
Abstract
Background
The lack of specificity and high degree of false positive and false negative rates when using mammographic screening for detecting early-stage breast cancer is a critical issue. Blood-based molecular assays that could be used in adjunct with mammography for increased specificity and sensitivity could have profound clinical impact. Our objective was to discover and independently verify a panel of candidate blood-based biomarkers that could identify the earliest stages of breast cancer and complement current mammographic screening approaches.
Methods
We used affinity hydrogel nanoparticles coupled with LC-MS/MS analysis to enrich and analyze low-abundance proteins in serum samples from 20 patients with invasive ductal carcinoma (IDC) breast cancer and 20 female control individuals with positive mammograms and benign pathology at biopsy. We compared these results to those obtained from five cohorts of individuals diagnosed with cancer in organs other than breast (ovarian, lung, prostate, and colon cancer, as well as melanoma) to establish IDC-specific protein signatures. Twenty-four IDC candidate biomarkers were then verified by multiple reaction monitoring (LC-MRM) in an independent validation cohort of 60 serum samples specifically including earliest-stage breast cancer and benign controls (19 early-stage (T1a) IDC and 41 controls).
Results
In our discovery set, 56 proteins were increased in the serum samples from IDC patients, and 32 of these proteins were specific to IDC. Verification of a subset of these proteins in an independent cohort of early-stage T1a breast cancer yielded a panel of 4 proteins, ITGA2B (integrin subunit alpha IIb), FLNA (Filamin A), RAP1A (Ras-associated protein-1A), and TLN-1 (Talin-1), which classified breast cancer patients with 100% sensitivity and 85% specificity (AUC of 0.93).
Conclusions
Using a nanoparticle-based protein enrichment technology, we identified and verified a highly specific and sensitive protein signature indicative of early-stage breast cancer with no false positives when assessing benign and inflammatory controls. These markers have been previously reported in cell-ECM interaction and tumor microenvironment biology. Further studies with larger cohorts are needed to evaluate whether this biomarker panel improves the positive predictive value of mammography for breast cancer detection.
Similar content being viewed by others


Background
Invasive ductal carcinoma (IDC) is the most common type of breast cancer, accounting for 80% of all breast cancers and affecting women at any age. For asymptomatic individuals, annual or biennial mammographic screening is the primary tool for prevention and early diagnosis of breast cancers. However, several trials have called into question the efficacy of mammographic screening to reduce mortality. False positives and false negatives represent a clear limitation of mammography screening. The incidence of IDC, and its associated mortality, has not decreased in the past 10 years despite large-scale mammographic screening efforts [1]. Specifically, in current mammographic image-based screening approaches, lack of sensitivity to detect many early-stage breast cancer is concomitant with increased frequency of unnecessary biopsies (biopsies of benign lesions) [2], over-diagnosis, and over-treatment [1, 3, 4]. It has been proposed that the consequences of over-treatment, such as complications from surgery, cardiotoxicity, and cardiovascular disease have offset the expected reduction in mortality resulting from mammographic screening efforts [3].
Despite technological advances in screening mammography between 2005 and 2013 [5], breast cancer detection rates only marginally increased (31.5 to 34.7%). Surprisingly, an increase in abnormal interpretations was recorded during the same time span (from 8 to 12.6%), alongside a decrease in positive predictive values (PPV) of biopsy recommendations and performed biopsies (31.5% down to 27.5% and 39.5% down to 30.4%, respectively) [6]. Moreover, current screening approaches reportedly have a low negative predictive value (NPV), missing approximately 20% of aggressive subtypes of IDC [7]. In these subtypes specifically, high breast density, a well-known risk factor for aggressive breast cancer, can reduce the ability of screening methods to detect cancer and lead to false negative results [8].
The availability of a robust, minimally invasive, and clinically actionable blood-based test to support and confirm mammography breast cancer screening results would be of great utility to increase both PPV and NPV especially for early-stage IDC detection. In particular, identification of a robust blood-based molecular signature of breast cancer could complement imaging-based screening approaches and would likely reduce false positive and false negative results when used together. So far, however, no blood-based clinical assay based on a single protein or panel of proteins has demonstrated sufficient diagnostic specificity and sensitivity to detect early-stage breast cancer in a clinical setting [9, 10].
Our study objectives aimed at identifying blood-based low molecular weight (LMW) proteins and protein fragments that show the potential to be used in parallel to mammography screening as serum biomarkers for early-stage breast cancer. Our final goal was to perform discovery, qualification, and verification of a panel of breast cancer candidate biomarkers using two independent mass spectrometry method and independent sample sets.
Blood levels of early-stage cancer biomarkers are expected to be low. In this study, we employed hydrogel nanoparticles (HNs) as a cutting-edge sample preparation technology, which we have specifically engineered to capture and enrich low abundance and LMW proteins in biofluids such as blood [11,12,13,14]. Our biomarker discovery efforts focus on the LMW portion of the proteome because any cancer biomarker that originates in the affected organ must be able to effectively traverse the endothelial cell wall barrier of the vasculature that provides a size selection of biomarkers below the MW of albumin [11].
In order to demonstrate breast cancer specificity, in addition to our breast cancer cohort, our discovery efforts utilized blood samples taken from patient cohorts with other solid cancers, including colon, ovarian, prostate and lung cancer, and melanoma. Moreover, in order to analyze the performance of the selected candidate markers and minimize the evaluation of those that associate with inflammation and benign pathologies, the verification set included a series of benign and inflammatory controls. If further validated in the future, these markers could augment mammographic screening in a clinical setting.
Materials and methods
Materials
N-isopropylacrylamide (NIPAm), N,N′ methylene bisacrylamide (BIS), allylamine (AA), potassium persulfate (KPS), vinyl sulfonic acid (VSA), Cibacron Blue F3GA (CB), dithiothreitol (DTT), iodoacetamide (IAA), urea, tris-HCl, sodium thiocynate (NaSCN), tris-(2-carboxyethyl)phosphine (TCEP), and ammonium hydroxide were procured from Sigma-Aldrich (St. Louis, MO). All solvents (water, acetonitrile [ACN], formic acid) were LC-MS grade and obtained from Fisher Scientific (Waltham, MA). Red top glass vacutainer tubes for serum separation were purchased from Becton, Dickinson and Company (Franklin Lakes, NJ). Stromal cell-derived factor 1β (SDF-1β; MW 11 kDa) was from Antigenix America (Melville, NY), insulin-like growth factor 1 (IGF1; MW 22 kDa) was from AbD Serotec (Raleigh, NC), insulin-like growth factor-binding protein 7 (IGFBP7; MW 29 kDa) was from PreproTech (Rocky Hill, NJ), and chicken lysozyme was from Sigma-Aldrich. All synthetic peptides were procured from Thermo Fisher.
Study cohorts and sample collection
Discovery set
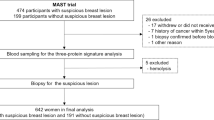
Our discovery study set used serum samples obtained from six cancer patient cohorts (breast, lung, colon, melanoma, prostate, and ovary) maintained by the Italian National Serum Bank hosted at Ospedale Maggiore Policlinico of Milan (Fig. 1). Each cohort included 20 cancer patients (cases) and 20 benign controls (total N = 240 samples). The controls were matched to each cancer cohort by median age and range, smoking habits, benign pathologies including those that could confound imaging results, and gender. The breast cancer cohort included female patients with IDC at stage I, II, or III, according to the TNM system. All patients in the cohort had suspicious or highly suspicious abnormalities at mammography (BI-RADS score 4–5). Moreover, all patients were treatment-naïve at the time of collection with regard to chemotherapy, radiation, and surgery. Whole blood (8 mL) was collected in red top glass vacutainer tubes, clotted for at least 30 min at room temperature, and centrifuged at 1500 rcf for 10 min. The serum samples were transferred to pre-labeled cryo-tubes and stored promptly at − 80 °C. The samples were shipped on dry ice to the Italian National Serum Bank. All procedures were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All specimens and clinical data were collected under an IRB-approved protocol between 2005 and 2007. Characteristics of patients involved in the study are described in Table 1 (breast cancer cohort) and Supplementary Table S1 (other cancer cohorts).

Workflow for biomarker discovery and verification based on nanoparticle enrichment and shotgun and targeted mass spectrometry
Verification set
The verification cohort (N = 60) included blood samples taken only from patients diagnosed with IDC at stage I (T1N0M0) along with samples from control individuals that included women who had positive mammograms with benign pathology at biopsy and women with minor pathologies such as hypertension and high cholesterol (Table 1). We applied the following inclusion criteria to determine study eligibility: available mammogram data (suspicious or highly suspicious abnormalities at mammography, BI-RADS score 4–5), age 33–89 years, and available outcome data.
HN synthesis
HNs functionalized with Cibacron Blue (CB) and coated with a vinylsulfonic acid (VSA) containing shell were synthetized as previously described by Tamburro et al. [11]. Briefly, N-isopropylacrylamide (NIPAm)-allylamine (AA) particles were synthesized by precipitation polymerization and then covalently functionalized with CB by nucleophilic substitution. NIPAm (Sigma-Aldrich (0.89 g, 7.83 mmol, 7.83 mmol)) and N,N′ methylene bisacrylamide (BIS; Sigma-Aldrich (0.042 g, 0.27 mmol)) were dissolved in water, and the solution was filtered and purged with nitrogen at room temperature. AA was then added (0.051 g, 0.90 mmol), followed by potassium persulfate (KPS) (0.0070 g, 0.025 mmol) to activate polymerization. The reaction was maintained at 75 °C under nitrogen for 3 h. Poly (NIPAm-co-AA) particles were washed by centrifugation to eliminate unreacted monomer. CB dissolved in sodium carbonate was added (0.76 g, 0.90 mmol) to the suspension and stirred at room temperature under nitrogen for 48 h. The resulting poly (NIPAm/CB) particles were first washed with water, and then exposed to a new polymerization reaction. A solution containing 20 ml of poly (NIPAm/CB) particles, NIPAm (0.156 g, 1.38 mmol), BIS (0.013 g, 0.084 mmol), and VSA (26 μl, 0.344 mmol) was purged with nitrogen and then heated to 70 °C, and polymerization was induced by adding KPS (0.092 g, 0.328 mmol). The reaction was performed at 70 °C under nitrogen for 6 h. Newly synthetized poly (NIPAm/CB) core–poly(NIPAm-co-VSA) shell particles were washed in water by centrifugation (16,100 rcf, 50 min, 25 °C).
HN-mediated enrichment of low-abundance plasma serum proteins
HNs offer several advantages over other methods used to enrich low abundant serum proteins: (i) Low abundance protein capture and enrichment is not based on immunodepletion methods but on the use of high affinity dye based selective capture in the core of the HN particle with simultaneous size-based exclusion of abundant proteins that often act as a carrier for low abundant proteins that may be lost during the immunodepletion process; (ii) HNs have been proven to protect proteins from enzymatic degradation during sample preprocessing [15]; (iii) HNs has shown to perform better in enriching low molecular weight (LMW; MW ≤ 40 kDa) when compared to methods based on peptide enrichment, or ultrafiltration [16].
Serum samples from the six discovery cohorts were processed by incubation with HNs in a randomized order. The serum samples (0.5 mL) were thawed, centrifuged (7 min, 4 °C, 16,100 rcf), and diluted with 1 mL 50 mM Tris-HCl, pH 7.0. SDF-1β, IGF1, and IGFBP7 were spiked in each sample as internal standards for quality and process control.
In the discovery set, both case and control samples contained IGF1 at 2 μg/μL, whereas IGFBP7 was spiked at 2 μg/μL in the case samples and 0.2 μg/μL in the control samples, and SDF-1β was spiked at 40 ng/μL in the control samples and 4 ng/μL in the case samples. Each sample was then incubated with 0.5 mL HNs (HN-CB/VSA, 7 mg/mL dry weight) for 30 min at room temperature. The samples were centrifuged (16,100 rcf, 25 °C, 20 min) and washed with 0.25 M NaSCN followed by two washes with water. The proteins were eluted by incubating the particles for 15 min at room temperature with 600 μL elution buffer (70% acetonitrile, 10% NH4OH) followed by centrifugation (16,100 rcf, 25 °C, 10 min). The elution step was repeated twice, and the eluates were combined, dried in a vacuum concentrator, and stored at − 20 °C until use.
For verification, chicken lysozyme was spiked at 0.2 μg/mL and was used as internal standard. Three hundred microliters of serum was diluted with 600 μL of 50 mM Tris-HCl, pH 7.0, and incubated with 300 μL of HN-CB/VSA particles (15 min, 25 °C) on a shaker. The nanoparticles were centrifuged at 16,100 rcf (30 min, 25 °C) and rinsed twice with 500 μL of 50 mM Tris-HCl, pH 7.0. The supernatant was discarded and the pelleted particles were incubated in 500 μL elution buffer (500 mM NaCl, 5 mM EDTA, 2% sodium deoxycholate in 50 mM Tris, pH 8.8) on a rocking shaker (47 rcf, 30 min, 60 °C). The samples were spun down (16,100 rcf, 15 min, 25 °C), and the supernatants were collected and vacuum-dried.
Protein digestion and LC-MS/MS analysis
Discovery set
All 240 samples analyzed from the discovery set were analyzed by LC-MS/MS in a randomized order to minimize batch effect and run bias. Dried protein eluates were reconstituted in 8 M urea, reduced in 10 mM dithiothreitol (DTT, Sigma), alkylated in 50 mM iodoacetamide (IAA, Sigma), and digested overnight in trypsin using an enzyme-to-protein ratio of 1:25 (v/v) (Promega Corporation, Madison, WI) at 37 °C. Tryptic peptides were further purified by C18 ZipTips (Millipore, Burlington, MA) and separated on a C18 analytical column (0.2 × 50 mm, Michrom Bioresources, Inc., Auburn, CA) using a HPLC Surveyor MS pump plus and Micro AS autosampler (Thermo Fisher) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). After sample injection, the column was washed for 2 min with mobile phase A (0.1% formic acid) and the peptides were eluted using a linear gradient of 0% mobile phase B (0.1% formic acid, 80% acetonitrile) to 50% mobile phase B in 90 min at 500 nL/min, and then 100% mobile phase B for an additional 5 min. The mass spectrometer was operated in data-dependent mode where the top five most abundant molecular ions were dynamically selected for collision-induced dissociation (CID) using a normalized collision energy (CE) of 35%. Raw files were searched against the NCBI human database with SEQUEST (Thermo Fisher Scientific). The following settings were applied: two missing cleavages allowed, methionine oxidation as a variable modification and cysteine carbamidomethylation as a fixed modification. High-confidence peptide identifications were obtained by applying the following filter criteria to the search results: Xcorr versus charge ≥ 1.9, 2.2, 3.5 for 1+, 2+, 3+ ions; ΔCn > 0.1; probability of randomized identification ≤ 0.01. Downstream differential analysis was based on spectral counts, as computed by Scaffold (Proteome Software, Inc., Portland, OR). The scaffold settings were protein and peptide probability threshold of 95%, 1 peptide per protein, and 1% false discovery rate.
Verification set
Protein eluates were quantified for total protein yield and normalization using reverse phase protein array analysis as previously described [17], and then reduced (5 mM TCEP, 40 min, 60 °C) and alkylated (10 mM IAA, 37 °C) in the dark. The samples were then diluted 4-fold in 50 mM Tris-HCl (pH 7.0), and 250 ng/μL trypsin Gold (Promega) was added at an enzyme-to-protein ratio of 1:50 (v/v). After 3 h of incubation at 37 °C, fresh trypsin was added to a final enzyme-to-protein ratio of 1:25 (v/v) and incubation was continued overnight (8 h, 63 rcf, 37 °C). Heavy-labeled peptides, GYSLGNWVCAA[13C615N2(K)] and FESNFNTQATN[13C615N4(R)], corresponding to GYSLGNWVCAAK and FESNFNTQATNR (Supplementary Table S2) for chicken LYZ were spiked in to a final concentration of 1750 fmoles. The samples were acidified with 50% formic acid to a final concentration of 1%, sieved through an AcroPrep 96-well plate (Pall Corporation, Port Washington, NY) to remove precipitated deoxycholate, and desalted using 1 cc Sep-Pak C18 (Waters, Milford, MA) solid-phase extraction columns. Finally, the eluted peptides were vacuum-dried and stored at − 20 °C until LC-MRM analysis.
The dried peptides were reconstituted in 0.1% formic acid and analyzed using online liquid chromatography on a nanoACQUITY UPLC (Waters) coupled to a Xevo TQ-s triple quadrupole mass spectrometer (Waters) with nano ESI in positive mode. Chromatography separation was performed on a BEH C18, 1.7 μm, 0.1 × 100 mm analytical column (Waters) using an 83.5 min gradient from 3 to 90% mobile phase B (acetonitrile, 0.1% formic acid) and 97 to 10% mobile phase A (water, 0.1% formic acid) at a flow rate 0.5 μL/min. The following gradient conditions were used: 3 to 7% mobile phase B for 1 min, 7 to 25% mobile phase B for 1 to 72 min, 25 to 45% mobile phase B for 10 min, 45 to 90% mobile phase B for 0.5 min, 90% B for 0.5 min, and column equilibration on 3% B for 10 min. Mobile phase A blanks were injected alternatively after every sample to prevent any carryover, column clogging, and rise in column pressure during the data acquisition. The E. coli tryptic digests (250 ng, Waters) bracketed every five patient sample injections (Supplementary Table S3) to assess instrumental variance. The measured variance remained under 15% coefficient of variation (CV) for the area under the curve throughout the acquisition. Peaks were integrated, and area-under-the-curve (AUC) values were calculated using the Skyline 3.5.0.9319 software package [20].
LC-MRM optimization
Candidate peptides selected for LC-MRM verification are listed in Table 3. They were selected based on experimental (highest number of peptide identifications and intensities, with at least 3 highly abundant − y and − b ions) and computational (highest ESP scores [18], highest scored transitions using PeptideAtlas [19]) criteria. Although most peptides presented unique sequences, our panel contains several candidates with sequences shared by more than one protein or shared by their isoforms (Table 3). For each candidate marker, if available, several light and heavy peptides were synthesized following common empirical rules, such as size tolerance (7–15 amino acids) and absence of methionine of proline residues (Table 3, Supplementary Table S4). The LC-MRM assay was created using Skyline 3.5.0.9319 [20]. MRM transitions were optimized for charge state, cone voltage, and collision energy by direct infusion of synthetic light peptides at 5 μL/min, five-at-a-time at 200 nmoles in 10% acetonitrile, 0.1% formic acid, and a ramping collision energy from to 5 to 53 V. The most abundant precursor charge state within the mass range of m/z 400–1200 and − y type product ions were screened for each peptide. Of those, the three most abundant ions for each peptide with optimal cone voltage and collision energies were chosen (Supplementary Table S4). An equimolar mixture of all the synthetic peptides at 500 fmoles/peptide was subjected to unscheduled LC-MRM analysis (20 ms dwell time/transition) to identify optimal retention times with a window of ± 7.5 min. Heavy peptides corresponding to chicken LYZ were used for retention time alignment, determination of instrumental variance, and relative quantitation. Of these, the chicken LYZ peptide FESNFNTQATNR showed the least variation (CV = 18.07%) and was thus selected for normalization. Based on a known spike-in concentration and peak area ratio of light-to-heavy forms of the LYZ peptide, a normalization factor (concentration ratio of light-to-heavy/peak area ratio of light-to-heavy) was computed. This factor was applied for peptide normalization, and the normalized area for each candidate peptide was used for downstream data analysis.
Data analysis
Discovery set
For each discovery set (breast cancer and other solid tumors), the relative difference (Diff [%]) between spectral counts in cases and controls was determined as follows:
where
Proteins with Diff (%) ≥ 50% that were detected (≥ 1 spectral count) in at least 21% of cases or controls were considered candidate biomarkers. Peptides attributed to each biomarker were manually inspected to confirm raw spectral matching by SEQUEST. Candidate biomarkers found to be differentially abundant in other cancer cohorts were considered non-specific to breast cancer and removed from the candidate list. Internal spike-in process controls were monitored to assess case: control stoichiometry. Functional enrichment analysis comparing proteins identified in control or stage I, II, or III breast cancer samples was performed using Ingenuity Pathway Analysis (IPA) (QIAGEN, Redwood City, CA) using default IPA settings. Enriched pathways were filtered for significance (p < 0.05) and presence in > 50% of samples.
Verification set
We did not observe any batch effect in the verification dataset. The data obtained by MRM in the verification phase of the project were used to develop a statistical model to predict breast cancer. An initial statistical model (model 1) included all 41 candidate peptides corresponding to the 25 proteins of interest (Table 3). The probability for model entry was 0.05. A second logistic regression model (model 2) was computed with significant predictors from the first model entered simultaneously to obtain bootstrapped standard errors using 1000 samples: model sensitivity, specificity, PPV, NPV, and AUC. Data were analyzed using SPSS and STATA. Peptide significance (p < 0.05) was calculated by a Wilcoxon rank sum test and corrected for multiple testing using the Benjamini-Hochberg method (Supplementary Excel Table). Peptides were modeled in raw format and not transformed or dichotomized. Logistic regression models are reported using the observed coefficient and significance values with bootstrapped standard errors and confidence intervals.
Results
Discovery of IDC candidate biomarkers
To define differences in levels of circulating proteins predictive of organ-specific tumor growth, we analyzed six sets of serum samples from cancer patients (n = 20) and matched healthy controls (n = 20) using shotgun mass spectrometry. Proteins identified in IDC breast cancer patients (Table 1, Discovery set) were compared to proteins identified in patients affected by cancers of the colon, ovaries, lung, and prostate, as well as melanoma (Supplementary Table S1, Fig. 1).
For each organ-specific cancer cohort, we first evaluated relative differences between cases and controls for three spiked-in recombinant proteins—SDF-1β, IGF1, and IGFBP7, each of which served as internal quality control and was spiked in at a ratio case/control respectively of 1:10, 1:1, and 10:1. In the breast cancer cohort, the Diff (%) was − 124% for SDF-1β, 52% for IGF1, and 132% for IGFBP7, respectively, confirming that the approach can detect a 10-fold difference in protein levels, described by Diff (%) > 120%. Candidate biomarkers were selected in each discovery set based on a low stringency cut-off of 50%.
Fifty-six proteins showed a Diff (%) ≥ 50% between the IDC cases and matched benign controls (Table 2). Those proteins span a range of molecular weights from 10 to 532 kDa (median = 50 kDa). Proteins with a molecular weight between 20 and 60 kDa were preponderant (Supplementary Fig. 1A). The average absolute concentration in blood has not been reported for most of the 56 candidate markers. Therefore, we refer to their estimated abundance in parts per million, as reported by the PaxDB database (H. sapiens - Plasma (Integrated)) [21].Most have abundances in plasma below 5 ppm (Supplementary Fig. 1B).
Table 2 contains annotations for each of the 56 proteins. According to the gene ontology (GO) annotation, 37 (65%) of the 56 candidates are extracellular proteins and 14 (18%) are membrane proteins. Half of the annotations refer to roles in cell motility, such as cell-to-cell junctions, focal adhesions, and reorganization of the actin cytoskeleton (Supplementary Excel Table, “Gene ontology (GO)”). Seventeen of 56 (~ 30%) are known secreted proteins (Supplementary Excel Table “Subcellular location [CC]”). Two proteins, AKT3 and PITX2, were not previously reported in blood using mass spectrometry. Overall, 160 candidate serum biomarkers were identified across all 6 cohorts. Of those, 33 proteins were differentially abundant in IDC only (Fig. 2, Table 2).

Venn diagram representing the number of specific and common candidate biomarkers identified in the study cohorts
Functional enrichment analysis for proteins identified in each disease subgroup in the breast cancer cohort (stages I, II, III, and control; Table 1, Discovery Set) yielded 38, 46, 36, and 49 pathways respectively, with significant overlap between groups (Supplementary Fig. 2). Enrichment of Breast Cancer Regulation by Stathmin1 in breast cancer stages I and II was partly driven by 6 of the tubulin biomarker candidates. Estrogen-Dependent Breast Cancer Signaling was enriched in the control and stage III. The Role of Tissue Factor in Cancer was enriched in > 50% of samples only at stage I. Four of the seven proteins driving enrichment of The Role of Tissue Factor in Cancer are also candidate biomarkers: RAC-gamma serine/threonine-protein kinase (AKT3), Cofilin1 and 2 (CFL1, CFL2), and integrin beta 3 (ITGB3).
Independent verification of candidate biomarkers and diagnostic models
Twenty-four proteins among the 56 found in the discovery phase were selected for verification in an independent cohort using LC-MRM. Of the 24 candidates, 41 proteotypic peptides were selected for MRM analysis (Table 3). The peptides were quantified in the verification cohort of 19 breast cancer patients at stage I (T1N0M0) and in 41 controls including serum samples obtained from women with inflammatory diseases and benign breast pathology findings (Table 1, Validation Set). All 41 peptides were shown to be more abundant in sera from breast cancer patients, and 11 were statistically significant (p value ≤ 0.01) (Supplementary Excel Table, sheet: “Wilcoxon test”). We then performed logistic regression analysis to determine if marker combinations showed superior sensitivity and specificity compared to single markers. Model 1 was built using three proteins previously identified as potential biomarkers of breast cancer in blood and/or tissue: Cofilin 1 (CFL1, LGGSAVISLEGKPL), Alpha-2-HS-glycoprotein (AHSG, HTFMGVVSLGSPSGEVSHPR), and Filamin A (FLNA, SPFSVAVSPSLDLSK). CFL1 was the only one that showed a significant difference between IDC cases and controls (Fig. 3). However, CFL1 was also differentially abundant in our discovery set in lung cancer and therefore not specific to IDC. Using all three proteins, model 1 yielded a sensitivity of 89.47% and a specificity of 80.49%. For these predictors, PPV and NPV was 68% and 95.3%, respectively, with an AUC of 0.88 (Table 4, Fig. 4a). Model 2 included four candidate predictors specific for breast cancer in this study: Ras-related protein Rap-1A (RAP1A, LVVLGSGGVGK), Integrin alpha-IIb (ITGA2B, VYLFLQPR), FLNA (ANLPQSFQVDTSK), and Talin-1 (TLN1, LAQAAQSSVATITR). Using univariate analysis, only TLN1 was significantly different between cases and controls. However, the combination of the four markers in model 2 outperformed the single marker, achieving a sensitivity of 100% and specificity of 85.37%. PPV and NPV were 76% and 100% (p < 0.05), respectively, with an AUC of 0.93 (Table 4, Fig. 4b). In both models, the combined markers outperformed single markers (Supplementary Table S5). Two peptides from FLNA (ANLPQSFQVDTSK, SPFSVAVSPSLDLSK) were identified separately as predictors for the two models, but the correlation between their levels as measured by LC-MRM was low (Supplementary Fig. 3).

Serum protein levels measured by LC-MRM in the validation set. Normalized AUC values are shown for proteins in models 1 and 2 (cases = 19, white; controls = 41, gray). Adjusted p values were obtained using the Benjamini-Hochberg correction. ANLPQSFQVDTSK (FLNA, adjusted p value = 0.67); HTFMGVVSLGSPSGEVSHPR (AHSG, adjusted p value = 0.84); LAQAAQSSVATITR (TLN1, adjusted p value = 0.0019); LGGSAVISLEGKPL (CFL1, adjusted p value = 8.29E−5); LVVLGSGGVGK (RAP1A, p value = 0.34); SPFSVAVSPSLDLSK (FLNA, p value = 0.58) and VYLFLQPR (ITGA2B, p value = 0.69)

Receiver operator curve analysis of 19 cases and 41 controls by multivariate logistic regression of individual or combined peptides. Model 1 (a) includes peptides LGGSAVISLEGKPL (CFL1), HTFMGVVSLGSPSGEVSHPR (AHSG), and SPFSVAVSPSLDLSK (FLNA). Model 2 (b) includes peptides LVVLGSGGVGK (RAP1A), VYLFLQPR (ITGA2B), ANLPQSFQVDTSK (FLNA), and LAQAAQSSVATITR (TLN1)
Discussion
The development of a minimally-invasive molecular assay using blood-borne/circulating biomarkers to support mammography screening is highly desirable to increase PPV for early-stage breast cancer detection while limiting the number of unnecessary biopsy of benign conditions that mammography cannot currently discriminate from frank early malignancy.
In this study, we uncovered 56 candidate protein biomarkers of IDC breast cancer (and/or protein fragments) in serum. When we compared the 56 candidate proteins with proteins increased in patient sera across the other cancer cohorts, 32 were altered in IDC but not in prostate, ovarian, colon or lung cancer, or melanoma or in their matched benign/inflammatory control cohorts.
The strategy of protein enrichment by HNs showed to be effective for the identification of rare/low abundance serum proteins. Proteins known to circulate in blood (plasma/serum) at low concentrations such as CCL14 (~ 5 ng/mL [22]), BMP1 (~ 50 ng/mL [23]), and heparanase (~ 100 pg/mL [24]) were identified.
Many of the proteins upregulated in the serum of breast cancer patients had never been previously observed in serum using LC-MS/MS, such as paired-like homeodomain transcription factor 2 isoform b (PITX2), AKT3 kinase isoform 2 (AKT3), tissue inhibitor of metalloproteinase 3 precursor (TIMP3) [25, 26], and Tropomyosin alpha-4 chain (TMP4). TIMP3 and TMP4 are relevant to breast cancer biology and both were reported to be more abundant in tumor interstitial fluid collected from triple negative breast cancer patients compared to normal interstitial fluid [27]. Queries using ProteomicsDB (https://www.ProteomicsDB.org) and The Human Protein Atlas (2020-07-27) showed that none of the identified proteins were exclusive to breast cancer cell lines or breast tissue (data not shown). However, even if our discovery study across multiple cancers suggested that the proteins are more abundant in blood of individuals with breast cancer, as compared to cancers in other organs, we did not expect them to be specific to breast cancer tumors. Indeed, our study shows a high degree of specificity of those markers as circulating proteins in breast cancer patients.
According to the NCI-funded Breast Cancer Surveillance Consortium (HHSN261201100031C, downloaded on 2019/02/06), sensitivity and specificity calculated over more than two million screening mammography examinations are 85.6% and 90.5%, respectively.
Our model 1 discriminated cases and controls with a sensitivity of 89.5% and a specificity of 80.5%, outperforming the specificity of the single markers reported previously. In particular, plasma levels of the Filamin-A (FLNA) 280 kDa variant predict metastatic breast cancer with 96.7% sensitivity but only 67.8% specificity [28]. In model 2, the individual markers achieved very high specificity (> 90%), but low sensitivity. However, the combination of the four markers dramatically increased the sensitivity (100%), while still maintaining adequate specificity (85%).
Our study supports the results of previous breast cancer biomarker studies and highlights a significant presence in the serum of IDC patients of proteins derived from the extracellular matrix and associated to cell proliferation, migration, adhesion, and metastasis.
Using the “Analysis of protein sets” function of Proteomics DB (Supplementary Fig. 4), revealed four proteins included in model 2 (ITGA2B, FLNA, RAP1A, and TLN1) are widely expressed across different type of tissues. However, ITGA2B shows to be particularly enriched in platelet, non-small cell lung cancer cells (ProteomicsDB), and basophils (Human Protein Atlas). The presence of circulating ITGA2B may indicate an ongoing process of tumor cell-induced platelet aggregation [29].
We observed increased levels of AHSG in serum of breast cancer patients. In a previous study, anti-Alpha-2-HS-glycoprotein (AHSG) antibodies were detected in 33 of 36 patients with breast cancer (91.7%) [30]. The two studies support the hypothesis that changes at the level of expression, but possibly also to location, PTMs or structure, occur to AHSG during breast cancer, such as to induce the production of autoantibodies.
Cofilin 1 (CFL1) is an intracellular actin-modulating protein associated with EGF-stimulated chemotaxis [31] and invadopodia localization in breast cancer cell invasion [32]. Although overexpression of CFL1 in breast cancer tissue has been associated with poor prognosis and survival [33], to our knowledge serum levels of CFL1 were never assessed. Talin 1 (TLN1) is a cytoskeletal protein that functions in extravasation and breast cancer cell migration [34, 35]. High levels of both TLN1 and CFL1 are reported in the secretome of breast cancer cell lines (including, metastatic, triple-negative, MCF7 ER-positive, and triple-negative) [36], confirming their release into the ECM. In addition to increased TLN1 abundance, we observed an increased abundance of integrin and RAS oncogene family members in IDC patients, suggesting that an ongoing dynamic remodeling of the cytoskeleton involving integrins and active regulation of adhesion molecules by TLN1 is detectable in serum due to an increase level of these proteins.
Integrin alpha-IIb (ITG2AB) plays a role in breast cancer metastasis through its role in matrix cross-linking processes [37]. ITG2AB polymorphisms are associated with increased breast cancer risk [38]. RAP1A, a member of the RAS oncogene family, regulates signaling associated with proliferation, adhesion, and migration mediated by beta1 integrin levels [34, 35, 39].
FLNA was found to be a predictor in both models, but with two different peptides. Both cover a region common to isoforms 1 and 2 and are found in the secreted form of FLNA (280 kDa) N-terminal side to the calpain cleavage site. Levels of both peptides showed relatively low correlation with each other (Pearson correlation coefficient = 0.51) (Supplementary Fig. 3). In targeted mass spectrometry experiments, this phenomenon is attributed to variability in digestion efficiency, the presence of missed cleavages, protein modifications, different isoforms, or differential enzymatic degradation. However, SPFSVAVSPSLDLSK may be modified by phosphoserine (pS968) [36, 40] while no modifications likely occur in ANLPQSFQVDTSK. Moreover, ANLPQSFQVDTSK levels correlate positively with a third peptide YGGQPVPNFPSK (Pearson correlation coefficient = 0.96), which is likely unmodified and was not a significant factor in any of our diagnostic models (Supplementary Fig. 3). Interestingly, SPFSVAVSPSLDLSK and ANLPQSFQVDTSK contribute to different models: SPFSVAVSPSLDLSK participates in the first model, which comprises proteins specific to both breast cancer sera and homeostatic response pathways, while ANLPQSFQVDTSK participates in the second, which includes only breast specific proteins and favors a strong motility component.
We postulate that the breast cancer protein signature described here reflects the changing ECM and stromal composition of the breast cancer tissue microenvironment during early tumorigenic processes. As observed in rodent models, breast tissue is characterized by high stromal content and is particularly rich in fibrillar collagens and matricellular proteins [25, 28] involved in the activation of adhesion signals [37, 41] [42] and enhancing invasion and metastasis [43]. ECM remodeling occurs during the early stages of IDC and results in leakage of proteins and protein fragments into the circulation. Shen et al. observed that breast tumors shed cancer-specific peptides and products of proteolytic activity into circulation [44]. Our data reinforces the hypothesis that this shedding of ECM components into the circulation even at the earliest stages of malignancy can be used to design a specific and sensitive biomarker panel to improve detection of breast cancer.
Although this study uses an innovative nanoparticle-based protein capture technique that focuses on the LMW portion of the proteome to identify candidate protein/protein fragments and peptides as serum biomarkers of breast cancer, as well as a unique collection of multi-tumor serum sets and matched benign conditions, a separate discovery and verification set including semiquantitative MRM-based verification of selected candidates, there are some limitations to our study.
The use of spectral counting in our discovery cohort may have limited the accuracy of the analysis of semi-quantitative label-free data. Many other approaches for spectral counting and ion intensity normalization [45, 46] have been explored. Although our candidates were independently verified using an LC-MRM method, we recognize that use of semi-quantitative label-free data employed in the discovery phase may be a limitation of the study. Other alternative nanoparticle-based sample processing techniques provide broad proteomic coverage of blood proteins [47]. Unlike our HN sample enrichment method, these approaches do not specifically enrich the extra-vascular content contained in the LMW low abundant blood proteome.
Relevant to FLNA peptides, we emphasize that the HN-CB/VSA serum pre-processing is aimed at analysis of low molecular weight proteins. Therefore, we cannot rule out that circulating fragments belonging to FLN1 or other high molecular weight proteins might be enriched by HN and further digested to tryptic peptides after elution.
Moreover, while our discovery and verification sets were carefully constructed to maximize chances of identification of specific and sensitive markers for early breast cancer detection, the study sets were inherently unbalanced and do not reflect the frequency of occult/non-detected breast cancer in the general population. However, the number and type of samples chosen for this study was consistent with the objectives of discovery, qualification and verification of potential breast cancer biomarkers. Additional validation studies will require not only a larger population of individuals comprised of different BI-RADS scores and molecular subtypes [48], but also a thorough and stringent validation of our MRM methods, following the Tier for validation suggested by the National Institutes of Health, the National Cancer Institute (CPTAC - Clinical Proteomic Tumor Analysis Consortium), and National Heart, Lung, and Blood Institute (Proteomics Centers) [49].
In conclusion, these findings would require much more intensive validation in blinded, independent study sets in order to judge the potential for clinical impact. Our results, taken together, justify such further validation.
Conclusion
Our objective was to use a series of innovative sample processing and proteomic approaches coupled to a unique sample study set discover and validate a panel of candidate serum proteins that could potentially be used to detect early-stage breast cancer as an adjunct with mammography. We developed a semi-quantitative MRM assay that employs a simple method of protein enrichment, representing a robust foundation suitable for future validation studies. We also discovered a panel of proteins, which were validated in an independent cohort from largely early-stage (T1a) breast cancers vs serum taken from women with benign/inflammatory conditions as a control set, with a sensitivity and specificity profile that could have clinical impact when combined with mammography. Further studies on larger cohorts of individuals who were subjected to mammography will be required to clarify if our proposed protein panel can complement the diagnostic performance of mammography.
Availability of data and materials
The data used in the present study are available from the corresponding author upon reasonable request.
Abbreviations
- SDF-1β:
-
Stromal cell-derived factor 1β
- IGFBP7:
-
Insulin-like growth factor binding protein 7
- IGF1:
-
Insulin-like growth factor 1
- MRM:
-
Multiple reaction monitoring
- PPV:
-
Positive predictive value
- NPV:
-
Negative predictive value
- ECM:
-
Extracellular matrix
References
Shieh Y, Eklund M, Sawaya GF, Black WC, Kramer BS, Esserman LJ. Population-based screening for cancer: hope and hype. Nat Rev Clin Oncol. 2016;13(9):550–65.
Hofvind S, Ponti A, Patnick J, Ascunce N, Njor S, Broeders M, et al. False-positive results in mammographic screening for breast cancer in Europe: a literature review and survey of service screening programmes. J Med Screen. 2012;19(Suppl 1):57–66.
Løberg M, Lousdal ML, Bretthauer M, Kalager M. Benefits and harms of mammography screening. Breast Cancer Res. 2015;17:63.
Joy JE, Penhoet EE, Petitti DB, National Cancer Policy Board (U.S.). Committee on New Approaches to Early Detection and Diagnosis of Breast Cancer., National Research Council (U.S.). Policy and Global Affairs., National Research Council (U.S.). Board on Science Technology and Economic Policy. Saving women’s lives: strategies for improving breast cancer detection and diagnosis. Washington, D.C.: National Academies Press; 2005. xxi, 361 p. p.
Lehman CD, Wellman RD, Buist DS, Kerlikowske K, Tosteson AN, Miglioretti DL, et al. Diagnostic accuracy of digital screening mammography with and without computer-aided detection. JAMA Intern Med. 2015;175(11):1828–37.
Sprague BL, Arao RF, Miglioretti DL, Henderson LM, Buist DS, Onega T, et al. National performance benchmarks for modern diagnostic digital mammography: update from the Breast Cancer Surveillance Consortium. Radiology. 2017;283(1):59–69.
Hoff SR, Abrahamsen AL, Samset JH, Vigeland E, Klepp O, Hofvind S. Breast cancer: missed interval and screening-detected cancer at full-field digital mammography and screen-film mammography-- results from a retrospective review. Radiology. 2012;264(2):378–86.
Wang AT, Vachon CM, Brandt KR, Ghosh K. Breast density and breast cancer risk: a practical review. Mayo Clin Proc. 2014;89(4):548–57.
NationalCancerInstitute(NCI). Early Detection Research Network [Available from: https://edrn.nci.nih.gov/biomarkers]. Accessed 2 July 2020.
Di Seri M, Manna A, Sciò M, Marchei GG, De Benedetto A, Reale MG. Evaluation of the new marker CA 549 versus CA 15-3 in neoplastic and non-neoplastic disease of the breast. J Nucl Med Allied Sci. 1990;34(4 Suppl):49.
Tamburro D, Fredolini C, Espina V, Douglas TA, Ranganathan A, Ilag L, et al. Multifunctional core-shell nanoparticles: discovery of previously invisible biomarkers. J Am Chem Soc. 2011;133(47):19178–88.
Fredolini C, Meani F, Luchini A, Zhou W, Russo P, Ross M, et al. Investigation of the ovarian and prostate cancer peptidome for candidate early detection markers using a novel nanoparticle biomarker capture technology. AAPS J. 2010;12(4):504–18.
Conti A, Fredolini C, Tamburro D, Magagnoli G, Zhou W, Liotta LA, et al. Identification of novel candidate circulating biomarkers for malignant soft tissue sarcomas: correlation with metastatic progression. Proteomics. 2016;16(4):689–97.
Longo C, Gambara G, Espina V, Luchini A, Bishop B, Patanarut AS, et al. A novel biomarker harvesting nanotechnology identifies Bak as a candidate melanoma biomarker in serum. Exp Dermatol. 2011;20(1):29–34.
Kim B, Araujo R, Howard M, Magni R, Liotta LA, Luchini A. Affinity enrichment for mass spectrometry: improving the yield of low abundance biomarkers. Expert Rev Proteomics. 2018;15(4):353–66.
Capriotti AL, Caruso G, Cavaliere C, Piovesana S, Samperi R, Laganà A. Comparison of three different enrichment strategies for serum low molecular weight protein identification using shotgun proteomics approach. Anal Chim Acta. 2012;740:58–65.
Espina V, Wulfkuhle JD, Calvert VS, Petricoin EF 3rd, Liotta LA. Reverse phase protein microarrays for monitoring biological responses. Methods Mol Biol. 2007;383:321–36.
Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27(2):190–8.
Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, et al. The PeptideAtlas project. Nucleic Acids Res. 2006;34(Database issue):D655–8.
MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–8.
H.sapiens - Plasma (Integrated). [cited March 2017]. Available from: https://pax-db.org/dataset/9606/171/.
Jacobs ES, Keating SM, Abdel-Mohsen M, Gibb SL, Heitman JW, Inglis HC, et al. Cytokines Elevated in HIV Elite Controllers Reduce HIV Replication. J Virol . 2017;91(6):e02051–16.
Grgurevic L, Macek B, Healy DR, Brault AL, Erjavec I, Cipcic A, et al. Circulating bone morphogenetic protein 1-3 isoform increases renal fibrosis. J Am Soc Nephrol. 2011;22(4):681–92.
Shafat I, Ilan N, Zoabi S, Vlodavsky I, Nakhoul F. Heparanase levels are elevated in the urine and plasma of type 2 diabetes patients and associate with blood glucose levels. PLoS One. 2011;6(2):e17312.
Goddard ET, Hill RC, Barrett A, Betts C, Guo Q, Maller O, et al. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int J Biochem Cell Biol. 2016;81(Pt A):223–32.
Giussani M, Merlino G, Cappelletti V, Tagliabue E, Daidone MG. Tumor-extracellular matrix interactions: identification of tools associated with breast cancer progression. Semin Cancer Biol. 2015;35:3–10.
Gromov P, Gromova I, Bunkenborg J, Cabezon T, Moreira JM, Timmermans-Wielenga V, et al. Up-regulated proteins in the fluid bathing the tumour cell microenvironment as potential serological markers for early detection of cancer of the breast. Mol Oncol. 2010;4(1):65–89.
Kolonin MG, Evans KW, Mani SA, Gomer RH. Alternative origins of stroma in normal organs and disease. Stem Cell Res. 2012;8(2):312–23.
Zarà M, Canobbio I, Visconte C, Canino J, Torti M, Guidetti GF. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell Signal. 2018;48:45–53.
Fernández-Grijalva AL, Aguilar-Lemarroy A, Jave-Suarez LF, Gutiérrez-Ortega A, Godinez-Melgoza PA, Herrera-Rodríguez SE, et al. Alpha 2HS-glycoprotein, a tumor-associated antigen (TAA) detected in Mexican patients with early-stage breast cancer. J Proteome. 2015;112:301–12.
Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer. 2007;7(6):429–40.
Huang X, Pan Q, Sun D, Chen W, Shen A, Huang M, et al. O-GlcNAcylation of cofilin promotes breast cancer cell invasion. J Biol Chem. 2013;288(51):36418–25.
Maimaiti Y, Tan J, Liu Z, Guo Y, Yan Y, Nie X, et al. Overexpression of cofilin correlates with poor survival in breast cancer: a tissue microarray analysis. Oncol Lett. 2017;14(2):2288–94.
McSherry EA, Brennan K, Hudson L, Hill AD, Hopkins AM. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast Cancer Res. 2011;13(2):R31.
Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule a interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20(7):1916–25.
Ziegler YS, Moresco JJ, Yates JR, Nardulli AM. PLoS One. 2016;11(6):e0158296.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906.
Langsenlehner U, Renner W, Yazdani-Biuki B, Eder T, Wascher TC, Paulweber B, et al. Integrin alpha-2 and beta-3 gene polymorphisms and breast cancer risk. Breast Cancer Res Treat. 2006;97(1):67–72.
Zhang YL, Wang RC, Cheng K, Ring BZ, Su L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol Med. 2017;14(1):90–9.
Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62.
Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20(4):360–7.
Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH, et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Mater. 2014;13(10):970–8.
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4(1):38.
Shen Y, Tolić N, Liu T, Zhao R, Petritis BO, Gritsenko MA, et al. Blood peptidome-degradome profile of breast cancer. PLoS One. 2010;5(10):e13133.
Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13(9):2513–26.
Al Shweiki MR, Mönchgesang S, Majovsky P, Thieme D, Trutschel D, Hoehenwarter W. Assessment of label-free quantification in discovery proteomics and impact of technological factors and natural variability of protein abundance. J Proteome Res. 2017;16(4):1410–24.
Blume JE, Manning WC, Troiano G, Hornburg D, Figa M, Hesterberg L, et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat Commun. 2020;11(1):3662.
Paulovich AG, Whiteaker JR, Hoofnagle AN, Wang P. The interface between biomarker discovery and clinical validation: the tar pit of the protein biomarker pipeline. Proteomics Clin Appl. 2008;2(10–11):1386–402.
Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13(3):907–17.
Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D50.
Acknowledgements
The authors appreciate the generous support of the Department of Life Sciences at George Mason University. Nancy Linford, PhD, provided editing services. Seema Plaisier, PhD, provided bioinformatics analyses. The authors very much acknowledge the support from Professor Enrico Garaci and the team at the Istituto Superiore di Sanità, Rome, Italy. Mrs. Veronica Michetti, Drs. Paolo Roazzi, and Fabio Galati at Istituto Superiore di Sanità, Rome, Italy, for samples database creation and data entry. Drs. Francesca Poli, Lucilla Lecchi, Viviana Sioli and Silvia Giovanelli at Biobank POLI-MI Department of Transfusion Medicine and Hematology Fondazione IRCCS Ca′ Granda, Ospedale Maggiore Policlinico di Milano, Milano, Italy, for sample storage, biobank management, and shipments, are kindly acknowledged.
Declarations
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [50] partner repository with the dataset identifier PXD020129.
Funding
This work was supported by institutional funding the Translational Genomics Research Institute and George Mason University. This work was partly supported by the Italian Istituto Superiore di Sanità in the framework of the Italy/USA cooperation agreement between the U.S. Department of Health and Human Services, George Mason University, and the Italian Ministry of Public Health. Research reported in this publication included work performed in the mass spectrometry core supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
CF, LL, EP, CB, SM, FF, EG, RDM, and PP contributed to the experimental design. CF, LP, DT, WZ, KAT, MR, and TT performed the sample preparation. WZ, PR, RM, TT, and KVP performed the data acquisition. CF, PP, and KGM performed the data analysis. CF, KVP, KC, KGM, EFP, and PP wrote the manuscript with contributions from all the authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research protocol was approved by the Ethic Committee of each Clinical Center involved, and all patients provided appropriate informed consent.
Consent for publication
All authors approved the manuscript and consented to its publication.
Competing interests
EFP, AL, and LL are co-inventors on the HN technology owned by George Mason University and might receive royalties as provided by US LAW. EFP, AL, and LL are co-founders of Ceres Nanosciences, Inc. the exclusive licensee to the HN technology, with less than 3% equity ownership interests. EFP and LL act as consultants to the Company.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Cancer cohort characteristics. Table S2. Peptides used as internal standards in this study. Table S3. E.Coli peptides used to evaluate instrumental variance in the LC-MRM validation assay. Table S4. Candidate peptide transitions employed for the LC-MRM validation assay. Table S5. Logistic regression analysis and validation of candidate peptide markers. Figure S1. Frequency distribution and histograms of molecular weight (A) and abundance (B) for unique plasma proteins identified across cases and controls. Enrichment of low abundance, low molecular weight proteins using nanoparticle capture is shown. Figure S2. Overlap of pathways identified by functional enrichment analysis across control and IDC cases (stages I-III). Enriched pathways were filtered for p < 0.05 and relevance in > 50% of samples. Figure S3. Correlation analysis of Filamin A peptides AUC in models 1 and 2. Pearson’s correlation coefficients are shown for each correlation pair. Analysis excludes outlier cases 22 and 44. Starred correlations: p < 0.001. Figure S4. Expression heatmap generated using the ProteomicsDB online analytical tool (https://www.proteomicsdb.org/).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fredolini, C., Pathak, K.V., Paris, L. et al. Shotgun proteomics coupled to nanoparticle-based biomarker enrichment reveals a novel panel of extracellular matrix proteins as candidate serum protein biomarkers for early-stage breast cancer detection. Breast Cancer Res 22, 135 (2020). https://doi.org/10.1186/s13058-020-01373-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-020-01373-9