
Abstract
Disordered chromatin remodeling regulation has emerged as an essential driving factor for cancers. Imitation switch (ISWI) family are evolutionarily conserved ATP-dependent chromatin remodeling complexes, which are essential for cellular survival and function through multiple genetic and epigenetic mechanisms. Omics sequencing and a growing number of basic and clinical studies found that ISWI family members displayed widespread gene expression and genetic status abnormalities in human cancer. Their aberrant expression is closely linked to patient outcome and drug response. Functional or componential alteration in ISWI-containing complexes is critical for tumor initiation and development. Furthermore, ISWI-non-coding RNA regulatory networks and some non-coding RNAs derived from exons of ISWI member genes play important roles in tumor progression. Therefore, unveiling the transcriptional regulation mechanism underlying ISWI family sparked a booming interest in finding ISWI-based therapies in cancer. This review aims at describing the current state-of-the-art in the role of ISWI subunits and complexes in tumorigenesis, tumor progression, immunity and drug response, and presenting deep insight into the physiological and pathological implications of the ISWI transcription machinery in cancers.
Similar content being viewed by others
Background
Normal gene transcription is fundamental for cell physiology. The gene transcription program is executed by transcription complexes (TCs). Chromatin remodeling complexes (CRCs) are multisubunit TCs containing a series of ATP-dependent remodeling enzymes, which act as ‘molecular motors’ that couple ATP hydrolysis to the perturbation of histone-DNA contacts with respect to individual nucleosome core particles [1]. Based on the sequence homology of the catalytic ATPase and the accessory subunits, CRCs are divided into four main subfamilies: switch/sucrose nonfermentable (SWI/SNF), chromodomain-helicase DNA-binding protein (CHD), inositol-requiring mutant 80 (INO80) and imitation switch (ISWI) [1,2,3]. Generally, ISWI complexes help the initial histone–DNA complexes (pre-nucleosomes) to mature into canonical octameric nucleosomes and spacing of nucleosomes at relatively fixed distances [4, 5], and are involved in multiple aspects of cell physiology, such as transcriptional regulation [6,7,8,9], DNA damage response, repair and recombination [10,11,12,13].
To date, a growing number of preclinical and clinical studies have highlighted that ISWI complexes play critical pathological roles in tumorigenesis, tumor development, tumor immunity and drug response. ISWI subunits display multiple functions in affecting tumor cell phenotypes via regulation of oncogenic gene transcription. Somatic mutations, copy number changes or translocations have been identified that may produce gain/loss-of-function properties of ISWI subunits. Deregulation of ISWI complexes by abnormal expression or activity disrupts the normal interplay between ISWI subunits and TFs or facilitates the activity of oncogenic ISWI-containing TCs, which is expected to upset gene regulatory networks. Here, we provide unique insight into the implications of ISWI complexes and subunits in cancer.
The ISWI complex: types and composition
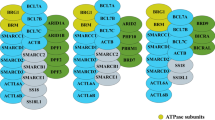
ISWI family is one of the best conserved ATPase families. It possesses highly conserved SWI2/SNF2 family ATPase domain, belonging to the superfamily of DEAD/H-helicases, that provides the motor for chromatin remodeling and a characteristic HAND-SANT-SLIDE domains with DNA binding activity [4]. Chromatin remodeling complexes containing the ISWI ATPase, including NURF, CHRAC and ACF, were, originally identified in Drosophila homologs, and later shown to be highly conserved in many other organisms, such as yeast and mammals [14]. In general, the CHRAC and ACF complexes seem to function in assisting nucleosome sliding [15]. NURF acts particularly in the epigenetic regulation, such as the regulation of higher-order chromatin structure [4]. The ISWI complexes display various variants over different species. For example, there are two ISWI catalytic subunit variants (Isw1 and Isw2) in Saccharomyces cerevisiae, forming 4 different complexes via association with different subunits [16]. Isw1 forms an Isw1a complex with Ioc3, which prevents the binding of basal Pol II transcription machinery to the promoter and inhibits transcription initiation [16]. In addition, Isw1 forms an Isw1b complex together with Ioc2 and Ioc4 subunits, which play a regulatory role in Pol II transcription elongation and termination [4]. Isw2 forms a complex with Itcl, Dpb4 and Dls1, which regulates the spacing of nucleosome series and play a remodeling function [4]. In Drosophila, it contains only one ISWI ATPase, which is a constituent of three complexes: dNURF, dACF and dCHRAC [17]. dNURF promotes H1 loading onto chromosomes in vivo and directly facilitated some genes-mediated transcription from chromatin templates, such as GAL4 [4, 18]. dNURF can be recruited by the transcriptional repressor dKen to repress STAT responsive-genes, blocking activation until signal thresholds are reached [19]. dACF is able to assist the assembly of chromatin with regular nucleosome spacing and is capable of catalyzing considerable ACF-dependent motility of entire chromatosomes within fully loaded arrays [20]. In mammals, a specific ISWI complex is composed of one ATPase subunit (SMARCA5 or SMARCA1) and one to three noncatalytic subunits [21]. Specially, both human ATPases, SMARCA1 and SMARCA5, form stable complexes with all regulatory subunits, which expands the ISWI complex members up to 16, including RSF-1/RSF-5 (SMARCA1/5 and RSF1), ACF-1/ACF-5 (SMARCA1/5 and BAZ1A), CHRAC-1/CHRAC-5 (SMARCA5/1, BAZ1A, CHRAC1 and POLE3), WICH-1/WICH-5 (SMARCA1/5 and BAZ1B), NoRC-1/NoRC-5 (SMARCA1/5 and BAZ2A), NuRF-1/NuRF-5 (SMARCA1/5, BPTF, RBBP7 and RBBP4), CERF-1/CERF-5 (SMARCA1/5 and CECR2) and BRF-1/BRF-5 (SMARCA1/5, BAZ2B) [14, 22] (Fig. 1A). Different subunits possess different functional domains and play distinct roles in complexes, which is summarized in Fig. 1B and Table 1.

Classification of ISWI family. Molecular components, functional domains, subcellular localization and targeting inhibitors for the ISWI family. A Sixteen different types of ISWI complexes are shown [14, 22, 48]. They harbor either SMARCA1 or SMARCA5 as ATPase subunits and 1–3 noncatalytic subunits. B Schematic representation of functional domains, protein subcellular localization, number of nucleotides and targeting inhibitors for each ISWI protein member
Genetic alterations in ISWI members are common in cancer and correlated with prognosis
The pan-cancer analysis of TCGA dataset and a growing number of studies revealed that ISWI subunits are abnormally expressed in human cancers (Fig. 2A and B). Whole genome, exome and transcriptome sequencing identified aberrant genetic states for ISWI subunits, including somatic mutations, abnormal copy numbers and gene fusions in various tumor types (Tables 2, 3 and 4). The genetic abnormality is a main factor determining the levels of some ISWI subunits in a particular type of cancer, and contributing to tumor phenotypes. For example, BPTF gene copy number is frequently amplified in human tumors, particularly in melanoma [23], neuroblastomas and lung cancers [24]. Specifically, in 67% of the BPTF-positive lung tumors cases, gain of the 17q24.3 locus was associated with poor prognosis (grade III). Knock-down of excessive BPTF negates the pre-malignant phenotype of highly proliferating lung embryonic fibroblasts cells [24].

Gene expression analysis of ISWI family members in human cancers. A Fold changes in gene expression of ISWI members across a variety of tumor types compared to the normal control, based on the TCGA dataset (only tumor types with more than 10 normal samples and tumor samples were selected). FPKM data were used as the expression profile, and the R language limma package was used to conduct variance analysis. Red indicates upregulation, and blue indicates downregulation (P < 0.05 after correction) in tumors. LogFC = log2 (average (tumor)/average (normal)). Full names for cancer abbreviations are shown in the abbreviation table. B A schematic diagram shows the most significant change in the mRNA expression levels of ISWI member genes in different types of cancer based on TCGA dataset analysis or literature summaries. The most significantly upregulated or downregulated genes are marked in red and in blue, respectively
Some ISWI subunits have been found to be closely related to patient prognosis. For example, serum RSF1 DNA levels are obviously upregulated in lung cancer patients and closely associated with TNM stage and lymph node metastasis [25]. In HER2+ breast tumors, high levels of BAZ1A are associated with detrimental relapse-free survival (RFS) and extremely poor overall survival (OS) [26]. The BAZ1A gene within the 14q12-q13 amplicon is frequently amplified in esophageal squamous cell carcinoma (ESCC), while this region harbors some oncogenic genes whose amplification leads to the development and progression of various types of tumors, such as small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [27]. The levels of BAZ2A are upregulated and associated with poor prognosis and recurrence in various tumors, such as prostate cancer [28]. Interestingly, high levels of BAZ2A correlate with the deletion of the PTEN gene in primary prostate cancer, which can be an indicator of poor prognosis [29]. BPTF is suggested to be a pan prognostic marker in various cancers. BPTF, which is highly expressed in NSCLC tumor tissues, is positively associated with advanced clinical stage, more lymph nodes and distant metastasis [30, 31]. In particular, it is strongly relevant to the risk for lung adenocarcinoma (LUAD) with EGFR mutations [32]. Moreover, BPTF, amplified in human breast tumors, is linked to shorter metastasis-free survival (MFS) [33, 34].
Roles of ISWI subunits and ISWI-containing transcription complexes in cancer development
The ATPase subunit of the major mammalian ISWI complex interacts with different proteins, forming multiprotein complexes that in some cases have many subunits. ISWI ATPases also interact with a variety of DNA-binding factors and cofactors, which are involved in malignant transformation and tumor progression. In contrast, noncatalytic subunits are critical for the functional diversity of ISWI complexes and perform specialized functions in cancers.
SMARCA1
SMARCA1 is broadly expressed in primary human tissues. However, multiple types of genetic abnormalities, including mutation, amplification and deletion lead to changes in SMARCA1 expression in tumors (Tables 2, 3 and 4). SMARCA1 plays an oncogenic or a tumor suppressor role, depending on tumor type. In breast, lung and cervical cancer, the oncogenic effects of SMARCA1 involve cell survival and cell cycle progression [35]. Inhibition of SMARCA1 increases the activity of caspase 9 by upregulating the expression of Apaf-1, which subsequently activates the caspase cascade [35]. In contrast, as a tumor suppressor, SMARCA1 is frequently silenced in gastric cancer due to aberrant DNA methylation. SMARCA1 knockdown promotes the growth of gastric tumor cells, accompanied by downregulation of genes related to cellular homeostasis [36]. Similarly, SMARCA1 is strongly expressed in normal melanocytes but is widely absent in malignant melanoma. Forced expression of SMARCA1 in melanoma cells inhibits cell proliferation and metastasis by attenuating Wnt/β-catenin signaling [37].
It has been found that SMARCA1 regulates normal stem cell or cancer stem cell (CSC) characteristics by interplaying with other types of cofactors. For example, WASH, an actin nucleating factor of the WASP family, assists the recruitment of SMARCA1-NURF complex to the c-Myc promoter and enhances gene transcription through VCA domain-dependent nuclear actin nucleation, which maintains the differentiation potential of long-term hematopoietic stem cells (LT-HSCs) to mature blood lineages [38] (Fig. 3A). Additionally, the SMARCA1-NURF complex was recruited to the OCT4 promoter by TF ZIC2 and initiated OCT4 transcription via elevated chromatin accessibility and H3K4 me3 levels of the OCT4 locus, which maintained the self-renewal of liver CSCs [39]. Correspondingly, high expression of the SMARCA1-NURF complex, ZIC2 and OCT4 are positively correlated with the clinicopathological stages of hepatocellular carcinoma (HCC) [39] (Fig. 3B).

Representative models of ISWI-IF/cofactor interplay. ISWI family proteins regulate cell phenotypes via association with TF/cofactor, in which the ISWI–TF/cofactor interplay is critical for optimal TF activity. A WASH recruits the NURF complex to the c-Myc promoter through VCA domain-dependent nuclear actin nucleation, which initiates c-Myc expression and maintains the self-renewal and differentiation potential of LT-HSCs [38]. B In liver CSCs. ZIC2 recruits the NURF complex to the OCT4 promoter, accompanied by enrichment of H3K4me3 and increased chromatin accessibility of OCT4, which activates OCT4 transcription and drives the self-renewal of liver CSCs [39]. C BPTF functions as a crucial cofactor of c-MYC required for tumorigenesis. The BPTF requirement for target recognition by c-MYC depends on the epigenetic context: it is dispensable for c-MYC to bind with H3K4me3-rich ‘high-affinity’ promoters and is also necessary for c-MYC to bind with ‘low-affinity’ sequences. BPTF leads to increased c-MYC recruitment to target DNA and regulates chromatin accessibility at c-MYC target promoters, thus increasing c-MYC target gene transcription and driving tumorigenesis [99,100,101]
SMARCA5
SMARCA5 and SMARCA1 share a high degree of amino acid sequence homology but appear to have different functions, as judged, for example, by their expression profiles and involvement in structurally and functionally different remodeling complexes [40]. SMARCA5 is frequently overexpressed in breast cancer [41], ovarian cancer [42], HCC [43] and acute myeloid leukemia (AML) [44, 45]. In breast cancer, the SMARCA5 expression level is positively correlated with tumor size, TNM stage and poor overall survival. Knockdown of SMARCA5 inhibits cell proliferation by arresting the G1 to S phase transition and suppresses MMP2-mediated invasion [41]. In ovarian cancer, the interplay between Rsf-1 and SMARCA5 contributes to tumor cell survival and growth. As a partnership of SMARCA5, induction of Rsf-1 expression not only facilitates translocation of SMARCA5 into nuclei where it colocalizes with Rsf-1 but also enhances SMARCA5 protein expression [42]. In HCC, SMARCA5 is able to promote cell survival and proliferation by increasing the protein level of β-catenin and enhancing its nuclear accumulation [43]. SMARCA5-containing fusion proteins caused by chromosomal translocations also contribute to carcinogenesis. For example, Sumegiet et al. identified that a chromosomal translocation t(4;22)(q31;q12) causes an in-frame fusion of the first 7 exons of EWSR1 to the last 19 exons of SMARCA5 in extraskeletal Ewing sarcoma/primitive neuroectodermal tumors. NIH3T3 cells expressing the EWSR1–SMARCA5 fusion exhibited anchorage-independent growth and formed colonies in soft agar, indicating this chimeric protein has tumorigenic potential [46].
SMARCA5 plays an important role in hematological malignancies. SMARCA5 was upregulated in CD34+ AML progenitors, and loss of SMARCA5 inhibited AML cell proliferation [44, 45]. Moreover, the binding site of SMARCA5-CTCF complex at the key hematopoietic regulator PU.1 was methylated in AML, which blocked the complex binding. Upon demethylation by 5-azacitidine (AZA) treatment, the occupancy of SMARCA5-CTCF complex was partly restored, which led to the downregulation of PU.1 transcription and induced myeloid differentiation, indicating that the occupancy of the SMARCA5-CTCF complex at the specific hematopoietic gene is critical for normal hematopoiesis [45] (Fig. 4A).

ISWI proteins mediate the chemotherapy response and chemical resistance. Some chemotherapy response- and chemoresistance-related genes are regulated by ISWI complexes. The transition of ISWI occupancy by changing DNA methylation levels or targeting ISWI protein/TF interactions may be two treatment strategies for pharmacological intervention. A In AML, the binding of the SMARCA5-CTCF complex at the PU.1 gene is blocked due to DNA methylation. Upon treatment by AZA-mediated DNA demethylation, the SMARCA5-CTCF complex is recruited to the enhancer of the PU.1 gene and inhibits its expression [45]. B The RSF1-SMARCA5 complex functions as a coactivator for NF-κB, consequently augmenting the expression of NF-κB-dependent chemoresistance-related genes, further resulting in paclitaxel resistance in ovarian cancer cells [56]. C Breast cancer cells with estrogen receptor positivity. After EB1089 (vitamin D analog) treatment, the occupancy of vitamin D receptor (VDR) and BAZ1B on CYP19A1 (encoding the enzyme aromatase, which can catalyze the conversion of androgens to estrogens) promoter are altered with the recruitment of VDR and dissociation of BAZ1B, which results in the inhibition of CYP19A1 transcription and contributes to the EB1089 treatment response by arresting the transformation of androgens to estrogens [70, 71]. D In HCC, the recruitment of the NuRD complex by SALL4 to the promoter of some tumor suppressors via an interaction of SALL4 with RBBP4. FFW (a highly potent SALL4-RBBp4 antagonist peptide) treatment abolishes the binding of SALL4 to RBBP4, which leads to the reactivation of tumor suppressors [111]
It should be noted that although SMARCA5 and SMARCA1 have overlapping expression patterns in some cases, the activities of SMARCA1- and SMARCA5-containing complexes likely are not fully redundant. For instance, the spatial and temporal expression patterns of SMARCA1 and SMARCA5 are distinct throughout murine development and in the adult animals, indicating that they may have non-overlapping functions [40]. Through analysis of TCGA database, the SMARCA5 and SMARCA1 gene have divergent patterns of expression in cancers, such as colon adenocarcinoma (COAD), LUAD and stomach adenocarcinoma (STAD) (Fig. 2A), suggesting that they have differential roles or even opposing roles. Indeed, SMARCA5-null embryos die during the peri-implantation stage due to hypoproliferation of the inner cell mass and trophoectoderm, while SMARCA1-null mice survive normally, but show hyperproliferation of cortical progenitors, resulting in an enlarged brain [47]. Therefore, changing the balance of SMARCA5 and SMARCA1 levels could be a potential therapeutic strategy after confirming their opposing expression levels and functions in one given tumor.
RSF complex
RSF1 functions as a nuclear protein with histone chaperon function and interacts with SMARCA5 or SMARCA1 to form RSF complexes that is involved in nucleosome assembly and ATPase-dependent chromatin remodeling [48] (Fig. 1A). The formation of RSF-5 complex is conducive to the nuclear import of SMARCA5, and stabilize SMARCA5 levels through preventing SMARCA5 protein degradation [49]. RSF1 is overexpressed in various tumor tissues based on recent studies and the TCGA database, including breast cancer, HCC, Glioma, etc. [50,51,52,53,54,55] (Fig. 2A). Meanwhile, overexpression of it is associated with poor overall survival, advanced clinical features and drug resistance in many types of cancer [50,51,52,53,54,55,56,57]. Mutations and abnormal copy numbers have also been found in a variety of tumors, such as acral melanoma, STAD, bladder cancer, etc. (Tables 2 and 3). As a binding partner, SMARCA5 is also overexpressed in the above-mentioned RSF1-overexpressing tumors, as reflected by recent reports [42, 50, 55] and the TCGA database analysis (Fig. 2A).
RSF1 was identified as an amplified gene that activated NF-κB–dependent gene expression such as XIAP (antiapoptotic gene) and PTGS2 (anti-inflammatory gene) for paclitaxel resistance by functioning as a coactivator for the NF-κB-SMARCA5 complex in ovarian tumor cells [56] (Fig. 4B). In some case, the functional characteristics of RSF1 for malignant transformation are associated with p53 expression or its mutation status. For example, in non-transformed cells, upregulation of RSF1 induces an ATM/p53-dependent DNA damage response, which leads to growth arrest and apoptosis in cells with wild-type p53. Inactivation of p53 by knockout of p53 alleles or p53 mutation reverses the growth inhibitory effects of RSF1 and favors outgrowth of cell clones [48, 58]. In contrast, upregulation of an cyclin E1-RSF1-SMARCA5 complex promotes tumorigenicity in the presence of p53mut RK3E cells, while tumorigenesis was not detected if they were expressed in a p53wt background [59]. In addition, Sehdev et al. found that p53 mutations probably represent a very early molecular genetic change in the development of high-grade serous carcinoma, which initiates a cascade of molecular changes, including upregulation of RSF1 [60]. These studies suggest that RSF1 may act as a driver gene for tumorigenesis, confer on cells with a p53 mutation background certain selective advantages with respect to neighbouring cells.
ACF complex
BAZ1A forms ACF complexes together with SMARCA1/5 or CHRAC complexes with SMARCA1/5, CHRAC1 and POLE3 (Fig. 1A). The ACF complex catalyzes both the relaxation of chromatin structure and the deposition of histones into extended periodic nucleosome arrays [61]. Within the ACF complex, BAZ1A stimulates SMARCA5 activity, regulates SMARCA5 remodeling properties and enhances the efficiency of nucleosome sliding during DNA replication and transcription, while SMARCA5 maintains the stability of the BAZ1A protein [11, 62]. Furthermore, CHRAC1 and POLE3 interaction facilitates nucleosome sliding and assembly mediated by the BAZ1A-CHRAC complex [63].
In normal cells, BAZ1A is involved in DNA double-strand breaks (DSBs) and DSB repair. Upon DNA damage, BAZ1A and SMARCA5 accumulate rapidly at DSBs and contribute to nonhomologous end-joining (NHEJ) repair of DSBs via recruitment of the KU70/80 complex and formation of the CHRAC complex. Inhibition of BAZ1A or SMARCA5 causes cells to become extremely sensitive to X-rays and chemical treatments with unrepaired DSBs [62]. The ACF complex is also associated with cellular senescence. Li et al. found that disruption of the ACF complex either by inhibition of BAZ1A or SMARCA5 resulted in the upregulation of the target gene SMAD3, which in turn activated p21 gene transcription and senescence-associated phenotypes of tumor cells [64].
Through TCGA database analysis, we found that BAZ1A, CHRAC1 and POLE3 are simultaneously up-regulated in a variety of tumors, such as esophageal carcinoma (ESCA), liver hepatocellular carcinoma (LIHC), STAD, breast invasive carcinoma (BRCA), etc. (Fig. 2A). Specially, various kinds of tumors, such as adrenocortical carcinoma, gastric cancer, melanoma, etc. display high frequency of abnormal copy numbers in at least two components of CHRAC complexes (Table 3). These data suggest that CHRAC complexes potentially play a driving role in cancers. Indeed, studies found that the BAZ1A gene within the 14q12-q13 amplicon is frequently amplified in ESCC, while this region harbors some oncogenic genes [27]. CHRAC1 gene, amplified on chromosome 8q24.3, is confirmed to be a driver gene regulating the proliferation/survival of clonogenic breast cancer cells [65]. In prostate cancer, POLE3 is upregulated by aberrant copy number amplification in cisplatin-resistant testicular germ cell tumors harboring the 9q32-q33.1 gain [66]. Knockout of the POLE3 gene increased the sensitivity of cells to an ATR inhibitor, a PARP inhibitor, and camptothecin [67].
BAZ1B
BAZ1B, a tyrosine-protein kinase belonging to the bromodomain family, was originally identified as a hemizygously deleted gene in Williams syndrome [68]. BAZ1B interacts with SMARCA1/5 to form WICH complexes (Fig. 1A). BAZ1B binds specifically to acetylated histones and plays a critical role in chromatin assembly, RNA polymerase I and III gene regulation, vitamin D metabolism, and DNA repair. Acetylation or phosphorylation (S158) of BAZ1B increases intrinsic kinase activities [69]. BAZ1B is overexpressed in breast and colorectal carcinoma cancer tissues, as well as in ESCA, STAD, etc. from TCGA database (Fig. 2A). Moreover, BAZ1B possesses high frequency of mutation in cutaneous cancer and lung carcinoma as well as high frequency of copy numbers amplification in breast cancer, gastric cancer and etc. (Tables 2 and 3).
In breast cancer, BAZ1B acts as an activator of a CYP19A1 gene that encodes the enzyme aromatase and ERα genes [70]. Treatment with the vitamin D analog EB1089 effectively inhibits aromatase-dependent growth of breast cancer cells by dissociating BAZ1B from the CYP19A1 promoter in a vitamin D receptor (VDR)-dependent manner, suggesting that BAZ1B is a potential drug target in breast cancer [70, 71] (Fig. 4C). In lung cancer, Meng et al. found that BAZ1B acts as an oncogene to promote tumor aggressiveness by inducing epithelial–mesenchymal transition (EMT) via activation of the PI3K/Akt and IL-6/STAT3 pathways [72].
BAZ1B functions not only in intranuclear transcription regulation but also in intercellular communication. Liu et al. found that the KRASG12 mutant induces the release of BAZ1B into the extracellular space by activating NRG3 transcription that can transport BAZ1B [73]. A BAZ1B-NRG3 complex in the extracellular space activates a series of oncogenic pathways such as the RAS-MAPK, NOTCH1 and JAK pathways, in normal colon cells carrying KRASWT and endows them with the ability to express NRG3 and release BAZ1B-NRG3 complexes, thereby promoting the transformation of surrounding normal cells in a cascaded manner [73]. BAZ1B is also involved in drug response. Blockade of extracellular BAZ1B restores the cetuximab sensitivity of colon cancer cells with mutant KRAS [73, 74]. Moreover, BAZ1B silencing synergistically potentiates the anti-growth effects of bortezomib in myeloma [75]. Phosphorylation of histones by BAZ1B is a key epigenetic mechanism that mediates UV and drug responses. In breast, prostate and bone tumor cells, BAZ1B, INTS3 and RUNX2 form UV-responsive complexes with the serine-139-phosphorylated isoform of H2AX (γH2AX). This complex supports histone displacement, DNA unwinding and stabilization of single-stranded DNA to mount an integrated response to DNA damage [76]. Like BAZ1A, BAZ1B has high-frequency mutations in skin melanoma cells (Table 2), which provides an intriguing link to UV-sensitivity. Upregulation of BAZ1B by imatinib is involved in apoptosis of CML cells by phosphorylation of H2AX at Tyr142 [77]. Moreover, BAZ1B acts as a key mediator that connects Ras/ERK signaling and phosphorylation of H2AX (H2AXY142ph). Ras-ERK1/2 induces BAZ1B degradation by increasing MDM2 expression to downregulate H2AXY142ph, which promotes cell growth and metastasis in gastric cancer [78].
NORC complex
NORC complexes, composed of BAZ2A and SMARCA1/5, recruit promoter-bound TTF-I, pRNA, and acetylated H4K16 to ribosomal DNA (rDNA) through BAZ2A’s TAM bromodomain domain [79, 80]. In human, rDNA instability is observed in cancers and premature aging syndromes. The NORC complex is critical for preventing cellular senescence through interaction with SIRT7, which maintains rDNA heterochromatin [81]. Depletion of BAZ2A unleashes rDNA instability, with excision and loss of rDNA gene copies, which in turn induced acute senescence [82].
BAZ2A are up-regulated in multiple tumors, such as prostate cancer, HCC, and chronic lymphocytic leukaemia (CLL) [28, 83, 84]. Moreover, BAZ2A are overexpressed in ESCA and STAD with high frequency of mutations according to TCGA database (Fig. 2A and Table 2). BAZ2A has been found to act as an oncogenic partner of TFs [85,86,87]. Upregulation of BAZ2A in HCC promoted the EMT progression of HCC cells by enhancing the interaction between β-catenin and TCF7L2 through its interaction with TCF7L2 [83]. Moreover, a fusion of ETV6 with the BAZ2A intron sequence generated by a cytogenetically cryptic rearrangement between 12p13 and 12q13 occurred in a pediatric case of pre-B acute lymphoblastic leukemia (ALL), which encodes a truncated form of ETV6 that leads to a pathogenic effect [88]. In a mouse prostate cancer organoid model, high BAZ2A expression is critical for the initiation of prostate cancer of luminal origin mediated by Pten-loss whereas it is dispensable once Pten-loss mediated transformation is established, which suggest that BAZ2A-mediated epigenetic second events sensitize for key genetic events that drive tumor development [89]. Mechanismly, BAZ2A binds to a class of inactive enhancers that are marked by H3K14ac via its bromodomain and represses the expression of genes implicated in aggressive and dedifferentiated prostate cancer [90].
CERF complex
The CERF complex remodels chromatin via nucleosome-dependent ATPase activity and is involved in DNA double strand break repair [91, 92]. Recently, CECR2 together with nine other genes, including ARHGAP21, ENSA, GPATCH8, KIAA1109, MGMT, PCDHB13, SELM, SPAG9 and WDR6, have been reported to be selected as the optimal gene combination that could be associated with the prognosis of patients with glioma [93]. Furthermore, a non-BET BRD inhibitor, NVS-CECR2–1, is able to kill SW48 colon tumor cells by targeting CECR2, suggesting that it may be an oncogene in some types of tumors [94]. However, the molecular and clinical implications of CECR2 in cancer remain largely unknown.
NURF complex
The NURF complex catalyzes ATP-dependent nucleosome sliding. Within the complex, BPTF, recognizes histone loci of methylation (H3K4me2/3) and acetylation (H4K12/16/20) by its PHD finger domain and bromodomain, respectively, and thus promotes gene transcription [31]. RBBP4 and RBBP7 are members of the WD-40 protein family and originally found associated with retinoblastoma proteins [95]. Both proteins are components of several multi-protein complexes that contain histone deacetylases (HDACs) and are involved in chromatin remodeling, histone post-translational modifications and regulation of gene expression, suggesting diverse functions for RBBP4/7 [95, 96].
BPTF
Genetic disorders of the BPTF gene are commonly seen in various cancer tumors. According to TCGA database, BPTF is upregulated in STAD, ESCA, LIHC, LUAD and downregulated mainly in kidney Chromophobe (KICH), thyroid carcinoma (THCA), kidney renal clear cell carcinoma (KIRC), prostate adenocarcinoma (PRAD), etc. (Fig. 2A). Moreover, BPTF possess both high frequency of mutation and abnormal copy number in angiosarcoma, COAD, ESCA, LIHC, LUAD, melanoma and urothelial carcinoma (Tables 2 and 3) [23, 24]. Particularly, BPTF was found to be the most frequently mutated gene observed (28.6%) in whole exome sequencing of Lacrimal Gland Adenoid Cystic Carcinoma (LGACC) [97]. Moreover, a NUP98-BPTF gene fusion in which a C-terminal chromatin recognition module of BPTF is fused to the N-terminal moiety of NUP98 has been identified in primary refractory acute megakaryoblastic leukemia (AMKL) that contributes to refining the NUP98 rearrangement subgroup of pediatric AMKL [98].
BPTF functions as a cofactor of oncogenic TFs. For example, BPTF-NURF complex is essential for c-MYC recruitment, chromatin accessibility and remodeling [99,100,101] (Fig. 3C). Knockdown of BPTF impairs tumor development with inactivation of the c-MYC and/or c-Myc target genes transcriptional program in preneoplastic pancreatic acinar cells and high-grade glioma [99, 101]. Correspondingly, BPTF expression is positively correlated with c-MYC gene signatures [99,100,101]. Particularly, the epigenetic environment is important for the genomic locations of BPTF. In bladder cancer, H2A.Z nucleosomes are enriched for the active histone modification H3K4me2/me3, which facilitates to recruit BPTF to H2A.Z target genes [102, 103]. The BPTF-c-Myc signaling axis is suggested to be a driving factor. For example, In the Eμ-Myc transgenic mouse model of aggressive B-cell lymphoma, BPTF allele is sufficient to delay lymphomagenesis, which display decreased c-MYC levels and pathway activity [104], suggesting that interruption of the c-Myc-BPTF-NURF complex interaction is a potential strategy for the treatment of c-Myc driven tumors.
RBBP7
Through the TCGA database analysis, RBBP4/7 and BPTF is simultaneously overexpressed in ESCA, LIHC, STAD etc. and downregulated in KIRC, KICH, kidney renal papillary cell carcinoma (KIRP) and prostate adenocarcinoma (PRAD) (Fig. 2A). RBBP4/7 expression were also consistent with HCC severity and prognosis together with BPTF [39]. Moreover, RBBP4 or RBBP7 and BPTF possess high frequency of abnormal copy number in angiosarcoma, breast cancer, pancreatic cancer, prostate cancer and gastric cancer (Table 3), and are mutated in colorectal adenocarcinoma, lymphoma, melanoma, etc. (Table 2).
A dual role of RBBP7 in tumor metastasis has been found to be closely related to recruitment of different RBBP7-containing complexes to EMT-related genes. For example, in cervical cancer, RBBP7 and BAF155 can be differentially recruited by NKX6.1 to suppress the vimentin gene and activate the E-cadherin gene, thus suppressing the EMT program [105] (Fig. 5A). In lung cancer, the region between − 428 and − 377 of the E-cadherin promoter contains a binding site for RBBP7, where RBBP7 functions as a transcriptional activator of the E-cadherin gene by binding to its promoter region, thereby repressing EMT progression [106] (Fig. 5B). Furthermore, RBBP7, as a corepressor, interacts normally with HNF1B to repress the SLUG gene and EMT program. However, in prostate cancer, upregulation of EZH2 suppresses the levels of the RBBP7/HNF1B transcriptional complex via direct inhibition of HNF1B expression, promoting SLUG transcription [107] (Fig. 5C). Additionally, in breast cancer, the transcription factor TWIST recruits an RBBP7/4-MTA2/Mi-2/HDAC1/HDAC2 complex to the proximal regions of the E-cadherin promoter for transcriptional repression via an interaction with RBBP7, which promotes breast cancer cell invasion and metastasis [108] (Fig. 5D).

ISWI-mediated EMT regulation is critical for tumor progression. ISWI proteins function as cofactors together with specific TFs to modulate EMT-related genes in a context-dependent manner. A In cervical cancer, NKX6.1 directly represses vimentin by interacting with the RBBP7 corepressor, accompanied by an increased H3K27me3 level. Meanwhile, NKX6.1 directly activates E-cadherin by interacting with the BAF155 coactivator with an increased H3K9 acetylation level [105]. B In lung cancer cells, RBBP7 acts as a transcriptional activator of the E-cadherin gene by binding to its promoter region, thereby repressing EMT progression [106]. C Normally, RBBP7, as a corepressor, interacts with HNF1B to repress SLUG transcription and EMT progression. In prostate cancer, upregulation of EZH2 suppresses the levels of the RBBP7/HNF1B transcriptional complex via direct inhibition of HNF1B expression, promoting SLUG transcription and EMT progression [107]. D RBBP7, as a corepressor, suppresses E-cadherin by interacting with TWIST and recruiting the complex to proximal regions of the E-cadherin promoter, thus inducing EMT [108]
RBBP4
RBBP4-containing complexes are involved in drug resistance. RBBP4 interacts with CBP/p300 to form a chromatin modifying complex that binds to MGMT, RAD51 and selected DNA repair genes. RBBP4 deletion enhanced temozolomide (TMZ) sensitivity, induced synthetic lethality to PARP inhibition and increased DNA damage signaling in response to TMZ in glioblastoma [109]. Release of RBBP4 complex occupancy or targeting the interactions between RBBP4 and oncogenic TFs provides increased opportunities for tumor intervention. For example, a BCL11A peptide inhibitor that blocks the recruitment of RBBP4-BCL11A complexes to BCL11A-targeted genes decreases aldehyde dehydrogenase-positive breast cancer stem cells (BCSCs) [110]. FFW functions as a specific SALL4–RBBP4 inhibitory peptide by targeting RBBP4 in HCC, releasing the expression of tumor suppressor PTEN [111] (Fig. 4D). In neuroblastoma cells, RBBP4 interacts with ARMC12 to facilitate the enrichment of PRC2 and H3K27me3 on tumor suppressive genes such as CADM1, EGLN3 and SMAD9, resulting in transcriptional repression. While a cell-penetrating inhibitory peptide blocks the interaction between ARMC12 and RBBP4, inhibiting aggressive cell behaviors [112].
However, although some ISWI dependent functions of RBBP4/7 as components of NURF complexes have been uncovered [38, 39, 113,114,115,116,117,118]. It should be noted that RBBP4 and RBBP7 not only participate in ISWI complexes, but also in several multi-protein transcription complexes [95, 110, 119]. Specially, ISWI participates in different larger protein complexes [120,121,122]. At present, a large proportion of studies mentioned above focus on the biological functions and/or mechanisms of RBBP4 and RBBP7 alone, which can not make a conclusion that RBBP4 or RBBP7 play those roles in some given tumors in a ISWI or non-ISWI dependent manners. It is possible that both ISWI or non-ISWI dependent mechanisms simultaneously contribute to the whole impact of RBBP4 or RBBP7.
BRF complexes
BAZ2B interacts with SMARCA1/5 to form BRF-1 and BRF-5 complexes, which induces remodeling of the DNA-bound mononucleosomes [22]. Crystal structure studies of purified BAZ2B protein show that its PHD domain interacts with unmodified histone H3K4 while the bromodomain interacts with the acetylated histone marks on H3K14 and H3K16 [123]. According to the TCGA database, BAZ2B is down-regulated in a variety of tumors, such as KICH, lung squamous cell carcinoma (LUSC), BRCA, etc. (Fig. 2A) Moreover, BAZ2B possess highly frequency mutations and abnormal copy number in some tumors, such as melanoma, bladder cancer, COAD, HCC, etc. (Table 2 and Table 3) However, its pathogenic mechanisms still remain unclear, and need to be further explored.
The ISWI and non-coding RNA functional connection
Recent studies showed that non-coding RNAs regulate chromatin modification and gene expression through their interactions with the ISWI complexes, which affect tumorigenesis and development in different ways. Non-coding RNAs can directly bind to the subunit of the ISWI complexes and serve as a guide to anchor the ISWI complexes. Moreover, Non-coding RNAs can be incorporated into the ISWI complexes and function as a scaffold to assemble the complex for chromatin remodeling. For example, in gastric tumor cells, the circ-DONSON recruits the SMARCA1-NURF complex to the SOX4 promoter by directly interacting with SMARCA1, which facilitates tumor cell development via enrichment of the active markers H3K27ac and H3K4me3 on the promoter and activation of SOX4 transcription [115] (Fig. 6A). In colorectal cancer, the SMARCA1-NURF complex is recruited by lnc-DLEU1 to the KPNA3 promoter and initiates KPNA3 expression via H3K27ac enrichment, which promotes tumor cell proliferation and migration [116] (Fig. 6B). SMARCA1-noncoding RNA complexes also play a vital role in tumor-initiating cells (TICs) and their properties. lncHOXA10 recruits the SMARCA1-NURF complex to the HOXA10 promoter and activates gene transcription, promoting the self-renewal of liver TICs [117] (Fig. 6C). Similarly, lncGata6 guides the localization of the SMARCA1-NURF complex to the Ehf promoter, which subsequently induces Lgr4/5 expression and activation of Wnt signaling in intestinal stem cells (ISCs) [118] (Fig. 6D). ISWI proteins are also involved in the noncoding RNA regulatory network. For example, RSF1 functions as an effector in lncRNA-induced drug resistance. In NPC, NEAT1/let-7a-5p axis regulates the cisplatin resistance by targeting RSF1 [124]. In ESCC, NSUN2-methylated lncRNA (NMR) directly bind to BPTF and potentially elevate MMP3 and MMP10 expression by the ERK1/2 pathway by recruiting BPTF to chromatin [125].

Representative models of ISWI-noncoding RNA interplay in cancer. ISWI family proteins are implicated in the regulation of oncogene transcription involving their interplay with noncoding RNAs. Generally, noncoding RNA recruits ISWI proteins to alter the chromatin environment and histone modification, thereby affecting oncogene transcription. A In gastric tumors, circ-DONSON recruits the SNF2 L-NURF complex to the SOX4 promoter by interacting with SMARCA1, which enriches the transcriptionally active markers H3K27ac and H3K4me3 and increases SOX4 promoter accessibility and transcription [115]. B In colorectal cancer, lncRNA DLEU1 recruits the SNF2 L-NURF complex to the KPNA3 promoter and promotes gene expression through the enrichment of histone modification H3K27ac, which further facilitates malignant behaviors [116]. C LncHOXA10 guides the localization of the SNF2 L-NURF complex to the HOXA10 promoter by directly interacting with SMARCA1, which further activates HOXA10 transcription and promotes liver tumor-initiating cell self-renewal [117]. D lncGata6 recruits the NURF complex to the Ehf promoter and induces gene expression by directly binding with SMARCA1, which further promotes Lgr4/5 (ISC cell-specific marker) expression and activation of Wnt signaling and thus maintains ISC stemness or promotes tumorigenesis [118]
In particular, recent studies found that circRNAs transcribed from the ISWI genes were involved in the etiology of cancer. cSMARCA5, a circRNA derived from exons 15 and 16 of the SMARCA5 gene (hsa_circ_0001445) was upregulated in prostate cancer. Knockdown of cSMARCA5 significantly repressed the cell cycle and promoting tumor apoptosis [126]. The presence of cSMARCA5 inhibits the growth and metastasis of HCC by acting as a sponge of miR-17-3p and miR-181b-5p to upregulate TIMP3 [127, 128]. Interestingly, cSMARCA5 and SMARCA5 displayed opposite expression in HCC. Both products of the SMARCA5 gene and cSMARCA5 are associated with poor prognosis, synergistically promoting the progression of HCC [43, 127, 128]. Circ-BPTF also plays an important role in tumor progression. For example, a circular RNA (hsa_circ_0000799) derived from BPTF exons attenuates the anti-oncogenic effect of miR-31-5p and consequently enhances RAB27A expression in bladder cancer [129].
Impact of ISWI proteins on tumor immunity
The levels of tumor-infiltrating immune cells within the tumor microenvironment (TME) and immune checkpoints on immune cells and tumor cells are two key factors determining the antitumor immune response. A correlation analysis in 33 tumor types based on the TCGA database showed that the gene levels of some ISWI members are strongly correlated with immune checkpoint gene levels and/or the tumor-infiltrating immune cell ratio within the TME, suggesting that ISWI members play a key role in the transcriptional regulation of immune-related genes and pathways (Fig. 7). To date, BPTF, POLE3, RSF1, BAZ1B and CECR2 have been found to be involved in immune cell development and activity and the regulation of immune-related genes.

The mRNA expression levels of ISWI members correlate with immune checkpoint gene levels and immune cell infiltration. The most significant correlation between ISWI members and immune checkpoints at the gene level or between the gene level of ISWI and immune cell infiltration in the 33 tumors from TCGA database are shown in the pie chart. Spearman’s method was used to calculate the correlation between ISWI family members and immune checkpoints (A-C) or immune cell infiltration (D) in 33 tumors. The CIBERSORT algorithm was used to calculate the proportion of different types of cells. Red indicates a positive correlation, blue indicates a negative correlation, and gray indicates no significant correlation. Cancer is abbreviated as follows: Esophageal carcinoma (ESCA), Liver hepatocellular carcinoma (LIHC), Rectum adenocarcinoma (READ), Colon adenocarcinoma (COAD), Lung adenocarcinoma (LUAD), Stomach adenocarcinoma (STAD), Head and Neck squamous cell carcinoma (HNSC), Bladder Urothelial Carcinoma (BLCA), Breast invasive carcinoma (BRCA), Lung squamous cell carcinoma (LUSC), Uterine Corpus Endometrial Carcinoma (UCEC), Kidney renal clear cell carcinoma (KIRC), Thyroid carcinoma (THCA), Kidney Chromophobe (KICH), Kidney renal papillary cell carcinoma (KIRP), Prostate adenocarcinoma (PRAD)
The BPTF-NURF complex plays vital roles in normal thymocyte development. This complex facilitates the differentiation of CD4+ and CD8+ cells to mature T cells by activating the transcription of key thymocyte maturation-specific genes (e.g., Egr1, Ikaros, IL-2, and IL7ra) in a T-cell antigen receptor signaling-dependent manner [130]. For example, BPTF facilitates the recruitment of the NURF complex to the promoter region of the Egr1 gene via interaction with TF SRF [2, 130]. Aberrantly high levels of the BPTF/NURF complex are supposed to promote cancer immune escape by impacting multiple types of immune cell activity. Deletion of BPTF activates a stimulatory molecule and inhibits an inhibitory antigen on the surface of mouse breast cancer and skin melanoma cells, inducing a T-cell mediated immune response [131]. In BPTF knockout mouse models of breast cancer and melanoma, BPTF depletion enhances antigen processing and CD8+ T cell cytotoxicity [131]. Mechanistically, BPTF knockout promotes the expression of the immunoproteasome subunits Psmb8 and Psmb9 and the antigen transporters Tap1 and Tap2, resulting in enhanced antigenicity and T-cell antitumor immunity [131]. Tumor cells with BPTF deficiency display increased CD8+ cell infiltration and CD8+ cell cytotoxicity, including the release of perforin, granzyme and IFN-γ and subsequent induction of the JAK/STAT and Fas/TRAIL pathways [132]. BPTF also stimulates heparanase expression, which reduces cell surface heparan sulfate proteoglycan and natural cytotoxicity receptor co-ligand abundance, therefore inhibiting NK cell antitumor activity in breast cancer cell lines [33]. Moreover, BPTF is vital for self-tolerance and immune homeostasis by stabilizing Treg function and Foxp3 expression in a cell-intrinsic manner [133]. Lack of BPTF in Foxp3-expressing Treg cells caused defective suppressive function of Treg cells, reduced Foxp3 expression, and increased lymphocyte infiltration in the nonlymphoid organs, ultimately leading to aberrant immune activation and an autoimmune syndrome [133].
In addition to BPTF, POLE3, RSF1 and BAZ1B also play vital roles in T cell-mediated immunity via regulation of cell cycle progression, antigen-specific cytotoxic T lymphocyte (CTL) responses and metabolic pathways. C-terminal mutants of POLE3 cause a cell-autonomous, stage-specific interruption of T and B cell development and interfere with the S-phase of cell cycle progression in lymphocytes, leading to severe peripheral lymphopenia [134]. RSF1-transduced dendritic cells induced a CTL response to produce IFN-γ and IL-12 against ovarian cancer cells in vitro, suggesting that RSF1-transduced dendritic cells may be a potential adjuvant immunotherapy [135]. BAZ1B functions as an amino acid sensor to modulate T cell antitumor immunity, cytokine production and survival [136]. Upregulated L-arginine levels have been reported to induce global metabolic changes, including a conversion from glycolysis to oxidative phosphorylation in activated T cells, and increase the generation of central memory-like cells with higher survival capacity and antitumor activity [136]. Interestingly, BAZ1B, PSIP1, and TSN could sense and modulate the L-arginine-dependent reprogramming of T cells toward increased survival capacity during the above process [136,137,138].
CECR2 has been found to be involved in the regulation of tumor immunity via macrophages. In this respect, upregulation of CECR2 in metastatic breast cancer is positively related to M2 macrophages and increases tumor metastasis by promoting M2 macrophage polarization to create an immunosuppressive microenvironment [139]. Mechanistically, CECR2 formed a complex with p65 through its bromodomain to activate the expression of the NF-κB target genes CSF1 and CXCL1, which are critical for macrophage-mediated immune suppression at metastatic sites [139]. Correspondingly, the inhibition of CECR2 by targeting bromodomain arrests immunosuppression by macrophages and inhibits breast cancer metastasis [139].
ISWI complexes as potential targets for cancer therapy
The emergence of ISWI members as oncology targets has spurred significant drug discovery efforts with the goal of identifying small molecule inhibitors that target their functional domains for therapeutic applications [140]. The bromodomain of ISWI is an attractive target for drug design. Bromodomains are readers of acetyl marks in histone tails or nonhistone proteins. From the perspective of bromodomain structure, the bromodomain structure possesses a hydrophobic acetylated lysine-binding pocket, which is optimal for the interaction of the charge-neutralized acetylated lysine and has comparatively low strength of protein-protein interaction, thus making this domain particularly targetable by small molecules that interfere with this interaction [141]. Several potent inhibitors are being developed for bromodomains present in the ISWI complexes. NVS-CECR2–1, the first selective inhibitor targeting the CECR2 bromodomain, inhibits chromatin binding of CECR2 bromodomain and displaces CECR2 from chromatin within cells. NVS-CECR2–1 exhibits cytotoxic activity against various human cancer cells mainly through inducing cell apoptosis [94]. GSK2801 is a selective and cell-active acetyl-lysine competitive inhibitor of BAZ2A/B bromodomains. Although GSK2801 has little effect on growth arrest as a single agent, it shows a strong synergistic effect on triple-negative breast cancer (TNBC) in combination with the BET bromodomain inhibitor (BETi) JQ1 [142]. Their synergistic inhibition on bromodomains induces apoptosis of TNBC by a combinatorial suppression of ribosomal DNA transcription and ETS-regulated genes. Arylurea (AU1) was the first small molecule selective inhibitor of the BPTF bromodomain and was selective for BPTF over BRD4 with moderate potency in an in vitro assay. AU1 treatment alters chromatin accessibility, decreases target gene c-MYC chromatin occupancy, weakens proliferative capacity, and leads to G1 arrest in mouse breast cancer cells [143].
The availability of crystal structures of ISWI subunits provided a unique strategy to develop their selective antagonists. To date, the crystal structures of ISWI subunits in some species have been elucidated to different resolution [144,145,146,147]. For example, Yan et al. crystalized a construct of ISWI containing the catalytic core from M. thermophila called MtISWI with 2.4 Å resolution, whose sequence is about 68, 68, and 58% identical to those of ISW1 and ISW2 of Saccharomyces cerevisiae and human SNF2h, respectively [148]. Besides, Chittori et al. determined a cryo-EM structure of the complex formed between nucleosome and the ATPase domain of the Chaetomium thermophilum ISWI [149]. In human, Armache et al. presented cryo-EM structures of the full-length form of the human ISWI remodeler at 3.4 Å, showing structures of SNF2h-nucleosome complexes with ADP-BeFx [150]. Tallant C et al. identified the high-resolution crystal structures of PHD zinc finger and BRD from of human BAZ2A and BAZ2B in complex with H3 and/or H4 histones from 1.6 Å to 1.99 Å [79].
Conclusions and perspectives
As key chromatin remodeling complexes, ISWI variably functions as a part of different larger protein complexes, and different kinds of tumor cells have diverse expression arrays of ISWI components, each with discrete functions. Different ISWI subunits have unique regulatory roles and determine ISWI complex functions, which confer a complex ability to regulate a variety of cellular events in normal and malignant cells (Table 5). ISWI actions in cancer are gene- or context-dependent, while cooperation with different TFs or TCs may produce distinct tumor properties. The abnormal activity and expression of ISWI subunits or the occurrence of aberrant composition in ISWI-containing complexes could lead to malignant phenotypes by upsetting gene regulatory networks. In some cases, the function of oncogenic TFs and fusion proteins is reliant on direct interactions with ISWI-containing complexes, where the ISWI family is critical for the optimal oncogenic activity of the complexes. Many factors, such as mutation, copy number variations (CNVs) or aneuploidy, induce imbalances in ISWI subunit stoichiometry in cancers. In general, too few of any one subunit limits the number of assembled complexes that can satisfy biological functions. Conversely, excess subunits outside their designated complexes are often nonfunctional and may have adverse effects. Therefore, stoichiometric imbalances of key “driver” components of ISWI complexes in a given tumor may be a key etiology. Balancing or restoring the normal expression levels and/or function of components within ISWI complex or between ISWI and other protein complexes promises exciting therapeutic insights. ISWI bromodomain inhibitors have shown synergistic or additive effects with numerous chemotherapeutic agents. In preclinical and clinical settings. ISWI subunits have recently emerged as immunoregulators that modulate immune cell phenotypes or the expression of immune checkpoints. Therefore, the combination of ISWI inhibitors with immune checkpoint inhibitors may be considered a major breakthrough in the treatment of malignancies.
Given that epigenetic regulation of the ISWI family globally affects the gene regulatory network, an ensemble of key cancer-driving ISWI subunits has been identified. Future studies need to identify more regulatory factors that dysregulate ISWI family expression and determine how their misexpression contributes to the pathogenesis of malignancies. Identification of more crystal structures of ISWI subunits and epigenetic mechanisms, thereby targeting specific ISWI domains, ISWI-mediated pathways and the TF/ISWI interface, may contribute to the development of novel inhibitors in ISWI-associated malignancies.
Availability of data and materials
Not applicable.
Abbreviations
- AZA:
-
5-azacitidine
- ALL:
-
Acute lymphoblastic leukemia
- AMKL:
-
Acute megakaryoblastic leukemia
- AML:
-
Acute myeloid leukemia
- AU1:
-
Arylurea
- BETi:
-
BET bromodomain inhibitors
- BRCA:
-
Breast invasive carcinoma
- BRD:
-
Bromodomain
- CSCs:
-
Cancer stem cells
- CRCs:
-
Chromatin remodeling complexes
- CHD:
-
Chromodomain-helicase DNA-binding protein
- CLL:
-
Chronic lymphocytic leukaemia
- COAD:
-
Colon adenocarcinoma
- CTL:
-
Cytotoxic T lymphocytes
- DSBs:
-
Double-strand breaks
- EMT:
-
Epithelial–mesenchymal transition
- ESCA:
-
Esophageal carcinoma
- ESCC:
-
Esophageal squamous cell carcinoma
- HCC:
-
Hepatocellular Carcinoma
- ISWI:
-
Imitation switch
- INO80:
-
Inositol-requiring mutant 80
- ISCs:
-
Intestinal stem cells
- KICH:
-
Kidney Chromophobe
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LGACC:
-
Lacrimal gland adenoid cystic carcinoma
- LIHC:
-
Liver hepatocellular carcinoma
- LT-HSCs:
-
Long term-hematopoietic stem cells
- LADC:
-
Lung adenocarcinoma
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- MFS:
-
Metastasis-free survival
- NPC:
-
Nasopharyngeal carcinoma
- NRG3:
-
Neuregulin 3
- NHEJ:
-
Nonhomologous end-joining
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PRAD:
-
Prostate adenocarcinoma
- RFS:
-
Relapse free survival
- RCC:
-
Renal cell carcinoma
- SCLC:
-
Small cell lung cancer
- STAD:
-
Stomach adenocarcinoma
- SWI/SNF:
-
Switch/sucrose non-fermentable
- TMZ:
-
Temozolomide
- THCA:
-
Thyroid carcinoma
- TCs:
-
Transcription complexes
- TFs:
-
Transcription factors
- TNBC:
-
Triple-negative breast cancer
- TICs:
-
Tumor initiating cells
- TME:
-
Tumor microenvironment
- UBC:
-
Urothelial bladder cancer
- VDR:
-
Vitamin D receptor
References
Wang Z, Wang P, Li Y, Peng H, Zhu Y, Mohandas N, et al. Interplay between cofactors and transcription factors in hematopoiesis and hematological malignancies. Signal Transduct Target Ther. 2021;6:24.
Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development (Cambridge, England). 2016;143:2882–97.
Wang P, Wang Z, Liu J. Role of HDACs in normal and malignant hematopoiesis. Mol Cancer. 2020;19:5.
Tyagi M, Imam N, Verma K, Patel AK. Chromatin remodelers: we are the drivers!! Nucleus. 2016;7:388–404.
Erdel F, Schubert T, Marth C, Längst G, Rippe K. Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc Natl Acad Sci U S A. 2010;107:19873–8.
Ma Y, Liu X, Liu Z, Wei S, Shang H, Xue Y, et al. The chromatin remodeling protein Bptf promotes posterior neuroectodermal fate by enhancing Smad2-activated wnt8a expression. J Neurosci. 2015;35:8493–506.
Judd J, Duarte FM, Lis JT. Pioneer-like factor GAF cooperates with PBAP (SWI/SNF) and NURF (ISWI) to regulate transcription. Genes Dev. 2021;35:147–56.
Barisic D, Stadler MB, Iurlaro M, Schübeler D. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature. 2019;569:136–40.
Aydin ÖZ, Marteijn JA, Ribeiro-Silva C, Rodríguez López A, Wijgers N, Smeenk G, et al. Human ISWI complexes are targeted by SMARCA5 ATPase and SLIDE domains to help resolve lesion-stalled transcription. Nucleic Acids Res. 2014;42:8473–85.
Aydin ÖZ, Vermeulen W, Lans H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014;13:3016–25.
Oppikofer M, Sagolla M, Haley B, Zhang HM, Kummerfeld SK, Sudhamsu J, et al. Non-canonical reader modules of BAZ1A promote recovery from DNA damage. Nat Commun. 2017;8:862.
Vidi PA, Liu J, Salles D, Jayaraman S, Dorfman G, Gray M, et al. NuMA promotes homologous recombination repair by regulating the accumulation of the ISWI ATPase SNF2h at DNA breaks. Nucleic Acids Res. 2014;42:6365–79.
Klement K, Luijsterburg MS, Pinder JB, Cena CS, Del Nero V, Wintersinger CM, et al. Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J Cell Biol. 2014;207:717–33.
Goodwin LR, Picketts DJ. The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol Cell Neurosci. 2018;87:55–64.
Chioda M, Vengadasalam S, Kremmer E, Eberharter A, Becker PB. Developmental role for ACF1-containing nucleosome remodellers in chromatin organisation. Development. 2010;137:3513–22.
Mellor J, Morillon A. ISWI complexes in Saccharomyces cerevisiae. Biochim Biophys Acta. 2004;1677:100–12.
Bouazoune K, Brehm A. ATP-dependent chromatin remodeling complexes in Drosophila. Chromosom Res. 2006;14:433–49.
Corona DF, Siriaco G, Armstrong JA, Snarskaya N, McClymont SA, Scott MP, et al. ISWI regulates higher-order chromatin structure and histone H1 assembly in vivo. PLoS Biol. 2007;5:e232.
Kwon SY, Xiao H, Glover BP, Tjian R, Wu C, Badenhorst P. The nucleosome remodeling factor (NURF) regulates genes involved in Drosophila innate immunity. Dev Biol. 2008;316:538–47.
Maier VK, Chioda M, Rhodes D, Becker PB. ACF catalyses chromatosome movements in chromatin fibres. EMBO J. 2008;27:817–26.
Witkowski L, Foulkes WD. In brief: picturing the complex world of chromatin remodelling families. J Pathol. 2015;237:403–6.
Oppikofer M, Bai T, Gan Y, Haley B, Liu P, Sandoval W, et al. Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 2017;18:1697–706.
Dar AA, Nosrati M, Bezrookove V, de Semir D, Majid S, Thummala S, et al. The role of BPTF in melanoma progression and in response to BRAF-targeted therapy. J Natl Cancer Inst. 2015;107:djv034.
Buganim Y, Goldstein I, Lipson D, Milyavsky M, Polak-Charcon S, Mardoukh C, et al. A novel translocation breakpoint within the BPTF gene is associated with a pre-malignant phenotype. PLoS One. 2010;5:e9657.
Hou Y-L, Chen H, Ge M-J, Li F-Z, Xue C-J, Wu Y-F, et al. Quantification of serum HBXAP DNA in lung cancer patients by quantitative fluorescent polymerase chain reaction. Mol Biol Rep. 2013;40:4091–6.
Pérez-Pena J, Páez R, Nieto-Jiménez C, Sánchez VC, Galan-Moya EM, Pandiella A, et al. Mapping Bromodomains in breast cancer and association with clinical outcome. Sci Rep. 2019;9:5734.
Yasui K, Imoto I, Fukuda Y, Pimkhaokham A, Yang ZQ, Naruto T, et al. Identification of target genes within an amplicon at 14q12-q13 in esophageal squamous cell carcinoma. Genes, Chromosomes Cancer. 2001;32:112–8.
Gu L, Frommel SC, Oakes CC, Simon R, Grupp K, Gerig CY, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet. 2015;47:22–30.
Pietrzak K, Kuzyakiv R, Simon R, Bolis M, Bär D, Aprigliano R, et al. TIP5 primes prostate luminal cells for the oncogenic transformation mediated by PTEN-loss. Proc Natl Acad Sci U S A. 2020;117:3637–47.
Dai M, Lu J-J, Guo W, Yu W, Wang Q, Tang R, et al. BPTF promotes tumor growth and predicts poor prognosis in lung adenocarcinomas. Oncotarget. 2015;6:33878–92.
Dai M, Hu S, Liu C-F, Jiang L, Yu W, Li Z-L, et al. BPTF cooperates with p50 NF-κB to promote COX-2 expression and tumor cell growth in lung cancer. Am J Transl Res. 2019;11:7398–409.
Shiraishi K, Okada Y, Takahashi A, Kamatani Y, Momozawa Y, Ashikawa K, et al. Association of variations in HLA class II and other loci with susceptibility to EGFR-mutated lung adenocarcinoma. Nat Commun. 2016;7:12451.
Mayes K, Elsayed Z, Alhazmi A, Waters M, Alkhatib SG, Roberts M, et al. BPTF inhibits NK cell activity and the abundance of natural cytotoxicity receptor co-ligands. Oncotarget. 2017;8:64344–57.
Koedoot E, Fokkelman M, Rogkoti V-M, Smid M, van de Sandt I, de Bont H, et al. Uncovering the signaling landscape controlling breast cancer cell migration identifies novel metastasis driver genes. Nat Commun. 2019;10:2983.
Ye Y, Xiao Y, Wang W, Wang Q, Yearsley K, Wani AA, et al. Inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol Cancer Res. 2009;7:1984–99.
Takeshima H, Niwa T, Takahashi T, Wakabayashi M, Yamashita S, Ando T, et al. Frequent involvement of chromatin remodeler alterations in gastric field cancerization. Cancer Lett. 2015;357:328–38.
Eckey M, Kuphal S, Straub T, Rümmele P, Kremmer E, Bosserhoff AK, et al. Nucleosome remodeler SNF2L suppresses cell proliferation and migration and attenuates Wnt signaling. Mol Cell Biol. 2012;32:2359–71.
Xia P, Wang S, Huang G, Zhu P, Li M, Ye B, et al. WASH is required for the differentiation commitment of hematopoietic stem cells in a c-Myc-dependent manner. J Exp Med. 2014;211:2119–34.
Zhu P, Wang Y, He L, Huang G, Du Y, Zhang G, et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J Clin Invest. 2015;125:3795–808.
Lazzaro MA, Picketts DJ. Cloning and characterization of the murine imitation switch (ISWI) genes: differential expression patterns suggest distinct developmental roles for Snf2h and Snf2l. J Neurochem. 2001;77:1145–56.
Jin Q, Mao X, Li B, Guan S, Yao F, Jin F. Overexpression of SMARCA5 correlates with cell proliferation and migration in breast cancer. Tumour Biol. 2015;36:1895–902.
Sheu JJ-C, Choi JH, Yildiz I, Tsai F-J, Shaul Y, Wang T-L, et al. The roles of human sucrose nonfermenting protein 2 homologue in the tumor-promoting functions of Rsf-1. Cancer Res. 2008;68:4050–7.
Wang Y, Qin J, Liu Q, Hong X, Li T, Zhu Y, et al. SNF2H promotes hepatocellular carcinoma proliferation by activating the Wnt/β-catenin signaling pathway. Oncol Lett. 2016;12:1329–36.
Stopka T, Zakova D, Fuchs O, Kubrova O, Blafkova J, Jelinek J, et al. Chromatin remodeling gene SMARCA5 is dysregulated in primitive hematopoietic cells of acute leukemia. Leukemia. 2000;14:1247–52.
Dluhosova M, Curik N, Vargova J, Jonasova A, Zikmund T, Stopka T. Epigenetic control of SPI1 gene by CTCF and ISWI ATPase SMARCA5. PLoS One. 2014;9:e87448.
Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol. 2011;24:333–42.
Stopka T, Skoultchi AI. The ISWI ATPase Snf2h is required for early mouse development. Proc Natl Acad Sci U S A. 2003;100:14097–102.
Sheu JJ-C, Guan B, Choi J-H, Lin A, Lee C-H, Hsiao Y-T, et al. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem. 2010;285:38260–9.
Min S, Choi YW, Yun H, Jo S, Ji JH, Cho H. Post-translational regulation of the RSF1 chromatin remodeler under DNA damage. Mol Cells. 2018;41:127–33.
Choi JH, Sheu JJ-C, Guan B, Jinawath N, Markowski P, Wang T-L, et al. Functional analysis of 11q13.5 amplicon identifies Rsf-1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res. 2009;69:1407–15.
He J, Fu L, Li Q. Rsf-1 regulates malignant melanoma cell viability and chemoresistance via NF-κB/Bcl-2 signaling. Mol Med Rep. 2019;20:3487–98.
Zhang X, Fu L, Xue D, Zhang X, Hao F, Xie L, et al. Overexpression of Rsf-1 correlates with poor survival and promotes invasion in non-small cell lung cancer. Virchows Arch. 2017;470:553–60.
Tai H-C, Huang H-Y, Lee S-W, Lin C-Y, Sheu M-J, Chang S-L, et al. Associations of Rsf-1 overexpression with poor therapeutic response and worse survival in patients with nasopharyngeal carcinoma. J Clin Pathol. 2012;65:248–53.
Wu D, Nie X, Ma C, Liu X, Liang X, An Y, et al. RSF1 functions as an oncogene in osteosarcoma and is regulated by XIST/miR-193a-3p axis. Biomed Pharmacother. 2017;95:207–14.
Zhao X-C, An P, Wu X-Y, Zhang L-M, Long B, Tian Y, et al. Overexpression of hSNF2H in glioma promotes cell proliferation, invasion, and chemoresistance through its interaction with Rsf-1. Tumour Biol. 2016;37:7203–12.
Yang Y-I, Ahn J-H, Lee K-T, Shih I-M, Choi J-H. RSF1 is a positive regulator of NF-κB-induced gene expression required for ovarian cancer chemoresistance. Cancer Res. 2014;74:2258–69.
Chen X, Sun X, Guan J, Gai J, Xing J, Fu L, et al. Rsf-1 influences the sensitivity of non-small cell lung cancer to paclitaxel by regulating NF-κB pathway and its downstream proteins. Cell Physiol Biochem. 2017;44:2322–36.
Kshirsagar M, Jiang W, Shih I-M. DNA damage response is prominent in ovarian high-grade serous carcinomas, especially those with Rsf-1 (HBXAP) overexpression. J Oncol. 2012;2012:621685.
Sheu JJ-C, Choi JH, Guan B, Tsai F-J, Hua C-H, Lai M-T, et al. Rsf-1, a chromatin remodelling protein, interacts with cyclin E1 and promotes tumour development. J Pathol. 2013;229:559–68.
Sehdev AS, Kurman RJ, Kuhn E, Shih I-M. Serous tubal intraepithelial carcinoma upregulates markers associated with high-grade serous carcinomas including Rsf-1 (HBXAP), cyclin E and fatty acid synthase. Mod Pathol. 2010;23:844–55.
Ito T, Levenstein ME, Fyodorov DV, Kutach AK, Kobayashi R, Kadonaga JT. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev. 1999;13:1529–39.
Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R, et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell. 2010;40:976–87.
Kukimoto I, Elderkin S, Grimaldi M, Oelgeschläger T, Varga-Weisz PD. The histone-fold protein complex CHRAC-15/17 enhances nucleosome sliding and assembly mediated by ACF. Mol Cell. 2004;13:265–77.
Li X, Ding D, Yao J, Zhou B, Shen T, Qi Y, et al. Chromatin remodeling factor BAZ1A regulates cellular senescence in both cancer and normal cells. Life Sci. 2019;229:225–32.
Mahmood SF, Gruel N, Chapeaublanc E, Lescure A, Jones T, Reyal F, et al. A siRNA screen identifies RAD21, EIF3H, CHRAC1 and TANC2 as driver genes within the 8q23, 8q24.3 and 17q23 amplicons in breast cancer with effects on cell growth, survival and transformation. Carcinogenesis. 2014;35:670–82.
Piulats JM, Vidal A, García-Rodríguez FJ, Muñoz C, Nadal M, Moutinho C, et al. Orthoxenografts of testicular germ cell tumors demonstrate genomic changes associated with cisplatin resistance and identify PDMP as a resensitizing agent. Clin Cancer Res. 2018;24:3755–66.
Su D, Feng X, Colic M, Wang Y, Zhang C, Wang C, et al. CRISPR/CAS9-based DNA damage response screens reveal gene-drug interactions. DNA Repair. 2020;87:102803.
Lu X, Meng X, Morris CA, Keating MT. A novel human gene, WSTF, is deleted in Williams syndrome. Genomics. 1998;54:241–9.
Liu Y, Zhang YY, Wang SQ, Li M, Long YH, Li YF, et al. WSTF acetylation by MOF promotes WSTF activities and oncogenic functions. Oncogene. 2020;39:5056–67.
Lundqvist J, Kirkegaard T, Laenkholm A-V, Duun-Henriksen AK, Bak M, Feldman D, et al. Williams syndrome transcription factor (WSTF) acts as an activator of estrogen receptor signaling in breast cancer cells and the effect can be abrogated by 1α,25-dihydroxyvitamin D(3). J Steroid Biochem Mol Biol. 2018;177:171–8.
Lundqvist J, Hansen SK, Lykkesfeldt AE. Vitamin D analog EB1089 inhibits aromatase expression by dissociation of comodulator WSTF from the CYP19A1 promoter-a new regulatory pathway for aromatase. Biochim Biophys Acta. 1833;2013:40–7.
Meng J, Zhang X-T, Liu X-L, Fan L, Li C, Sun Y, et al. WSTF promotes proliferation and invasion of lung cancer cells by inducing EMT via PI3K/Akt and IL-6/STAT3 signaling pathways. Cell Signal. 2016;28:1673–82.
Liu Y, Wang S-Q, Long Y-H, Chen S, Li Y-F, Zhang J-H. KRASG12 mutant induces the release of the WSTF/NRG3 complex, and contributes to an oncogenic paracrine signaling pathway. Oncotarget. 2016;7:53153–64.
Hennig EE, Mikula M, Rubel T, Dadlez M, Ostrowski J. Comparative kinome analysis to identify putative colon tumor biomarkers. J Mol Med (Berlin, Germany). 2012;90:447–56.
Zhu YX, Tiedemann R, Shi C-X, Yin H, Schmidt JE, Bruins LA, et al. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood. 2011;117:3847–57.
Yang S, Quaresma AJC, Nickerson JA, Green KM, Shaffer SA, Imbalzano AN, et al. Subnuclear domain proteins in cancer cells support the functions of RUNX2 in the DNA damage response. J Cell Sci. 2015;128:728–40.
Y-j Z, Lu C-r, Cao Y, Luo Y, Bao R-f, Yan S, et al. Imatinib induces H2AX phosphorylation and apoptosis in chronic myelogenous leukemia cells in vitro via caspase-3/Mst1 pathway. Acta Pharmacol Sin. 2012;33:551–7.
Dong C, Sun J, Ma S, Zhang G. K-ras-ERK1/2 down-regulates H2A.X(Y142ph) through WSTF to promote the progress of gastric cancer. BMC Cancer. 2019;19:530.
Tallant C, Valentini E, Fedorov O, Overvoorde L, Ferguson FM, Filippakopoulos P, et al. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure. 2015;23:80–92.
Anosova I, Melnik S, Tripsianes K, Kateb F, Grummt I, Sattler M. A novel RNA binding surface of the TAM domain of TIP5/BAZ2A mediates epigenetic regulation of rRNA genes. Nucleic Acids Res. 2015;43:5208–20.
Paredes S, Angulo-Ibanez M, Tasselli L, Carlson SM, Zheng W, Li T-M, et al. The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability. J Biol Chem. 2018;293:11242–50.
Yang L, Song T, Chen L, Soliman H, Chen J. Nucleolar repression facilitates initiation and maintenance of senescence. Cell Cycle. 2015;14:3613–23.
Li C, Wu W, Ding H, Li Q, Xie K. The transcription factor 7 like 2-binding protein TIP5 activates β-catenin/transcription factor signaling in hepatocellular carcinoma. Mol Med Rep. 2018;17:7645–51.
Hanlon K, Rudin CE, Harries LW. Investigating the targets of MIR-15a and MIR-16-1 in patients with chronic lymphocytic leukemia (CLL). PLoS One. 2009;4:e7169.
Santoro R, Li J, Grummt I. The nucleolar remodeling complex NoRC mediates heterochromatin formation and silencing of ribosomal gene transcription. Nat Genet. 2002;32:393–6.
Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65.
Guetg C, Scheifele F, Rosenthal F, Hottiger MO, Santoro R. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol Cell. 2012;45:790–800.
Panagopoulos I, Strömbeck B, Isaksson M, Heldrup J, Olofsson T, Johansson B. Fusion of ETV6 with an intronic sequence of the BAZ2A gene in a paediatric pre-B acute lymphoblastic leukaemia with a cryptic chromosome 12 rearrangement. Br J Haematol. 2006;133:270–5.
Pietrzak K, Kuzyakiv R, Simon R, Bolis M, Bär D, Aprigliano R, et al. TIP5 primes prostate luminal cells for the oncogenic transformation mediated by PTEN-loss. Proc Natl Acad Sci U S A. 2020;117:3637–47.
Peña-Hernández R, Aprigliano R, Carina Frommel S, Pietrzak K, Steiger S, Roganowicz M, et al. BAZ2Amediated repression via H3K14ac-marked enhancers promotes prostate cancer stem cells. EMBO Rep. 2021:e53014. https://doi.org/10.15252/embr.202153014.
Elliott J, Norton KA, Niri FH, McDermid HE. Reported DNA repair protein CECR2, which is associated with neural tube defects in mice, is not required for double-strand break repair in primary neurospheres. DNA Repair (Amst). 2020;94:102876.
Lee SK, Park EJ, Lee HS, Lee YS, Kwon J. Genome-wide screen of human bromodomain-containing proteins identifies Cecr2 as a novel DNA damage response protein. Mol Cells. 2012;34:85–91.
Zhang Y, Zhu J. Ten genes associated with MGMT promoter methylation predict the prognosis of patients with glioma. Oncol Rep. 2019;41:908–16.
Park SG, Lee D, Seo HR, Lee SA, Kwon J. Cytotoxic activity of bromodomain inhibitor NVS-CECR2-1 on human cancer cells. Sci Rep. 2020;10:16330.
Abbey M, Trush V, Gibson E, Vedadi M. Targeting human retinoblastoma binding protein 4 (RBBP4) and 7 (RBBP7). bioRxiv. 2018. https://doi.org/10.1101/303537.
Balboula AZ, Schultz RM. RBBP4 and RBBP7 regulate histone deacetylation during oocyte maturation in mouse. Biol Reprod. 2012;87:306.
Sant DW, Tao W, Field MG, Pelaez D, Jin K, Capobianco A, et al. Whole exome sequencing of lacrimal gland adenoid cystic carcinoma. Invest Ophthalmol Vis Sci. 2017;58:BIO240–6.
Roussy M, Bilodeau M, Jouan L, Tibout P, Laramée L, Lemyre E, et al. NUP98-BPTF gene fusion identified in primary refractory acute megakaryoblastic leukemia of infancy. Genes, Chromosomes Cancer. 2018;57:311–9.
Richart L, Carrillo-de Santa Pau E, Río-Machín A, de Andrés MP, Cigudosa JC, VJS-A L, et al. BPTF is required for c-MYC transcriptional activity and in vivo tumorigenesis. Nat Commun. 2016;7:10153.
Richart L, Real FX, Sanchez-Arevalo Lobo VJ. c-MYC partners with BPTF in human cancer. Mol Cell Oncol. 2016;3:e1152346.
Green AL, DeSisto J, Flannery P, Lemma R, Knox A, Lemieux M, et al. BPTF regulates growth of adult and pediatric high-grade glioma through the MYC pathway. Oncogene. 2020;39:2305–27.
Balbás-Martínez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Márquez M, Vazquez M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. 2013;45:1464–9.
Kim K, Punj V, Choi J, Heo K, Kim J-M, Laird PW, et al. Gene dysregulation by histone variant H2A.Z in bladder cancer. Epigenetics Chromatin. 2013;6:34.
Richart L, Felipe I, Delgado P, Andrés MP, Prieto J, Pozo ND, et al. Bptf determines oncogenic addiction in aggressive B-cell lymphomas. Oncogene. 2020;39:4884–95.
Li HJ, Yu PN, Huang KY, Su HY, Hsiao TH, Chang CP, et al. NKX6.1 functions as a metastatic suppressor through epigenetic regulation of the epithelial-mesenchymal transition. Oncogene. 2016;35:2266–78.
Li M, Aliotta JM, Asara JM, Wu Q, Dooner MS, Tucker LD, et al. Intercellular transfer of proteins as identified by stable isotope labeling of amino acids in cell culture. J Biol Chem. 2010;285:6285–97.
Wang J, He C, Gao P, Wang S, Lv R, Zhou H, et al. HNF1B-mediated repression of SLUG is suppressed by EZH2 in aggressive prostate cancer. Oncogene. 2020;39:1335–46.
Fu J, Qin L, He T, Qin J, Hong J, Wong J, et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275–89.
Kitange GJ, Mladek AC, Schroeder MA, Pokorny JC, Carlson BL, Zhang Y, et al. Retinoblastoma binding protein 4 modulates temozolomide sensitivity in glioblastoma by regulating DNA repair proteins. Cell Rep. 2016;14:2587–98.
Moody RR, Lo M-C, Meagher JL, Lin C-C, Stevers NO, Tinsley SL, et al. Probing the interaction between the histone methyltransferase/deacetylase subunit RBBP4/7 and the transcription factor BCL11A in epigenetic complexes. J Biol Chem. 2018;293:2125–36.
Liu BH, Jobichen C, Chia CSB, Chan THM, Tang JP, Chung TXY, et al. Targeting cancer addiction for SALL4 by shifting its transcriptome with a pharmacologic peptide. Proc Natl Acad Sci U S A. 2018;115:E7119–28.
Li D, Song H, Mei H, Fang E, Wang X, Yang F, et al. Armadillo repeat containing 12 promotes neuroblastoma progression through interaction with retinoblastoma binding protein 4. Nat Commun. 2018;9:2829.
Dar AA, Majid S, Bezrookove V, Phan B, Ursu S, Nosrati M, et al. BPTF transduces MITF-driven prosurvival signals in melanoma cells. Proc Natl Acad Sci U S A. 2016;113:6254–8.
Koludrovic D, Laurette P, Strub T, Keime C, Le Coz M, Coassolo S, et al. Chromatin-Remodelling complex NURF is essential for differentiation of adult melanocyte stem cells. PLoS Genet. 2015;11:e1005555.
Ding L, Zhao Y, Dang S, Wang Y, Li X, Yu X, et al. Circular RNA circ-DONSON facilitates gastric cancer growth and invasion via NURF complex dependent activation of transcription factor SOX4. Mol Cancer. 2019;18:45.
Liu T, Han Z, Li H, Zhu Y, Sun Z, Zhu A. LncRNA DLEU1 contributes to colorectal cancer progression via activation of KPNA3. Mol Cancer. 2018;17:118.
Shao M, Yang Q, Zhu W, Jin H, Wang J, Song J, et al. LncHOXA10 drives liver TICs self-renewal and tumorigenesis via HOXA10 transcription activation. Mol Cancer. 2018;17:173.
Zhu P, Wu J, Wang Y, Zhu X, Lu T, Liu B, et al. LncGata6 maintains stemness of intestinal stem cells and promotes intestinal tumorigenesis. Nat Cell Biol. 2018;20:1134–44.
Glancy E, Ciferri C, Bracken AP. Structural basis for PRC2 engagement with chromatin. Curr Opin Struct Biol. 2021;67:135–44.
Dirscherl SS, Krebs JE. Functional diversity of ISWI complexes. Biochem Cell Biol. 2004;82:482–9.
Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996;10:2117–30.
Emelyanov AV, Vershilova E, Ignatyeva MA, Pokrovsky DK, Lu X, Konev AY, et al. Identification and characterization of ToRC, a novel ISWI-containing ATP-dependent chromatin assembly complex. Genes Dev. 2012;26:603–14.
Arumugam K, Shin W, Schiavone V, Vlahos L, Tu X, Carnevali D, et al. The master regulator protein BAZ2B can reprogram human hematopoietic lineage-committed progenitors into a multipotent state. Cell Rep. 2020;33:108474.
Liu F, Tai Y, Ma J. LncRNA NEAT1/let-7a-5p axis regulates the cisplatin resistance in nasopharyngeal carcinoma by targeting Rsf-1 and modulating the Ras-MAPK pathway. Cancer Biol Ther. 2018;19:534–42.
Li Y, Li J, Luo M, Zhou C, Shi X, Yang W, et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 2018;430:57–66.
Kong Z, Wan X, Zhang Y, Zhang P, Zhang Y, Zhang X, et al. Androgen-responsive circular RNA circSMARCA5 is up-regulated and promotes cell proliferation in prostate cancer. Biochem Biophys Res Commun. 2017;493:1217–23.
Yu J, Xu Q-G, Wang Z-G, Yang Y, Zhang L, Ma J-Z, et al. Circular RNA cSMARCA5 inhibits growth and metastasis in hepatocellular carcinoma. J Hepatol. 2018;68:1214–27.
Liu Q, Cai Y, Xiong H, Deng Y, Dai X. CCRDB: a cancer circRNAs-related database and its application in hepatocellular carcinoma-related circRNAs. Database. 2019;2019:baz063.
Bi J, Liu H, Cai Z, Dong W, Jiang N, Yang M, et al. Circ-BPTF promotes bladder cancer progression and recurrence through the miR-31-5p/RAB27A axis. Aging. 2018;10:1964–76.
Landry JW, Banerjee S, Taylor B, Aplan PD, Singer A, Wu C. Chromatin remodeling complex NURF regulates thymocyte maturation. Genes Dev. 2011;25:275–86.
Mayes K, Alkhatib SG, Peterson K, Alhazmi A, Song C, Chan V, et al. BPTF depletion enhances T-cell-mediated antitumor immunity. Cancer Res. 2016;76:6183–92.
Peterson K. Investigating the role of Bptf in Immunoediting in breast cancer and melanoma; 2015.
Wu B, Wang Y, Wang C, Wang GG, Wu J, Wan YY. BPTF is essential for T cell homeostasis and function. J Immunol. 2016;197:4325–33.
Siamishi I, Iwanami N, Clapes T, Trompouki E, O'Meara CP, Boehm T. Lymphocyte-specific function of the DNA polymerase epsilon subunit Pole3 revealed by neomorphic alleles. Cell Rep. 2020;31:107756.
Sun L, Kong B, Sheng X, Sheu JJ, Shih Ie M. Dendritic cells transduced with Rsf-1/HBXAP gene generate specific cytotoxic T lymphocytes against ovarian cancer in vitro. Biochem Biophys Res Commun. 2010;394:633–8.
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829-842.e813.
Wu D. Innate and adaptive immune cell metabolism in tumor microenvironment. Adv Exp Med Biol. 2017;1011:211–23.
Wei J, Raynor J, Nguyen TL, Chi H. Nutrient and metabolic sensing in T cell responses. Front Immunol. 2017;8:247.
Zhang M, Liu Z, Aoshima K, An Y, Aoshima A, Chan L, Lang S, Sun H, Rutter S, Booth C, et al: CECR2 drives breast cancer metastasis by suppressing macrophage inflammatory responses.2020.
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. 2019;4:62.
Pérez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics. 2017;12:323–39.
Bevill SM, Olivares-Quintero JF, Sciaky N, Golitz BT, Singh D, Beltran AS, et al. GSK2801, a BAZ2/BRD9 Bromodomain inhibitor, synergizes with BET inhibitors to induce apoptosis in triple-negative breast cancer. Mol Cancer Res. 2019;17:1503–18.
Frey WD, Chaudhry A, Slepicka PF, Ouellette AM, Kirberger SE, Pomerantz WCK, et al. BPTF maintains chromatin accessibility and the self-renewal capacity of mammary gland stem cells. Stem cell reports. 2017;9:23–31.
Grüne T, Brzeski J, Eberharter A, Clapier CR, Corona DFV, Becker PB, et al. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell. 2003;12:449–60.
Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, et al. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature. 2011;472:448–53.
Harrer N, Schindler CEM, Bruetzel LK, Forné I, Ludwigsen J, Imhof A, et al. Structural architecture of the nucleosome remodeler ISWI determined from cross-linking, mass spectrometry, SAXS, and modeling. Structure. 2018;26:282–294.e6.
Yan L, Wu H, Li X, Gao N, Chen Z. Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling. Nat Struct Mol Biol. 2019;26:258–66.
Yan L, Wang L, Tian Y, Xia X, Chen Z. Structure and regulation of the chromatin remodeller ISWI. Nature. 2016;540:466–9.
Chittori S, Hong J, Bai Y, Subramaniam S. Structure of the primed state of the ATPase domain of chromatin remodeling factor ISWI bound to the nucleosome. Nucleic Acids Res. 2019;47:9400–9.
Armache JP, Gamarra N, Johnson SL, Leonard JD, Wu S, Narlikar GJ, et al. Cryo-EM structures of remodeler-nucleosome intermediates suggest allosteric control through the nucleosome. eLife. 2019;8:e46057.
Acknowledgements
Not applicable.
Funding
This work was supported by the grants from National Key Research and Development Program of China (Grant number 2108YFA0107800); National Natural Science Foundation of China [Grant numbers 81920108004, 81770107, 81702722 and 81470362]; National Postdoctoral Program for Innovative Talents [Grant number BX201700292]; Natural Science Foundation of Hunan Province [Grant number 2018JJ3703]; Science and Technology Key Project of Hunan Province [Grant number 2018SK21212]. The Fundamental Research Funds for the Central Universities of Central South University [Grant number 2021zzts0562, 2020zzts427].
Author information
Authors and Affiliations
Contributions
ZW designed this study. ZW, YL, HG, PW and YZ drafted the manuscript. JL and HP revised this manuscript. YL, YZ, YC and HL drew the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All of the authors are aware of and agree to the content of the paper and their being listed as a co-author of the paper.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Gong, H., Wang, P. et al. The emerging role of ISWI chromatin remodeling complexes in cancer. J Exp Clin Cancer Res 40, 346 (2021). https://doi.org/10.1186/s13046-021-02151-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02151-x