et al (2006) The stability and anti-apoptotic function of A1 are controlled by its C terminus. J Biol Chem 281:13663–13671. doi:https://doi.org/10.1074/jbc.M600266200
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell Biol 7:683–694. doi:https://doi.org/10.1016/s1097-2765(01)00214-3
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7:673–682. https://doi.org/10.1016/s1097-2765(01)00213-1
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058. doi:https://doi.org/10.1126/science.288.5468.1053
Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302:1036–1038. doi:https://doi.org/10.1126/science.1090072
Dötsch V, Bernassola F, Coutandin D, Candi E, Melino G (2010) p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2:a004887. doi:https://doi.org/10.1101/cshperspect.a004887
Levrero M, De Laurenzi V, Costanzo A, Gong J, Wang JY, Melino G (2000) The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci 113:1661–1670. doi:https://doi.org/10.1242/jcs.113.10.1661
You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M et al (2006) FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med 203:1657–1663. doi:https://doi.org/10.1084/jem.20060353
Lurlaro R, Muñoz-Pinedo C (2016) Cell death induced by endoplasmic reticulum stress. FEBS J 283. doi:https://doi.org/10.1111/febs.13598
Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM et al (2005) Slug antagonizes p53- mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 123:641–653. doi:https://doi.org/10.1016/j.cell.2005.09.029
Fricker M, O’Prey J, Tolkovsky A et al (2010) Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis 1. doi:https://doi.org/10.1038/cddis.2010.38
Ploner C, Kofler R, Villunger A (2008) Noxa: at the tip of the balance between life and death. Oncogene 27:S84–S92. doi:https://doi.org/10.1038/onc.2009.46
Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH (2004) BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1 alpha. J Exp Med 199:113–124. doi:https://doi.org/10.1084/jem.20030613
Lallemand C, Blanchard B, Palmieri M, Lebon P, May E, Tovey MG (2007) Single-stranded RNA viruses inactivate the transcriptional activity of p53 but induce NOXA-dependent apoptosis via post-translational modifications of IRF-1, IRF-3 and CREB. Oncogene 26:328–338. doi:https://doi.org/10.1038/sj.onc.1209795
Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, Pan Z, Sun Y (2017) Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res 23:1104–1116. doi:https://doi.org/10.1158/1078-0432.CCR-16-1585
Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A, Cohen GM (2012) NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death Differ 19:1424–1434
Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10:1201–1204. doi:https://doi.org/10.1016/S0960-9822(00)00728-4
Muthalagu N, Junttila MR, Wiese KE, Wolf E, Morton J, Bauer B, Evan GI, Eilers M, Murphy DJ (2014) BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep 8:1347–1353. doi:https://doi.org/10.1016/j.celrep.2014.07.057
Hughes R, Kristiansen M, Lassot I, Desagher S, Mantovani R, Ham J (2011) NF-Y is essential for expression of the proapoptotic bim gene in sympathetic neurons. Cell Death Differ 18:937–947
Wildey GM, Patil S, Howe PH (2003) Smad3 potentiates transforming growth factor beta (TG Fbeta)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J Biol Chem 278:18069–18077. doi:https://doi.org/10.1074/jbc.M211958200
Yano T, Ito K, Fukamachi H, Chi XZ, Wee HJ, Inoue K, Ida H, Bouillet P, Strasser A, Bae SC, Ito Y (2006) The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta-induced apoptosis. Mol Cell Biol 26:4474–4488. doi:https://doi.org/10.1128/MCB.01926-05
Harris CA, Johnson EM Jr (2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276:37754–37760
Sionov RV, Vlahopoulos SA, Granot Z (2015) Regulation of Bim in Health and Disease. Oncotarget 6:23058–23134. doi:https://doi.org/10.18632/oncotarget.5492
Herold MJ, Rohrbeck L, Lang MJ, Grumont R, Gerondakis S, Tai L, Bouillet P, Kaufmann T, Strasser A (2013) Foxo-mediated Bim transcription is dispensable for the apoptosis of hematopoietic cells that is mediated by this BH3-only protein. EMBO Rep 14:992–998. doi:https://doi.org/10.1038/embor.2013.152
San José-Eneriz E, Agirre X, Jiménez-Velasco A, Cordeu L, Martín V, Arqueros V, Gárate L, Fresquet V, Cervantes F, Martínez-Climent JA, Heiniger A, Torres A, Prósper F, Roman-Gomez J (2009) Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur J Cancer 45:1877–1889. doi:https://doi.org/10.1016/j.ejca.2009.04.005
Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P et al (2009) Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes & Dev 23:2806–2811. doi:https://doi.org/10.1101/gad.1872909
Molitoris JK, McColl KS, Distelhorst CW (2011) Glucocorticoid-mediated repression of the oncogenic microRNA cluster miR-17~ 92 contributes to the induction of Bim and initiation of apoptosis. Mol Endocrinol 25:409–420. doi:https://doi.org/10.1210/me.2010-0402
Petrocca F, Vecchione A, Croce CM (2008) Emerging role of miR-106b-25/mir-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res 68:8191–8194. doi:https://doi.org/10.1158/0008-5472.CAN-08-1768
Labi V, Peng S, Klironomos F, Munschauer M et al (2019) Context-specific regulation of cell survival by a miRNA-controlled BIM rheostat. Genes & Dev 7. doi:https://doi.org/10.1101/gad.330134.119
Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Nat Acad Sci USA 100:2432–2437. doi:https://doi.org/10.1073/pnas.0438011100
Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ et al (2003) JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 38:899–914. doi:https://doi.org/10.1016/s0896-6273(03)00355-6
Puthalakath H, Huang DCS, O’Reilly LA, King SM, Strasser A (1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3:287–296. doi:https://doi.org/10.1016/s1097-2765(00)80456-6
O’Reilly LA, Kruse EA, Puthalakath H, Kelly PN, Kaufmann T, Huang DCS, Strasser A (2009) MEK/ERK-Mediated Phosphorylation of Bim Is Required to Ensure Survival of T and B Lymphocytes during Mitogenic Stimulation. J Immunol 18 3:261–269. doi:https://doi.org/10.4049/jimmunol.0803853
Schmelzle T, Mailleux AA, Overholtzer M et al (2007) Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A 104:3787–3792. doi:https://doi.org/10.1073/pnas.0700115104
Labi V, Erlacher M, Kiessling S et al (2008) Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J Exp Med 205:641–655. doi:https://doi.org/10.1084/jem.20071658
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17:393–403. doi:https://doi.org/10.1016/j.molcel.2004.12.030
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17:525–535. doi:https://doi.org/10.1016/j.molcel.2005.02.003
Willis SN, Chen L, Dewson G, Wei A, Naik E et al (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes & Dev 19:1294–1305. doi:https://doi.org/10.1101/gad.1304105
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH et al (2016) BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165:421–433. doi:https://doi.org/10.1016/j.cell.2016.02.026
Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L et al (2018) Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173:1217–1230. doi:https://doi.org/10.1016/j.cell.2018.04.036
Hanahan D (2022) Hallmarks of Cancer: New Dimensions. Cancer Discov 12:31–46. doi:https://doi.org/10.1158/2159-8290.CD-21-1059
Reed JC (2008) Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood 111:3322–3330. doi:https://doi.org/10.1182/blood-2007-09-078162
Perciavalle RM, Opferman JT (2013) Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol 23:22–29. doi:https://doi.org/10.1016/j.tcb.2012.08.011
Magdalena K, Pavel K (2020) BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancer 12:938. doi:https://doi.org/10.3390/cancers12040938
Adams CM, Clark-Garvey S, Porcu P, Eischen CM (2019) Targeting the Bcl-2 Family in B Cell Lymphoma. Front Oncol 8:636. doi:https://doi.org/10.3389/fonc.2018.00636
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J et al (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463:899–905. doi:https://doi.org/10.1038/nature08822
Tsujimoto Y, Cossman J, Jaffe E, Croce C (1985) Involvement of the Bcl-2 gene in human follicular lymphoma. Science 228:1440–1443. doi:https://doi.org/10.1126/science.3874430
Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, Valente LJ, Robati M, Tai L, Fairlie WD, Lee EF, Lindstrom MS, Wiman KG, Huang DCS, Bouillet P, Rowe M, Rickinson AB, Herold MJ, Strasser A (2014) Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes & Dev 28:58–70. doi:https://doi.org/10.1101/gad.232009.113
Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM (1984) Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science 224:1403–1406. doi:https://doi.org/10.1126/science.6610211
Cleary ML, Sklar J (1985) Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA 82:7439–7443. doi:https://doi.org/10.1073/pnas.82.21.7439
Hanada M, Delia D, Aiello A, Stadmauer E, Reed JC (1993) Bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 82:1820–1828
Robertson LE, Plunkett W, McConnell K, Keating MJ, McDonnell TJ (1996) Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia 10:456–459
Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S et al (1998) Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood 91:3379–3389
Ikegaki N, Katsumata M, Minna J, Tsujimoto Y (1994) Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res 54:6–8. doi:https://doi.org/10.1016/0169-5002(96)00568-5
Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nun ̃ez G (1993) Expression of the apoptosis-suppressing protein bcl-2 in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol 143:1543–1550
Strasser A, Harris AW, Bath ML, Cory S (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348:331–333
Strasser A, Harris AW, Cory S (1991) Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67:889–899. doi:https://doi.org/10.1016/0092-8674(91)90362-3
Strasser A, Harris AW et al (1994) DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell 79:329–339. doi:https://doi.org/10.1016/0092-8674(94)90201-1
Weiler M, Bähr O, Hohlweg U et al (2006) BCL-xL: time-dependent dissociation between modulation of apoptosis and invasiveness in human malignant glioma cells. Cell Death Differ 13:1156–1169. doi:https://doi.org/10.1038/sj.cdd.4401786
Martin SS, Ridgeway AG, Pinkas J et al (2004) A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 23:4641–4645. doi:https://doi.org/10.1038/sj.onc.1207595
Peeters SDPWM, Hovenga S, Rosati S, Vellenga E (2005) Bcl-xl expression in multiple myeloma. Med Oncol 22:183–190. doi:https://doi.org/10.1385/MO:22:2:183
Leiter U, Schmid RM, Kaskel P, Peter RU, Krähn G (2000) Antiapoptotic bcl-2 and bcl-xL in advanced malignant melanoma. Arch Dermatol Res 292:225–232. doi:https://doi.org/10.1007/s004030050479
Cragg MS, Kuroda J, Puthalakath H, Huang DCS, Strasser A (2007) Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 4:1681–1689. doi:https://doi.org/10.1371/journal.pmed.0040316
Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL (2008) Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest 118:3651–3659. doi:https://doi.org/10.1172/JCI35437
Oltersdorf T, Elmore S, Shoemaker A et al (2005) An inhibitor of Bcl-2 family proteins induces regression of. solid tumours Nature 435:677–681. doi:https://doi.org/10.1038/nature03579
Xerri L, Parc P, Brousset P, Schlaifer D, Hassoun J, Reed JC et al (1996) Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br J Haematol 92:900–906. doi:https://doi.org/10.1046/j.1365-2141.1996.423958.x
Sejic N, George LC, Tierney RJ, Chang C et al (2020) BCL-XL inhibition by BH3-mimetic drugs induces apoptosis in models of Epstein-Barr virus–associated T/NK-cell lymphoma. Blood Adv 4:4775–4787. doi:https://doi.org/10.1182/bloodadvances.2020002446
Ramesh P, Lannagan TRM, Jackstadt R et al (2021) BCL-XL is crucial for progression through the adenoma-to-carcinoma sequence of colorectal cancer. Cell Death Differ 28:3282–3296. doi:https://doi.org/10.1038/s41418-021-00816-w
Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS, Choi KM et al (2006) Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res 66:4991–4995. doi:https://doi.org/10.1158/0008-5472.CAN-05-4254
Wilson JW, Nostro MC, Balzi M, Faraoni P, Cianchi F, Becciolini A et al (2000) Bcl-w expression in colorectal adenocarcinoma. Br J Cancer 82:178–185. doi:https://doi.org/10.1054/bjoc.1999.0897
Adams CM, Kim AS, Mitra R, Choi JK, Gong JZ, Eischen CM (2017) BCL-W has a fundamental role in B cell survival and lymphomagenesis. J Clin Invest 127:635–650. doi:https://doi.org/10.1172/JCI89486
Diepstraten ST, Chang C, Tai L, Gong JN, Lan P, Dowell AC, Taylor GS, Strasser A, Kelly GL (2020) BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines. Blood Adv 4:356–366. doi:https://doi.org/10.1182/bloodadvances.2019000541
Campbell KJ, Dhayade S, Ferrari N et al (2018) MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis 9. doi:https://doi.org/10.1038/s41419-017-0035-2
Akgul C (2009) Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci 66:1326–1336. doi:https://doi.org/10.1007/s00018-008-8637-6
Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K et al (2004) VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood 104:2886–2892. doi:https://doi.org/10.1182/blood-2004-05-1760
Kobayashi S, Werneburg NW, Bronk SF, Kaufmann SH, Gores GJ (2005) Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 128:2054–2065. doi:https://doi.org/10.1053/j.gastro.2005.03.010
Kelly GL, Strasser A (2020) Toward Targeting Antiapoptotic MCL-1 for Cancer Therapy. Annu Rev of Cancer Biol 4:299–313. doi:https://doi.org/10.1146/annurev-cancerbio-030419-033510
Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD et al (2001) MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 97:3902–3909. doi:https://doi.org/10.1182/blood.v97.12.3902
Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL, Cory S (2010) Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood 116:3197–3207. doi:https://doi.org/10.1182/blood-2010-04-281071
Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ et al (2012) Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes & Dev 26:120–125. doi:https://doi.org/10.1101/gad.182980.111
Grabow S, Delbridge AR, Valente LJ, Strasser A (2014) MCL-1 but not BCL-XL is critical for the development and sustained expansion of thymic lymphoma in p53-deficient mice. Blood 124:3939–3946. doi:https://doi.org/10.1182/blood-2014-09-601567
Munkhbaatar E, Dietzen M, Agrawal D et al (2020) MCL-1 gains occur with high frequency in lung adenocarcinoma and can be targeted therapeutically. Nat Commun 11. doi:https://doi.org/10.1038/s41467-020-18372-1
Fernández-Marrero Y, Spinner S, Kaufmann T, Jost PJ (2016) Survival control of malignant lymphocytes by anti-apoptotic MCL-1. Leukemia 30:2152–2159. doi:https://doi.org/10.1038/leu.2016.213
Kotschy A, Szlavik Z, Murray J et al (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538:477–482. doi:https://doi.org/10.1038/nature19830
Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, Whittington DA, Huang X, Poppe L, Cheng AC, Cardozo M (2018) AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies AMG 176 alone and combined in hematologic cancer models. Cancer Discov 8:1582–1597. doi:https://doi.org/10.1158/2159-8290.CD-18-0387
Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E et al (2018) Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 9. doi:https://doi.org/10.1038/s41467-018-07551-w
Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE et al (2008) Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol 28:5391–5402. doi:https://doi.org/10.1128/MCB.00907-07
Zantl N, Weirich G, Zall H et al (2007) Frequent loss of expression of the pro-apoptotic protein Bim in renal cell carcinoma: evidence for contribution to apoptosis resistance. Oncogene 26:7038–7048. doi:https://doi.org/10.1038/sj.onc.1210510
Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ et al (2010) Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood 116:2531–2542. doi:https://doi.org/10.1182/blood-2010-02-268003
Karst AM, Dai DL, Martinka M, Li G (2005) PUMA expression is significantly reduced in human cutaneous melanomas. Oncogene 24:1111–1116. doi:https://doi.org/10.1038/sj.onc.120837
Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M (2005) Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24:1348–1358. doi:https://doi.org/10.1038/sj.onc.1208300
Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA et al (2007) Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 109:271–280. doi:https://doi.org/10.1182/blood-2006-06-026500
Sinicrope FA, Rego RL, Okumura K, Foster NR, O’Connell MJ, Sargent DJ et al (2008) Prognostic impact of bim, puma, and noxa expression in human colon carcinomas. Clin Cancer Res 14:5810–5818. doi:https://doi.org/10.1158/1078-0432.CCR-07-5202
Miquel C, Borrini F, Grandjouan S, Aupérin A, Viguier J, Velasco V, Duvillard P, Praz F, Sabourin J-C (2005) Role of bax mutations in apoptosis in colorectal cancers with microsatellite instability. Am J Clin Pathol 23:562–570. doi:https://doi.org/10.1309/JQ2X-3RV3-L8F9-TGYW
Sakuragi N, Salah-eldin AE, Watari H, Itoh T, Inoue S, Moriuchi T, Fujimoto S (2002) Bax, Bcl-2, and p53 Expression in Endometrial Cancer. Gynecol Oncol 86:288–296. doi:https://doi.org/10.1006/gyno.2002.6742
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P et al (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6:1389–1399. doi:https://doi.org/10.1016/s1097-2765(00)00136-2
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328. doi:https://doi.org/10.1016/s1535-6108(03)00244-7
Bouillet P, Metcalf D, Huang DCS et al (1999) Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286:1735–1738. doi:https://doi.org/10.1126/science.286.5445.1735
Erlacher M, Labi V, Manzl C et al (2006) Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 203:2939–2951. doi:https://doi.org/10.1084/jem.20061552
Egle A, Harris AW, Bouillet P, Cory S (2004) Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA 101:6164–6169. doi:https://doi.org/10.1073/pnas.0401471101
Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK et al (2009) Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ 16:684–696. doi:https://doi.org/10.1038/cdd.2008.195
Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW (2004) Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci USA 101:9333–9338. doi:https://doi.org/10.1073/pnas.0403286101
Michalak EM, Vandeberg CJ, Delbridge ARD et al (2010) Apoptosis-promoted tumorigenesis: γ-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes & Dev 24:1608–1613. doi:https://doi.org/10.1101/gad.1940110
Valente LJ, Gray GHD, Michalak EW et al (2013) p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3:1339–1345
Janic A, Valente LJ, Wakefield MJ et al (2018) DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 24:947–953. doi:https://doi.org/10.1038/s41591-018-0043-5
Geng M, Wang L, Li P (2013) Correlation between chemosensitivity to anticancer drugs and Bcl-2 expression in gastric cancer. Int J Clin Exp Pathol 6:2554–2559
Beale P, Rogers P, Boxall F et al (2000) BCL-2 family protein expression and platinum drug resistance in ovarian carcinoma. Br J Cancer 82:436–440. doi:https://doi.org/10.1054/bjoc.1999.0939
Zhao Y, Zhang CL, Zeng BF, Wu XS, Gao TT, Oda Y (2009) Enhanced chemosensitivity of drug-resistant osteosarcoma cells by lentivirus-mediated Bcl-2 silencing. Biochem Biophys Res Commun 390:642–647. doi:https://doi.org/10.1016/j.bbrc.2009.10.020
Kim DW, Kim KO, Shin MJ, Ha JH, Seo SW, Yang J, Lee FY (2009) siRNA-based targeting of antiapoptotic genes can reverse chemoresistance in P-glycoprotein expressing chondrosarcoma cells. Mol Cancer 8:28. doi:https://doi.org/10.1186/1476-4598-8-28
Minn AJ, Rudin CM, Boise LH, Thompson CB (1995) Expression of Bcl-XL can confer a multidrug resistance phenotype. Blood 86:1903–1910
Huang DCS, Cory S, Strasser A (1997) Bcl-2, Bcl-xL and adenovirus protein E1B19kD are functionally equivalentin their ability to inhibit cell death. Oncogene 14:405–414. doi:https://doi.org/10.1038/sj.onc.1200848
Horita M, Andreu EJ, Benito A, Arbona C, Sanz C, Benet I et al (2000) Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J Exp Med 191:977–998. doi:https://doi.org/10.1084/jem.191.6.977
Williams J, Lucas PC, Griffith KA, Choi M, Fogoros S, Hu YY, Liu JR (2005) Expression of Bcl-xL in ovarian carcinoma is associated with chemoresistance and recurrent disease. Gynecol Oncol 96:287–295. doi:https://doi.org/10.1016/j.ygyno.2004.10.026
Michels J, Obrist F, Vitalee I, Lissa D, Garcia P, Behnam-Motlagh P, Kohno K, Wu GS, Brenner C, Castedo M, Kroemer G (2014) MCL-1 dependency of cisplatin-resistant cancer cells. Biochem Pharmacol 92:55–61. doi:https://doi.org/10.1016/j.bcp.2014.07.029
Martin AP, Miller A, Emad L, Rahmani M, Walker T, Mitchell C, Hagan MP, Park MA, Yacoub A, Fisher PB, Grant S (2008) Lapatinib resistance in HCT116 cells is mediated by elevated MCL-1 expression and decreased BAK activation and not by ERBB receptor kinase mutation. Mol Pharmacol 74:807–822. doi:https://doi.org/10.1124/mol.108.047365
Hussain SR, Cheney CM, Johnson AJ, Lin TS, Grever MR, Caligiuri MA, Lucas DM, Byrd JC (2007) Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin Cancer Res 13:2144–2150. doi:https://doi.org/10.1158/1078-0432.CCR-06-2294
Stam RW, Den Boer ML, Schneider P, de Boer J, Hagelstein J, Valsecchi MG, de Lorenzo P et al (2010) Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukemia. Blood 115:1018–1025. doi:https://doi.org/10.1182/blood-2009-02-205963
Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, Pawel BR, Hogarty MD (2009) Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther 8:1587–1595. doi:https://doi.org/10.4161/cbt.8.16.8964
Osaki S, Tazawa H, Hasei J, Yamakawa Y, Omori T, Sugiu K, Komatsubara T, Fujiwara T, Sasaki T, Kunisada T, Yoshida A (2016) Ablation of MCL1 expression by virally induced microRNA-29 reverses chemoresistance in human osteosarcomas. Sci Rep 6. doi:https://doi.org/10.1038/srep28953
Happo L, Cragg MS, Phipson B, Jon MH et al (2010) Maximal killing of lymphoma cells by DNA damage–inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 116:5256–5267. doi:https://doi.org/10.1182/blood-2010-04-280818
Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A (2006) Bim and Bad mediate imatinib-induced killing of Bcr/Abl + leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A 103:14907–14912. doi:https://doi.org/10.1073/pnas.0606176103
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S et al (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68:3421–3428. doi:https://doi.org/10.1158/0008-5472
Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG et al (2007) Programmed Anuclear Cell Death Delimits Platelet Life Span. Cell 128. doi:https://doi.org/10.1016/j.cell.2007.01.037
Wilson WH, O’Connor OA, Czuczman MS et al (2010) Navitoclax, a targeted high affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 11:1149–1159. doi:https://doi.org/10.1016/S1470-2045(10)70261-8
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND et al (2013) ABT- 199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19:202–208. doi:https://doi.org/10.1038/nm.3048
Roberts AW, Davids MS, Pagel JM, Kahl BS et al (2016) Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 374:311–322. doi:https://doi.org/10.1056/NEJMoa1513257
Seymour JF, Ma S, Brander DM et al (2017) Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol 18:230–240. doi:https://doi.org/10.1016/S1470-2045(17)30012-8
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL et al (2016) Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 6:1106–1117. doi:https://doi.org/10.1158/2159-8290.CD-16-0313
Thol F, Ganser A (2020) Treatment of Relapsed Acute Myeloid Leukemia. Curr Treat Options in Oncol 21. https://doi.org/10.1007/s11864-020-00765-5
Ma S, Seymour JF, Brander DM, Kipps TJ, Choi MY, Anderson MA, Humphrey K et al (2021) Efficacy of venetoclax plus rituximab for relapsed CLL: 5-year follow-up of continuous or limited- duration therapy. Blood 138:836–846. doi:https://doi.org/10.1182/blood.2020009578
Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM et al (2013) Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol 9:390–397. doi:https://doi.org/10.1038/nchembio.1246
Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, Czabotar PE (2014) Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett 5:1088–1093. doi:https://doi.org/10.1021/ml5001867
Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LDNP, Xiao Y, Ma XM et al (2015) Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 7:279ra240. doi: https://doi.org/10.1126/scitranslmed.aaa4642
Leverson J, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P et al (2015) Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 6:e1590. doi:https://doi.org/10.1038/cddis.2014.561
Merino D, Whittle JR, Vaillant F, Serrano A et al (2017) Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med 9. doi:https://doi.org/10.1126/scitranslmed.aam7049
Yasuda Y, Ozasa H, Kim YH et al (2020) MCL1 inhibition is effective against a subset of small-cell lung cancer with high MCL1 and low BCL-XL expression. Cell Death Dis 11. doi:https://doi.org/10.1038/s41419-020-2379-2
Nangia V, Siddiqui FM, Caenepeel S, Timonina D, Bilton SJ, Phan N, Gomez-Caraballo M, Archibald HL, Li C, Fraser C, Rigas D et al (2018) Exploiting MCL-1 dependency with combination MEK + MCL-1 inhibitors leads to induction of apoptosis and tumor regression in KRAS mutant non-small cell lung cancer. Cancer Discov 8:1598–1613. doi:https://doi.org/10.1158/2159-8290.CD-18-0277
Clinicaltrials Safety Tolerability, Pharmacokinetics and Efficacy of AMG 397 in Subjects with Multiple Myeloma, NHL, and AML
Wang H, Guo M, Wei H et al (2021) Targeting MCL-1 in cancer: current status and perspectives. J Hematol Oncol 14. doi:https://doi.org/10.1186/s13045-021-01079-1
Wang Xi, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT (2013) Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes & Dev 27:1351–1364. doi:https://doi.org/10.1101/gad.215855.113
Grabow S, Kueh AJ, Ke F, Vanyai HK, Sheikh BN, Dengler MA, Chiang W, Eccles S, Smyth IM, Jones LK, De Sauvage FJ (2018) Subtle changes in the levels of BCL-2 proteins cause severe craniofacial abnormalities. Cell Rep 24:3285–3295. doi:https://doi.org/10.1016/j.celrep.2018.08.048
Moujalled DM, Pomilio G, Ghiurau C et al (2019) Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 33:905–917. doi:https://doi.org/10.1038/s41375-018-0261-3
Olie RA, Zangemeister-Wittke U (2001) Targeting tumor cell resistance to apoptosis induction with antisense oligonucleotides: progress and therapeutic potential. Drug Resist Updat 4:9–15. doi:https://doi.org/10.1054/drup.2001.0181
Zhang XD, Gillespie SK, Borrow JM, Hersey P (2004) The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther 3:425–435. doi:https://doi.org/10.1158/1535-7163.425.3.4
Cragg MS, Harris C, Strasser A, Scott CL (2009) Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 9:321–326. doi:https://doi.org/10.1038/nrc2615
Matthews GM, Newbold A, Johnstone RW (2012) Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res 116:165–197. doi:https://doi.org/10.1016/B978-0-12-394387-3.00005-7
Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM (2007) Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia 21:1773–1782. doi:https://doi.org/10.1038/sj.leu.2404760
Kharas MG, Fruman DA (2005) ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res 65:2047–2053
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Stambolic V, Suzuki A, De La Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95:29–39. doi:https://doi.org/10.1016/S0092-8674(00)81780-8
Boros LG, Lee WN, Cascante M (2002) Imatinib and chronic-phase leukemias. N Engl J Med 67–68. doi:https://doi.org/10.1056/NEJM200207043470116
Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, Hallin J et al (2011) PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther 10:2189–2199. doi:https://doi.org/10.1158/1535-7163.MCT-11-0185
Rodrigues DA, Sagrillo FS, Fraga CA (2019) Duvelisib: a 2018 novel FDA-approved small molecule inhibiting phosphoinositide 3-kinases. Pharmaceuticals 12:69. doi:https://doi.org/10.3390/ph12020069
Loar P, Wahl H, Kshirsagar M, Gossner G, Griffith K, Liu JR (2010) Inhibition of glycolysis enhances cisplatin-induced apoptosis in ovarian cancer cells. Am J Obstet Gynecol 202:371–e371. doi:https://doi.org/10.1016/j.ajog.2009.10.883
Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S et al (2010) Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 29:1641–1652. doi:https://doi.org/10.1038/onc.2009.448
Ekoff M, Kaufmann T, Engstorm M, Motoyama N et al (2007) The BH3-only protein Puma plays an essential role in cytokine deprivation induced apoptosis of mast cells. Blood 110:3209–3217. doi:https://doi.org/10.1182/blood-2007-02-073957
Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC (2008) Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Chem 283:36344–36353. doi:https://doi.org/10.1074/jbc.M803580200
Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB (2002) Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol 3:932–939
Vallabhapurapu S, Karin M (2009) Regulation and function of NFkappaB transcription factors in the immune system. Annu Rev Immunol 27:693–733. https://doi.org/10.1146/annurev.immu-nol.021908.132641
Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G (1999) NFkappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A 96:9136–9141. doi:https://doi.org/10.1073/pnas.96.16.9136
Schmidt E (1999) The role of c-myc in cellular growth control. Oncogene 18:2988–2996. doi:https://doi.org/10.1038/sj.onc.1202751
Fletcher S, Prochownik EV (2015) Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta 1849:525–543. doi:https://doi.org/10.1016/j.bbagrm.2014.03.005
Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA et al (2008) Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14:447–457. 10.1016/j. ccr.2008.10.018
Nguyen HV, Vandenberg CJ, Ng AP, Robati MR, Anstee NS, Rimes J, Hawkins ED, Cory S (2020) Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 135:1019–1031. doi:https://doi.org/10.1182/blood.2019003014
Michalak EM, Villunger A, Adams JM, Strasser A (2008) In several cell types the tumour suppressor, p53, induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 15:1019–1029. doi:https://doi.org/10.1038/cdd.2008.16
Falschlehner C, Emmerich CH, Gerlach B, Walczak H (2007) TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol 39:1462–1475. doi:https://doi.org/10.1016/j.biocel.2007.02.007
Funding
Research in the labs of the authors is supported by fellowships and grants from the Australian National Health and Medical Research Council (NHMRC) (Program Grant GNT1113133 to AS, Research Fellowship GNT1116937 to AS, Project Grants GNT1143105 to AS, Ideas Grants GNT 2002618 and GNT2001201 to GLK, Synergy Grants GNT 2011139 to GLK and GNT 2010275 to AS), the Leukemia & Lymphoma Society of America (Specialized Center of Research [SCOR] grant no. 7015-18 to AS, and GLK), Victorian Cancer Agency (MCRF Fellowship 17028 to GLK and ECRF Fellowship 21006 to STD), CASS foundation (STD), DK is supported by Walter and Eliza Hall Johnson PhD Scholarship and Melbourne Research Scholarship (University of Melbourne), the estate of Anthony (Toni) Redstone OAM (AS and GLK), the Craig Perkins Cancer Research Foundation (GLK), the Dyson Bequest (GLK) and the Harry Secomb Foundation (GLK). Work in the laboratories of the authors was made possible through Victorian State Government Operational Infrastructure Support (OIS) and Australian Government NHMRC Independent Research Institute Infrastructure Support (IRIIS) Scheme.
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
DK wrote the article in consultation with STD, AS and GLK. The article was reviewed and revised by all authors.
Corresponding author
Ethics declarations
Competing interests Statement
All authors are employees of WEHI which receives milestone and royalty payments related to venetoclax. AS and GLK have received research funding from Servier. AS is a consultant for Genentech and Servier.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaloni, D., Diepstraten, S.T., Strasser, A. et al. BCL-2 protein family: attractive targets for cancer therapy. Apoptosis 28, 20–38 (2023). https://doi.org/10.1007/s10495-022-01780-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01780-7