
Abstract
Sepsis-associated encephalopathy is a severe neurologic syndrome characterized by a diffuse dysfunction of the brain caused by sepsis. This review provides a concise overview of diagnostic tools and management strategies for SAE at the acute phase and in the long term. Early recognition and diagnosis of SAE are crucial for effective management. Because neurologic evaluation can be confounded by several factors in the intensive care unit setting, a multimodal approach is warranted for diagnosis and management. Diagnostic tools commonly employed include clinical evaluation, metabolic tests, electroencephalography, and neuroimaging in selected cases. The usefulness of blood biomarkers of brain injury for diagnosis remains limited. Clinical evaluation involves assessing the patient's mental status, motor responses, brainstem reflexes, and presence of abnormal movements. Electroencephalography can rule out non-convulsive seizures and help detect several patterns of various severity such as generalized slowing, epileptiform discharges, and triphasic waves. In patients with acute encephalopathy, the diagnostic value of non-contrast computed tomography is limited. In septic patients with persistent encephalopathy, seizures, and/or focal signs, magnetic resonance imaging detects brain injury in more than 50% of cases, mainly cerebrovascular complications, and white matter changes. Timely identification and treatment of the underlying infection are paramount, along with effective control of systemic factors that may contribute to secondary brain injury. Upon admission to the ICU, maintaining appropriate levels of oxygenation, blood pressure, and metabolic balance is crucial. Throughout the ICU stay, it is important to be mindful of the potential neurotoxic effects associated with specific medications like midazolam and cefepime, and to closely monitor patients for non-convulsive seizures. The potential efficacy of targeted neurocritical care during the acute phase in optimizing patient outcomes deserves to be further investigated. Sepsis-associated encephalopathy may lead to permanent neurologic sequelae. Seizures occurring in the acute phase increase the susceptibility to long-term epilepsy. Extended ICU stays and the presence of sepsis-associated encephalopathy are linked to functional disability and neuropsychological sequelae, underscoring the necessity for long-term surveillance in the comprehensive care of septic patients.
Similar content being viewed by others
Introduction
Sepsis-associated encephalopathy (SAE) is a severe neurologic syndrome characterized by a diffuse dysfunction of the brain caused by sepsis, a life-threatening condition resulting from the dysregulated response of the body to an infection. At the acute phase, patients with SAE typically present with an acute onset of encephalopathy, ranging from delirium to coma [1]. The pathophysiology of sepsis-associated encephalopathy is complex and involves multiple mechanisms that collectively contribute to brain dysfunction and injury [2]. One of the primary mechanisms is the release of pro-inflammatory cytokines, which leads to the disruption of the blood–brain barrier (BBB), causing an influx of immune cells and inflammatory mediators into the brain. This inflammation triggers the activation of microglia, the immune cells of the brain, which further release cytokines and reactive oxygen species, leading to oxidative stress and neuronal damage. Other important acute phase mechanisms they notably include cerebral hypoxia, metabolic disturbances, microvascular and BBB alterations, and neurotransmitter imbalances. SAE can be possibly triggered or aggravated by secondary causes, including systemic insults, renal or hepatic dysfunction, environmental factors, and the use of neurotoxic agents. Although SAE is classically seen as a fully reversible pathophysiological process due to systemic inflammation, there is increasing evidence suggesting that sepsis may be associated with structural brain injury and neurologic sequelae in the long term [3, 4].
In the present article, we review recent findings in the field of SAE focusing on its epidemiology, diagnosis, and management at the acute phase. We also provide an update on long-term effects of SAE observed in sepsis survivors.
Epidemiology and short-term outcomes
SAE is most frequently defined as an acute encephalopathy occurring during sepsis or septic shock, and not attributable to any other cause than sepsis itself [5]. SAE is thought to be the most common cause of encephalopathy in the intensive care unit (ICU) [6]. In a landmark study conducted in 50 non-sedated ICU septic patients, SAE, defined by a score on the Glasgow coma scale (GCS) < 15 was observed in 54% of patients [7]. In the most recent cohorts, where SAE was defined as sepsis associated with a GCS < 15 or delirium features, reported incidences were 53% in a French ICU multicenter cohort [8], and up to 68% in a cohort of septic patients from United Sates databases MIMIC-IV and eICU [9]. In a recent large multicenter study, sepsis-associated delirium had a median duration among affected participants of 3 (interquartile range 2–6) days [10]. With these definitions, however, SAE remain a broad syndrome with severity ranging from mild delirium to deep coma, impacting patient prognosis accordingly. It has been well demonstrated that occurrence of SAE is independently associated with short-term mortality [7,8,9]. Severity of SAE is also correlated to mortality, patients with GCS 3 to 8 having the worst prognosis (HR 3.37, 95% CI 2.82–4.03) [8]. Interestingly, even mild alterations of mental status, defined by a score on the GCS of 13 or 14 are independently associated with an increased risk of death (HR 1.38, 95% CI 1.09–1.38).
Risk factors and clinical presentation
Most of the available data on acute encephalopathy in the ICU has been generated from studies conducted in the general population. Few specific epidemiological studies have been conducted in patients with sepsis, and risk factors for SAE identified in these studies are described in Table 1 [7, 8, 11, 12]. These studies are biased by the lack of consensual definitions for SAE and the use of different sepsis criteria. Therefore, large multicenter epidemiological studies specific to SAE are needed.
Clinical evaluation of SAE is challenging in the ICU because neurologic assessment can be confounded by several factors, including fever, metabolic derangements, and the use of hypnotic agents in mechanically ventilated patients. SAE manifests as a rapid change from baseline cognitive status or level of consciousness, and presents with a wide range of symptoms, from mild delirium (19%) to coma (40%) [1, 8]. Coma or hypoactive delirium is the most common presentation of SAE, whereas agitation (~10% of cases) and dysautonomia are less frequent [8]. Convulsive seizures (~2% of cases) and focal signs (~1% of cases) are uncommon and should trigger investigations to rule out brain injury. Thus, underdiagnosis of SAE is probable in the absence of systematic screening with validated tools [13]. Conversely, SAE may be the first sign of early sepsis, and any new-onset encephalopathy must prompt clinicians to screen their patients for infection.
Neuroimaging
Data on the usefulness of brain CT studies in patients with SAE is limited. In a meta-analysis conducted in adults with acute non-traumatic encephalopathy, CT abnormal findings were observed in 11% of cases [14]. In medical ICU patients, most common acute findings diagnosed from non-contrast head CT studies included infarction (5% of cases) and hemorrhage (4% of cases) [15]. In patients presenting with acute coma or any other clinical sign suggesting brainstem involvement, angio-CT should be performed to rule out acute basilary artery occlusion. Acute basilary occlusion may represent up to 10% of unexplained non-traumatic coma, with more than 40% of cases misdiagnosed with non-contrast head CT [16].
Brain magnetic resonance imaging (MRI) is indicated in presence of focal signs, brainstem symptoms, new-onset seizures, and in case of persistent encephalopathy in the absence of common confounders (i.e. metabolic/toxic factors and sedation). It is recommended to include a diffusion-weighted imaging (DWI) sequence in the MRI protocol, which is the most sensitive sequence for detection of cerebral ischemia and inflammatory changes. MRI alterations diagnosed at the acute phase of SAE include parenchymal lesions and atrophy, that are reported in about 55% and 16% of cases, respectively [17, 18]. Ischemic lesions are diagnosed in 14–27% of SAE patients presenting persistent encephalopathy, focal signs or seizures during ICU stay [17, 19]. Infarct patterns can be multiple (67%), large (43%), and/or junctional (29%) and are independently associated with disseminated intravascular coagulation and lower platelet counts [20]. Ischemic lesions result from both macrocirculatory compromise like low blood pressure and disrupted autoregulation of cerebral blood flow, as well as microcirculatory changes such as damaged blood vessel linings and increased blood clotting. These factors collectively lead to cerebrovascular damage.
White matter lesions (WML) are observed in a significant percentage (14–81%) of SAE patients presenting with persistent encephalopathy [19, 21]. These lesions share a periventricular distribution pattern similar to the brain changes seen in small vessel disease linked to hypertension. Posterior reversible encephalopathy syndrome is reported in 9% of SAE cases [21]. WML are associated with vasogenic edema, and are mostly located in the superior frontal sulci or parieto-occipital sulci.
Brain atrophy is more pronounced in patient with SAE than in healthy controls [18]. This atrophy appears to be diffuse, with a significant reduction in the total volumes of the cerebral cortex, white matter, and hippocampus [22]. Cerebral atrophy is more pronounced in patients with higher APACHE II and SOFA scores and is associated with worse neurologic outcomes [17].
Acute neuroimaging changes have prognostic significance as predictors of disability and survival in the first year following SAE. In a single-center study, ischemic stroke was found to be independently associated with increased ICU mortality and poor functional status at 6 months [20]. In another study, acute neuroimaging abnormalities (i.e., infarction, hemorrhage and/or edema) were independently associated with ICU mortality, 1-year mortality following ICU discharge, and disability. Greater brain atrophy has been correlated with worse cognitive performance at 12 months [23]. In a recent study, the presence of PRES was not associated with worse outcomes [21].
Electroencephalography and evoked potentials
EEG
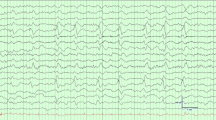
EEG can be a valuable tool in the positive diagnosis of SAE, for excluding non-convulsive status epilepticus, and for prognostication (Fig. 1). In non-sedated patients, EEG is indicated in patients with altered mental status (ranging from delirium to coma), seizures or stereotyped abnormal movements (especially myoclonus). In mechanically ventilated patients, EEG recordings can be obtained if there is delayed awakening, typically in case of persistent unresponsiveness after 48 h after sedation discontinuation [24].

Aspect, prevalence, and prognostic value of EEG patterns in sepsis-associated encephalopathy. (Prevalence and prognosis data from Azabou et al. [31], Berisavac et al. [32], Gilmore et al. [36], Hosokawa et al. [30], Benghanem et al. [37], Velissaris et al. [28]). ⊥These EEG patterns observed without concomitant sedation were associated with mortality. Definitions: Background frequency is described as δ (0.2–3.5 Hz), θ (4–7.5 Hz), α (8–13 Hz) or β (14–30 Hz) bands. Low voltage (200ms). Sporadic triphasic waves: rare slow wave with an initial negative deflection (upward) followed by a positive component (downward) and then negative again; when associated to encephalopathy, they are ample diffuse slow waves, frequently prominent in the fronto-central regions. Periodic discharges: abundant periodic abnormalities (spike or wave, with a return to the EEG background between abnormalities), during >50% of the recording. Rhythmic discharges: abundant rhythmic discharges (spike or wave, without return to the EEG background between abnormalities) during >50% of the recording. Electrographic seizure: rhythmic discharges at >2.5 Hz for ≥10 seconds or any pattern with definite spatio-temporal evolution and lasting ≥10 seconds
One of the main challenges is the impact of sedation on EEG background, which is dependent on the dosage and the specific sedative used. Sedatives can lead to a dose-dependent slowing of the EEG background. Benzodiazepines often produce diffuse rapid rhythms (> 13Hz), whereas propofol and barbiturates may result in low voltage, discontinuous patterns at moderate doses, and burst suppression or suppression patterns at higher doses [25, 26] (Fig. 1).
EEG is sensitive but not specific for assessing SAE, as similar abnormal patterns can appear in different encephalopathies. So, understanding the patient's clinical situation and considering other possible diagnoses is vital when interpreting EEG results. Moreover, there is variability in how neurophysiologists interpret EEG and the American Clinical Neurophysiology Society (ACNS) introduced specific terms for critical care EEG to address this issue [27].
Early EEG abnormalities may precede clinical neurologic impairment and correlate with the severity of encephalopathy [28, 29]. In recent studies, EEG background is described as slow with a theta or delta dominant rhythm in 10 to 50% and 30 to 60% of patients, respectively (Fig. 1) [28, 30,31,32,33]. Amplitude and continuity can also be affected, from low voltage and discontinuous background (10–60% of cases) to burst suppression or suppression (3–8% of cases) [31, 32, 34]. The more severe EEG patterns are mostly observed in sedated patients, and thus the proportion of EEG abnormalities related to sedation versus SAE is not easy to assess. Diffuse triphasic waves are observed in 6–20% patients [30,31,32], and periodic discharges are reported in 5–20% patients [31, 32]. The discrepancies in the prevalence of EEG abnormalities between studies could be explained by the type/duration of EEG recording (standard vs continuous), the time course and severity of sepsis, and the lack of standardization in interpretation.
The pathophysiology of electrographic seizures (ESz) in SAE remains debated, but could be related to the increased neuronal excitotoxicity and epileptogenic factors, including neurotoxic antimicrobials, metabolic disturbances, and severe acute kidney injury. Most ESz are non-convulsive, highlighting the interest of continuous EEG recording [35]. ESz are observed in 0–30% of cases [31, 32, 34, 36].
Some EEG patterns are associated with delirium, including slow delta rhythm, absence of EEG reactivity, discontinuity, and presence/burden periodic discharges (PDs) [31, 34, 37]. PDs may contribute to brain hypoxia and might be considered as a cause of secondary brain injury [34]. Conversely, rapid beta activity is associated with a reduced risk of delirium [38].
Some EEG patterns are also associated with ICU mortality. The absence of reactivity has been shown to be independently associated with ICU and 1 year mortality [31, 32, 36]. A recent prospective study highlighted that triphasic waves, slow delta background and suppressed EEG were the most frequent patterns observed within 24h prior to death [32]. The score on the Synek scale, which was developed in anoxic and trauma patients, is also associated with mortality in septic patients, similarly to PDs presence/burden [31, 34]. In a general population of medical ICU patients remaining unresponsive after sedation interruption, a pattern consisting of a reactive standard electroencephalography with a background frequency greater than 4 Hz was associated with reduced mortality [24]. It is important to highlight that the capacity of these EEG patterns to predict delirium or mortality is only moderate on an individual patient level.
Evoked potentials
Evoked potentials (EPs) are neural responses time-locked to some stimulus and differ from EEG signals as they are stimulus-induced. EPs reflect the combined activity of many neurons firing together and necessitate averaging multiple sensory or auditory stimulations. These components are labeled based on their polarity (negative as "N" and positive as "P") and their latency (measured in milliseconds) from the stimulus. [39]. Somato-sensory evoked potential (SSEP) are the most commonly used EP in the ICU, mostly for neuroprognostication, and the bilateral absence of N20 is recognized as the most robust marker of poor outcome in comatose patients after cardiac arrest [40]. Other types of EPs include as brainstem auditory evoked potentials (BAEPs) and long latency event-related potentials (ERPs) with mismatch negativity (MMN) and P300 responses [41]. The interest of EPs for the diagnostic and prognostic of SAE remains debated. A prospective cohort of septic patients suggested that subcortical (i.e., N20–N23 interlatency) and cortical (N20–N70 interlatency) pathways of SSEP were impaired in 34% and 84% of patients, respectively, these late latencies being correlated with the APACHE III score [42]. The intracranial conduction time (ICCT, namely P14–N20 latency) assessed by SSEP and the intrapontine conduction time (IPCT) assessed by BAEP could be interesting makers in predicting ICU mortality and delirium, in deeply sedated critically ill patients. One study suggested that ICCT impairment was associated with ICU mortality (OR 2.69, 95%CI 1.05–6.85), and that IPCT was only delayed in delirious patients. These ICCT and IPCT impairments could be considered as early indicators of brain and brainstem dysfunction [43]. In deeply sedated critically ill patients, a greater MMN amplitude was observed in patients who awakened compared to those who did not [44]. The utilization of EPs is hindered by several factors, including a restricted availability of devices in the ICU setting, difficulties in interpretation, and a moderate prognostic value in the sepsis population.
Blood biomarkers
Blood biomarkers associated with neuronal injury, specifically neuron-specific enolase (NSE) and neurofilament light (NfL), as well as biomarkers linked to glial injury, such as protein S100 beta (PS100), were evaluated in sepsis patients to anticipate the onset of SAE and predict outcomes. NSE is the most accessible biomarker, and the prevalence of elevated NSE levels (i.e. > 12.5µg/L) in sepsis varies between 28 and 53% [45, 46]. Previous studies showed a modest increase in NSE concentrations during sepsis, with median serum levels of 6.6 [IQR 4.1–13.8] µg/L [45], 18.8 [IQR 13.9 –30.5] µg/L [34], and 30.33 [IQR 19.6–46.5] µg/L [47].
In a single-center study, a NSE threshold > 24.15µg/L (AUC 0.66) had a specificity of 83% and a sensitivity of 54.2% for the diagnosis of SAE [48]. One prospective cohort study found no correlation between NSE levels and mortality at day 28 [46]. Conversely, two prospective cohorts found an association between NSE levels and ICU or hospital mortality (NSE at day 4 > 25.94µg/L, AUC 0.75 [47]; NSE > 24.15µg/L, AUC 0.59 [48]). Another retrospective study demonstrated that abnormal NSE levels on ICU admission were associated with a 23.3% risk of death, and each doubling of NSE level was linked to a 7.3% increased risk of death [45]. One study suggested that NSE levels > 12.5µg/L were associated with a 29.3% risk of delirium, and each doubling of NSE level was associated with an additional 5.2% risk of delirium [5].
The prognostic value of glial injury biomarkers was examined in several studies, highlighting conflicting results. Previous prospective cohort studies indicated that high PS100 levels were associated with hospital mortality (PS100 > 0.131 μg/L, AUC 0.73) [48], but also with hypoactive delirium [46]. However, two other studies did not find any association between PS100 levels and outcomes, including occurrence of SAE and altered cognition in the long term [49, 50].
One study investigated the link between serum NfL levels and SAE outcomes [19]. Among sepsis patients, serum NfL levels increased over time, contrasting with stable levels in non-sepsis patients. Notably, SAE patients had notably higher plasma NfL values, and these values were connected to the severity of SAE. Elevated plasma NfL levels were also associated with poorer long-term functional outcomes.
One prospective study found that serum concentrations of Glial Fibrillary Acidic Protein (GFAP), a protein expressed by astrocytes, were higher in SAE patients compared to non-SAE patients [51]. Serum GFAP concentrations > 0.536 ng/ml predicted mortality (AUC 0.77), and higher GFAP levels were associated with worse long-term outcomes. Serum concentrations of microRNAs (mRNAs) have also been considered as diagnostic and prognostic biomarkers of SAE, although their use in clinical practice remains limited [52]. Of note, biomarker profiles differ between patients with SAE, sepsis, and delirium, implying that the underlying pathways associated with SAE are distinct from those associated with delirium and sepsis [53].
Using blood biomarkers for SAE diagnosis faces challenges due to undefined optimal assessment timing, uncertainty about the precise SAE onset, inconclusive findings about prognostic benefits and definitive thresholds, and limited access to certain biomarkers, impeding their routine clinical use. The profile of biomarkers differs between SAE, sepsis, and delirium patients, suggesting that pathways related to SAE are different from those related to delirium and sepsis itself.
Management
General measures
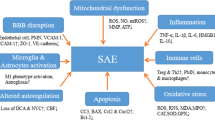
Multidisciplinary care is frequently necessary for the comprehensive management of SAE. Prompt identification and treatment of the infection are vital, typically involving the administration of antibiotics and supportive measures such as fluid resuscitation and vasopressors. Moreover, it is essential to control factors contributing to secondary brain injury, which includes maintaining adequate levels of oxygenation and blood pressure, addressing metabolic imbalances, and detecting/treating seizures. Delirium prevention is of paramount importance and implementation of the ABCDEF bundle is associated with improved survival and a reduction in the number of days of delirium and coma [54]. A simplified algorithm for the diagnosis of SAE is presented in Fig. 2.

A simplified algorithm for the diagnosis of sepsis-associated encephalopathy. CSF cerebrospinal fluid; EEG Electroencephalography; ICU Intensive Care Unit; TSH thyroid-stimulating hormone
To prevent secondary insults resulting from agitation, or dysautonomia in severe cases, the use of sedatives (propofol, dexmedetomidine) or antipsychotics may be necessary. In two randomized controlled trials conducted in patients with sepsis, the use of dexmedetomidine compared to standard sedation did not result in lower rates of delirium or coma [55, 56]. The administration of midazolam should be avoided due to its independent association with encephalopathy [57,58,59]. Renal failure can contribute to the accumulation of various substances, including antimicrobials and hypnotics. Therefore, it is crucial to systematically monitor serum concentrations of drugs with potential neurotoxicity (such as beta-lactam antibiotics, calcineurin inhibitors, and antifungals) [60]. A multimodal approach for the management of SAE is presented in Fig. 3.

A multimodal approach for the management of sepsis-associated encephalopathy. EEG Electroencephalography; SAE Sepsis-Associated Encephalopathy
Systemic causes of secondary brain injury
Systemic causes of secondary brain injury are frequent at sepsis onset and are linked to poorer outcomes [8]. These causes encompass hypo- and hyperglycemia, hypercapnia, and hypernatremia, as identified in prior studies [8]. However, special consideration should be given to hypoxemia and hyperoxia, anemia, hypothermia, and hyperthermia, as the failure to manage these factors adequately could theoretically result in additional brain injury. Current sepsis guidelines do not provide specific recommendations for managing systemic causes of secondary brain injury [61]. We propose that targeted neurocritical care should align with the recommendations outlined in the international sepsis guidelines [61], the post resuscitation care guidelines [40] and the neurocritical care guidelines for brain injury [62]. Proposed targets are described in Table 2.
Antibiotics
Three distinct phenotypes of antibiotic-associated encephalopathy have been identified: First, an acute-onset encephalopathy commonly accompanied by clinical seizure (mostly stereotyped clonus or myoclonus) or non-epileptic myoclonus, typically manifesting within days of antibiotic administration (commonly associated with cephalosporins and penicillin); second, an encephalopathy characterized by psychosis that arises within days of antibiotic administration (commonly associated with quinolones, macrolides, and procaine penicillin); and third, a subacute encephalopathy linked to cerebellar signs and MRI abnormalities that develop weeks after initiating antibiotic therapy (commonly associated with metronidazole) [70]. The most frequently observed EEG abnormalities include non-specific signs of encephalopathy, such as diffuse slowing, and generalized PDs displaying triphasic morphology.
Cefepime remains the most frequently reported molecule associated with neurologic events, with renal dysfunction being the primary risk factor for cefepime-induced neurotoxicity [71,72,73]. The median time for the development of neurotoxicity after initiating cefepime is 4 days. Patients commonly present with altered mental status (93%), myoclonus (37%), and/or non-convulsive seizures (28%). If neurotoxicity is suspected, serum cefepime concentration should be monitored and EEG should be systematically performed to rule out ESz, and to assess for PDs/triphasic waves. A trough serum cefepime level exceeding 20 mg/L increases the risk of neurotoxicity. Symptoms usually improve with dose reduction or discontinuation of cefepime, with a median time to improvement of 3 days. In a retrospective study, neurotoxic side effects were not observed when the trough concentration of cefepime was below 7.7 mg/L [72]. In contrast, neurologic adverse events were always present when levels exceeded 38.1 mg/L.
Long term outcomes and recovery
Although most studies on long term outcomes have focused on the general ICU population, current data suggest that sepsis survivors experience a wide range of cognitive, psychiatric, physical, and social impairment after ICU discharge [74]. A general description is provided in Fig. 4. In a secondary analysis of international randomized trials, one third of adults with sepsis had died after six months and one third were no longer able to perform daily living activities [75].

Complications associated with sepsis-associated encephalopathy at the acute phase and in the long term
Cognitive impairment
Inflammatory processes are thought to participate in early brain alterations but also in long-term cognitive impairment [76]. In a prospective cohort of patients followed during nine years, sepsis episodes were associated with an increased risk of developing dementia [77]. A multicenter prospective cohort study found that a longer duration of sepsis-associated delirium was associated with altered cognitive function at 3 and 12 months [10]. Moreover, hippocampal atrophy has been described in sepsis survivors with cognitive impairment [33]. A systematic review found CNS infection, length of hospitalization and depressive symptoms to be risk factors for post-sepsis cognitive impairment [78], while data from a large randomized controlled trial highlighted older age, longer ICU stay and mechanical ventilation to be associated with a higher risk of cognitive alterations [79]. There is controversial evidence from general critically ill and septic patients that blood biomarkers could predict cognitive impairment. In general ICU patients, IL-6 and IL-10 levels were associated with poorer cognitive performances after ICU discharge [80], but acute phase plasmatic inflammation and coagulation markers did not appear to be good predictors of cognitive dysfunction [81]. One report highlighted the association of higher E-selectin and S100B levels with worse global cognition at 3 and 12 months after respiratory failure or shock [82]. High serum levels of NSE and interferon-γ were associated with poor cognitive performance after ICU discharge [78].
Seizures and epilepsy
Sepsis survivors face a higher long-term risk of seizures than other hospitalized patients. In a large cohort, the annual incidence of seizure after sepsis was 1.29%, with incidence rate ratios of 4.98 and 4.33, compared to the general population and hospitalized patients without sepsis, respectively [83]. Among sepsis survivors, younger patients and those with chronic kidney disease appear to be at higher risk of epilepsy [84]. Taken together, these findings suggest may be an unrecognized epilepsy risk factor leading to permanent neurologic sequelae.
Neuropsychological consequences
Sepsis survivors experience long-term emotional and behavioral changes, including depressive symptoms, anxiety and post-traumatic stress disorder (PTSD) [2]. A study of sepsis survivors found that 12% of patients had PTSD at 6 months after ICU discharge, often with delayed onset [85]. After ICU discharge, the severity of depressive symptoms was found to be associated with chronic pain or post-traumatic stress [86]. Patients were also found to experience anxiety, fatigue, and sleep disturbance. Providing primary care interventions for 12 months to sepsis survivors after ICU discharge reduced PTSD symptoms, but did not improve psychic quality of life compared to usual care [87].
Functional disability
Compared to mechanically ventilated patients of similar acuity and length of stay without sepsis, patients with sepsis have an increased risk of mortality and a similar risk of new disability at 6 months [88]. Critically ill patients with sepsis have higher healthcare resource use and costs but similar survival and health-related quality of life compared to matched patients without sepsis [89].
ICU-acquired weakness is another frequent complication associated with sepsis resulting from alterations of small nerve fibers [90]. Typical presentation include fatigue, muscle weakness, during ICU stay or after discharge. ICU-acquired weakness likely represents an additional indicator of long term morbidity and mortality [91].
Conclusion
SAE is a complex condition that requires a multidisciplinary approach for its diagnosis and management. Timely identification and treatment of the underlying infection are paramount, along with effective control of systemic factors that may contribute to secondary brain injury. Upon admission to the ICU, maintaining appropriate levels of oxygenation, blood pressure, and metabolic balance is crucial. Throughout the ICU stay, it is important to be mindful of the potential neurotoxic effects associated with specific medications like midazolam and cefepime, and to closely monitor patients for non-convulsive seizures. A multimodal approach based on clinical evaluation, neuroimaging and bedside available non-invasive tools may have important prognostic implications both at the acute phase and in the long term. The potential efficacy of targeted neurocritical care during the acute phase in optimizing patient outcomes deserves to be further investigated.
SAE may lead to permanent neurologic sequelae. Seizures occurring in the acute phase increase the susceptibility to long-term epilepsy. Extended ICU stays and the presence of sepsis-associated encephalopathy are linked to functional disability and neuropsychological sequelae, underscoring the necessity for long-term surveillance in the comprehensive care of septic patients.
Availability of data and materials
Not applicable.
Abbreviations
- BAEP:
-
Brainstem auditory evoked potentials
- BBB:
-
Blood brain barrier
- CT:
-
Computed tomography
- DWI:
-
Diffusion weighted imaging
- EEG:
-
Electroencephalogram
- EPs:
-
Evoked potentials
- ERPs:
-
Event related potentials
- ESz:
-
Electrographic seizures
- GFAP:
-
Glial fibrillary acidic protein
- PDs:
-
Periodic discharges
- ICU:
-
Intensive care unit
- ICCT:
-
Intracranial conduction time
- IPCT:
-
Intrapontine conduction time
- MMN:
-
Mismatch negativity
- MRI:
-
Magnetic resonance imaging
- Nfl:
-
Neuro-filament light
- NSE:
-
Neuron serum enolase
- PS100:
-
Protein S100
- PTSD:
-
Post traumatic stress disorder
- SAE:
-
Sepsis-associated encephalopathy
- WML:
-
White matter lesions
References
Slooter AJC, Otte WM, Devlin JW, Arora RC, Bleck TP, Claassen J, et al. Updated nomenclature of delirium and acute encephalopathy: statement of ten Societies. Intensive Care Med. 2020;46:1020–2.
Heming N, Mazeraud A, Verdonk F, Bozza FA, Chrétien F, Sharshar T. Neuroanatomy of sepsis-associated encephalopathy. Crit Care. 2017;21:65.
Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Françoise G. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33.
Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–94.
Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–66.
Bleck TP, Smith MC, Pierre-Louis SJ, Jares JJ, Murray J, Hansen CA. Neurologic complications of critical medical illnesses. Crit Care Med. 1993;21:98–103.
Eidelman LA, Putterman D, Putterman C, Sprung CL. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996;275:470–3.
Sonneville R, de Montmollin E, Poujade J, Garrouste-Orgeas M, Souweine B, Darmon M, et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 2017;43:1075–84.
Lu X, Qin M, Walline JH, Gao Y, Yu S, Ge Z, et al. Clinical phenotypes of sepsis-associated encephalopathy: a retrospective cohort study. Shock. 2023;59:583–90.
Girard TD, Thompson JL, Pandharipande PP, Brummel NE, Jackson JC, Patel MB, et al. Clinical phenotypes of delirium during critical illness and severity of subsequent long-term cognitive impairment: a prospective cohort study. Lancet Respir Med. 2018;6:213–22.
Chen J, Shi X, Diao M, Jin G, Zhu Y, Hu W, et al. A retrospective study of sepsis-associated encephalopathy: epidemiology, clinical features and adverse outcomes. BMC Emerg Med. 2020;20:77.
Jin G, Wang S, Chen J, Hu W, Zhu Y, Xi S. Identification of sepsis-associated encephalopathy risk factors in elderly patients: a retrospective observational cohort study. Turk J Med Sci. 2022;52:1513–22.
Gusmao-Flores D, Salluh JIF, Chalhub RÁ, Quarantini LC. The confusion assessment method for the intensive care unit (CAM-ICU) and intensive care delirium screening checklist (ICDSC) for the diagnosis of delirium: a systematic review and meta-analysis of clinical studies. Crit Care. 2012;16:R115.
Acharya R, Kafle S, Shrestha DB, Sedhai YR, Ghimire M, Khanal K, et al. Use of computed tomography of the head in patients with acute atraumatic altered mental status: a systematic review and meta-analysis. JAMA Netw Open. 2022;5:e2242805.
Chokshi FH, Sadigh G, Carpenter W, Kang J, Duszak R, Khosa F. Altered mental status in ICU patients: diagnostic yield of noncontrast head CT for abnormal and communicable findings. Crit Care Med. 2016;44:e1180–5.
Esquevin A, Raoult H, Ferré J-C, Ronzière T, Stamm A, Perennes M, et al. Systematic combined noncontrast CT–CT angiography in the management of unexplained nontraumatic coma. Am J Emerg Med. 2013;31:494–8.
Orhun G, Esen F, Özcan PE, Sencer S, Bilgiç B, Ulusoy C, et al. Neuroimaging findings in sepsis-induced brain dysfunction: association with clinical and laboratory findings. Neurocrit Care. 2019;30:106–17.
Orhun G, Tüzün E, Bilgiç B, Ergin Özcan P, Sencer S, Barburoğlu M, et al. Brain volume changes in patients with acute brain dysfunction due to sepsis. Neurocrit Care. 2020;32:459–68.
Ehler J, Petzold A, Wittstock M, Kolbaske S, Gloger M, Henschel J, et al. The prognostic value of neurofilament levels in patients with sepsis-associated encephalopathy: a prospective, pilot observational study. PLoS ONE. 2019;14:e0211184.
Polito A, Eischwald F, Maho A-L, Polito A, Azabou E, Annane D, et al. Pattern of brain injury in the acute setting of human septic shock. Crit Care. 2013;17:R204.
Orhun G, Sencer S, Tüzün E, Bebek N, Ergin Özcan P, Barburoğlu M, et al. Posterior reversible encephalopathy in sepsis-associated encephalopathy: experience from a single center. Neurocrit Care. 2022;36:372–86.
Yuan M, Yan D-Y, Xu F-S, Zhao Y, Zhou Y, Pan L-F. Effects of sepsis on hippocampal volume and memory function. World J Emerg Med. 2020;11:223–30.
Morandi A, Rogers BP, Gunther ML, Merkle K, Pandharipande P, Girard TD, et al. The relationship between delirium duration, white matter integrity, and cognitive impairment in intensive care unit survivors as determined by diffusion tensor imaging: the VISIONS prospective cohort magnetic resonance imaging study*. Crit Care Med. 2012;40:2182–9.
Legouy C, Girard-Stein L, Wanono R, de Montmollin E, Vellieux G, Bouadma L, et al. Association of standard electroencephalography findings with mortality and command following in mechanically ventilated patients remaining unresponsive after sedation interruption. Crit Care Med. 2021;49:e423–32.
Yppärilä H, Nunes S, Korhonen I, Partanen J, Ruokonen E. The effect of interruption to propofol sedation on auditory event-related potentials and electroencephalogram in intensive care patients. Crit Care. 2004;8:R483-490.
Sutter R, Kaplan PW, Valença M, De Marchis GM. EEG for diagnosis and prognosis of acute nonhypoxic encephalopathy: history and current evidence. J Clin Neurophysiol. 2015;32:456–64.
Hirsch LJ, Fong MWK, Leitinger M, LaRoche SM, Beniczky S, Abend NS, et al. American Clinical Neurophysiology Society’s standardized critical care EEG terminology: 2021 version. J Clin Neurophysiol. 2021;38:1–29.
Velissaris D, Pantzaris N-D, Skroumpelou A, Polychronopoulos P, Karamouzos V, Pierrakos C, et al. Electroencephalographic abnormalities in sepsis patients in correlation to the calculated prognostic scores: a case series. J Transl Int Med. 2018;6:176–80.
Pantzaris N-D, Platanaki C, Tsiotsios K, Koniari I, Velissaris D. The use of electroencephalography in patients with sepsis: a review of the literature. J Transl Int Med. 2021;9:12–6.
Hosokawa K, Gaspard N, Su F, Oddo M, Vincent J-L, Taccone FS. Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Crit Care. 2014;18:674.
Azabou E, Magalhaes E, Braconnier A, Yahiaoui L, Moneger G, Heming N, et al. Early standard electroencephalogram abnormalities predict mortality in septic intensive care unit patients. PLoS ONE. 2015;10:e0139969.
Berisavac II, Padjen VV, Ercegovac MD, Beslać-Bumbaširević LG, Stanarčević PD, Stefanović-Budimkić MS, et al. Focal epileptic seizures, electroencephalography and outcome of sepsis associated encephalopathy: a pilot study. Clin Neurol Neurosurg. 2016;148:60–6.
Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84:62–9.
Ferlini L, Maenhout C, Crippa IA, Quispe-Cornejo AA, Creteur J, Taccone FS, et al. The association between the presence and burden of periodic discharges and outcome in septic patients: an observational prospective study. Crit Care. 2023;27:179.
Rossetti AO, Schindler K, Sutter R, Rüegg S, Zubler F, Novy J, et al. Continuous vs routine electroencephalogram in critically Ill adults with altered consciousness and no recent seizure: a multicenter randomized clinical trial. JAMA Neurol. 2020;77:1225–32.
Gilmore EJ, Gaspard N, Choi HA, Cohen E, Burkart KM, Chong DH, et al. Acute brain failure in severe sepsis: a prospective study in the medical intensive care unit utilizing continuous EEG monitoring. Intensive Care Med. 2015;41:686–94.
Benghanem S, Cariou A, Diehl J-L, Marchi A, Charpentier J, Augy J-L, et al. Early clinical and electrophysiological brain dysfunction is associated with ICU outcomes in COVID-19 critically Ill patients with acute respiratory distress syndrome: a prospective bicentric observational study. Crit Care Med. 2022;50:1103–15.
Nielsen RM, Urdanibia-Centelles O, Vedel-Larsen E, Thomsen KJ, Møller K, Olsen KS, et al. Continuous EEG monitoring in a consecutive patient cohort with sepsis and delirium. Neurocrit Care. 2020;32:121–30.
André-Obadia N, Zyss J, Gavaret M, Lefaucheur J-P, Azabou E, Boulogne S, et al. Recommendations for the use of electroencephalography and evoked potentials in comatose patients. Neurophysiol Clin. 2018;48:143–69.
Nolan JP, Sandroni C, Böttiger BW, Cariou A, Cronberg T, Friberg H, et al. European Resuscitation Council and European Society of Intensive Care Medicine guidelines 2021: post-resuscitation care. Intensive Care Med. 2021;47:369–421.
Benghanem S, Pruvost-Robieux E, Bouchereau E, Gavaret M, Cariou A. Prognostication after cardiac arrest: how EEG and evoked potentials may improve the challenge. Ann Intensive Care. 2022;12:111.
Zauner C, Gendo A, Kramer L, Funk GC, Bauer E, Schenk P, et al. Impaired subcortical and cortical sensory evoked potential pathways in septic patients. Crit Care Med. 2002;30:1136–9.
Azabou E, Rohaut B, Heming N, Magalhaes E, Morizot-Koutlidis R, Kandelman S, et al. Early impairment of intracranial conduction time predicts mortality in deeply sedated critically ill patients: a prospective observational pilot study. Ann Intensive Care. 2017;7:63.
Azabou E, Rohaut B, Porcher R, Heming N, Kandelman S, Allary J, et al. Mismatch negativity to predict subsequent awakening in deeply sedated critically ill patients. Br J Anaesth. 2018;121:1290–7.
Anderson BJ, Reilly JP, Shashaty MGS, Palakshappa JA, Wysoczanski A, Dunn TG, et al. Admission plasma levels of the neuronal injury marker neuron-specific enolase are associated with mortality and delirium in sepsis. J Crit Care. 2016;36:18–23.
Nguyen DN, Spapen H, Su F, Schiettecatte J, Shi L, Hachimi-Idrissi S, et al. Elevated serum levels of S-100beta protein and neuron-specific enolase are associated with brain injury in patients with severe sepsis and septic shock. Crit Care Med. 2006;34:1967–74.
Zhang L-T, Xu X, Han H, Cao S-M, Li L-L, Lv J, et al. The value of NSE to predict ICU mortality in patients with septic shock: a prospective observational study. Medicine (Baltimore). 2022;101:e30941.
Yao B, Zhang L-N, Ai Y-H, Liu Z-Y, Huang L. Serum S100β is a better biomarker than neuron-specific enolase for sepsis-associated encephalopathy and determining its prognosis: a prospective and observational study. Neurochem Res. 2014;39:1263–9.
Piazza O, Russo E, Cotena S, Esposito G, Tufano R. Elevated S100B levels do not correlate with the severity of encephalopathy during sepsis. Br J Anaesth. 2007;99:518–21.
van den Boogaard M, Kox M, Quinn KL, van Achterberg T, van der Hoeven JG, Schoonhoven L, et al. Biomarkers associated with delirium in critically ill patients and their relation with long-term subjective cognitive dysfunction; indications for different pathways governing delirium in inflamed and noninflamed patients. Crit Care. 2011;15:R297.
Wu L, Ai M-L, Feng Q, Deng S, Liu Z-Y, Zhang L-N, et al. Serum glial fibrillary acidic protein and ubiquitin C-terminal hydrolase-L1 for diagnosis of sepsis-associated encephalopathy and outcome prognostication. J Crit Care. 2019;52:172–9.
Osca-Verdegal R, Beltrán-García J, Pallardó FV, García-Giménez JL. Role of microRNAs as biomarkers in sepsis-associated encephalopathy. Mol Neurobiol. 2021;58:4682–93.
Tomasi CD, Vuolo F, Generoso J, Soares M, Barichello T, Quevedo J, et al. Biomarkers of delirium in a low-risk community-acquired pneumonia-induced sepsis. Mol Neurobiol. 2017;54:722–6.
Barnes-Daly MA, Phillips G, Ely EW. Improving hospital survival and reducing brain dysfunction at seven California Community Hospitals: implementing PAD guidelines via the ABCDEF bundle in 6064 patients. Crit Care Med. 2017;45:171–8.
Kawazoe Y, Miyamoto K, Morimoto T, Yamamoto T, Fuke A, Hashimoto A, et al. Effect of dexmedetomidine on mortality and ventilator-free days in patients requiring mechanical ventilation with sepsis: a randomized clinical trial. JAMA. 2017;317:1321–8.
Hughes CG, Mailloux PT, Devlin JW, Swan JT, Sanders RD, Anzueto A, et al. Dexmedetomidine or propofol for sedation in mechanically ventilated adults with sepsis. N Engl J Med. 2021;384:1424–36.
Pandharipande P, Shintani A, Peterson J, Pun BT, Wilkinson GR, Dittus RS, et al. Lorazepam is an independent risk factor for transitioning to delirium in intensive care unit patients. Anesthesiology. 2006;104:21–6.
Zaal IJ, Devlin JW, Hazelbag M, Klein Klouwenberg PMC, van der Kooi AW, Ong DSY, et al. Benzodiazepine-associated delirium in critically ill adults. Intensive Care Med. 2015;41:2130–7.
Singh TD, O’Horo JC, Day CN, Mandrekar J, Rabinstein AA. Cefepime is associated with acute encephalopathy in critically Ill patients: a retrospective case-control study. Neurocrit Care. 2020;33:695–700.
Abdul-Aziz MH, Alffenaar J-WC, Bassetti M, Bracht H, Dimopoulos G, Marriott D, et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: a position paper. Intensive Care Med. 2020;46:1127–53.
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021;47:1181–247.
Cook AM, Morgan Jones G, Hawryluk GWJ, Mailloux P, McLaughlin D, Papangelou A, et al. Guidelines for the acute treatment of cerebral edema in neurocritical care patients. Neurocrit Care. 2020;32:647–66.
Asfar P, Meziani F, Hamel J-F, Grelon F, Megarbane B, Anguel N, et al. High versus low blood-pressure target in patients with septic shock. N Engl J Med. 2014;370:1583–93.
Jouan Y, Seegers V, Meziani F, Grelon F, Megarbane B, Anguel N, et al. Effects of mean arterial pressure on arousal in sedated ventilated patients with septic shock: a SEPSISPAM post hoc exploratory study. Ann Intensive Care. 2019;9:54.
Asfar P, Schortgen F, Boisramé-Helms J, Charpentier J, Guérot E, Megarbane B, et al. Hyperoxia and hypertonic saline in patients with septic shock (HYPERS2S): a two-by-two factorial, multicentre, randomised, clinical trial. Lancet Respir Med. 2017;5:180–90.
Schortgen F, Clabault K, Katsahian S, Devaquet J, Mercat A, Deye N, et al. Fever control using external cooling in septic shock: a randomized controlled trial. Am J Respir Crit Care Med. 2012;185:1088–95.
Schortgen F, Charles-Nelson A, Bouadma L, Bizouard G, Brochard L, Katsahian S. Respective impact of lowering body temperature and heart rate on mortality in septic shock: mediation analysis of a randomized trial. Intensive Care Med. 2015;41:1800–8.
Dupuis C, Sonneville R, Adrie C, Gros A, Darmon M, Bouadma L, et al. Impact of transfusion on patients with sepsis admitted in intensive care unit: a systematic review and meta-analysis. Ann Intensive Care. 2017;7:5.
Hirano Y, Miyoshi Y, Kondo Y, Okamoto K, Tanaka H. Liberal versus restrictive red blood cell transfusion strategy in sepsis or septic shock: a systematic review and meta-analysis of randomized trials. Crit Care. 2019;23:262.
Bhattacharyya S, Darby RR, Raibagkar P, Gonzalez Castro LN, Berkowitz AL. Antibiotic-associated encephalopathy. Neurology. 2016;86:963–71.
Payne LE, Gagnon DJ, Riker RR, Seder DB, Glisic EK, Morris JG, et al. Cefepime-induced neurotoxicity: a systematic review. Crit Care. 2017;21:276.
Boschung-Pasquier L, Atkinson A, Kastner LK, Banholzer S, Haschke M, Buetti N, et al. Cefepime neurotoxicity: thresholds and risk factors: a retrospective cohort study. Clin Microbiol Infect. 2020;26:333–9.
Maan G, Keitoku K, Kimura N, Sawada H, Pham A, Yeo J, et al. Cefepime-induced neurotoxicity: systematic review. J Antimicrob Chemother. 2022;77:2908–21.
Herridge MS, Azoulay É. Outcomes after critical illness. Hardin CC, editor. N Engl J Med. 2023;388:913–24.
Yende S, Austin S, Rhodes A, Finfer S, Opal S, Thompson T, et al. Long-term quality of life among survivors of severe sepsis: analyses of two international trials. Crit Care Med. 2016;44:1461–7.
Sharshar T, Bozza F, Chrétien F. Neuropathological processes in sepsis. Lancet Neurol. 2014;13:534–6.
Peters Van Ton AM, Meijer-van Leijsen EMC, Bergkamp MI, Bronkhorst EM, Pickkers P, De Leeuw F-E, et al. Risk of dementia and structural brain changes following nonneurological infections during 9-year follow-up*. Crit Care Med. 2022;50:554–64.
Calsavara AJC, Nobre V, Barichello T, Teixeira AL. Post-sepsis cognitive impairment and associated risk factors: a systematic review. Aust Crit Care. 2018;31:242–53.
Kosilek RP, Schmidt K, Baumeister SE, Gensichen J, SMOOTH Study Group. Frequency and risk factors of post-intensive care syndrome components in a multicenter randomized controlled trial of German sepsis survivors. J Crit Care. 2021;65:268–73.
Maciel M, Benedet SR, Lunardelli EB, Delziovo H, Domingues RL, Vuolo F, et al. Predicting long-term cognitive dysfunction in survivors of critical illness with plasma inflammatory markers: a retrospective cohort study. Mol Neurobiol. 2019;56:763–7.
Brummel NE, Hughes CG, Thompson JL, Jackson JC, Pandharipande P, McNeil JB, et al. Inflammation and coagulation during critical illness and long-term cognitive impairment and disability. Am J Respir Crit Care Med. 2021;203:699–706.
Hughes CG, Patel MB, Brummel NE, Thompson JL, McNeil JB, Pandharipande PP, et al. Relationships between markers of neurologic and endothelial injury during critical illness and long-term cognitive impairment and disability. Intensive Care Med. 2018;44:345–55.
Reznik ME, Merkler AE, Mahta A, Murthy SB, Claassen J, Kamel H. Long-term risk of seizures in adult survivors of sepsis. Neurology. 2017;89:1476–82.
Antaya TC, Allen BN, Richard L, Shariff SZ, Saposnik G, Burneo JG. Epilepsy risk among survivors of intensive care unit hospitalization for sepsis. Neurology. 2020;95:e2271–9.
Wintermann G-B, Brunkhorst FM, Petrowski K, Strauss B, Oehmichen F, Pohl M, et al. Stress disorders following prolonged critical illness in survivors of severe sepsis. Crit Care Med. 2015;43:1213–22.
Boede M, Gensichen JS, Jackson JC, Eißler F, Lehmann T, Schulz S, et al. Trajectories of depression in sepsis survivors: an observational cohort study. Crit Care. 2021;25:161.
Schmidt KF, Schwarzkopf D, Baldwin L-M, Brunkhorst FM, Freytag A, Heintze C, et al. Long-term courses of sepsis survivors: effects of a primary care management intervention. Am J Med. 2020;133:381-385.e5.
Hodgson CL, Higgins AM, Bailey M, Barrett J, Bellomo R, Cooper DJ, et al. Comparison of 6-month outcomes of sepsis versus non-sepsis critically ill patients receiving mechanical ventilation. Crit Care. 2022;26:174.
Thompson K, Taylor C, Jan S, Li Q, Hammond N, Myburgh J, et al. Health-related outcomes of critically ill patients with and without sepsis. Intensive Care Med. 2018;44:1249–57.
Axer H, Grimm A, Pausch C, Teschner U, Zinke J, Eisenach S, et al. The impairment of small nerve fibers in severe sepsis and septic shock. Crit Care. 2016;20:64.
Van Aerde N, Meersseman P, Debaveye Y, Wilmer A, Gunst J, Casaer MP, et al. Five-year impact of ICU-acquired neuromuscular complications: a prospective, observational study. Intensive Care Med. 2020;46:1184–93.
Funding
None.
Author information
Authors and Affiliations
Contributions
Manuscript conception and design: RS. literature search: All authors. Analysis and interpretation: RS, SB, LJ, EDM, MD, AG, MT, JFT. Drafting: RS, SB, LJ, EDM, MD, AG, MT. Revision for important intellectual content: all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
R. Sonneville reports grants from the French Ministry of Health and LFB, outside the submitted work. Other authors report no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sonneville, R., Benghanem, S., Jeantin, L. et al. The spectrum of sepsis-associated encephalopathy: a clinical perspective. Crit Care 27, 386 (2023). https://doi.org/10.1186/s13054-023-04655-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-023-04655-8