
Abstract
Background
One of the 11 recognized mucopolysaccharidosis (MPS) diseases is Sanfilippo. It is autosomal recessive in its mode of transmission. There are four subtypes of Sanfilippo (A, B, C, and D). The most worldwide prevalent subtypes of mucopolysaccharidosis type III (MPS III) are A and B followed by C and D subtypes. To estimate the frequency of MPS IIIA among MPS III patients, we diagnose and compare their clinical features with those of MPS IIIB and also compare the prevalence of MPS IIIB versus MPS IIIA among diagnosed cases at the Biochemical Genetic Department at NRC. For every case that was referred, the quantitative determination of urine Glycosaminoglycans (GAGs) was assessed. Two-dimensional electrophoresis (2DE) of GAGs extracted from urine was performed on all cases with high urinary GAG levels. Both N-sulphoglucosamine sulphohydrolase (MPS IIIA) and N-alpha-acetylglucosaminidase (MPS IIIB) enzyme activity were determined fluorometrically.
Results
From November 2019 to May 2022, 535 cases were referred to the National Research Centre’s Biochemical Genetics Department. 233 (43%) MPS cases were diagnosed with high urinary GAG levels for their ages. 73 (31.3%) MPS III cases were diagnosed by 2DE out of the 233 MPS cases. Plasma N-alpha-acetylglucosaminidase enzyme assay was insufficient in 36 (49.3%) patients (Sanfilippo type B), while N-sulphoglucosamine sulphohydrolase enzyme activity was deficient in 15 (20.6%) patients. The other 22 (30.1%) patients are either Sanfilippo type C or D.
Conclusion
N-sulphoglucosamine sulphohydrolase enzyme activity was measured for the first time in Egypt. Thirty-one percent of all diagnosed MPS cases during the last 3 years were MPS type III, making Sanfilippo the most common MPS type among the referred cases to our Biochemical Genetics Department. MPS IIIA accounts for 20.6% of MPSIII cases in this study. Still, MPS type IIIB is the commonest type among diagnosed patients.
Similar content being viewed by others
Background
Sanfilippo syndrome, MPS III is one of the lysosomal storage disorders which is a neurodegenerative disease. It is caused by enzymatic defects in heparan sulphate (HS) catabolism. Sanfilippo has four subtypes A, B, C, and D; each has its own genetic mutation that results in N-sulphoglucosamine sulphohydrolase, α-N-acetylglucosaminidase, α-glucosaminide-N-Ac-transferase, and glucosamine-N-sulphatase enzyme deficiency, respectively. The most common subtypes are MPSIIIA and B [1]. Any of these enzymes’ deficiencies cause subsequent HS accumulation in lysosomes of affected tissues and organs [2].
Neurological manifestations such as cognitive decline, hyperactivity, aggressiveness, sleep disturbances, and/or epilepsy are the main symptoms of Sanfilippo. These are associated with significant abnormalities of the central nervous system (CNS), including neurological manifestations, brain atrophy, and spinal cord compression [3]. Delayed milestones and neurological manifestations as hyperactive behaviors are the first symptoms of the disease and start at the age of 2 or 3 years [4].
The first laboratory investigations of MPS III after clinical suspicion are quantitative and qualitative 2DE GAG measurement in urine that demonstrates excessive HS excretion [5]. Specific enzyme measurement in plasma or leukocytes is the 2nd step in diagnosis; N-sulphoglucosamine sulphohydrolase (SGSH, EC 3.10.1.1) for MPS IIIA, N-acetyl-α-glucosaminidase (NAGLU, EC 3.2.1.50) for MPS IIIB, heparan-α-glucosaminide N-acetyl transferase (EC 2.3.1.78) for MPS IIIC, and N-acetyl glucosamine 6-sulphatase (EC 3.1.6.14) for MPS IIID. The final step is molecular variant identification for the gene encoding the deficient enzyme [6].
Signs and symptoms of Sanfilippo disorder progression are variable partially due to the amount of residual enzyme activity dictated by the molecular variant. The clinical course is affected by genotype which in some times not as expected may be due to epigenetic factors [7].
The neuro-pathological manifestations (main feature) of Sanfilippo are due to Lysosomal HS accumulation in the brain [8]. In this study, we measured the enzyme activity for Sanfilippo A and B. MPS III A was 20.6% and MPS III B 49.3%, while the rest cases, 30.1% were suspected to be MPS III C or D. This study should establish Sanfilippo type A enzyme assay method and try to give impact about the percent of these cases in Egypt in relation to Sanfilippo B, C, and D.
Using enzyme replacement therapy is under trial but without medical approval till now. Supportive treatments are only to delay multisystem complications, but curative therapies that prevent heparan sulfate accumulation are not found till now [9].
Subjects and methods
Subjects and ethics
Five hundred fifty-three cases were referred to the Biochemical Genetic Department, National Research Centre, during the period from November 2019 to May 2022. Their age ranged between 9 months to 17 years. A control group of matching age and sex to the study group was used in the study. All participants’ parents gave written informed consent after a full explanation of the study. The ethical approval was obtained from the Medical Ethical Committee at the NRC and the Faculty of Medicine, Al Azhar University (Girls), Cairo, Egypt (The Faculty of Medicine, Al Azhar University (Girls), Approval no. 201910199).
Complete history taking and pedigree construction were recorded for each patient.
Methods
Mentioned in detail with supplementary data (Additional file 1).
Statistical analysis
All data were coded and organized, and then, SPSS program version 16 was used for statistical analysis. Shapiro–Wilk test was used to test the data normality and if it is normally distributed also to describe the nonparametric data in numbers. Differences between groups were analyzed for significance using a one-way ANOVA with tests of homogeneity of variances. Correlations were performed using case summaries, chi-squared test of homogeneity, and independent sample effect sizes with Levene’s test for equality of variances. The comparison between the two variables was calculated by Pearson’s rho coefficient(r). The p value < 0.05 was considered significant.
Results
According to enzyme assay results, 36 (49.3%) cases had deficient alpha-N-acetylglucosaminidase enzyme activity, 15 (20.6%) cases had deficient N-sulphoglucosamine sulphohydrolase enzyme, and 22 (30.1%) patients had a normal level of both enzymes. These 22 cases are diagnosed provisionally as MPS III C or D for further confirmation.
The study included 535 subjects referred to the Biochemical Genetic Department at Human Genetics and Genome Research Institute, National Research Centre. They included 302 (56.4%) males and 233 (43.6%) females.
The age range within 535 cases was from 1 month to 20 years. The mean age of diagnosis in 48/73 (65.7%) MPS III cases was 5 years, 7/73 (9.6%) cases were early diagnosed at the age of 3 to 12 months, and 18 cases were diagnosed at the age of 6 to 17 years.
Four hundred thirty (80.4%) cases had consanguineous parents while 105 (19.6%) cases had non-consanguineous parents. The consanguinity rate in the present study was 64/73 (87.7%) in MPS III patients, 32/36 (88.9%) cases in MPS IIIB patients, 19/22 (86.4%) cases in MPS IIIC or D patients, and 13/15 (86.7%) cases in MPS IIIA patients (Fig. 2).
The 233 MPS diagnosed cases were 143 (61.4%) male and 90 (38.6%) female, and they were 2 months to 17 years old. One hundred eighty-three (78.5%) MPS patients had positive parental consanguinity against 50 (21.5%) patients with non-consanguineous parents.
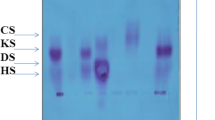
Out of 233 MPS cases, 73 MPS III cases were diagnosed due to the presence of heparan and heparan sulfate spots in their 2DE (Fig. 1). MPS III cases included 48 (65.8%) males and 25 (34.2%) females with age ranging from 3 months to 17 years. Sixty-four cases (87.7%) had consanguineous parents, while 9 cases (12.3%) had negative consanguineous marriages.

Two-dimensional electrophoresis pattern in normal and Sanfilippo subjects. CS chondroitin sulfate, H heparan, and HS heparan sulfate
Clinical findings
All the 535 referred cases were suspected to have MPS. Clinical examination of MPS III cases showed signs and symptoms represented in Figs. 3 and 4.
Manifestations of hyperactivity were found in 86.7% of MPS IIIA cases, 80.6% of MPS IIIB cases, and 59.1% of MPS IIIC or D cases (Fig. 4).
Nineteen patients (26%), 11 males, and 8 females had general ear problems (Table 2). Out of the 19 cases, 5 (26.3%) cases had MPS IIIA, 11 (57.9%) cases had MPS IIIB, and 3 (15.8%) cases had MPS IIIC or D (Fig. 4).
Cardiac anomalies were detected in 5 cases, 3 cases with MPS type IIIA and 2 cases with MPS IIIB (Figs. 3 and 4). Four cases out of 5 had mitral regurgitation, and one MPS IIIA case had aortic regurgitation.
It was found that 5 years are the mean age of diagnosis of 73 MPS III. Thirty-six patients were < 5 years (from 3 months to 4 years and 6 months), while 27 patients were > 5 years (from 4 and 6/12 to 17 years) and ten patients were 5 years old at the time of diagnosis.
Biochemical finding
Urinary glycosaminoglycans levels
Glycosaminoglycans were measured in the urine of all 535 MPS suspected cases. GAG level was high in 242 cases in relation to their age. Our reference values for urinary normal GAG levels according to Dong et al. [10] are shown in Table 1.
Urinary GAG levels of 73 MPS III cases are presented in Table 2. According to age, the studied cases were classified into 3 groups, group I (0–12 months) which included 7 cases, group II (1–5 years) which included 38 cases, and group III (> 5 years) which included 28 cases. Nine months were the mean age among group I, 3 years among group II, and 8 years among group III. Group I showed 48.8 mg/mmol of creatinine as the mean level of urinary GAGs, while group II and III urinary GAG mean levels were 47.4 and 27 mg/mmol of creatinine, respectively. Urinary GAG levels (means ± SD) of MPS IIIA and MPS IIIB cases were 47.5 ± 0.15 and 25.6 ± 0.27 mg/mmol of creatinine, respectively, as shown in Table 3.
Two-dimensional electrophoresis in urine
Two-dimensional electrophoretic separation of urinary GAGs was done on 242 cases that showed high levels of urinary GAGs. Two hundred thirty-three cases showed abnormal patterns of MPS types, but 73 patients showed heparan and heparan sulfate spots (MPS III pattern) (Fig. 1) and nine cases with high urinary GAGs and normal 2DE pattern.
Alpha-N-acetylglucosaminidase enzyme assay
Alpha-N-glucosaminidase enzyme activity was measured in all 73 MPS III after being diagnosed by 2DE. Thirty-six patients had low enzyme activity which confirmed the diagnosis of MPS IIIB (Table 4).
N-sulphoglucosamine sulphohydrolase enzyme assay
N-sulphoglucosamine sulphohydrolase enzyme activity was measured in the 37 cases that showed normal α-N-acetylglucosaminidase enzyme activity. Fifteen (40%) patients had low enzyme activity (Table 5). The remaining 22 cases with normal α-N-acetylglucosaminidase (Table 4) and N-sulphoglucosamine sulphohydrolase (Table 5) enzyme levels were accordingly diagnosed as either MPS IIIC or D for further enzymatic study.
Sanfilippo A cases showed almost the same parental consanguinity as that of the 73 Sanfilippo cases as 13 (86.7%) MPS III cases had consanguineous parents. Comparison within three diagnosed MPS III subtypes (A, B, and C or D) according to the mean level of α-N-acetylglucosaminidase enzyme assay showed significant differences as represented in Fig. 5.
IQ test
IQ test was done on 50/73 (68.5%) MPS III patients, and the average level was (< 18 months). Out of the 23 cases who did not undergo IQ evaluation, 8 cases stopped following up, 6 cases had bone marrow transplants, and 9 cases could not complete the test due to hyperactivity or severe regression stage of the disease. 11/50 (22%) patients were MPS IIIA, 23/50 (46%) MPS IIIB, and 16/50 (32%) MPS III types C or D (Fig. 4).
Discussion
Sanfilippo disease is an autosomal recessive disorder caused by a genetic mutation in one of the responsible four enzymes for heparan sulfate degradation. Accordingly, there are 4 subtypes of MPS III A, B, C, and D. Accumulated heparan and heparan sulfate in various body organs especially the brain results in MPS III signs and symptoms [11].
In the present study, MPS-suspected subjects were fewer than those investigated in Fateen et al.’s studies (1448 and 1294), respectively. This could be due to a short study period (3 years versus 6 and 11 years, respectively). In these studies, MPS III cases were 28% in Fateen et al. and 15% in Fateen et al., while it was higher in this study. Another earlier study has discussed the Egyptian inborn errors of metabolism (IEM) over 15 years of experience and lysosomal storage diseases (LSD) had the highest percent of diagnosed cases (69.4%) of all IEM. Out of LSD cases, 48.9% had one of the MPS disorders and 17.3% had MPS III [12].
All these studies proved that mucopolysaccharidosis disorders are the commonest subtype of LSD in Egypt, and of all MPS types, MPS III had the highest incidence. This was explained by a high rate of consanguineous marriages in Egypt [13]. Also, late or misdiagnosed cases cause the absence of genetic counseling for affected families that allow the repetition of many cases in the same family.
A Tunisian MPS study in the period of 1970–2005 showed 96/132 MPS cases and 30 (31.2%) MPS III making it the most common MPS type in Tunisia [13]. Another Saudi Arabia study detected 49/189 (25.9%) inborn error of metabolism (IEM) cases, 15/49 (30.6%) MPS cases, and 2/15 (13.3%) MPS III cases [14]. In Egypt, MPS has a higher incidence due to the larger population number and also due to the presence of diagnostic facilities at our Biochemical Genetic Department lab for more than two decades.
The high rate of consanguinity within Arab populations may be due to deeply rooted cultural traditions [15,16,17]. The consanguinity rate in the present study (Fig. 2) matches the latest Fateen et al., study consanguinity rate of 76%, which is very high helping the accumulation of deleterious genes in the families [15, 17]. These shows the importance of awareness campaigns for the population and the affected families’ genetic counseling.

Consanguinity percent among parents of MPS IIIA, B, and C or D patients
Male to female ratio in MPS cases was 1.9:1 which is common among Arab populations especially in rural areas. This agrees with other studies that recorded high ratios as in Fateen et al. study the male-to-female ratio was 1.7:1, 1.4:1 in Fateen et al. and 1.2:1 in Ben Turkia et al. study [12, 13, 15].
In our study, the mean age of diagnosis of MPS III cases was 5 years and the 7 early diagnosed cases as they were already siblings of affected patients. The late diagnosis of 18 cases may be because they were the first cases diagnosed in the family or they were attenuated phenotypes of Sanfilippo. In relation to Fateen et al., they also noticed a broad range in the age of onset and diagnosis of MPS cases (1 day to 20 years), while Rouse et al. showed that the 1st phase of MPS III may start from 2 to 6 years [12, 18]. Rouse et al. said that this delay in diagnosis usually accompanies MPS III cases due to a general lack of knowledge of MPS III disease [18].
IQ test (Stanford-Binet IV Intelligence Test) done on MPS III patients’ average level was (< 18 months) (Fig. 4). In the study of Valester et al., 66 MPS III cases had low IQ tests, 27 cases had an IQ level below 3 months, and 39 cases with an IQ level from 6 months to 6 and half years. They included 32/66–48.5% patients with MPS IIIA, 22/66–33.3% patients with MPS IIIB, and 12/66–18.2% patients with MPS IIIC. Their age ranged from 1 to 77 years old while in the present study, patients’ age ranged from 9 months to 17 years. IQ levels among Valester et al. study were higher than that in our study (< 18 months). Both differences in IQ level and age range could be explained by ethnic and mutational variations that affect disease phenotype and subsequently the age of onset [7].
In the present study, aggressive behaviors and hyperactivity were often found in MPS IIIA cases (86.7%) followed by MPS IIIB cases (80.6%) and less found in MPS IIIC or D cases (59.1%) (Fig. 4). This result agrees with Coutinho et al. publication; Sanfilippo A is the severe phenotype followed by Sanfilippo B while Sanfilippo C and D show milder symptoms [19].
Abnormal facies (broad forehead, thick lips, wide-spaced teeth, and frontal bossing) were detected by clinical examination among of MPS III cases showed while the other 13 cases were younger to develop course facies (Figs. 3 and 4).

Signs and symptoms of the study group

Signs and symptoms among different MPS III types
Poor documentation of auditory function in MPS III may be owing to the difficulty of the collection of audiometric data from patients with MPS III due to their behavioral problems [20]. Conductive issues also appear early in life, as otitis media has a reported rate of 91%, and tympanostomy tubes are placed in most patients before 5 years [21]. In the present study, general ear problems were found within 33.3% of MPS IIIA cases, 30.6% of MPS IIIB cases, and 13.6% of MPS IIIC or D cases. This differs from previously reported rates of hearing loss which are 100% (3/3) in MPS IIIB [21], 87% (48/55) in MPS IIIA [22], and 25% (1/4) in MPS IIID [23]. This may explain the point of view of Zafeiriou et al. in considering MPS IIIB as the severe form of all MPS III subtypes [21]. Jansen et al. result was more or less expected as MPS IIID is the mild form of Sanfilippo disease though it is the rare subtype [23].
MPS III is associated with cardiac anomalies as MPS I and II [24, 25]. The incidence of cardiac involvement in the present study is lower than that reported by Andrade et al. (60% of MPS IIIA cases, 30% of MPS IIIB cases, 4% of MPS IIIC cases, and 6% of MPS IIID cases had cardiac affection) [26]. Even though MPS III is the least MPS type that causes cardiac affection such as cardiomyopathy and valvular injury increases with age as aortic valve abnormality and mitral valve stenosis [18]. This may explain the lower cardiac affection in our MPS IIIA cases as we had few cases over the age of 10 years.
In the present study, urinary GAGs were high in 242 cases out of 535 referred patients, and 233 were diagnosed as MPS cases due to abnormal 2DE. Nine (3.7%) cases had high GAG level, but normal 2DE that occurs in many disorders with dysplasia [15, 27].
In Fateen (b) et al.’s study, the mean urinary GAG level for 43 MPS III patients was 34.4 mg/mmol creatinine. This is more or less similar to the mean urinary GAGs of our study MPS III urinary GAGs. The MPS III urinary GAG results were divided into 3 groups according to their age, 1–12 months, 1–5 years, and > 5 years, and their mean were 48.8, 47.4, and 27 mg/mmol creatinine, respectively (Table 2) [28]. Also, the mean of MPS IIIA cases urinary GAGs (25.6 mg/mmol creatinine) was lower than that of MPS IIIB cases (47.5 mg/mmol creatinine) (Table 3).
Seventy-three (73/233–31.3%) cases were diagnosed as MPS III due to the presence of heparan and heparan sulfate spots in 2DE. This is considered a quite high percentage when compared to other studies, MPS III formed 31.2% of all MPS cases in Turkia et al., 17.3% in Fateen(b) et al., and 28% in Fateen et al. [13, 15, 28]. Fateen (b) et al.’s study diagnosed 61 cases as Sanfilippo, and only 22 cases were diagnosed as MPS IIIB by enzyme assay while the rest of the cases remained undefined as MPS IIIA, C, or D due to the absence of MPS IIIA enzyme measuring method [28]. The same was found in Fateen et al. who diagnosed 134 MPS IIIB cases out of 177 cases of Sanfilippo and mentioned that the absence of MPS III A, C, and D was a limitation in their study [15].
Thirty-six (36/73–49.3%) patients had low alpha-N-acetylglucosaminidase enzyme activity and were diagnosed with MPS IIIB. Al-Sannaa et al.’s study reported 3/3–100% MPS IIIB cases, while 6/7–85.7% MPS IIIB cases were detected by Al-Jasmi et al.’s study and 22/61–36.1% MPS IIIB cases were diagnosed by Fateen et al. [11, 13, 26]. Meaning that according to our study, Al-Sannaa et al.’s study, Al-Jasmi et al.’s study, and Fateen et al.’s study, MPS IIIB is the commonest subtype of Sanfilippo followed by MPS IIIA through the Mediterranean region [15, 29, 30].
In the present study, 15/73–20.5% of patients had low N-sulphoglucosamine sulphohydrolase enzyme activity (MPS IIIA patients). No MPS IIIA was detected in some studies [29, 30], while in Estonia, all cases were MPS IIIA [31]. This could be explained by the high consanguinity rate among MPS IIIA patient (13/15 patients, 86.7%) cases accompanied by ethnic variations.
N-sulphoglucosamine sulphohydrolase enzyme activity was different than that of previously reported results in Egyptian cases. Cases with deficient MPS IIIA enzyme had a mean range of 0.25 ± 0.19 nmol/mg prot/17 h. Patients with normal MPS IIIA enzyme activity (MPS IIIC or D cases) had mean enzyme activity of 12.8 ± 0.23 nmol/mg prot/17 h. The range of normal cases is slightly higher in our study 3.4–42.6 nmol/mg prot/17 h. In comparison to Karpova et al.’s study, the normal range of this study was slightly higher than that of Karpova et al. The mean range of affected MPS IIIA patients in both studies is almost the same [32].
This could be explained by Egyptian ethnic variations. In the near future, molecular studies of MPS IIIA cases will provide more explanation about the difference in the enzyme level and phenotypic variations.
When the results of alpha-N-acetylglucosaminidase enzyme activities for MPS III groups (A, B, and C or D) were compared, a significant difference was detected (Fig. 5). This characteristic difference was also found in Karpova et al.’s results [32]. This allows easy prediction of MPS IIIA cases whenever finding a case with high alpha-N-acetylglucosaminidase enzyme activities.

Comparison between the levels of α-glucosaminidase enzyme activity in three groups of MPS III: A, B, and C or D
This study provided us with the easy, rapid diagnosis of MPS IIIA cases. Soon it will allow easy detection of the common Egyptian genetic mutations and its correlation with the phenotypic variation. MPS IIIA molecular study is now under search and very soon will allow molecular prenatal diagnosis via amniocentesis or cell-free DNA [18, 33]. Also, molecular studies may provide a seed for the promising MPS III cases of gene therapy [18].
MPS III has no enzyme replacement therapy (ERT) despite its availability for MPS I, II, IV, and VI as it is not able to bypass the blood–brain barrier and cure the neurological manifestations of Sanfilippo disease [15]. The availability of MPS III therapy either ERT or gene therapy will be an exciting milestone in encouraging the patients to seek diagnosis and raise the physician’s awareness.
Conclusion
From this study, we concluded that MPS IIIA and B cannot be differentiated clinically as heparan sulfate accumulation causes almost the same symptoms in both types. Two-dimensional electrophoresis was a very good diagnostic tool for MPS type III in general, but the specific enzyme assay can differentiate and diagnose the four subtypes A, B, C, and D. Also, the mean age of diagnosis was 5 years, and most of the early diagnosed cases had previous family history of MPS III cases. More attention should be given to Sanfilippo disorder and its early diagnosis. MPS type III represents 31.3% of all MPS diagnosed cases in a period of 3 years of study, which is a very high percentage. We could notice a high incidence of MPS IIIA which was not diagnosed before as the only enzyme that could be measured for the last 25 years in Egypt was alpha-N-acetylglucosaminidase (MPS IIIB) enzyme activity. Out of MPS type III cases, 87.7% of them were to consanguine parents. Counseling of the families is an important aspect of the genetic specialist and our practice—to increase the awareness of the families and explain the course and nature of the disease. Prenatal diagnosis is offered to all diagnosed families as well as carrier detection to the other siblings. Awareness should be raised by the medics regarding the problems caused by consanguineous marriages: medical and genetic.
No enzyme replacement therapy is available yet for MPS type III as it is mainly a neurologic disorder; still, MPS IIIB is the commonest type in the Mediterranean region 36 (49.3%) cases versus 15 (20.6%) cases of MPS IIIA and 22 (30%) cases of MPS III types C or D.
Availability of data and materials
The article contains all the data generated during this study.
Abbreviations
- MPS:
-
Mucopolysaccharidosis
- MPS III:
-
Mucopolysaccharidosis type III
- GAGs:
-
Glycosaminoglycans
- 2DE:
-
Two-dimensional electrophoresis
- HS:
-
Heparan sulphate
- CNS:
-
Central nervous system
- SGSH:
-
N-Sulphoglucosamine sulphohydrolase
- NAGLU:
-
N-Acetyl-α-glucosaminidase
- EDTA:
-
Ethylene-di-amine-tetra-acetic acid
- IEM:
-
Inborn error of metabolism
- ERT:
-
Enzyme replacement therapy
References
He´ron B, Mikaeloff Y, Froissart R et al (2011) Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet Part A. 155:58–68
Neufeld EF and Muenzer J. The mucopolysaccharidoses, in: C.R. Scriver, A.L. Beaudet, W.S. Sly, D. Valle (Eds.), The Metabolic and Molecular Bases of Inherited Disease, 3McGraw-Hill, NewYork, 2001; 3421–3452.
Muschol N, Koehn A, Von Cossel K et al (2023) A phase I/II study on intracerebroventricular tralesinidase alfa in patients with Sanfilippo syndrome type B. J Clin Invest 133(2):e165076
Muschol N, Giugliani R, Jones SA et al (2022) Sanfilippo syndrome: consensus guidelines for clinical care. Orphanet J Rare Dis 17:391
D’Avanzo F, Rigon L, Zanetti A, Tomanin R (2020) Mucopolysaccharidosis type II: one hundred years of research, diagnosis, and treatment. Int J Mol Sci 21(4):1258
Arunkumar N, Langan TJ, Stapleton M et al (2020) Newborn screening of mucopolysaccharidoses: past, present, and future. J Hum Genet 65(7):557–567
Valstar MJ, Neijs S, Bruggenwirth HT et al (2010) Mucopolysaccharidosis typeIII A: clinical spectrum and genotype-phenotype correlations. Ann Neurol 68(6):876–887
Wilkinson FI, Holley RJ, Langford Smith KJ et al (2012) Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One 7(4):e35787
Seker YB, Davison J, Jones SA et al (2021) Novel therapies for mucopolysaccharidoses type III. J Inherit Metab Dis 44(1):129–147
De Dong JG, Wevers RA, Liebrand-van SR (1992) Measuring urinary glycosaminoglycans in the presence of protein: an improved screening procedure for mucopolysaccharidosis based on dimethyleneblue. Clin Chem 38:803–807
Winner LK, Rogers M, Snel MF et al (2023) Biomarkers for predicting disease course in Sanfilippo syndrome: an urgent unmet need in childhood-onset dementia. J Neurochem 3(166):481–496
Fateen EM, Gouda AS, Ibrahim MM, et al (a) (2014) Fifteen years’ experience: Egyptian metabolic lab. Egypt J Med Hum Genet 15:379–385
Ibrahim DMA, Ali OSM, Nasr H et al (2023) Biochemical and mutational analyses of HEXA in a cohort of Egyptian patients with infantile. Tay-Sachs diseaseExpansion of the mutation spectrum. Orphanet J Rare Dis 18:52
Ben Turkia H, Tebib N, Azzouz H et al (2009) Incidence of mucopolysaccharidoses in Tunisia. La tuni Med 87:782–785
Fateen E, Abdallah ZY, Nazim WS et al (2021) Mucopolysaccharidoses diagnosis in the era of enzyme replacement therapy in Egypt. Heliyon 8(7):e07830
Temtamy SA and Loutife A (1970) Surgical aspects of cleft lip-cleft palate problems in Egypt. CL Palate J 578–581
Temtamy SA, Aglan MS and Meguid NA (2010) Genetic disorders in the Egyptians, in: A.S. Teebi (Ed.). Genetic Disorders Among Arab Population. Springer
Rouse CJ, Jensen VN, Heldermon CD (2023) Mucopolysaccharidosis type IIIB: a current review and exploration of the AAV therapy landscape. Neural Regen Res 19(2):355–359
Coutinho MF, Lacerda L, Alves S (2012) Glycosaminoglycan storage disorders: a review. Biochem Res Int 2012:1–16
Andrade F, Aldamiz-Echevarria L, Llarena M et al (2015) Sanfilippo syndrome: overall review. Pediatr Int 57:331–338
Zafeiriou DI, Savvopoulou-Augoustidou PA, Sewell A et al (2001) Serial magnetic resonance imaging findings in mucopolysaccharidoses III B (Sanfilippo’s syndrome B). Brain Dev 23:385–389
Jansen AC, Cao H, Kaplan P et al (2007) Sanfilippo Syndrome Type D: natural history and identification of 3 novel mutations in the GNS gene. Arch Neurol 64:1629–1634
Buhrman D, Thakkar K, Poe M et al (2013) Natural history of Sanfilippo syndrome type A. J Inherit Metab Dis 37(3):431–437
Fesslova V, Coru P, Sersale G et al (2009) The natural course and the impact of the therapies of cardiac involvement in mucopolysaccharidosis. Cardiol, Young 19:170–178
Muschol N, Koehn A, Von Cossel K et al (2023) A phase I/II study on intracerebroventricular tralesinidase alfa in patients with Sanfilippo syndrome type B. J Cin Invest 133(2):e165076
Braunlin E, Wang R (2016) Cardiac issues in adults with the mucopolysaccharidoses: current knowledge and emerging needs. Heart 102(16):1257–1262
Tomatsu S, Okamura K, Maeda H et al (2005) Keratan sulfate levels in mucopolysaccharidoses and mucolipidosis. J Inherit Metab Dis 28:187–202
Fateen EM, Ibrahim MM, Gouda AS, et al (b) (2014) Biochemical diagnosis of mucopolysaccharidoses over 11 years: the Egyptian experience. Egypt J Med Hum Genet 3(1):16–23
Alfadhel M, Benmeakel M, Hossain MA, Al Mutairi F, Al Othaim A, Alfares AA, Al Balwi M, Alzaben A, Eyaid W (2016) Thirteen-year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J. Rare Dis 11:126
Al-Jasmi FA, Tawfig N, Berniah A et al (2013) Prevalence and novel mutations of lysosomal storage disorders in United Arab Emirates: LSD in UAE. JIMD Rep 10:1–9
Krabbi K, Joost K, Zordania R et al (2012) The livebirth prevalence of mucopolysaccharidoses in Estonia. Genet Test Mol Biomarkers 16:846–849
Karpova EA, Voznyi YaV, Keulemans JL et al (1996) A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type A (MPS IIIA). J. Inherit. Metab. Dis 19(3):278–285
Breveglieri G, D’Aversa E, Finotti A et al (2019) Non-invasive prenatal testing using fetal DNA. Mol Diagn Ther 23:291–299
Harmatz P, Ketteridge D, Giugliani R et al (2005) Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 115:e681–e689
de Jong JG, Wevers RA, Laarakkers C, Poorthuis BJ et al (1989) Dimethylmethylene blue-based spectrophotometry of glycosaminoglycans in untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin Chem 35(7):1472–1477
Applrgarth ER, Elango EM, Priya S, Maya SR (1997) Diagnostic electrophoresis of glycosami-noglycans: a screening method for mucopolysaccharidosis. Indian J Pediatr 56:505–510
Tessitore A, Villani GRD, Di Domenico C et al (2000) Molecular defects in the a-N-acetylglucosaminidase gene in Italian Sanfilippo type B patients. Hum Genet 107:568–576
Cooper A, Hatton CF, Thornley M et al (1988) Human β-mannosidase deficiency Biochemical findings in plasma, fibroblasts, white cells and urine. J Inher Metab Dis 11:17–29
Lowry OH, Rosebrough NJ, Farr AL et al (1951) Protein measurement with folin phenol reagent. J Biol Chem 193:265–275
Rintz E, Podlacha M, Cyske Z et al (2023) Activities of (poly) phenolic antioxidants and other natural autophagy modulators in the treatment of Sanfilippo disease: remarkable efficacy of resveratrol in cellular and animal models. Neurotherapeutics 20:254–271
Auray-Blais C, Bhérer P, Gagnon R et al (2011) Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I. II and VI Mol Genet Metab 102:49–56
Al-Sannaa NA, Al-Abdulwahed HY, Al-Ghamdi MS (2017) Lysosomal storage disorders (LSDs): the prevalence in the Eastern Province of Saudi Arabia. Int J Neurol Dis 1(2):38–43
Shawky R, Zaki E, Fateen E et al (2008) Profile of Egyptian patients with mucopolysaccharidoses. Egypt J Med Hum Genet 9:11–21
Shawky RM, Abdel Monim MT, el Sebai AA et al (2001) Cardiac and ocular manifestations in Egyptian patients with mucopolysaccharidoses. East. Mediterr. Health J 7(6):981–991
Selim L, Abdelhamid N, Salama E et al (2016) Cardiovascular abnormalities in Egyptian children with mucopolysaccharidoses. J. Clin. Diagn. Res 10(11):SC05–SC08
Nijmeijer SCM, Van den Born LI, Kievit AJA et al (2019) The attenuated end of the phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to a non-neuronopathic phenotype. Orphanet J Rare Dis 14(1):1–10
Acknowledgements
The authors are grateful to all members of the Biochemical Genetics Department, National Research Centre, and also would like to express their appreciation to all participating MPS patients who accepted to be part of this research. Without their cooperation, this work would not have been accomplished.
Funding
This work was totally funded by the authors and did not receive any financial assessment by public or commercial funding agencies.
Author information
Authors and Affiliations
Contributions
Ekram M. Fateen developed the concept and designed the experiments, analyzed and interpreted the data, supervised the structure and design of the manuscript, and revised the manuscript. Seham M. S El Nakeeb supervised the structure and design of the manuscript, revised the manuscript, and approved the final version. Soha S. Nosier performed most of the experiments; contributed reagents, materials, analysis tools, or data; analyzed and interpreted the data; and drafted the manuscript. Mona Ibrahim contributed to the data, supervised the structure and design of the manuscript, and revised the manuscript. Mona El-Gammal performed the clinical examination of all patients, supervised the structure and design of the manuscript, and revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval was obtained from the Ethics Committee of the National Research Centre. Written consent including full information was obtained from the participants’ guardians.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Biochemical study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nosier, S.S., El Nakeeb, S.M.S., Ibrahim, M.M. et al. Biochemical diagnosis of Sanfilippo disorder types A and B. J Genet Eng Biotechnol 21, 112 (2023). https://doi.org/10.1186/s43141-023-00586-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00586-7