
Abstract
Mucopolysaccharidoses (MPS) are rare inherited disorders caused by a deficit of the lysosomal hydrolases involved in the degradation of mucopolysaccharides, also known as glycosaminoglycans (GAGs). They are all monogenic defects, transmitted in an autosomal recessive way, except for MPS type II which is X-linked. The enzymatic deficit causes a pathologic accumulation of undegraded or partially degraded substrates inside lysosomes as well as in the extracellular compartment. MPS generally present with recognizable signs and symptoms to raise a clinical suspicion. However, although they have individual peculiarities, often signs and symptoms may overlap between different MPS types. Therefore, a deeper evaluation of specific disease biomarkers becomes necessary to reach an appropriate diagnosis. This paper stresses the central role of the laboratory in completing and confirming the clinical suspicion of MPS according to a standardized procedure: first, a biochemical evaluation of the patient samples, including qualitative/quantitative urinary GAG analysis and a determination of enzyme activities, and then the molecular diagnosis. We also encourage a constant and close communication between clinicians and laboratory personnel to address a correct and early MPS diagnosis.
Similar content being viewed by others


Background
The degradation of the glycosaminoglycans (GAGs or mucopolysaccharides), a major component of the extracellular matrix, joint fluid, and connective tissue, takes place in the lysosomes. Under physiological conditions, the main GAG chains — dermatan sulphate (DS), heparan sulphate (HS), keratan sulphate (KS), and chondroitin sulphate (CS) — are degraded by 11 lysosomal hydrolases through the sequential removal of monosaccharides followed by the removal of sulphate groups, resulting in the complete degradation of the polysaccharide to its individual components.
The deficit of any one of the 11 acid hydrolase activities gives rise to the progressive accumulation of GAGs in most tissues and organ systems, as well as in urine. Figure 1 illustrates the stepwise degradation of the main GAG chains by specific enzymes, as well as the resulting 11 distinct types of mucopolysaccharidoses (MPS) depending on the enzyme deficiency. Table 1 summarises the various enzyme defects and the type(s) of accumulated GAGs for each specific type/subtype of MPS. The 11 genes involved in the different subgroups of MPS have already been identified and characterized (Table 2), as well as the hereditary transmission model: all MPS have an autosomal recessive transmission with the exception of MPS II, which exhibits an X-linked pattern.

Stepwise degradation of the main glycosaminoglycan chains, heparan sulphate (HS), dermatan sulphate (DS), and keratan sulphate (KS). The enzymes involved in the pathway are shown in black. Defective enzyme activity leading to the different types of mucopolysaccharidosis (MPS) is indicated in blue. Note that the degradation pathway of chondroitin sulphates, being similar to that of DS, is not shown
Laboratory diagnosis
Biochemical assay
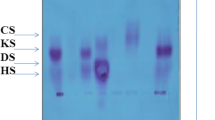
Preliminary diagnostic analyses range from semiquantitative spot tests [1] to quantitative assays [2, 3] followed by qualitative identification [4] of increased urinary GAGs.
It should be noted that none of these methods can be considered diagnostic per se since false negative/positive results are sometimes obtained. A relatively high incidence of false negative urinary screening has been reported for HS in MPS III patients with mild/intermediate clinical phenotypes [5] (Filocamo, personal data). KS does not form a reaction product with any of these routinely used methods; hence, quantitative GAG assessment in Morquio syndrome (MPS IV) is unreliable. An enzyme-linked immunosorbent assay (ELISA) technique has been shown to quantify KS accurately in the urine and blood of patients with Morquio syndrome type A [6]. It is also known that several factors can interfere with the measurement of urinary GAGs and lead to false positive results. For instance, the presence of the anticoagulant heparin, and blood or haemoglobin, as well as several diseases other than MPS (leukaemia, rheumatic arthritis, diabetes, obesity, etc.), may also lead to an abnormally high GAG excretion in the urine [7, 8].
Moreover, assays using tandem mass spectrometry (MS/MS) have been established to measure GAG levels in serum or plasma and, more recently, on dried blood spot (DBS). MS/MS, allowing for the simultaneous measurement of several GAGs, provides a sensitive, specific, and reproducible GAG analysis making it potentially useful for the screening, prognosis, and monitoring of any therapeutic effect in MPS patients [9, 10].
Enzymatic assays
The first step in the diagnostic process includes specific enzymatic assays in a variety of cells. Leukocytes, cultured lymphoblasts, or fibroblasts are generally used, with the actual choice depending upon the characteristics of the enzyme to be assessed as well as the corresponding method of detection. Enzyme activities are generally determined by fluorogenic (4-methylumbelliferyl) or, rarely, chromogenic (p-nitrophenyl) substrates.
Dried blood spot (DBS)
DBS testing is a good option for new-born screening (NBS) for various lysosomal storage disorders including some MPS [11,12,13].
Although this approach is helpful for certain disorders, positive DBS results need to be validated by conventional testing, in other words enzymatic assay in cellular extracts and/or molecular analysis. It is important for clinicians to understand how to interpret results from DBS screening as in the near future it is likely that NBS programmes will include DBS tests. For comprehensive reviews on this topic the reader can consult a relevant chapter in this same supplement.
Pseudodeficiency
Generally, a deficient activity of one of the 11 lysosomal enzymes involved in GAG degradation is associated with an MPS. However, there are individuals who show greatly reduced enzymatic activity but remain clinically healthy. This condition, termed enzymatic pseudodeficiency (Pd), is due to polymorphic genetic variants and can affect some of these lysosomal hydrolases. The potential presence of an enzymatic Pd poses a limitation for enzymatic tests and should be investigated whenever the results from an enzymatic assay do not concur with the clinical phenotype of the patient. The currently known enzymatic pseudodeficiencies are reported below, on a disease-by-disease basis.
Molecular analysis
Molecular testing, available for all MPS, follows the enzymatic diagnosis and serves to refine the diagnosis; it can also be extremely helpful in facilitating familial genetic counselling. In fact, once the genotype of an individual affected patient has been ascertained, genetic counselling should address the possible prediction of the potential clinical phenotype, indications for the most appropriate therapy, if any, and the prospect for identification of any at-risk carriers in the family.
Figure 2 graphically represents the conventional diagnostic flowchart.

Diagnostic flow chart for mucopolysaccharidoses (MPS). The diagnosis of the MPS can be performed starting with a screening of the urine followed by quantitative assay of urinary glycosaminoglycans (GAGs) and, if available, by qualitative evaluation of the GAGs accumulated that can steer the enzymatic assay. Once the enzymatic defect has been determined, the molecular analysis will serve to identify the causative genomic variant. Note that keratan sulphate (KS) does not form a reaction product with any of the routinely methods reported here; hence, quantitative GAG assessment in Morquio syndrome (MPS IV) requires other techniques or the two enzymatic assays. Differential laboratory diagnosis includes multiple sulphatase deficiency and mucolipidosis II and III, in which GAGs accumulate because of their primitive defect involving some of the enzymes of the GAG degradation pathway. HS heparan sulphate, DS dermatan sulphate
Next-generation sequencing
The use of massively parallel sequencing analysis, known as next-generation sequencing (NGS), is becoming increasingly common as a fast and low-cost sequencing approach. It includes whole-genome sequencing (WGS), whole-exome sequencing (WES), and the so-called “targeted resequencing approach” with focused “gene panels” which are designed to include genomic regions known to underlie diseases grouped by clinical affinity. In the near future, it is possible that gene analysis using NGS techniques will replace preliminary tests based on enzyme activities or metabolite concentrations. This would offer several advantages, mainly in terms of speeding up the diagnosis in patients where a differential analysis is difficult to perform.
However, the NGS-based technology is not without limitations. It should be taken into consideration that this technology is “error-prone” and that the presence of variants of unknown significance (VUS) can make data analysis extremely complicated [14, 15]. Nor should we overlook the ethical issue that could arise because of potential incidental findings obtained from NGS-based technology.
Finally, it should be stressed that, for orphan diseases, it is often difficult to build a base of molecular knowledge as mutations tend to be private and the molecular classification of each novel variant necessarily implies additional laboratory investigations such as functional studies based on enzyme and metabolite assays, which are both cumbersome and costly. Without such a validation, it is hard to decide whether a new variant is disease-causing or benign and would risk falling into the VUS category.
Genetic counselling
All MPS are inherited as autosomal recessive disorders, except MPS II which is X-linked and therefore only affects males except in extremely rare cases. Once a definitive diagnosis is achieved, genetic counselling can be offered to patients and their families. Such counselling should explain the mode of inheritance and the importance of testing at-risk family members, it should yield an estimate of recurrence risks, and provide information about prenatal genetic tests and available therapeutic approaches.
Prenatal diagnosis is performed on the most appropriate samples, which usually include fresh or cultured chorionic villus samples obtained by villocentesis, usually performed at approximately 12 weeks gestation, or cultured amniotic fluid obtained via amniocentesis at approximately 16 weeks gestation. The choice of the test, enzymatic and/or molecular, is based on the characteristics of the defect to be investigated. Prenatal molecular testing requires the genotype of the family index case to be known. The availability of both tests (enzymatic and molecular) enormously increases the reliability of the diagnosis. Hence, biochemical and molecular genetic testing are usually complementary for the prenatal diagnosis of MPS. However, special considerations are required for the prenatal diagnosis of Hunter syndrome, the only X-linked MPS. Since in a female carrier a skewed distribution of the cells expressing the mutant allele might yield results resembling those of an affected male fetus, the usual procedure is to determine fetal sex and only if the karyotype is 46,XY would fetal cells be biochemically analysed antenatally.
Biobanking
Since our understanding of genes, mutations, and diseases is continually improving, consideration should be given to biobanking samples and data from affected individuals, as well as from their parents and first-degree relatives, for future diagnostic and research purposes [16].
More comprehensive information on diagnosis and management of individual MPS is provided in some recently published guidelines [17,18,19,20,21,22].
Molecular genetics: Trying to understand the genotype-phenotype relationship
The elucidation of the molecular mechanisms underlying the MPS began in the 1990s. The process of cloning the diverse genes encoding the lysosomal enzymes involved in the GAG degradative pathways lasted until 2006 with the identification of the gene responsible for MPS IIIC. Although the large variety of mutations responsible for any MPS has often hampered the establishment of clear genotype–phenotype correlations, some general conclusions can be drawn in relation to recurrent mutations and are usually based on prior observations in large series of patients. However, even in the presence of a recurrent mutation, some patients carrying the same molecular lesion may present with different clinical phenotypes, suggesting that other variants at other gene loci and/or some environmental factors can modulate the clinical phenotype. This may be particularly important in the case of parents’ consanguinity, which is quite common in some ethnic groups due to geographical reasons or to the practice of arranged marriages [23,24,25].
Table 2 summarises the characteristics and types of mutations of the 11 genes responsible for the different MPS, as reported in the Human Gene Mutation Database [26].
MPS I
The gene encoding α-l-iduronidase (IDUA; MIM #252800) maps to chromosome 4p16.3 and contains 14 exons encoding for a polypeptide of 653 amino acids [27]. A deficiency of this enzyme results in the lack of degradation of DS and HS and their progressive accumulation. To date, at least 257 IDUA mutations are known, spread along the entire coding and splicing regions (Table 2). MPS I is a highly heterogeneous disorder with a wide spectrum of clinical manifestations [28, 29]. Overall, most mutations are ‘private’ with very few pan-ethnic mutations, the two most common being the p.Trp402X and p.Gln70X mutations found to be associated with a severe phenotype, and the p.Pro533Arg associated with an intermediate-severe phenotype; the p.Gly51Asp and p.Pro496Arg mutations have been found until now only in Italian patients, associated with a severe phenotype [30].
MPS I diagnosis can be hampered by the occurrence of α-l-iduronidase Pd. Although only the variant p.Ala300Thr has been reported so far as being associated with enzymatic Pd [31], other variants (p.Ala79Thr, p.His82Gln, p.Asp223Asn, and p.Val322Glu) are under investigation as candidates for Pd [32, 33]. Therefore, as NBS for MPS I has begun in some countries, it is imperative to follow-up a real deficient α-l-iduronidase result with urinary GAG analysis and IDUA gene sequencing prior to initiating treatment for MPS I.
MPS II
The iduronate 2-sulfatase gene (IDS; MIM #300823) contains nine exons and spans ~ 24 kb on Xq28. The 1650 bp open reading frame of the IDS gene is predicted to encode a 550-amino acid polypeptide [34]. The enzyme cleaves the sulphate group in position 2 of HS and DS; thus, its deficit results in the diffused pathological deposit of the both GAGs in most body districts. No cases with significant residual enzymatic activity have so far been described. A pseudogene, IDS2, presenting more than 88% homology with IDS exons 2 and 3, as well as introns 2, 3, and 7, is located 20 kb telomeric to the functional gene but in the opposite orientation [34]. The presence of the pseudogene renders molecular diagnosis more difficult to perform, since appropriate primers need to be designed to selectively avoid amplification of the homologous pseudogene sequences. To date, 628 different mutations in the IDS gene have been reported (Table 2). Among these, gross alterations including insertions, deletions, and gene-pseudogene recombinational rearrangements represent about 12% (76/628) of all known IDS gene lesions, 29% are small deletions, insertions and indels and 9% (59/628) are splicing mutations, while about 50% (313/628) are missense/nonsense variants. The de novo occurrence of IDS gene mutations has sometimes been demonstrated by family studies in large MPS II cohorts. Whilst germline mosaicism has been assumed in some cases, this has only rarely been formally demonstrated [35]. No high-frequency recurring mutations have been reported which has tended to constrain studies of the genotype-phenotype relationship. However, an approximate relationship may be based on the observation that, while missense variations may be associated with both severe and attenuated phenotypes, nonsense variants, splicing transcriptional defects, gross rearrangements, or deletions/insertions are more commonly associated with severe phenotypes. A recent study conducted on 65 Japanese families showed that missense variants were mainly associated with attenuated phenotypes (87.5%) and less frequently with severe phenotypes (29.3%) [36]. Additionally, identical mutations have been found in patients with both mild and severe disease, implying the possible contribution of other genetic or environmental modifiers on the phenotype [35, 37, 38]. Rare female MPS II patients have also been reported, mostly due to the non-random inactivation of the X chromosome (XCI) [27, 39,40,41,42]. Very rarely, a female MPS II patient may be due to a balanced reciprocal translocation involving one X chromosome, coupled with a skewed XCI of the normal X chromosome [43].
MPS IIIA
The HS sulphatase gene (SGSH; MIM #605270) spanning about 11 kb in length, comprises 8 exons. The most prominent of the three demonstrated transcripts (3.1, 4.3, and 7.1 kb) in most tissues, that of 3.1 kb, encodes a 502-amino acid sulphamidase protein [44]. The deficit of the heparan N-sulphatase enzyme results in the accumulation of the HS in organs and tissues. To date, of the 145 mutations reported in Table 2, only general conclusions have been drawn for p.Arg245His, p.Gln380Arg, p.Ser66Trp, and p.Val361SerfsX52 mutations, which have been found to be related to the development of a severe phenotype, while p.Gly122Arg, p.Arg206Pro, p.Ser298Pro, p.Ile322Ser, and p.Glu369Lys mutations have been reported as being associated with an attenuated phenotype [45]. Overall, no pan-ethnic recurrent mutations have been reported, although some mutations predominate in different ethnogeographic groups, reflecting possible founder effects. In particular, p.Arg74Cys and p.Val361SerfsX52 are frequent in Poland (56%) and Spain (45%), p.Arg245His in Germany (35%) and Holland (57%), and p.Ser66Trp in Italy (29%) [46,47,48].
MPS IIIB
The gene encoding α-N-acetyl-d-glucosaminidase (NAGLU; MIM #609701) is approximately 8.5 kb in length and comprises 6 exons. Its transcript of 2.7 kb encodes a protein of 743 amino acids [49, 50]. The defect of α-N-acetyl-d-glucosaminidase enzyme causes the accumulation of the HS in organs and tissues. A total of 166 NAGLU mutations have been described (Table 2). All the reported mutations occur at low frequencies, indicating an extensive NAGLU mutational heterogeneity which is likely to be responsible for the wide clinical spectrum of MPS IIIB. Some predictions have been made for p.Phe48Lys, p.Gly69Ser, p.Ser612Gly, and p.Arg643Cys mutations that appear to be related to a less severe clinical phenotype [51]. However, common mutations have not been identified in MPS IIIB patients.
A potential complication reported in MPS IIIB is the simultaneous presence of the two polymorphisms p.Ser141Ser and p.Arg737Gly producing a pseudodeficiency allele that leads to a reduced level of α-N-acetyl-d-glucosaminidase activity (Filocamo, personal data).
MPS IIIC
The heparin acetyl CoA:α-glucosaminide-N-acetyltransferase gene (HGSNAT; MIM #610453) contains 18 exons generating a 1908-base pair cDNA that encodes a 635-amino acid protein. The deficit of acetyl CoA:α-glucosaminide-acetyltransferase leads to the accumulation of HS in organs and tissues. To date, 68 mutations have been described for the HGSNAT gene (Table 2). Only two mutations (p.Arg344Cys and p.Ser518Phe) have been reported with a higher frequency in the Dutch population [52]. Despite the identification of several MPS IIIC-causing mutations, it has proven difficult to establish a clear genotype-phenotype correlation, except in a few cases such as mutations p.Gly262Arg and p.Ser539Cys from two patients that were associated with an attenuated phenotype [53, 54].
MPS IIID
The α-N-acetylglucosamine-6-sulphate sulphatase gene (GNS; MIM #607664) consists of 14 exons spanning approximately 46 kb of DNA; the predicted protein has 552 amino acids [55, 56]. The defect of N-acetylglucosamine-6-sulphatase causes the accumulation of HS in organs and tissues. Only 25, mostly private, disease-causing mutations in MPS IIID patients have been reported to date, thereby hampering genotype-phenotype correlation studies (Table 2).
MPS IVA
In MPS IVA, or Morquio A, the deficient activity of N-acetylgalactosamine-6-sulphatase (GALNS) causes accumulation of the GAGs KS and CS in multiple tissues with skeletal and connective tissue abnormalities [57].
The GALNS gene, encoding the lysosomal enzyme N-acetylgalactosamine-6-sulphatase (GALNS; E.C. 3.1.6.4; MIM #612222), maps to chromosome 16q24.3, has a length of about 50 kb, and is organized into 14 exons [58]. The GALNS cDNA is 2339 bp in length with a 1566-bp open reading frame encoding a 522-amino acid protein [58, 59]. The GALNS protein is found as a homodimer, but it has also been described in a multiprotein complex with other lysosomal enzymes [60].
To date, at least 333 different GALNS gene mutations causing Morquio A disease have been described (see the Human Gene Mutation Database [26]). Most of them are ‘private’, with only a few being common. Genotype-phenotype correlations have been reported for some of the recurrent mutations, representing founder alleles in certain population groups [61]. Thus, the p.Ile113Phe [62] and p.Thr312Ser missense variants recur in patients of British-Irish origin and a milder phenotype has been associated with the residual activity of the mutant p.Thr312Ser protein [63].
In the GALNS gene, large deletions and/or rearrangements and a uniparental disomy have also been reported [64,65,66]. Deep intronic mutations have also been detected (Morrone, personal data). So far, no pseudodeficiency has been described in the GALNS gene.
MPS IVB
The β-galactosidase gene (GLB1; E.C.3.2.1.23; MIM #611458) maps to chromosome 3p22.3, spans more than 60 kb, and is organized into 16 exons [67]. The GLB1 gene gives rise to two alternately spliced mRNAs: a transcript of 2.5 kb encoding the lysosomal enzyme β-galactosidase and a transcript of 2.0 kb encoding the elastin-binding protein (EBP). Both GLB1 transcripts form protein complexes. β-galactosidase lysosomal enzyme or GLB1 is stabilised in a lysosomal multienzyme complex with protective protein/cathepsin A (PPCA), neuraminidase (NEU1), and N-acetyl galactosamine-6-sulphatase (GALNS) [60]. EBP is required for the assembly of tropoelastin monomers into elastic fibres on the cell surface where it forms a complex with PPCA and NEU1 [60, 68, 69].
Mutations in the GLB1 gene are responsible for two allelic disorders: the neurodegenerative GM1 gangliosidosis (non-MPS) disorder (MIM 230500) and the rare MPS IVB (Morquio B) syndrome (MIM 253010) [70]. In contrast to GM1 gangliosidosis, patients with Morquio B retain neurological functions, but develop generalized skeletal dysplasia, keratan sulphaturia, and corneal clouding [71]. However, the clinical demarcation between GM1 and Morquio B at an early stage of diagnosis can be very difficult and obscured by a late-onset mental regression and by the presence of keratan sulphaturia in juvenile GM1 gangliosidosis forms.
The GLB1 mutations underlying Morquio B syndrome affect the catabolism of KS but have little effect on GM1 gangliosides.
To date, 215 GLB1 gene mutations have been reported (see the Human Gene Mutation Database [26]); among these, 22 were also identified in patients with Morquio B but only a few can be related to the specific phenotype [72,73,74]. The c.817_818del TG insCT leading to p.Trp273Leu is one of the most common mutations leading to a Morquio B phenotype [72].
Some GLB1 mutations have been identified in both GM1 and Morquio B, and the same genetic assessment has been shown in patients who exhibited different symptoms, further complicating possible prognoses in these individuals [72,73,74]. The common p.Arg201His detected at the heterozygous level has been correlated with the juvenile GM phenotype but it has also been detected in a 15-year-old Morquio B patient harbouring the mutation at the homozygous level [73]. Thus, phenotypes in compound GLB1 heterozygous genotypes sometimes remain difficult to predict [75, 76]. In addition, the GLB1 gene polymorphic variants p.Arg521Cys, p.Ser532Gly, and p.Arg595Trp have been reported to be associated with beta-galactosidase enzymatic pseudodeficiency [77].
MPS VI
The N-acetylgalactosamine 4-sulphatase (arylsulphatase B) gene (ARSB; MIM #611542), located in 5q14.1, spans a region of about 206 kb and contains 8 exons encoding a 533-amino acid glycoprotein [78]. ARSB deficit causes a pathological accumulation of undegraded DS in most organ systems. Presently, 197 different alterations have been reported, the vast majority of which are missense/nonsense lesions (Table 2). Since no common mutations have been reported up to now, the potential for studies of genotype-phenotype correlation in MPS VI is somewhat limited. Only one pseudodeficiency allele was reported a few years ago, p.Thr212Ile, in a family for which very low ARSB enzyme activity was described in relation to normal clinical findings [79]. Interestingly, the p.Ser384Asn (S384 N), one of the most common polymorphisms of the gene [80], had been traded for several years as a pathogenetic variant and correlated with a severe phenotype, leading to a series of incorrect molecular diagnoses of the ARSB gene [81].
MPS VII
The gene encoding β-glucuronidase (GUSB; MIM #611499) is 21 kb long and contains 12 exons. Two types of cDNAs arise through alternative splicing, resulting in an exon 6 corresponding to the 153-bp deletion in the shorter of the two types [82]. The presence of unprocessed multiple pseudogenes requires particular attention in diagnostic mutation analysis. To date, 64 different mutations have been reported in the GUSB gene (Table 2). Another complication reported in MPS VII diagnosis is the presence of the missense polymorphism p.Asp152Asn producing a pseudodeficiency allele that leads to greatly reduced levels of beta-glucuronidase activity (27% of the control) without apparent deleterious consequences [83].
MPS IX
The gene encoding hyaluronidase (HYAL1; MIM #607071) contains 3 exons and spans 3.5 kb [84]. HYAL1, together with HYAL2 and HYAL3, constitutes a multigene family for lysosomal hyaluronidase. To date, only one patient in the original report, and three others belonging to a second family have been described, yielding a total of three different mutations responsible for the disease (Table 2) [85, 86].
Conclusions
Although MPS share many clinical features among different enzyme deficiencies, they can also show a wide spectrum of clinical severity within each enzyme deficiency. Hence, even in the presence of typical clinical signs and symptoms, the choice of samples and diagnostic tests can be different for each MPS and require a multidisciplinary approach, including laboratory specialists and clinicians.
Increased urinary excretion of GAGs may only support the clinical suspicion of MPS and it is necessary to follow this with specific genetic testing including enzymatic and/or molecular analyses, performed on suitable samples, as leukocytes and/or cultured cell lines. While both analyses must be considered complementary for the definitive diagnosis of MPS and the genetic counselling, the molecular analysis is essential for carrier detection, and can sometimes predict prognosis and support therapeutic choices. The recently proposed NGS techniques will very soon complement the conventional testing, in particular for those patients where a differential diagnosis is difficult to perform.
Abbreviations
- CS:
-
Chondroitin sulphate
- DBS:
-
Dried blood spot
- DS:
-
Dermatan sulphate
- GAG:
-
Glycosaminoglycan
- HS:
-
Heparan sulphate
- KS:
-
Keratan sulphate
- MPS:
-
Mucopolysaccharidosi(e)s
- MS:
-
Mass spectrometry
- NBS:
-
New-born screening
- NGS:
-
Next-generation sequencing
- Pd:
-
Pseudodeficiency
References
Berry HK. Screening for mucopolysaccharide disorders with the berry spot test. Clin Biochem. 1987;20:365–71.
Bitter T, Muir HM. A modified uronic acid carbazole reaction. Anal Biochem. 1962;4:330–4.
Coppa GV, Catassi C, Gabrielli O, Giorgi PL, Dall'Amico R, Naia S, et al. Clinical application of a new simple method for the identification of mucopolysaccharidoses. HelvPaediatr Acta. 1987;42:419–23.
Humbel R, Chamoles NA. Sequential thin layer chromatography of urinary acidic glycosaminglycans. Clin Chim Acta. 1972;40:290–3.
Wijburg FA, Węgrzyn G, Burton BK, Tylki-Szymańska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013;102:462–70.
Tomatsu S, Okamura K, Taketani T, Orii KO, Nishioka T, Gutierrez MA, et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004;55:592–7.
Whitley CB, Ridnour MD, Draper KA, Dutton CM, Neglia JP. Diagnostic test for mucopolysaccharidoses: I. direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin Chem. 1989;35:374–9.
Piraud M, Maire I, Mathieu M. Pitfalls of screening for mucopolysaccharidoses by the dimethylmethylene blue test. Clin Chem. 1993;39:163–4.
Kubaski F, Osago H, Mason RW, Yamaguchi S, Kobayashi H, Tsuchiya M, et al. Glycosaminoglycans detection methods: applications of mass spectrometry. Mole Genet Metab. 2017;120:67–77.
Kubaski F, Suzuki Y, Orii K, Giugliani R, Church HJ, Mason RW, et al. Glycosaminoglycan levels in dried blood spots of patients with mucopolysaccharidoses and mucolipidoses. Mol Genet Metab. 2017;120:247–54.
Elliott S, Buroker N, Cournoyer JJ, Potier AM, Trometer JD, Elbin C, et al. Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Mol Genet Metab. 2016;118:304–9.
Navarrete-Martínez JI, Limón-Rojas AE, Gaytán-García MJ, Reyna-Figueroa J, Wakida-Kusunoki G, Delgado-Calvillo MDR, et al. Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: three-year findings from a screening program in a closed Mexican health system. Mol Genet Metab. 2017;121:16–21.
Burlina AB, Polo G, Salviati L, Duro G, Zizzo C, Dardis A, et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in north East Italy. J Inherit Metab Dis. 2017. https://doi.org/10.1007/s10545-017-0098-3.
Meacham F, Boffelli D, Dhahbi J, Martin DI, Singer M, Pachter L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451.
Fernández-Marmiesse A, Morey M, Pineda M, Eiris J, Couce ML, Castro-Gago M, et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis. 2014;9:59.
Filocamo M, Baldo C, Goldwurm S, Renieri A, Angelini C, Moggio M, et al. For telethon network of genetic biobanks staff. Telethon network of genetic biobanks: a key service for diagnosis and research on rare diseases. Orphanet J Rare Dis. 2013;8:129.
Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19–29.
Giugliani R, Federhen A, Rojas MV, Vieira T, Artigalás O, Pinto LL, et al. Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol. 2010;33:589–604.
Scarpa M, Almássy Z, Beck M, Bodamer O, Bruce IA, De Meirleir L, et al. Hunter syndrome European expert council. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72.
Wood T, Bodamer OA, Burin MG, D'Almeida V, Fietz M, Giugliani R, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012;106:73–82.
Giugliani R, Villarreal ML, Valdez CA, Hawilou AM, Guelbert N, Garzón LN, et al. Guidelines for diagnosis and treatment of hunter syndrome for clinicians in Latin America. Genet Mol Biol. 2014;37:315–29.
Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, et al. International guidelines for the management and treatment of Morquio a syndrome. Am J Med Genet Part A. 2015;167A:11–25.
Zanetti A, Onenli-Mungan N, Elcioglu N, Ozbek MN, Kör D, Lenzini E, et al. Molecular analysis of Turkish Maroteaux-Lamy patients and identification of one novel mutation in the arylsulfatase B (ARSB) gene. JIMD Rep. 2014;14:1–9.
Costa-Motta FM,·Bender F, Acosta A, Abé-Sandes K, Machado T, Bomfim T, et al. A community-based study of mucopolysaccharidosis type VI in Brazil: the influence of founder effect, endogamy and consanguinity. Human Hered 2014;77:189–196.
Cheema HA, Malik HS, Hashmi MA, Fayyaz Z, Mushtaq I, Shahzadi N. Mucopolysaccharidoses—clinical spectrum and frequency of different types. J Coll Physicians Surg Pak. 2017;27:80–3.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77.
Scott HS, Anson DS, Orsborn AM, Nelson PV, Clements PR, Morris CP, Hopwood JJ. Human alpha-L-iduronidase: cDNA isolation and expression. Proc Natl Acad Sci U S A. 1991;88:9695–9.
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet A, Sly WE, Valle D, editors. The metabolic and molecular bases of inherited disease. (8th Edn) New York: McGraw-hill; 2001. p. 3421–52.
Clarke LA. Mucopolysaccharidosis type I. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993–2007. Available at http://www.genetests.org (Updated 21 September 2007).
Bertola F, Filocamo M, Casati G, Mort M, Rosano C, Tylki-Szymanska A, et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum Mutat. 2011;32:E2189–210.
Aronovich EL, Pan D, Whitley CB. Molecular genetic defect underlying alpha-L-iduronidase pseudodeficiency. Am J Hum Genet. 1996;58:75–85.
Pollard LM, Braddock SR, Christensen KM, Boylan DJ, Smith LD, Heese BA, et al. Inborn errors of metabolism: from identification to treatment. “Three apparent pseudo-deficiency alleles in the IDUA gene identified by newborn screening”. Abstract 82. Boston, MA: American Society of Human Genetics 63rd Annual Meeting; 2013.
Pollard L, Braddoc S, Christensen K, Boylan D, Heese B. Diagnostic follow-up of 47 infants with a positive newborn screen for Hurler syndrome: identification of four recurrent IDUA sequence changes that significantly reduce enzyme activity. In: APHL meeting, Anaheim, CA. Proceedings of the 2014 APHl newborn screening and genetic testing symposium, Anaheim, CA, October 27–30, 2014.
Wilson PJ, Morris CP, Anson DS, Occhiodoro T, Bielicki J, Clements PR, Hopwood JJ. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A. 1990;87:8531–5.
Rathmann M, Bunge S, Steglich C, Schwinger E, Gal A. Evidence for an iduronate-sulfatase pseudogene near the functional hunter syndrome gene in Xq27.3-q28. Hum Genet. 1995;95:34–8.
Froissart R, Da Silva IM, Maire I. Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr 2007;96:71–77.
Kosuga M, Mashima R, Hirakiyama A, Fuji N, Kumagai T, Seo JH, et al. Molecular diagnosis of 65 families with mucopolysaccharidosis type II (hunter syndrome) characterized by 16 novel mutations in the IDS gene: genetic, pathological, and structural studies on iduronate-2-sulfatase. Mol Genet Metab. 2016;118:190–7.
Lualdi S, Pittis MG, Regis S, Parini R, Allegri A, Furlan F, et al. Multiple cryptic splice sites can be activated by IDS point mutations generating misspliced transcripts. J Mol Med. 2006;84:692–700.
Lualdi S, Tappino B, Di Duca M, Dardis A, Anderson CJ, Biassoni R, et al. Enigmatic in vivo iduronate-2-sulfatase (IDS) mutant transcript correction to wild-type in hunter syndrome. Hum Mutat. 2010;31:E1261–85.
Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32:270–2.
Manara R, Rampazzo A, Cananzi M, Salviati L, Mardari R, Drigo P, et al. Hunter syndrome in an 11-year old girl on enzyme replacement therapy with idursulfase: brain magnetic resonance imaging features and evolution. J Inherit Metab Dis. 2010;33(Suppl 3):S67–72.
Piña-Aguilar RE, Zaragoza-Arévalo GR, Rau I, Gal A, Alcántara-Ortigoza MA, López-Martínez MS, Santillán-Hernández Y. Mucopolysaccharidosis type II in a female carrying a heterozygous stop mutation of the iduronate-2-sulfatase gene and showing a skewed X chromosome inactivation. Eur J Med Genet. 2013;56:159–62.
Lonardo F, Di Natale P, Lualdi S, Acquaviva F, Cuoco C, Scarano F, et al. Mucopolysaccharidosis type II in a female patient with a reciprocal X;9 translocation and skewed X chromosome inactivation. Am J Med Genet A. 2014;164A:2627–32.
Wood TC, Thompson JN. Analysis of the heparin sulfamidase gene: mutation analysis in patients with Sanfilippo syndrome type A (MPS IIIA) and analysis of sulfamidase expression in normal adult brain. Am J Hum Genet. 2000;67(Suppl A):1635.
Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008;31:240–252.
Weber B, van de Kamp JJ, Kleijer WJ, Guo XH, Blanch L, van Diggelen OP, et al. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo a). J Inherit Metab Dis. 1998;21:416–22.
Bunge S, Ince H, Steglich C, Kleijer WJ, Beck M, Zaremba J, et al. Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo a). hum. Mutat. 1997;10:479–85.
Di Natale P, Balzano N, Esposito S, Villani GR. Identification of molecular defects in Italian Sanfilippo a patients including 13 novel mutations. Hum Mutat 1998;11:313–320.
Weber B, Blanch L, Clements PR, Scott HS, Hopwood JJ. Cloning and expression of the gene involved in Sanfilippo B syndrome (mucopolysaccharidosis III B). Hum Mol Genet. 1996;5:771–7.
Zhao HG, Li HH, Bach G, Schmidtchen A, Neufeld EF. The molecular basis of Sanfilippo syndrome type B. Proc Natl Acad Sci U S A. 1996;93:6101–5.
Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: diagnostic, clinical, and biological implications. Hum Mutat. 2001;18:264–81.
Feldhammer M, Durand S, Mrazova L, Boucher RM, Laframboise R, Steinfeld R, et al. Sanfilippo syndrome type C: mutation spectrum in the heparan sulfate acetyl-CoA:alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum Mutat. 2009;30:918–25.
Berger-Plantinga EG, Vanneste JA, Groener JE, van Schooneveld MJ. Adult-onset dementia and retinitis pigmentosa due to mucopolysaccharidosis III-C in two sisters. J Neurol 2004; 251:479–481.
Ruijter GJ, Valstar MJ, van de Kamp JM, van der Helm RM, Durand S, van Diggelen OP, et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008;93:104–11.
Robertson DA, Callen DF, Baker EG, Morris CP, Hopwood JJ. Chromosomal localization of the gene for human glucosamine-6-sulphatase to 12q14. Hum Genet. 1988;79:175–8.
Mok A, Cao H, Hegele RA. Genomic basis of mucopolysaccharidosis type IIID (MIM 252940) revealed by sequencing of GNS encoding N-acetylglucosamine-6-sulfatase. Genomics. 2003;81:1–5.
Harmatz P, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, et al. The Morquio a clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio a subjects. Mol Genet Metab. 2013;109:54–61.
Nakashima Y, Tomatsu S, Hori T, Fukuda S, Sukegawa K, Kondo N, et al. Mucopolysaccharidosis IV a: molecular cloning of the human N acetylgalactosamine-6-sulfatase gene (GALNS) and analysis of the 5′ flanking region. Genomics. 1994;20:99–104.
Tomatsu S, Fukuda S, Masue M, Sukegawa K, Fukao T, Yamagishi A, et al. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem Biophys Res Commun. 1991;181:677–83.
Pshezhetsky AV, Ashmarina M. Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog Nucleic Acid Res Mol Biol. 2001;69:81–114.
Wood TC, Harvey K, Beck M, Burin MG, Chien YH, Church HJ, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293–307.
Tomatsu S, Fukuda S, Cooper A Wraith JE, Rezvi GM, Yamagishi A, et al. Mucopolysaccharidosis IVA: identification of a common missense mutation I113F in the N-acetylgalactosamine-6-sulfate sulfatase gene. Am J Hum Genet. 1995;57:556–63.
Yamada N, Fukuda S, Tomatsu S, Muller V, Hopwood JJ, Nelson J, et al. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: nine novel mutations including T312S, a common allele that confers a mild phenotype. Hum Mutat. 1998;11:202–8.
Catarzi S, Giunti L, Papadia F, Gabrielli O, Guerrini R, Donati MA, et al. Morquio a syndrome due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Mol Genet Metab. 2012;105:438–42.
Morrone A, Tylee KL, Al-Sayed M, Brusius-Facchin AC, Caciotti A, Church HJ, et al, Molecular testing of 163 patients with Morquio A (mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol Genet Metab 2014;112:160–170; erratum in: Mol Genet Metab. 2014;113:237.
Caciotti A, Tonin R, Rigoldi M, Ferri L, Catarzi S, Cavicchi C, et al. Optimizing the molecular diagnosis of GALNS: novel methods to define and characterize Morquio-a syndrome-associated mutations. Hum Mutat. 2015;36:357–68.
Morreau H, Galjart NJ, Gillemans N, Willemsen R, van der Horst GTJ, d’Azzo A. Alternative splicing of β-galactosidase mRNA generates the classic lysosomal enzyme and a β-galactosidase-related protein. J Biol Chem 1989;264:29655–29663.
Hinek A. Biological roles of the non-integrin elastin/laminin receptor. Biol Chem. 1996;377:471–80.
Privitera S, Prody CA, Callhan JW, Hinek A. The 67kDa enzymatically inactive alternatively spliced variant of β-galactosidase is identical to the elastin/laminin-binding protein. J Biol Chem. 1998;273:6319–26.
Oshima A, Yoshida K, Shimmoto M, Fukuhara Y, Sakuraba H, Suzuki Y. Human beta-galactosidase gene mutations in Morquio B disease. Am J Hum Genet. 1991;49:1091–3.
Suzuki Y, Oshima A. Namba E. β-Galactosidase deficiency (β-galactosidosis) GM1 gangliosidosis and Morquio B disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 3775–809.
Paschke E, Milos I, Kreimer-Erlacher H, Hoefler G, Beck M, Hoeltzenbein M, et al. Mutation analyses in 17 patients with deficiency in acid beta-galactosidase: three novel point mutations and high correlation of mutation W273L with Morquio disease type B. Hum Genet. 2001;109:159–66.
Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1 gangliosidosis and Morquio B patients: possible common origin for the prevalent p.R59H mutation among gypsies. Hum Mutat. 2006;27:1060.
Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, et al. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta. 2011;1812:782–90.
Santamaria R, Blanco M, Chabás A, Grinberg D, Vilageliu L. Identification of 14 novel GLB1 mutations, including five deletions, in 19 patients with GM1 gangliosidosis from South America. Clin Genet. 2007;71:273–9.
Moore T, Bernstein JA, Casson-Parkin S, Cowan TM. β-Galactosidosis in patient with intermediate GM1 and MBD phenotype. JIMD Rep. 2013;7:77–9.
Gort L, Santamaria R, Grinberg D, Vilageliu L, Chabás A. Identification of a novel pseudodeficiency allele in the GLB1 gene in a carrier of GM1 gangliosidosis. Clin Genet. 2007;72:109–11.
Modaressi S, Rupp K, von Figura K, Peters C. Structure of the human arylsulfatase B gene. Biol Chem Hoppe Seyler 1993;374:327–335.
Sandberg S, Deanching M, Hoganson G, Wenger D, Whiley C. Pseudo-deficiency allele of the N-acetylgalactosamine-4-sulfatase gene identified in a family with Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI). Mol Genet Metab. 2008;93:S14–46.
Zanetti A, Ferraresi E, Picci L, Filocamo M, Parini R, Rosano C, et al. Segregation analysis in a family at risk for the Maroteaux-Lamy syndrome conclusively revelas c.1151G>a (p.S384N) as to be a polymorphism. Eur J Hum Genet. 2009;17(9):1160–4.
Karageorgos L, Brooks DA, Pollard A, Melville EL, Hein LK, Clements PR, et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007;28:897–903.
Miller RD, Hoffmann JW, Powell PP, Kyle JW, Shipley JM, Bachinsky DR, Sly WS. Cloning and characterization of the human beta-glucuronidase gene. Genomics. 1990;7:280–3.
Vervoort R, Islam MR, Sly W, Chaba A, Wevers R, de Jong J, et al. A pseudodeficiency allele (D152N) of the human beta-glucuronidase gene. Am J Hum Genet. 1995;57:798–804.
Frost GI, Heng HH, Scherer SW, Mohapatra G, Stern R. The hyaluronidase gene HYAL1 maps to chromosome 3p21.2-p21.3 in human and 9F1-F2 in mouse, a conserved candidate tumor suppressor locus. Genomics. 1998;48:63–70.
Triggs-Raine F, Salo TJ, Zhang H, Wicklow BA, Natowicz MR. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc Natl Acad Sci U S A. 1999;96:6296–300.
Imundo L, Leduc CA, Guha S, Brown M, Perino G, Gushulak L, et al. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J Inherit Metab Dis. 2001;34:1013–22.
Funding
This work was partially supported by unrestricted grants from “Cinque per mille e Ricerca Corrente, Ministero della Salute” to MF and PRIN 2012 Prot. 20122EK9SZ 003 to RT.
The publication costs for this paper in the IJP supplement were made possible with unconditional financial support from BioMarin, Sanofi Genzyme, and Shire. The sponsors had no input into the content of articles, which were independently prepared by the authors and have undergone the journal’s standard peer-review process.
Availability of data and materials
Not applicable as no datasets were generated during the current study.
About this supplement
This article has been published as part of Italian Journal of Pediatrics, Volume 44 Supplement 2, 2018: Mucopolysaccharidoses: state of the art. The full contents of the supplement are available online at https://ijponline.biomedcentral.com/articles/supplements/volume-44-supplement-2 .
Author information
Authors and Affiliations
Contributions
MF drafted the initial manuscript. All authors reviewed the literature on MPS I, and MPS IX (FB), MPS II and MPS VI (RT), MPS III A, B, C, D and MPS VII (MF), MPS IV A and B (AM). All authors critically revised and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Filocamo, M., Tomanin, R., Bertola, F. et al. Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know. Ital J Pediatr 44 (Suppl 2), 129 (2018). https://doi.org/10.1186/s13052-018-0553-2
Published:
DOI: https://doi.org/10.1186/s13052-018-0553-2