
Abstract
Cancer early detection and treatment response prediction continue to pose significant challenges. Cancer liquid biopsies focusing on detecting circulating tumor cells (CTCs) and DNA (ctDNA) have shown enormous potential due to their non-invasive nature and the implications in precision cancer management. Recently, liquid biopsy has been further expanded to profile glycoproteins, which are the products of post-translational modifications of proteins and play key roles in both normal and pathological processes, including cancers. The advancements in chemical and mass spectrometry-based technologies and artificial intelligence-based platforms have enabled extensive studies of cancer and organ-specific changes in glycans and glycoproteins through glycomics and glycoproteomics. Glycoproteomic analysis has emerged as a promising tool for biomarker discovery and development in early detection of cancers and prediction of treatment efficacy including response to immunotherapies. These biomarkers could play a crucial role in aiding in early intervention and personalized therapy decisions. In this review, we summarize the significant advance in cancer glycoproteomic biomarker studies and the promise and challenges in integration into clinical practice to improve cancer patient care.
Similar content being viewed by others

Introduction
Cancer continues to be a significant source of human suffering, with an annual toll of 9.9 million lives lost and an additional 18.1 million new cases reported each year [1]. Biomarker discovery holds a key to significant advancement in cancer prevention and treatment, with applications in risk estimation, disease screening and early detection, diagnosis, prognosis, therapeutic prediction, and disease monitoring. Cancers are heterogeneous at every level, including molecular and cellular variations, histopathology, and clinical manifestations, which makes biomarker discovery an essential need for the vision of precision medicine. Multi-omics integration, including next-generation sequencing (NGS) and comprehensive immunohistochemical (IHC) profiles, have enabled the development of targeted therapies (e.g., against EGFR, Her2/neu, ALK, BRAF, and others) and immune therapies. These personalized strategies leverage individual patients’ unique dynamic molecular and cellular changes to improve cancer survival [2].
Since the 1950s, the concept of cancer liquid biopsy involved examining blood protein biomarkers [3]. While many protein biomarkers have been developed and approved for cancer diagnosis and monitoring (e.g., PSA, CA125), improved specificity and sensitivity remain as critical challenges [4]. The modern concept of cancer liquid biopsy has shifted to the analysis of circulating tumor cells (CTCs) and circulating tumor-derived DNA (ctDNA) [5]. Highly sensitive and specific technologies based on multiplex PCR (mPCR) or next-generation sequencing (NGS) have rapidly evolved, enabling large-scale detection of genetic alterations in circulating nucleic acids, including gene mutations, fusions, deletions, amplifications, translocations, epigenetic changes, and DNA fragmentomics of ctDNA in liquid biopsy studies [6].
In addition to nucleic acid-based strategies, proteomic profiling of cancer liquid biopsies has gained significant traction [7]. Proteins directly govern normal cellular functions, and the aberrant protein expression, post-translational modification, subcellular localization, or function (caused by mutations and post-translational modifications) can drive oncogenesis and serve as targets for cancer therapies. Due to its versatility in detecting and quantitating biological molecules, mass spectrometry (MS) has superseded gel-based approaches of previous decades and is now the standard technology for proteomics analyses [8]. High-dimensional proteomic data offer unprecedented insights for biomarker discovery and clinical implementation [9]. Liquid biopsy protein profiles provide organ-specific information, surpassing DNA/RNA and aiding in tumor origin identification. Combining novel protein biomarkers with nucleic acids can significantly improve diagnostic accuracy.
Gaining insights into post-translational protein modifications substantially enhances the quantity of cancer-relevant information attainable from these proteins. Protein glycosylation, a common post-translational modification, involves attaching glycans to proteins primarily via N- (asparagine) or O- (serine/threonine) linkages [10]. It affects various physiological events, including protein folding and stability and trafficking, cell–cell interactions, differentiation, and the immune response [11]. Aberrant protein glycosylation is a hallmark of cancer, crucial in malignant transformation, tumor development, invasiveness, and metastatic disease [12,13,14]. It significantly influences cancer-immune system interactions, including immunosurveillance and immunoediting. Unique tumor-specific glycosylation patterns frequently manifest in the forms of increased branching of N-glycans, higher density of O-glycans, incomplete synthesis of glycans, neosynthesis, increased sialylation, and increased fucosylation [15, 16], which are promising targets for liquid biopsy for discrimination between the benign and cancerous cells. Despite the importance of glycosylation, only recently have these complex post-translational modifications been extensively studied, primarily owing to availability of new chemical and MS-based technologies.
This review provides a summary of glycobiology, its incorporation in cancer liquid biopsy for biomarker discovery, and the potential of novel technologies for analyzing the glycoproteome in clinical oncology. The recent breakthroughs in this field carry significant implications for personalized cancer care and enhanced clinical decision-making.
Enhancing liquid biopsies with glycoproteomic insights
Liquid biopsies
Liquid biopsy, the analysis of tumor-derived biomarkers in body fluids, offers a promising non-invasive approach to detect cancer and monitor treatment response. To date, most liquid biopsy research has focused on analyzing ctDNA [17, 18], CTCs [17, 19], or exosomes [20] isolated from blood. However, despite great potential, these types of liquid biopsies have shown limited utility in early cancer detection. This limitation mainly stems from technical challenges associated with sensitivity and specificity in light of tumor heterogeneity [21, 22].
Specifically, a key challenge for ctDNA- and CTC-based liquid biopsy is the rarity of these biomarkers, leading to detection sensitivity issues especially in early-stage disease, where ctDNA is present in fewer than 2 copies per mL of plasma [23]. CTCs shed from tumors can be present at concentrations as low as a few cells per mL of blood [24, 25] and isolating intact, viable CTCs from the background of billions of blood cells represents a significant technical hurdle [25, 26]. Additionally, detection of ctDNA of low mutant allele fractions remains a challenge [23]. Beyond rarity, circulating DNA bearing tumor-related signatures can also result from non-malignant processes or normal biological variation, causing decreased specificity for malignancy [21, 23, 27]. To our best knowledge, there are currently no diagnostic tests that address these limitations of DNA-based liquid biopsies, rendering the utility for screening population screening of “average risk” individuals uncertain. More work is needed to understand the (sub)populations that would benefit most from screening, or how liquid biopsies further guide ongoing clinical trials to identify the patient populations likely to respond to therapies.
Introduction of glycoproteins
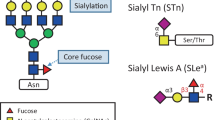
Enzymatic alterations of a protein after ribosomal synthesis are called post-translational modifications (PTMs) [28]. PTMs can impact structural and functional aspects of proteins, including protein stability, solubility, polarity, and folding [29, 30]. One of the most common and complex PTMs is protein glycosylation: the covalent attachment of glycans to a protein [31]. Most secreted and cell-surface proteins are glycosylated, and in many cases the glycan structures comprise the majority of the glycoprotein’s mass. The glycans on these extracellular proteins can affect the interactions of the protein with other proteins or the extracellular matrix, and they can also engage directly with glycan-binding receptors on other cells or in circulation. In this manner, glycosylation plays a key role both in normal and pathological processes such as cell trafficking, including tumor cell metastasis, and immune cell recognition of cells in the tumor microenvironment [32]. One of the most common forms of glycosylation is N-linked glycosylation, which comprises a glycan attached to the side chain nitrogen of an asparagine residue in an Asn-X-Ser or Asn-X-Thr sequon, where X can be any amino acid except proline [33, 34]. N-linked glycans share a common core structure on the reducing end consisting of five monosaccharide residues, two N-acetylglucosamines and three mannoses. The core structure is further extended with additional monosaccharides through glycosidic bonds, forming high mannose, complex, or hybrid type structures (Fig. 1a). Analysis of N-glycans is further simplified by the consensus sequence for a glycosylation site. Another form of glycosylation is O-linked glycosylation. In contrast to N-glycans, O-glycans represent a greater challenge for structural analysis as there is no unique consensus sequence for O-glycosylation. The difficulty of O-glycan analysis also lies in the lack of universal enzymes to release O-glycans. There are enzymes that can release mono- and disaccharides from proteins but not larger more complicated structures [35, 36]. Biosynthesis of N-Glycans and O-glycans include a series of competing enzymatic steps through nontemplate driven processes. Table 1 summarizes some key cancer-associated enzymes in the N- and O-glycosylation pathway. Each protein can have multiple glycosylation sites with different glycan structures (or glycoforms) at each site. N-linked glycans are attached to proteins co-translationally and concurrently with protein folding. Indeed, N-linked glycosylation is a key process that promotes correct folding and trafficking of proteins in the secretory pathway. Beyond their influence on the protein to which they are attached, N-linked glycans also contribute to a wide range of biological processes such as intra- and intercellular signaling and interactions with receptors on immune cells. Their central roles in multicellular biology have motivated considerable effort in the past two decades toward the comprehensive study of glycans and glycoproteins [35, 37]. Nonetheless, glycobiology has lagged other branches of molecular biology due to the significant complexity of these biopolymers compared to structurally simpler proteins and nucleic acids, which are linear, more easily sequenced, and amenable to direct genetic manipulation and amplification.

a Symbolic representation of N-linked glycan core and representative oligomannose, complex, and hybrid N-linked glycans; b Haptoglobin N-glycosylations sites. Symbols: blue square – N-acetylglucosamine (GlcNAc); yellow square – N-acetylgalactosamine (GalNAc); green circle – mannose; yellow circle – galactose; red triangle – fucose; purple diamond – N-acetylneuraminic acid (NeuAc, sialic acid)
An example of widely studied glycoproteins are haptoglobins (Hp), a hemoglobin-binding glycoprotein secreted by the liver (Fig. 1b). As a positive acute-phase response protein and the ninth most abundant protein in blood, Hp plays an important role in different biological processes. Hp’s primary physiological function is to bind free hemoglobin and suppress its oxidative activity [38]. As well, Hp has immune modulatory activity and can trigger the angiogenesis pathway. Structurally, Hp is a tetramer consisting of two heavy β-chain and two light chains: α1 and α2. Heavy and light chains of Hp are covalently linked to each other by disulfide bonds. There are four N-glycosylation sites on Hp’s 245-amino acid β-chain, positioned at Asn184, Asn207, Asn211 and Asn241. Hp’s α-chains possess no known glycosylation sites [39].
In recent years, many studies have focused on changes in Hp’s N-glycosylation pattern that are associated with different diseases, such as inflammatory disorders and malignancies. In cancer, the altered glycosylation of Hp is manifested at different levels such fucosylation (the addition of fucose residues to an underlying glycan), sialylation (the addition of sialic acid residues to an underlying glycan), branching, and the presence of so-called Lewis antigens (glycan structures with fucose attached to an N-acetyllactosamine core) [40]. Branching of N-glycans can be augmented by extensions of the core structure with GlcNAc residues through different linkage positions, leading to the production of bi-, tri‐ or tetra‐antennary glycans. Alterations in fucosylation, sialylation, and branching of Hp have been reported in different types of cancers [39]. For example, patients with hepatocellular carcinoma (HCC) display changes in α1-6 fucosylation and α2-6 sialylation of this abundant serum glycoprotein [41].
Neoplasia-induced hepatic reprogramming
As a sentinel organ filtering blood from the entire body, the liver is strategically positioned to detect, react, and potentially amplify the signals of distant tumors through changes in protein production and secretion. Tumorigenesis and cancer progression in the organs and tissues of human body can significantly impact the profile of hepatocytic synthesis and release of proteins including majority of circulating plasma proteins, leading to a “neoplasia-induced hepatic reprogramming” (Fig. 2) [51,52,53]. Thus, a comprehensive analysis of this ‘neoplasia-induced hepatic reprogramming’ via the hepatocyte-derived proteome represents a promising liquid biopsy approach to assess host response during oncogenesis and enable early cancer detection. For example, acute phase proteins including C-reactive protein (CRP), serum amyloid A (SAA), haptoglobin, and alpha-1-antitrypsin are dramatically upregulated in response to an inflammatory insult [39]. These hepatocyte-secreted proteins directly enter the bloodstream, serving as systemic biomarkers of a potentially local pathology [54].

Schematic overview of neoplasia-induced hepatic reprogramming. Top vessel provides a snapshot of homeostatic circulating glycoproteome. Mid vessel schematic depicts early hepatic reprogramming, including changes in glycosylation and abundance of circulating glycoproteins, of circulating liver-derived glycoproteome in pre-cancerous state. Bottom vessel depicts neoplasia-induced hepatic reprogramming with even further increase in glycosylation remodeling and abundance increase. At this stage tumor derived circulating DNA and circulating tumor cells can be detected in circulation
Beyond expression changes, aberrant glycosylation of hepatocyte-derived proteins occurs during malignancy [55, 56]. In fact, it is becoming increasingly recognized that the extent and type of glycosylation remodeling of acute phase proteins is unique for different pathologies, as opposed to a more general response as is the case of CRP in inflammatory state [57]. Glycans attached to proteins directly reflect cellular metabolism and homeostasis. For instance, the liver preferentially synthesizes specific glycoforms of alpha-1-acid glycoprotein in certain pathologies [58,59,60,61]. Increased branching, sialylation, and fucosylation across the plasma N-glycome are linked to tumor presence and progression [62,63,64].
While any individual protein change, including its glycosylation pattern, is unlikely to demonstrate sufficient specificity to serve as a biomarker on its own, global profiling of the plasma proteome enables high sensitivity detection of physiological states. For instance, mass spectrometry (MS) proteomics generates snapshots of hundreds to thousands of plasma proteins simultaneously and can provide detailed information on their glycosylation status. Coupled with machine learning algorithms, multivariate changes in hepatocyte-derived proteins discern subtle systemic effects of early oncogenesis. For example, large scale serum glycoproteomic profiling revealed unique signatures in clinical cohorts of malignant melanoma patients enabling immune checkpoint therapy response prediction [65]. A similar approach was used to detect advanced colonic adenomas as well as colorectal cancer (CRC) with high specificity and sensitivity [66]. Beyond MS, emerging proximity extension and aptamer assays directly quantify plasma proteins without relying on antibodies [67,68,69]. These technologies enable rapid, multiplexed measurement of hepatocyte-derived factors.
While detection sensitivity achieved by analyzing liver-derived serum glycoproteins seems high, more rigorous verification and validation through large, prospective cohorts is needed. Perhaps the most pressing question in liquid biopsy of circulating (glyco)proteins is determining specificity for tissue of disease origin, or the ability to detect actual tumor derived circulating (glyco)proteins. Nonetheless, increased awareness of the liver's role as a proxy amplification signal of systemic pathologies represents a promising strategy for liquid biopsy screening. We foresee that further integration of (glyco)proteomics, metabolomics and other complementary omics approaches offer exciting possibilities for unlocking the full potential of plasma-based liquid biopsies.
Glyco-analytical approaches
The potential of glycoproteins as cancer biomarkers has been recognized for more than four decades. However, outside of a handful of discrete molecules such as PSA and CA-125, their translation into clinical practice has been limited by inherent challenges, including the biological complexity of glycoproteins and limitations of analytical techniques [29]. The comprehensive analysis of glycoproteome requires the identification of the glycan structures as well as the proteins and sites to which they are attached. Over the past few years, multiple glyco-analytical approaches have been undertaken for the characterization of glycans and glycoproteins. Some of these approaches make use of monoclonal antibodies and lectins as highly specific glycan-binding proteins in ELISA formats [70, 71]. These lectin-antibody sandwich assays can be used to distinguish disease-associated from normal glycosylation patterns with a reasonable level of sensitivity and specificity. However, they have limited information content, which does not include the sites on a protein to which glycans are attached. Mass spectrometry (MS) techniques offer a wide range of additional capabilities and are now the key technology in identifying changes in protein glycosylation associated with disease. MS is a robust technique commonly used in cancer biomarker discovery due to its high sensitivity, compatibility with diverse biological matrices, potential for scaling, capability of providing structural information on very small amounts of biological samples, and large instrumental dynamic range. Over the past few years, MS-based methods which provide accurate mass and structural information have contributed significantly to the study of glycans and glycoproteins through glycomics (comprehensive characterization of glycan profile of biological samples) and glycoproteomics (comprehensive profiling of glycopeptides which provides both glycan and protein information) analyses.
Glycan analysis
Glycomics is the global analysis of glycans that are typically analyzed after cleavage from underlying protein (or lipid) scaffolds using enzymatic or chemical methods [32]. While glycomics are less complicated than glycoproteomics, such analyses provide information regarding aberrant glycosylation changes associated with disease. Mass-spectrometry based glycomics, in conjunction with exoglycosidases and database searches can provide valuable in-depth information about glycans. [72] N-Glycans are typically released from glycoproteins using an enzyme, peptide N-glycosidase F (PNGase F). PNGase F cleaves the linkage between the core GlcNAc and the asparagine residue in the NXT/S (X ≠ P) sequon [73]. PNGase F works for all classes of N-glycans, except the ones with α(1,3)-linked core fucose residues as observed in certain non-mammalian organisms. PNGase A is another enzyme that can be used to release all core fucosylated N-glycans with or without α (1,3)-linkage [74].
N-Glycan release is usually followed by a separation step to increase the specificity and sensitivity of glycan analysis prior to MS analysis. An efficient separation technique is often needed to characterize the structural heterogeneity of glycans, especially for complicated biological samples. Porous graphitized carbon (PGC), reversed phase (RPLC), and hydrophilic interaction chromatography (HILIC) are among the most common separation techniques that have been applied for glycan analysis [75,76,77,78]. Ruhaak et al. reviewed the key characteristics of each separation method [35].
Separated glycans are then identified using different MS-based techniques such as electrospray tandem mass spectrometry (ESI–MS/MS) or matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometry (MALDI-TOF MS/MS). Native mass spectrometry offers high sensitivity and ability to provide glycan structural information [79,80,81]. For glycan quantitation purposes, multiple reaction monitoring (MRM), which improves detection sensitivity by reducing chemical background noise and results in the identification of low abundance glycans, is among the most reliable MS-based methods [82]. While recent advancement in mass spectrometry-based methods and instrumentation have contributed significantly to the study of glycans, full structural analysis of glycans is still challenging. Different glycan structures might have the same mass and coelute on separation systems. Therefore, manual validation of a structural assignment from one technology using an orthogonal technology is required. Advancement of informatic tools in future can further alleviate this bottleneck.
From glycans to glycoproteins
A key question that often follows the identification of glycans by glycomics analysis is, which proteins carry the identified glycan structures? Protein glycosylation is known to be protein-specific, which means that different proteins can be glycosylated with different structures modifications even when they expressed in the same cell. Different factors such as common sequence motifs, protein structural conformation, and unique physicochemical patches surrounding the glycosylation site can potentially contribute to the protein-specific nature of glycosylation. However, the exact molecular basis for protein-specific glycosylation is still unknown. Glycoproteomics can provide both glycan and protein information.
Glycoprotein analysis
Glycoproteomics is based on global profiling of glycopeptides and consists of simultaneous identification of proteins and corresponding glycans [83]. Therefore, glycoproteomics links proteomic and glycomic analyses and ideally enables the identification of the detailed molecular features of all glycoproteins in a biological sample [84]. In such analyses, each glycan- or glycopeptide identified by MS is evaluated independently, or by grouping glycan structures that have similar structural properties into derived glycosylation traits. Most of the current MS-based glycosylation analyses have focused on the large-scale characterization of glycopeptides obtained by proteolytic digestion of complex samples [83]. Glycopeptide analysis by mass spectrometry is challenging due to several factors, for example, glycosylation tends to diminish the intensity of glycopeptide signals compared to signals from unmodified peptides. In addition, different glycoforms of the same peptide can dilute the intensity of the MS signal over several species [85]. Like glycomics analysis, different enrichment strategies are applied in glycoproteomics workflows to reduce competition for charge with the highly abundant and easily ionizable non-glycosylated peptides. Among common enrichment methods, multi-lectin affinity and HILIC are commonly used for untargeted glycoproteomics. On the other hand, immunoaffinity and single-lectin affinity are mostly applied to the targeted enrichment of one or a small group of glycoproteins [86]. Over the past few years, several groups have used various enrichment techniques to overcome some of the above-described issues associated with the analysis of glycoproteins [87,88,89]. Enriched glycopeptides are commonly separated using reversed-phase liquid chromatography (RPLC) prior to introduction to MS. RPLC separates glycopeptides based on the interaction of the stationary phase with the peptide backbone and not the glycan part. Therefore, it cannot separate different glycoforms of the same peptide backbone. Moreover, highly hydrophilic glycopeptides show poor retention on RPLC [90]. To address these limitations of RPLC, alternative separation methods such as capillary electrophoresis (CE) and ion mobility (IM) have been applied to separate glycopeptides [91, 92]. Untargeted discovery of cancer-associated glycopeptide biomarkers requires high resolution MS instrumentation, such as an orbitrap MS instrument, using data-dependent or data-independent acquisition (DDA or DIA) methods [93, 94]. Currently the best suited MS approach for the analysis of targeted cancer-related glycopeptides is multiple reaction monitoring (MRM). During MRM, predetermined precursor glycopeptides are selected for further collision-induced dissociation and the appearance of several diagnostic product ions are monitored. Therefore, MRM offers high analytical specificity and sensitivity. MRM analysis with addition of isotopically labeled internal standards enables absolute quantification of target glycopeptides [74, 87, 95].
Biomarker development
While with recent advancement of analytical technologies, there has been considerable improvements in understanding the role of glycosylation in cancer, there is still a large gap between our findings of glycoprotein biomarkers, often with diagnostic performance, and full clinical implementation of the cancer biomarkers. Development of biomarkers involves multiple steps. First, biomarkers should be discovered and validated using small sample sizes. Candidate biomarkers are then subjected to two types of validation following the discovery phase: 1) Analytical validation, which evaluates how accurately and reproducibly the analyte(s) of interest is measured within the patient samples; 2) Clinical validation, which aims to assess the robustness of the test result and its correlation with the clinical phenotype [96]. Clinical validation phase typically requires a relatively large and independent sample set to demonstrate the clinical validity and utility of the biomarker. Key considerations in biomarker discovery and validation study design include the patient population, prevalence of the disease, sample source (retro- vs. prospective), sample size, and sample type.
Tumor-originated protein biomarkers
Conventionally, cancer diagnostic tests are based on the levels of single biomarkers. Although most of the FDA-approved protein biomarkers for cancer are monitored based on their concentrations at the protein level only, the majority of these proteins are found or predicted to be glycosylated and their specific glycosylated forms associated with cancer progression have shown higher performance than protein levels alone in either clinical settings or research laboratories [74, 97]. The most common glycoprotein biomarkers and their cancer-associated glycoforms are listed in Table 2. Glyco-variant based assays for these protein biomarkers and their diagnostic power compared to conventional tests based on protein concentrations have been reviewed recently [29]. Analytical approaches for these glycosylated single protein biomarkers usually involve using an antibody to capture the protein, followed by glyco-profiling using lectin array to discover cancer-specific glycoforms, and then a selected lectin to detect the one specific group of glycoproteoforms associated with cancer [70, 98, 99]. For example, alpha-fetoprotein (AFP) is a well-established protein biomarker for hepatocellular carcinoma (HCC), and its core-fucosylated form AFP-L3 has been approved by FDA as a biomarker widely used in combination with the total AFP concentration for risk assessment of patients with chronic liver disease for development of HCC [100,101,102]. In a study of 689 patients with cirrhosis and/or chronic hepatitis B including 44 diagnosed HCC patients, the area under the receiver operating characteristic (ROC) curve (AUC) by combining AFP and AFP-L3 is 0.83 compared to 0.77 with AFP alone [103].
Another tumor-originated glycoprotein that has been extensively studied is prostate-specific antigen (PSA). PSA has remained the gold standard biomarker for prostate cancer screening although PSA testing has been controversial due to its poor performance [122, 123]. PSA has a single N-glycosylation site occupied by complex-type glycans. Detection of specific glycoforms such as ɑ2-3 sialylated, core-fucosylated, or LacdiNAc-modified glycans has shown potential as a novel tool for improving the clinical utility of PSA test [124,125,126,127,128]. Diagnostic performances of these glycoforms compared with total PSA alone are summarized in Table 3. In two small-scale studies with 100 or fewer subjects, ɑ2-3 sialylated PSA had much higher AUCs of 0.834 and 0.971 compared with the AUCs of total PSA at 0.506 and 0.806 respectively [129, 130]. In another relatively large-scale study, a total of 414 patient samples with 100 samples in the training set and 314 samples in the test set were analyzed using a magnetic bead-based immunoassay. With a sensitivity of 90.6%, the test set specificities were at 20.5% for total PSA and 64.2% for ɑ2-3 sialylated PSA [131]. Similar trends of increased performance were observed for core-fucosylated PSA and LacdiNAC-glycosylated PSA where AUCs are improved to 0.94 and 0.851, respectively [132, 133]. In more recent larger cohort studies, the trends of performance improvements are validated although not as significant. With a sensitivity of 90%, the specificity of core-fucosylated PSA was 36% for a cohort of 252 men, and that of LacdiNAc-glycosylated PSA was 48.6% for a cohort of 718 men [134, 135]. Findings from these independent studies have demonstrated the potential of glyco-PSA for prostate cancer detection. On the other hand, PSA has also been comprehensively studied by MS methods owing to its smaller protein size and simplicity of glycosylation profile with only one glycosylation site. In 2012, an interlaboratory study was conducted to compare MS-based analytical methods for the characterization of human seminal PSA and PSA-high isoelectric point isoform [136]. The collected datasets demonstrated the high heterogeneity of PSA glycosylation with 61 glycoforms observed by multiple laboratories. However, validation on large prospective cohorts is needed to demonstrate the clinical utility of these novel glycoforms as biomarkers.
Human carcinoembryonic antigen (CEA) is the most frequently used biomarker for colorectal cancer screening and monitoring. Unlike PSA, CEA has 28 potential N-glycosylation sites, making it an even more heterogeneous and variable target to characterize. Early studies revealed tumor-specific glycosylation of CEA and their interaction with dendritic cells indicating the potential of monitoring these glycoforms as novel biomarkers for early detection of CRC [137, 138]. Lectin array analysis of colorectal cancer samples showed the levels of fucosylated and mannosylated glycans are increased in tumor-associated CEA while branched and bisecting N-glycans are decreased [117]. The site-specific glycopeptide analysis of CEA remains challenging, but emerging LC–MS techniques have made exploratory studies possible. In one study, CEA proteins purified from human colon carcinoma and human liver metastases of colorectal carcinoma cells were characterized with 893 different N-glycopeptides and 128 unique N-glycan compositions identified from 21 out of 28 potential N-glycosylation sites. The site-specific glycosylation changes such as increased bisection and branching, incomplete galactosylation or LacNAc elongation on highly branched structures, moderate levels of sialylation, and extremely high levels of fucosylation were observed, providing another layer of potential in differentiating tumor-specific CEA [118]. Nonetheless, similar comprehensive profiles of serum CEA are still unavailable due to the low concentration of circulating CEA in blood. Besides CEA, several other glycoproteins of the carcinoembryonic antigen-related cell adhesion molecule (CEACAM) family are emerging as cancer biomarkers, such as CEACAM1, CEACAM5 (synonym of CEA) and CEACAM6 for pancreatic cancer [119, 120].
Cancer antigen 125 (CA125) for ovarian cancer as well as cancer antigen 15–3 (CA15-3) and cancer antigen 27–29 (CA27-29) for breast cancer are based on mucin proteins mucin-16 (MUC16) and mucin-1 (MUC1), respectively [139]. These mucin proteins are heavily glycosylated, and lectin-based studies have shown the associations of cancer progression with altered mucin glycosylation (Table 2). However, few studies have been conducted to reveal their site-specific molecular structures due to the challenges to process them for mass spectrometry analysis [140]. Comprehensive structural analyses have only been made possible in the past couple of years through the characterization and commercialization of mucinases such as StcE [141, 142]. The rise of mucinomics enabled by advancing sample preparation and MS techniques, although at its early stage, has provided the potential to unravel the complex relationship between mucins and cancer progression.
Despite the potential of glycosylation analysis of tumor-associated single protein biomarkers, the clinical applicability of these novel biomarkers remains challenging and the reported study scales have been small. Lectin assays have high sensitivity to detect overall glycosylation changes with high throughput. Nonetheless, the specificities of lectins are often broad and vaguely defined, which limits the improvement in assay performance when specific glycoforms are identified as more potent biomarkers [71, 143]. MS-based methodologies provide higher specificity that have made comprehensive site-specific profiling of these glycosylated protein biomarkers possible, but technical barriers towards clinical utility include the need of large sample volume to purify sufficient proteins, non-specific binding of interferences to antibodies during purification due to the low concentrations of these target proteins in serum, and the relatively high cost and low throughput compared to lectin assays. Therefore, to achieve breakthroughs in diagnostic performance, development of novel biomarkers with clinical applicability are necessary.
Glycoproteomics-based multi-marker assays
While single biomarkers can provide easily interpretable readouts to physicians and patients, multi-marker tests can often achieve higher diagnostic power [144, 145]. Recent advances in MS-based technologies have provided an opportunity to discover and validate a panel of glycoproteomic biomarkers for disease detection and monitoring. In most settings, biomarker discovery is performed on retrospective study samples while specimens and data collected from prospective trials are required for clinical validation. Therefore, new glycoproteomics-based multi-marker assays must prove their analytical applicability and validity for large and independent patient cohorts [146, 147].
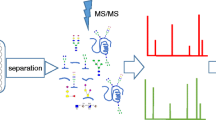
Analytical approaches for glycopeptide quantification have been extensively reviewed previously [148, 149]. In general, a small set of cases and controls are studied for biomarker discovery where the serum or plasma samples undergo multiple processing steps such as immunodepletion, proteolytic digestion, enrichment, and optionally isotope labeling and fractionation to achieve glycopeptide-rich samples. Then high-resolution MS instruments are used to identify and quantify as many glycopeptides as possible before glycopeptides with significant differences between the cases and controls are selected as biomarker candidates (Fig. 3). These glycopeptides are then quantified in a targeted manner across large patient cohorts for validation. Among different targeted approaches available, multiple reaction monitoring (MRM) is the gold standard MS method as it provides the highest sensitivity, high throughput, and simplicity of sample preparation and data interpretation.

Multi-marker assay development through biomarker discovery and validation workflows enabled by LC–MS and AI technologies
MRM methods for glycopeptides were initially developed for a handful of protein targets such as immunoglobulins and apolipoproteins [82, 87, 150, 151]. These methods were applied to various cancer cohorts. For example, truncated and galactosylated glycoforms from serum immunoglobulins were found to be associated with ovarian cancer and gastric cancer [152, 153], while fucosylated glycopeptides of haptoglobin and hemopexin were found to be elevated in HCC patients [154]. In 2019, we fully explored the potential of MRM for serum glycopeptide quantitation by expanding the method to quantify over 600 glycopeptides from 50 serum glycoproteins, and later, the method was further expanded to study the glycosylation of over 70 serum glycoproteins simultaneously [155, 156]. This workflow for targeted serum/plasma glycoproteomic analysis is illustrated in Fig. 3 in parallel with the biomarker discovery process. As the first step, serum samples from retrospective or prospective cohorts are transferred into 96-well plates for high-throughput preparation. To reduce variation introduced by sample pre-processing, the samples are handled only by a simple proteolytic digestion procedure and directly injected into a LC coupled with a triple quadrupole mass spectrometer (QQQ-MS) where the peptides and glycopeptides of interest are sequentially isolated, fragmented, and detected as chromatographic peaks. Due to the high heterogeneity of glycosylation and wide dynamic range of glycoforms, conventional peak integration software tools for proteomics are not readily applicable to glycopeptide MRM data. In 2020, an AI-based peak integration platform, PB-Net, was developed by training on a large dataset of over 170,000 expert annotated MRM peaks spanning a wide dynamic range, including both peptides and intact glycopeptides [157]. The peak areas with comparable accuracy as human integration are then exported for each analyte from all patient samples and used to build machine-learning models for cancer diagnosis. This MRM-based workflow has demonstrated its clinical applicability through several use cases including discovering novel glycoproteomic biomarkers for detecting NASH and HCC, differentiating symptomatic and asymptomatic COVID-19, as well as predicting clinical benefit from immune checkpoint inhibitor therapy for metastatic melanoma patients [65, 156, 158]. (Glycopeptides from a group of liver-derived glycoproteins, including α-2-macroglobulin, α-1-acid glycoprotein 1, haptoglobin, α-1-antitrypsin, and complement factor H were found to have pronounced unidirectional quantitative differences among controls, NASH, and HCC [156]. Besides these small scale pilot studies, this workflow has been applied to prospective clinical trials and the development of a laboratory-developed test (LDT) the reproducible measurements of intact glycopeptides by MRM, reliable data processing by AI-based platform, and novel machine-learning algorithms for data interpretation have started a new era for biomarker validation and cancer detection.
Glycopeptide biomarkers in clinical oncology
Given the improvement in glycoproteomic detecting technology, as well as machine learning and AI pipelines, glycopeptide biomarkers discovery and development emerge as very promising and clinically relevant field in clinical oncology. Broadly, these biomarkers play a critical role in clinical medicine by providing valuable insights into the various disease status and can be categorized into three distinct types: diagnostic, predictive, and prognostic biomarkers. Each type serves a specific purpose and offers unique insights into the management and treatment of cancer and other diseases.
Examples of diagnostic/screening glycopeptide markers in cancers
Cancer diagnostic/screening biomarkers hold the key to early disease detection and accurate diagnosis. Their primary function is to determine the presence of a disease in a noninvasive manner. By analyzing these biomarkers in blood, tissue, or other bodily fluids, clinicians can initiate timely interventions and treatments, greatly enhancing the patient's chances of improved outcomes. Also, the noninvasive nature of diagnostic biomarkers minimizes patient discomfort and allows for swift initiation of appropriate medical strategies. In the next section, we will give some examples of glycopeptide biomarkers and their clinical applicability.
Colorectal cancer
Despite its preventability by proper screening, CRC remains a major cause of cancer-related deaths [159]. Novel, more accurate, less invasive, and cost-effective screening methods are urgently needed. Recent research has highlighted specific biological changes during the transition from adenoma to carcinoma, particularly specific immune responses in the colonic crypt [160]. Abnormal protein glycosylation is a key player in driving these responses, evident in both colonic tissue and circulating glycoproteins (Fig. 4) [160].

During the adenoma to carcinoma sequence, glycosylation patterns within the colonic crypts undergo dynamic shifts (Fig. 4). This influence extends to circulating glycosylated acute-phase proteins and plasma-derived antibodies. Studies focusing on the N-glycome in CRC patients have illuminated a connection with branched and poly-LacNAc elongated N-glycans, which normalize after treatment [63]. Further investigations into N-glycan profiling reveal heightened fucosylation and sialylation on specific proteins, potentially aiding in the detection of CRC [161]. Moreover, disparities in IgG N glycome profiles have been observed between individuals with CRC and those without, offering potential diagnostic insights [162]. Intriguingly, matrix-assisted laser desorption/ionization-MS analysis of patient sera indicates correlations between multi-antennary, sialylated N-glycans and cancer progression, while biantennary core-fucosylated N-glycans show negative correlations [163]. This not only enhances the accuracy of CRC classification but also suggests possibilities for distinguishing advanced adenomas. Furthermore, glycan-binding proteins such as galectins, including galectin-2, -3, -4, and -8, have been identified at elevated levels in the serum of CRC patients, with galectin-2 demonstrating a positive association with high CRC mortality [164, 165].
The discussed advanced MS technologies and AI-driven data processing have enabled the study of the highly complex field of glycosylation, offering new and scalable possibilities for identifying circulating CRC biomarkers [157]. In a recent case–control study encompassing CRC, AA, and control serum samples, we used LC–MS glycoproteomic quantification and machine learning models with recurrent neural networks to facilitated chromatogram peak integration and molecular abundance quantification [66]. Through this approach, researchers identified 399 statistically significant differentially abundant glycopeptides/peptides when comparing CRC and advanced adenoma samples against healthy subjects. Subsequently, a multivariable classifier model, developed using a subset of six biomarkers, demonstrated strong performance in distinguishing CRC/advanced adenomas from healthy samples. The classifier yielded an Area under the Receiver Operating Characteristic curve (AuROC) of 0.96, reflecting its diagnostic accuracy. This model exhibited high sensitivity for detecting CRC across all stages (89.8%), including stages 1 to 4, as well as for advanced adenomas with and without high-grade dysplasia (90.9%). Additionally, it displayed promising specificity (89%) for normal findings [66].
This innovative glycoprotein-based strategy, empowered by artificial intelligence, showcases compelling clinical efficacy in detecting advanced adenomas and CRC. It offers unique insights into the early host response to polyp formation and immune escape, distinct from current liquid biopsy methods that require significant tumor materials. Prospective studies are underway to evaluate these glycoproteomic markers for AA/CRC detection (NCT05445570).
Lung cancer
Lung cancer is a leading cause of cancer-related deaths worldwide which is partly due to a frequent lack of symptoms and signs as well as reliable methods to screen for early-stage disease [166]. Lung cancers are often diagnosed at locally advanced or metastatic stage, the latter of which carries a five-year survival rate below 10% and more than 50% probability of mortality within a year after diagnosis. Current modality of screening involves low-dose computed tomography, but it is limited by the high-false positive rate, exposure to radiation and financial cost [167].
A few studies have investigated the role of glycoproteomic signatures in diagnosis of lung cancer. In one study, targeted glycoproteomic screening using hydrazide chemistry-based capture and enrichment showed varying abundance of 38 glycopeptides from 22 different proteins in patients with non-small cell lung cancer (NSCLC) versus matched controls [168]. A separate study used multiple-lectin affinity chromatography to identify differential levels of 38 serum glycoproteins in the sera of patients of lung adenocarcinoma [169]. In a study using hydrophilic interaction liquid chromatography (HILIC) and weak anion exchange high performance liquid chromatography (HPLC), significant differences in N-glycome were observed in patients with lung cancers of any histological types, with most significant alterations seen with di-sialylated and tri- and tetra-antennary glycans [170]. While the area under the curve (AUC) ranged from 0.640 to 0.811 for individual glycan peaks, it performed best with an AUC of 0.938 (85% sensitivity and 86% specificity) when all glycan data were considered. Similar observation was made with increased levels of fucosylated tri- and tetra-antennary structures using an alternative analytical methodology [171]. Notably, some changes were seen as early as in Stage I disease. In a separate study, Fang and colleagues investigated tumor specific N-glycosylation sites in Stage I lung adenocarcinoma using tandem mass tag labeling and liquid chromatography with tandem mass spectrometry. A total of 39 differential N-glycosylation sites were identified with implications in various biological pathways including cell migration, metabolism and immunity [172]. ITGB3-680 showed the highest diagnostic value with AUC of 99.2% and 95% of both sensitivity and specificity in comparison to the healthy control. Combination analysis of all N-glycosylation sites by machine learning model revealed 100% AUC in both training and testing groups. Similarly in small cell lung cancer, proteomic screening revealed significant alterations in fucosylated glycan patterns like that of PON1 with AUC of 0.91 [173]. Recently, similar glycoproteomic platform as used in CRC in the above session that combined mass spectrometry with a proprietary artificial-intelligence-based data processing engine that allows highly scalable interrogation of the glycoproteome in lung cancer was utilized to generate a glycoproteomic classifier that showed high sensitivity and specificity in separate lung cancer and normal tissues [174].
Ovarian cancer
Ovarian cancer ranks as the second most common gynecologic malignancy and claims the highest toll among gynecological cancers ranking 4th in leading causes of cancer-related death in US women [175]. While early-stage cancer can be treated effectively with surgery and (neo)adjuvant therapies, its diagnosis is hindered by subtle and nonspecific symptoms like pelvic pain, urinary changes, and abdominal bloating. Such symptoms intensify as the disease advances. Consequently, only 15–20% of cases are caught early, where the survival rate exceeds 90%, while most diagnoses occur at a later stage, yielding survival rates between 17 and 39% [176].
Circulating biomarkers, often involving previously discussed glycoproteins, are combined with other tests for diagnosing ovarian cancer. Elevated CA-125 levels, a glycosylated protein present in blood, can signal ovarian cancer, however, CA-125's performance as a diagnostic biomarker is constrained by its low sensitivity and specificity [177]. Notably, serum CA-125 remains unaltered in 21% of ovarian carcinomas, while elevated levels appear in various other conditions like endometriosis, uterine fibroids, menstruation, or pregnancy. Thus, CA-125 is more suited for monitoring cancer's progress and treatment response [178]. While tests combining CA-125 with other parameters have been developed, they are hindered by complexity and subpar performance. Hence, a critical need remains for highly sensitive and specific noninvasive diagnostic tools for ovarian cancer.
As previously discussed, a significant portion of existing cancer biomarkers are glycosylated proteins, with hyper-sialylation being a consistent glycan alteration observed in cancer. Hyper-sialylation involves the addition of sialic acid to the terminal end of glycoproteins by sialyltransferases. Of the over 20 human sialyltransferases identified, heightened expression of Golgi β-Galactoside α-2,6-Sialyltransferase 1 (ST6Gal1) appears to be linked to various cancers, including ovarian cancer [179,180,181]. ST6GAL1 modulates intracellular signaling to regulate tumor cell phenotype, in particular, ST6Gal-I upregulation in ovarian cancer cells seems to confer a cancer stem-like cell (CSC) phenotype [181]. Primary ovarian cancer cells from patient ascites or solid tumors sorted for α2-6 sialylation grew as spheroids, while cells lacking α2-6 sialylation remained as single cells and lost viability. In addition, sialylation of EGFR by ST6GAL1 induces receptor activation and modulates trafficking dynamics highly implicated in neoplastic mechanisms [179]. A mechanism for upregulated ST6Gal-I expression in ovarian cancer has recently been suggested whereby SOX2 and ST6GAL1 are coordinately amplified in cancer cells, with the Sox2 protein then binding the ST6GAL1 promoter to further augment ST6Gal-I expression [180].
Multiple glycoproteomic studies have been conducted in ovarian cancer [182,183,184]. In a recent study, a novel blood-based glycoproteomic approach combining mass spectrometry with machine learning was used to evaluate a glycopeptide classifier for the diagnosis of ovarian cancer [185]. Specific glycopeptide biomarkers were identified that effectively distinguish between individuals with benign pelvic masses and those with malignant ovarian cancer. The classifier exhibited impressive sensitivity and specificity rates of 83.5% and 90.1% in the training dataset and 86.7% and 86.7% in the testing dataset, respectively. Furthermore, it was reported that ovarian cancer patients had elevated levels of fucosylated markers, primarily originating from the liver. Individuals with advanced disease stages (FIGO stage III and IV) showed significantly higher levels of tri- and tetra-antennary glycopeptide markers containing fucose.
These data are encouraging and provide insights into the underlying mechanisms connecting ovarian cancer to the circulating glycoproteome. These results have the potential to guide the development of robust clinical tests for diagnosing and staging of ovarian cancer patients.
Selected examples of predictive biomarkers
Predictive biomarkers are instrumental in personalized medicine, tailoring treatment plans to the individual patient's characteristics. These biomarkers help determine how a patient is likely to respond to a particular therapy. A 'Companion Diagnostic or CDx' is a test for a predictive biomarker that classifies patients (e.g., tumors) into responders and non-responders, for a specified therapeutic agent. Companion diagnostics are designated as Class III medical devices by the FDA, because the test result equates directly to administration of a drug.
Predictive to immunotherapy
A limited number of studies have investigated the prognostic utility of glycoproteomic signatures in response to immune checkpoint inhibitors (ICI), which have revolutionized the treatment of various malignancies. A recent study explored predictive ICI biomarkers in melanoma using an advanced platform combining liquid chromatography/mass spectrometry and AI [65]. Glycoproteins were examined in pre-treatment plasma samples from metastatic melanoma patients undergoing different ICI therapies including first or second-line anti-PD-1 monotherapy (pembrolizumab or nivolumab) or anti-PD-1/anti-CTLA-4 combination therapy (nivolumab/ipilimumab). Biomarkers were identified for survival analysis, and patients were categorized by treatment response. Classifiers were developed using a discovery cohort, validated, and externally confirmed. In total, 143 glycoproteomic biomarkers distinguishing early treatment failure from sustained control were found. The subsequent classifier yielded a hazard ratio of 2.7 (p = 0.026) in the training set and 5.6 (p = 0.027) in an independent test set [65]. These markers demonstrated differential expression in patients with short overall survival as compared to those with favorable overall survival outcomes and therefore were used to generate classifiers that identified metastatic melanoma patients. In addition, a specific fucosylation signature in plasma N-glycoproteins of patients that do not achieve a durable response to ICI therapy was discovered.
Similarly, several abstracts have demonstrated highly encouraging preliminary findings in advanced NSCLC [186,187,188]. By utilizing a similar advanced glycoproteomics platform, the most recent study investigated 532 glycopeptide and peptide signatures representing 75 serum proteins in 123 individuals with unresectable stage 3 or 4 NSCLC prior to initiation of standard of care with pembrolizumab monotherapy or combination of pembrolizumab and chemotherapy. Multivariable model-based classifier consisting of 7 glycopeptides and other non-glycosylated peptide biomarkers was able to identify patients who are likely to benefit from ICI with > 95% sensitivity and 33% specificity. This classifier yielded a statistically significant hazard ratio (HR) of 3.6 for predicted benefit with median overall survival (OS) of 13.9 versus 4.2 months in the entire cohort based on its score above and below the cutoff. It similarly performed well with HR of 3.5 and median OS of 13.5 versus 4.5 months in the first-line treated patients. Altogether, these studies confirm the potential of the development of diagnostic tests to guide treatment decisions.
It is worth noting that programmed death-ligand 1 (PD-L1) is often found with significant N-linked glycan moieties in various primary cancers [189]. An important clinical implication of PD-L1 N-linked glycosylation is that it can hinder diagnostic antibodies from recognizing its antigenic regions. Indeed, pre-enzymatic digestion of tissue samples for de-glycosylation improves the binding affinity of PD-L1 antibodies and its signal intensity, which allows for more accurate quantification of PD-L1 expression to guide the therapeutic efficacy of PD-1/PD-L1 inhibitors [190]. Functionally, PD-L1 glycosylation enhances its stability via antagonizing interactions with glycogen synthase kinase 3β (GSK3β) and related phosphorylation-dependent proteasome degradation [191]. N-linked glycosylation is also critical for its suppression of T cell immunity via interaction with its cognate receptor programmed cell death protein 1 (PD-1) [191, 192]. Collectively, these studies highlight de-glycosylation of PD-L1 as not only a biomarker, but also a promising therapeutic target for cancer immunotherapy.
Conclusion and outlook
The field of cancer glycoproteomic biomarker studies is evolving rapidly with encouraging discoveries in cancer screening/early diagnosis, therapeutic prediction, and monitoring, among others. While these glycoproteomic signatures represent a promising possibility to serve as biomarkers, the field still faces challenges. First, it is still not fully clear if aberrant protein glycosylation patterns are unique to specific cancer types, pan-cancers or even more generalizable to other non-malignant pathologies including atypical hyperplasia, inflammation, infection, and other benign diseases. We need to better understand the association of the glycoprotein biology and cancer and other pathological processes. Further research is necessary to investigate the regulatory mechanisms underlying glycosylation sites and their biological consequences. Also, the role of neoplasia-induced hepatic reprogramming in the biomarker specificity remains to be determined. Secondly, we need to continue to advance with better technology and bioinformatics tools for ultrasensitive glycoproteomics to depict an in-depth and precise landscape of cancer. The development of small molecules or chip-based glycospecific techniques may also be necessary for their application in clinical settings given the poor affinity and imprecise specificities of lectin or antibody-based assays for occasional glycan epitopes. Finally, glycoproteomic biomarkers need to be translated into cancer care, which involves the leap from the proof of concept of retrospective small cohort studies to perspective and independent validation in both well-designed clinically investigations and real-world studies. Eventually, it needs to integrate with all clinical and pathological presentations, advanced imaging, and other tissue or liquid based omics, to provide a comprehensive but unique profile for precision care in cancer and risk management, diagnosis, and treatment.
In conclusion, years of advancing of glycoproteomic analyzing tools and algorithms, cancer biology and medicine, presents an emerging opportunity for scientists, physicians, industry and all involved, to work together towards further biomarker discovery and development, and ultimately to change the outcomes for cancer patients.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Raufaste-Cazavieille V, Santiago R, Droit A. Multi-omics analysis: Paving the path toward achieving precision medicine in cancer treatment and immuno-oncology. Front Mol Biosci. 2022;9:962743.
Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–83.
Hanash SM, Pitteri SJ, Faca VM. Mining the plasma proteome for cancer biomarkers. Nature. 2008;452:571–9.
Palmirotta R, Lovero D, Cafforio P, Felici C, Mannavola F, Pellè E, et al. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Ther Adv Med Oncol. 2018;10:1758835918794630.
Alix-Panabières C, Pantel K. Liquid biopsy: from discovery to clinical application. Cancer Discov. 2021;11:858–73.
Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol. 2021;18:297–312.
Ding Z, Wang N, Ji N, Chen Z-S. Proteomics technologies for cancer liquid biopsies. Mol Cancer. 2022;21:53.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–30.
Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–67.
Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21:729–49.
Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526–42.
Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–55.
Carvalho S, Catarino TA, Dias AM, Kato M, Almeida A, Hessling B, et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene. 2016;35:1619–31.
Taniguchi N, Kizuka Y. Glycans and Cancer. 2015. p. 11–51.
Rodrigues JG, Balmaña M, Macedo JA, Poças J, Fernandes Â, de-Freitas-Junior JCM, et al. Glycosylation in cancer: selected roles in tumour progression, immune modulation and metastasis. Cell Immunol. 2018;333:46–57.
Ignatiadis M, Lee M, Jeffrey SS. Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res. 2015;21:4786–800.
Dang DK, Park BH. Circulating tumor DNA: current challenges for clinical utility. J Clin Invest. 2022;132:1–10.
Alix-Panabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14:623–31.
Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, et al. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther. 2020;5:145.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J Clin Oncol. 2018;36:1631–41.
Cree IA, Deans Z, Ligtenberg MJL, Normanno N, Edsjö A, Rouleau E, et al. Guidance for laboratories performing molecular pathology for cancer patients. J Clin Pathol. 2014;67:923–31.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–38.
Zhou J, Kulasinghe A, Bogseth A, O’Byrne K, Punyadeera C, Papautsky I. Isolation of circulating tumor cells in non-small-cell-lung-cancer patients using a multi-flow microfluidic channel. Microsyst Nanoeng. 2019;5:8.
Deng Z, Wu S, Wang Y, Shi D. Circulating tumor cell isolation for cancer diagnosis and prognosis. EBioMedicine. 2022;83:104237.
Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat Methods. 2015;12:685–91.
Bronkhorst AJ, Ungerer V, Holdenrieder S. Early detection of cancer using circulating tumor DNA: biological, physiological and analytical considerations. Crit Rev Clin Lab Sci. 2019;57:253–69.
Ramazi S, Zahiri J. Posttranslational modifications in proteins: resources, tools and prediction methods. Database. 2021;2021:baab012.
Silva MLS. Capitalizing glycomic changes for improved biomarker-based cancer diagnostics. Explor Target Antitumor Ther. 2023;4:366–95.
Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discov Today. 2010;15:773–80.
Drake PM, Cho W, Li B, Prakobphol A, Johansen E, Anderson NL, et al. Sweetening the pot: adding glycosylation to the biomarker discovery equation. Clin Chem. 2010;56:223–36.
Trbojević-Akmačić I, Lageveen-Kammeijer GSM, Heijs B, Petrović T, Deriš H, Wuhrer M, et al. High-throughput glycomic methods. Chem Rev. 2022;122:15865–913.
Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–82.
Schiel JE. Glycoprotein analysis using mass spectrometry: unraveling the layers of complexity. Anal Bioanal Chem. 2012;404:1141–9.
Ruhaak LR, Xu G, Li Q, Goonatilleke E, Lebrilla CB. Mass spectrometry approaches to glycomic and glycoproteomic analyses. Chem Rev. 2018;118:7886–930.
Levery SB, Steentoft C, Halim A, Narimatsu Y, Clausen H, Vakhrushev SY. Advances in mass spectrometry driven O-glycoproteomics. Biochim Biophys Acta. 2015;1850:33–42.
Banazadeh A, Veillon L, Wooding KM, Zabet-Moghaddam M, Mechref Y. Recent advances in mass spectrometric analysis of glycoproteins. Electrophoresis. 2017;38:162–89.
Tamara S, Franc V, Heck AJR. A wealth of genotype-specific proteoforms fine-tunes hemoglobin scavenging by haptoglobin. Proc Natl Acad Sci U S A. 2020;117:15554–64.
Oh MJ, Lee SH, Kim U, An HJ. In-depth investigation of altered glycosylation in human haptoglobin associated cancer by mass spectrometry. Mass Spectrom Rev. 2023;42:496–518.
Fujimura T, Shinohara Y, Tissot B, Pang P-C, Kurogochi M, Saito S, et al. Glycosylation status of haptoglobin in sera of patients with prostate cancer vs. benign prostate disease or normal subjects. Int J Cancer. 2008;122:39–49.
Turner GA. Haptoglobin: a potential reporter molecule for glycosylation changes in disease. Adv Exp Med Biol. 1995;1995(376):231–8.
Dall’Olio F, Chiricolo M,. Sialyltransferases in cancer. Glycoconj J. 2001;18:841–50.
Noda K, Miyoshi E, Uozumi N, Yanagidani S, Ikeda Y, Gao C, et al. Gene expression of alpha1-6 fucosyltransferase in human hepatoma tissues: a possible implication for increased fucosylation of alpha-fetoprotein. Hepatology. 1998;28:944–52.
Liu Y-C, Yen H-Y, Chen C-Y, Chen C-H, Cheng P-F, Juan Y-H, et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A. 2011;108:11332–7.
Potapenko IO, Haakensen VD, Lüders T, Helland A, Bukholm I, Sørlie T, et al. Glycan gene expression signatures in normal and malignant breast tissue; possible role in diagnosis and progression. Mol Oncol. 2010;4:98–118.
Hiraiwa N, Yabuta T, Yoritomi K, Hiraiwa M, Tanaka Y, Suzuki T, et al. Transactivation of the fucosyltransferase VII gene by human T-cell leukemia virus type 1 Tax through a variant cAMP-responsive element. Blood. 2003;101:3615–21.
Matsuura N, Narita T, Hiraiwa N, Hiraiwa M, Murai H, Iwase T, et al. Gene expression of fucosyl- and sialyl-transferases which synthesize sialyl Lewisx, the carbohydrate ligands for E-selectin, in human breast cancer. Int J Oncol. 1998;12:1157–64.
Holmes EH, Hakomori S, Ostrander GK. Synthesis of type 1 and 2 lacto series glycolipid antigens in human colonic adenocarcinoma and derived cell lines is due to activation of a normally unexpressed beta 1–3N-acetylglucosaminyltransferase. J Biol Chem. 1987;262:15649–58.
Guo H, Nagy T, Pierce M. Post-translational glycoprotein modifications regulate colon cancer stem cells and colon adenoma progression in Apc(min/+) mice through altered Wnt receptor signaling. J Biol Chem. 2014;289:31534–49.
Yoshimura M, Nishikawa A, Ihara Y, Taniguchi S, Taniguchi N. Suppression of lung metastasis of B16 mouse melanoma by N-acetylglucosaminyltransferase III gene transfection. Proc Natl Acad Sci U S A. 1995;92:8754–8.
Niu L, Geyer PE, Wewer Albrechtsen NJ, Gluud LL, Santos A, Doll S, et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol Syst Biol. 2019;15:e8793.
Wang G, Li J, Bojmar L, Chen H, Li Z, Tobias GC, et al. Tumour extracellular vesicles and particles induce liver metabolic dysfunction. Nature. 2023;618:374–82.
Jiao Y, Xu P, Shi H, Chen D, Shi H. Advances on liver cell-derived exosomes in liver diseases. J Cell Mol Med. 2021;25:15–26.
Zhu J, Wu J, Yin H, Marrero J, Lubman DM. Mass spectrometric N-glycan analysis of haptoglobin from patient serum samples using a 96-well plate format. J Proteome Res. 2015;14:4932–9.
Mehta A, Herrera H, Block T. Glycosylation and liver cancer. Adv Cancer Res. 2015;126:257–79.
Zhu J, Warner E, Parikh ND, Lubman DM. Glycoproteomic markers of hepatocellular carcinoma-mass spectrometry based approaches. Mass Spectrom Rev. 2019;38:265–90.
Čaval T, Lin Y-H, Varkila M, Reiding KR, Bonten MJM, Cremer OL, et al. Glycoproteoform profiles of individual patients’ plasma alpha-1-antichymotrypsin are unique and extensively remodeled following a septic episode. Front Immunol. 2020;11:608466.
Keser T, Tijardović M, Gornik I, Lukić E, Lauc G, Gornik O, et al. High-throughput and site-specific N-glycosylation analysis of human alpha-1-acid glycoprotein offers a great potential for new biomarker discovery. Mol Cell Proteomics. 2021;20:100044.
Virág D, Kremmer T, Lőrincz K, Kiss N, Jobbágy A, Bozsányi S, et al. Altered glycosylation of human alpha-1-acid glycoprotein as a biomarker for malignant melanoma. Molecules. 2021;26:6003.
Yokobori T, Yazawa S, Asao T, Nakazawa N, Mogi A, Sano R, et al. Fucosylated α1-acid glycoprotein as a biomarker to predict prognosis following tumor immunotherapy of patients with lung cancer. Sci Rep. 2019;9:14503.
Yazawa S, Takahashi R, Yokobori T, Sano R, Mogi A, Saniabadi AR, et al. Fucosylated glycans in α1-acid glycoprotein for monitoring treatment outcomes and prognosis of cancer patients. PLoS ONE. 2016;11:e0156277.
Doherty M, Theodoratou E, Walsh I, Adamczyk B, Stöckmann H, Agakov F, et al. Plasma N-glycans in colorectal cancer risk. Sci Rep. 2018;8:8655.
de Vroome SW, Holst S, Girondo MR, van der Burgt YEM, Mesker WE, Tollenaar RAEM, et al. Serum N-glycome alterations in colorectal cancer associate with survival. Oncotarget. 2018;9:30610–23.
Dotz V, Wuhrer M. N-glycome signatures in human plasma: associations with physiology and major diseases. FEBS Lett. 2019;593:2966–76.
Pickering C, Aiyetan P, Xu G, Mitchell A, Rice R, Najjar YG, et al. Plasma glycoproteomic biomarkers identify metastatic melanoma patients with reduced clinical benefit from immune checkpoint inhibitor therapy. Front Immunol. 2023;14:1187332.
Desai K, Gupta S, May FP, Xu G, Shaukat A, Hommes DW, et al. Early detection of advanced adenomas and colorectal carcinoma by serum glycoproteome profiling. Gastroenterology. 2024;166:194-197.e2.
Chen M, Ren AH, Prassas I, Soosaipillai A, Lim B, Fraser DD, et al. Plasma protein profiling by proximity extension assay technology reveals novel biomarkers of traumatic brain injury—a pilot study. J Appl Lab Med. 2021;6:1165–78.
Wik L, Nordberg N, Broberg J, Björkesten J, Assarsson E, Henriksson S, et al. Proximity extension assay in combination with next-generation sequencing for high-throughput proteome-wide analysis. Mol Cell Proteomics. 2021;20:100168.
Pietzner M, Wheeler E, Carrasco-Zanini J, Kerrison ND, Oerton E, Koprulu M, et al. Synergistic insights into human health from aptamer- and antibody-based proteomic profiling. Nat Commun. 2021;12:6822.
Dwek MV, Jenks A, Leathem AJC. A sensitive assay to measure biomarker glycosylation demonstrates increased fucosylation of prostate specific antigen (PSA) in patients with prostate cancer compared with benign prostatic hyperplasia. Clin Chim Acta. 2010;411:1935–9.
Bojar D, Meche L, Meng G, Eng W, Smith DF, Cummings RD, et al. A useful guide to lectin binding: machine-learning directed annotation of 57 unique lectin specificities. ACS Chem Biol. 2022;17:2993–3012.
De Leoz MLA, Duewer DL, Fung A, Liu L, Yau HK, Potter O, et al. NIST interlaboratory study on glycosylation analysis of monoclonal antibodies: comparison of results from diverse analytical methods. Mol Cell Proteomics. 2020;19:11–30.
Shajahan A, Heiss C, Ishihara M, Azadi P. Glycomic and glycoproteomic analysis of glycoproteins-a tutorial. Anal Bioanal Chem. 2017;409:4483–505.
Kailemia MJ, Park D, Lebrilla CB. Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem. 2017;409:395–410.
Melmer M, Stangler T, Premstaller A, Lindner W. Comparison of hydrophilic-interaction, reversed-phase and porous graphitic carbon chromatography for glycan analysis. J Chromatogr A. 2011;1218:118–23.
Ruhaak LR, Deelder AM, Wuhrer M. Oligosaccharide analysis by graphitized carbon liquid chromatography-mass spectrometry. Anal Bioanal Chem. 2009;394:163–74.
Zhang C, Ye Z, Xue P, Shu Q, Zhou Y, Ji Y, et al. Evaluation of different N-glycopeptide enrichment methods for N-glycosylation sites mapping in mouse brain. J Proteome Res. 2016;15:2960–8.
Liu Z, Xu M, Zhang W, Miao X, Wang PG, Li S, et al. Recent development in hydrophilic interaction liquid chromatography stationary materials for glycopeptide analysis. Anal Methods. 2022;14:4437–48.
Balaguer E, Neusüss C. Glycoprotein characterization combining intact protein and glycan analysis by capillary electrophoresis-electrospray ionization-mass spectrometry. Anal Chem. 2006;78:5384–93.
Lingg N, Zhang P, Song Z, Bardor M. The sweet tooth of biopharmaceuticals: importance of recombinant protein glycosylation analysis. Biotechnol J. 2012;7:1462–72.
Han L, Costello CE. Mass spectrometry of glycans. Biochemistry (Mosc). 2013;78:710–20.
Song E, Pyreddy S, Mechref Y. Quantification of glycopeptides by multiple reaction monitoring liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2012;26:1941–54.
van der Burgt Y, Wuhrer M. The role of clinical glyco(proteo)mics in precision medicine. Mol Cell Proteomics. 2023;22:100565.
Varki A, Cummings R, Esko JD. Essentials of Glycobiology. 2022.
Wohlgemuth J, Karas M, Eichhorn T, Hendriks R, Andrecht S. Quantitative site-specific analysis of protein glycosylation by LC-MS using different glycopeptide-enrichment strategies. Anal Biochem. 2009;395:178–88.
Goumenou A, Delaunay N, Pichon V. Recent advances in lectin-based affinity sorbents for protein glycosylation studies. Front Mol Biosci. 2021;8:746822.
Hong Q, Ruhaak LR, Stroble C, Parker E, Huang J, Maverakis E, et al. A method for comprehensive glycosite-mapping and direct quantitation of serum glycoproteins. J Proteome Res. 2015;14:5179–92.
Ongay S, Boichenko A, Govorukhina N, Bischoff R. Glycopeptide enrichment and separation for protein glycosylation analysis. J Sep Sci. 2012;35:2341–72.
Xue Y, Xie J, Fang P, Yao J, Yan G, Shen H, et al. Study on behaviors and performances of universal N-glycopeptide enrichment methods. Analyst. 2018;143:1870–80.
Huang Y, Nie Y, Boyes B, Orlando R. Resolving isomeric glycopeptide glycoforms with hydrophilic interaction chromatography (HILIC). J Biomol Tech. 2016;27:98–104.
Mookherjee A, Guttman M. Bridging the structural gap of glycoproteomics with ion mobility spectrometry. Curr Opin Chem Biol. 2018;42:86–92.
Makrydaki E, Kotidis P, Polizzi KM, Kontoravdi C. Hitting the sweet spot with capillary electrophoresis: advances in N-glycomics and glycoproteomics. Curr Opin Biotechnol. 2021;71:182–90.
Macklin A, Khan S, Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteomics. 2020;17:17.
Ye Z, Vakhrushev SY. The role of data-independent acquisition for glycoproteomics. Mol Cell Proteomics. 2021;20:100042.
Miyamoto S, Stroble CD, Taylor S, Hong Q, Lebrilla CB, Leiserowitz GS, et al. Multiple reaction monitoring for the quantitation of serum protein glycosylation profiles: application to ovarian cancer. J Proteome Res. 2018;17:222–33.
Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res. 2015;4:256–69.
Kirwan A, Utratna M, O’Dwyer ME, Joshi L, Kilcoyne M. Glycosylation-based serum biomarkers for cancer diagnostics and prognostics. Biomed Res Int. 2015;2015:1–16.
Li F, Li C, Wang M, Webb GI, Zhang Y, Whisstock JC, et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics. 2015;31:1411–9.
Sato T, Furukawa K, Greenwalt DE, Kobata A. Most bovine milk fat globule membrane glycoproteins contain asparagine-linked sugar chains with GalNAc beta 1–>4GlcNAc groups. J Biochem. 1993;114:890–900.
FDA Center for Devices and Radiological Health. AFP-L3% Immunological Test Systems - Class II Special Controls Guidance Document for Industry and FDA Staff. 2005 Oct.
Kim H, Kim K, Jin J, Park J, Yu SJ, Yoon J-H, et al. Measurement of glycosylated alpha-fetoprotein improves diagnostic power over the native form in hepatocellular carcinoma. PLoS ONE. 2014;9:e110366.
Li D, Mallory T, Satomura S. AFP-L3: a new generation of tumor marker for hepatocellular carcinoma. Clin Chim Acta. 2001;313:15–9.
Choi J, Kim G, Han S, Lee W, Chun S, Lim Y. Longitudinal assessment of three serum biomarkers to detect very early-stage hepatocellular carcinoma. Hepatology. 2019;69:1983–94.
Dunbar C, Kushnir MM, Yang YK. Glycosylation profiling of the neoplastic biomarker alpha fetoprotein through intact mass protein analysis. J Proteome Res. 2023;22:226–34.
Zhou J-M, Wang T, Zhang K-H. AFP-L3 for the diagnosis of early hepatocellular carcinoma: a meta-analysis. Medicine. 2021;100:e27673.
Valmu L, Alfthan H, Hotakainen K, Birken S, Stenman U-H. Site-specific glycan analysis of human chorionic gonadotropin beta-subunit from malignancies and pregnancy by liquid chromatography–electrospray mass spectrometry. Glycobiology. 2006;16:1207–18.
Elliott MM, Kardana A, Lustbader JW, Cole LA. Carbohydrate and peptide structure of the alpha- and beta-subunits of human chorionic gonadotropin from normal and aberrant pregnancy and choriocarcinoma. Endocrine. 1997;7:15–32.
Terävä J, Tiainen L, Lamminmäki U, Kellokumpu-Lehtinen P-L, Pettersson K, Gidwani K. Lectin nanoparticle assays for detecting breast cancer-associated glycovariants of cancer antigen 15–3 (CA15-3) in human plasma. PLoS ONE. 2019;14:e0219480.
Choi JW, Moon B-I, Lee JW, Kim HJ, Jin Y, Kim H-J. Use of CA15-3 for screening breast cancer: an antibody-lectin sandwich assay for detecting glycosylation of CA15-3 in sera. Oncol Rep. 2018;40:145–54.
Chen W, Zhang Z, Zhang S, Zhu P, Ko JK-S, Yung KK-L. MUC1: structure, function, and clinic application in epithelial cancers. Int J Mol Sci. 2021;22:6567.
Scarà S, Bottoni P, Scatena R. CA 19–9: biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:247–60.
Canney PA, Wilkinson PM, James RD, Moore M. CA19-9 as a marker for ovarian cancer: alone and in comparison with CA125. Br J Cancer. 1985;52:131–3.
Duffy MJ. CA 19–9 as a marker for gastrointestinal cancers: a review. Ann Clin Biochem. 1998;35(Pt 3):364–70.
Bayoumy S, Hyytiä H, Leivo J, Talha SM, Huhtinen K, Poutanen M, et al. Glycovariant-based lateral flow immunoassay to detect ovarian cancer-associated serum CA125. Commun Biol. 2020;3:460.
Chen K, Gentry-Maharaj A, Burnell M, Steentoft C, Marcos-Silva L, Mandel U, et al. Microarray Glycoprofiling of CA125 improves differential diagnosis of ovarian cancer. J Proteome Res. 2013;12:1408–18.
Akita K, Yoshida S, Ikehara Y, Shirakawa S, Toda M, Inoue M, et al. Different levels of sialyl-Tn antigen expressed on MUC16 in patients with endometriosis and ovarian cancer. Int J Gynecol Cancer. 2012;22:531–8.
Zhao Q, Zhan T, Deng Z, Li Q, Liu Y, Yang S, et al. Glycan analysis of colorectal cancer samples reveals stage-dependent changes in CEA glycosylation patterns. Clin Proteomics. 2018;15:9.
Pont L, Kuzyk V, Benavente F, Sanz-Nebot V, Mayboroda OA, Wuhrer M, et al. Site-specific N-linked glycosylation analysis of human carcinoembryonic antigen by sheathless capillary electrophoresis-tandem mass spectrometry. J Proteome Res. 2021;20:1666–75.
Gebauer F, Wicklein D, Horst J, Sundermann P, Maar H, Streichert T, et al. Carcinoembryonic antigen-related cell adhesion molecules (CEACAM) 1, 5 and 6 as biomarkers in pancreatic cancer. PLoS ONE. 2014;9:e113023.
Zhang X, Han X, Zuo P, Zhang X, Xu H. CEACAM5 stimulates the progression of non-small-cell lung cancer by promoting cell proliferation and migration. J Int Med Res. 2020;48:300060520959478.
Beauchemin N, Arabzadeh A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013;32:643–71.
Hayes JH, Barry MJ. Screening for prostate cancer with the prostate-specific antigen test. JAMA. 2014;311:1143.
Thompson IM. Operating characteristics of prostate-specific antigen in men with an initial PSA level of 3.0 ng/mL or lower. JAMA. 2005;294:66.
Gilgunn S, Conroy PJ, Saldova R, Rudd PM, O’Kennedy RJ. Aberrant PSA glycosylation—a sweet predictor of prostate cancer. Nat Rev Urol. 2013;10:99–107.
Meany DL, Zhang Z, Sokoll LJ, Zhang H, Chan DW. Glycoproteomics for prostate cancer detection: changes in serum PSA glycosylation patterns. J Proteome Res. 2009;8:613–9.
Peracaula R. Altered glycosylation pattern allows the distinction between prostate-specific antigen (PSA) from normal and tumor origins. Glycobiology. 2003;13:457–70.
Prakash S, Robbins Ph. Glycotyping of prostate specific antigen. Glycobiology. 2000;10:173–6.
Wang C, Höti N, Lih T-SM, Sokoll LJ, Zhang R, Zhang Z, et al. Development of a glycoproteomic strategy to detect more aggressive prostate cancer using lectin-immunoassays for serum fucosylated PSA. Clin Proteomics. 2019;16:1–18.
Ferrer-Batallé M, Llop E, Ramírez M, Aleixandre R, Saez M, Comet J, et al. Comparative study of blood-based biomarkers, α2,3-sialic acid PSA and PHI, for high-risk prostate cancer detection. Int J Mol Sci. 2017;18:845.
Ishikawa T, Yoneyama T, Tobisawa Y, Hatakeyama S, Kurosawa T, Nakamura K, et al. An automated micro-total immunoassay system for measuring cancer-associated α2,3-linked sialyl N-glycan-carrying prostate-specific antigen may improve the accuracy of prostate cancer diagnosis. Int J Mol Sci. 2017;18:470.
Yoneyama T, Ohyama C, Hatakeyama S, Narita S, Habuchi T, Koie T, et al. Measurement of aberrant glycosylation of prostate specific antigen can improve specificity in early detection of prostate cancer. Biochem Biophys Res Commun. 2014;448:390–6.
Kaya T, Kaneko T, Kojima S, Nakamura Y, Ide Y, Ishida K, et al. High-sensitivity immunoassay with surface plasmon field-enhanced fluorescence spectroscopy using a plastic sensor chip: application to quantitative analysis of total prostate-specific antigen and GalNAcβ1–4GlcNAc-linked prostate-specific antigen for prostate cancer diagnosis. Anal Chem. 2015;87:1797–803.
Llop E, Ferrer-Batallé M, Barrabés S, Guerrero PE, Ramírez M, Saldova R, et al. Improvement of prostate cancer diagnosis by detecting PSA glycosylation-specific changes. Theranostics. 2016;6:1190–204.
Fujita K, Hatano K, Tomiyama E, Hayashi Y, Matsushita M, Tsuchiya M, et al. Serum core-type fucosylated prostate-specific antigen index for the detection of high-risk prostate cancer. Int J Cancer. 2021;148:3111–8.
Yoneyama T, Tobisawa Y, Kaneko T, Kaya T, Hatakeyama S, Mori K, et al. Clinical significance of the Lacdi <scp>NA</scp> c-glycosylated prostate-specific antigen assay for prostate cancer detection. Cancer Sci. 2019;110:2573–89.
Leymarie N, Griffin PJ, Jonscher K, Kolarich D, Orlando R, McComb M, et al. Interlaboratory study on differential analysis of protein glycosylation by mass spectrometry: the ABRF glycoprotein research multi-institutional study 2012. Mol Cell Proteomics. 2013;12:2935–51.
Saeland E, Belo AI, Mongera S, van Die I, Meijer GA, van Kooyk Y. Differential glycosylation of MUC1 and CEACAM5 between normal mucosa and tumour tissue of colon cancer patients. Int J Cancer. 2012;131:117–28.
van Gisbergen KPJM, Aarnoudse CA, Meijer GA, Geijtenbeek TBH, van Kooyk Y. Dendritic cells recognize tumor-specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell-specific intercellular adhesion molecule-3–grabbing nonintegrin. Cancer Res. 2005;65:5935–44.
Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874–85.
Rangel-Angarita V, Malaker SA. Mucinomics as the Next Frontier of Mass Spectrometry. ACS Chem Biol. 2021;16:1866–83.
Malaker SA, Riley NM, Shon DJ, Pedram K, Krishnan V, Dorigo O, et al. Revealing the human mucinome. Nat Commun. 2022;13:3542.
Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, et al. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci. 2019;116:7278–87.
Kletter D, Cao Z, Bern M, Haab B. Determining lectin specificity from glycan array data using motif segregation and glycosearch software. Curr Protoc Chem Biol. 2013;5:157–69.
Kim H, Park S, Jeong IG, Song SH, Jeong Y, Kim C-S, et al. Noninvasive precision screening of prostate cancer by urinary multimarker sensor and artificial intelligence analysis. ACS Nano. 2021;15:4054–65.
Yurkovetsky Z, Skates S, Lomakin A, Nolen B, Pulsipher T, Modugno F, et al. Development of a multimarker assay for early detection of ovarian cancer. J Clin Oncol. 2010;28:2159–66.
Füzéry AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10:13.
Ou F-S, Michiels S, Shyr Y, Adjei AA, Oberg AL. Biomarker discovery and validation: statistical considerations. J Thorac Oncol. 2021;16:537–45.
Delafield DG, Li L. Recent advances in analytical approaches for glycan and glycopeptide quantitation. Mol Cell Proteomics. 2021;20:100054.
Yin H, Zhu J. Methods for quantification of glycopeptides by liquid separation and mass spectrometry. Mass Spectrom Rev. 2023;42:887–917.
Hong Q, Lebrilla CB, Miyamoto S, Ruhaak LR. Absolute quantitation of immunoglobulin g and its glycoforms using multiple reaction monitoring. Anal Chem. 2013;85:8585–93.
Krishnan S, Shimoda M, Sacchi R, Kailemia MJ, Luxardi G, Kaysen GA, et al. HDL glycoprotein composition and site-specific glycosylation differentiates between clinical groups and affects IL-6 secretion in lipopolysaccharide-stimulated monocytes. Sci Rep. 2017;7:43728.
Ruhaak LR, Kim K, Stroble C, Taylor SL, Hong Q, Miyamoto S, et al. Protein-specific differential glycosylation of immunoglobulins in serum of ovarian cancer patients. J Proteome Res. 2016;15:1002–10.
Ruhaak LR, Barkauskas DA, Torres J, Cooke CL, Wu LD, Stroble C, et al. The serum immunoglobulin G glycosylation signature of gastric cancer. EuPA Open Proteom. 2015;6:1–9.
Darebna P, Novak P, Kucera R, Topolcan O, Sanda M, Goldman R, et al. Changes in the expression of N- and O-glycopeptides in patients with colorectal cancer and hepatocellular carcinoma quantified by full-MS scan FT-ICR and multiple reaction monitoring. J Proteomics. 2017;153:44–52.
Li Q, Kailemia MJ, Merleev AA, Xu G, Serie D, Danan LM, et al. Site-specific glycosylation quantitation of 50 serum glycoproteins enhanced by predictive glycopeptidomics for improved disease biomarker discovery. Anal Chem. 2019;91:5433–45.
Ramachandran P, Xu G, Huang HH, Rice R, Zhou B, Lindpaintner K, et al. Serum glycoprotein markers in nonalcoholic steatohepatitis and hepatocellular carcinoma. J Proteome Res. 2022;21:1083–94.
Wu Z, Serie D, Xu G, Zou J. PB-Net: Automatic peak integration by sequential deep learning for multiple reaction monitoring. J Proteomics. 2020;223:103820.
Pickering C, Zhou B, Xu G, Rice R, Ramachandran P, Huang H, et al. Differential peripheral blood glycoprotein profiles in symptomatic and asymptomatic COVID-19. Viruses. 2022;14:553.
Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. 2021;14:101174.
Chandrasekar D, Guerrier C, Alisson-Silva F, Dhar C, Caval T, Schwarz F, et al. Warning signs from the crypt: aberrant protein glycosylation marks opportunities for early colorectal cancer detection. Clin Transl Gastroenterol. 2023;14:e00592.
Qiu Y, Patwa TH, Xu L, Shedden K, Misek DE, Tuck M, et al. Plasma glycoprotein profiling for colorectal cancer biomarker identification by lectin glycoarray and lectin blot. J Proteome Res. 2008;7:1693–703.
Pan Y, Zhang L, Zhang R, Han J, Qin W, Gu Y, et al. Screening and diagnosis of colorectal cancer and advanced adenoma by Bionic Glycome method and machine learning. Am J Cancer Res. 2021;11:3002–20.
Gu Y, Duan B, Sha J, Zhang R, Fan J, Xu X, et al. Serum IgG N-glycans enable early detection and early relapse prediction of colorectal cancer. Int J Cancer. 2023;152:536–47.
Takei D, Harada K, Nouso K, Miyahara K, Dohi C, Matsushita H, et al. Clinical utility of a serum glycome analysis in patients with colorectal cancer. J Gastroenterol Hepatol. 2022;37:727–33.
Barrow H, Guo X, Wandall HH, Pedersen JW, Fu B, Zhao Q, et al. Serum galectin-2, -4, and -8 are greatly increased in colon and breast cancer patients and promote cancer cell adhesion to blood vascular endothelium. Clin Cancer Res. 2011;17:7035–46.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer Statistics, 2023. CA Cancer J Clin. 2023;73:17–48.
Mulshine JL, D’Amico TA. Issues with implementing a high-quality lung cancer screening program. CA Cancer J Clin. 2014;64:352–63.
Zeng X, Hood BL, Sun M, Conrads TP, Day RS, Weissfeld JL, et al. Lung cancer serum biomarker discovery using glycoprotein capture and liquid chromatography mass spectrometry. J Proteome Res. 2010;9:6440–9.
Heo S-H, Lee S-J, Ryoo H-M, Park J-Y, Cho J-Y. Identification of putative serum glycoprotein biomarkers for human lung adenocarcinoma by multilectin affinity chromatography and LC-MS/MS. Proteomics. 2007;7:4292–302.
Arnold JN, Saldova R, Galligan MC, Murphy TB, Mimura-Kimura Y, Telford JE, et al. Novel glycan biomarkers for the detection of lung cancer. J Proteome Res. 2011;10:1755–64.
Vasseur JA, Goetz JA, Alley WR, Novotny MV. Smoking and lung cancer-induced changes in N-glycosylation of blood serum proteins. Glycobiology. 2012;22:1684–708.
Fang K, Long Q, Liao Z, Zhang C, Jiang Z. Glycoproteomics revealed novel N-glycosylation biomarkers for early diagnosis of lung adenocarcinoma cancers. Clin Proteomics. 2022;19:43.
Ahn J-M, Sung H-J, Yoon Y-H, Kim B-G, Yang WS, Lee C, et al. Integrated glycoproteomics demonstrates fucosylated serum paraoxonase 1 alterations in small cell lung cancer. Mol Cell Proteomics. 2014;13:30–48.
Mitchell A, Pickering C, Xu G, Rice R, Castellanos A, Bhadra R, et al. Glycoproteomics as a powerful liquid biopsy-based screening tool for non-small cell lung cancer. J Clin Oncol. 2022;40:e21148–e21148.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer Statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Lu KH. Screening for ovarian cancer in asymptomatic women. JAMA. 2018;319:557–8.
Charkhchi P, Cybulski C, Gronwald J, Wong FO, Narod SA, Akbari MR. CA125 and ovarian cancer: a comprehensive review. Cancers. 2020;12:3730.
Dochez V, Caillon H, Vaucel E, Dimet J, Winer N, Ducarme G. Biomarkers and algorithms for diagnosis of ovarian cancer: CA125, HE4, RMI and ROMA, a review. J Ovarian Res. 2019;12:28.
Ankenbauer KE, Rao TC, Mattheyses AL, Bellis SL. Sialylation of EGFR by ST6GAL1 induces receptor activation and modulates trafficking dynamics. J Biol Chem. 2023;299:105217.
Dorsett KA, Jones RB, Ankenbauer KE, Hjelmeland AB, Bellis SL. Sox2 promotes expression of the ST6Gal-I glycosyltransferase in ovarian cancer cells. J Ovarian Res. 2019;12:93.
Schultz MJ, Holdbrooks AT, Chakraborty A, Grizzle WE, Landen CN, Buchsbaum DJ, et al. The tumor-associated glycosyltransferase ST6GaL-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 2016;76:3978–88.
O’Flaherty R, Muniyappa M, Walsh I, Stöckmann H, Hilliard M, Hutson R, et al. A robust and versatile automated glycoanalytical technology for serum antibodies and acute phase proteins: ovarian cancer case study. Mol Cell Proteomics. 2019;18:2191–206.
Pan J, Hu Y, Sun S, Chen L, Schnaubelt M, Clark D, et al. Glycoproteomics-based signatures for tumor subtyping and clinical outcome prediction of high-grade serous ovarian cancer. Nat Commun. 2020;11:6139.
Hu Y, Pan J, Shah P, Ao M, Thomas SN, Liu Y, et al. Integrated proteomic and glycoproteomic characterization of human high-grade serous ovarian carcinoma. Cell Rep. 2020;33:108276.
Serie D, Moser K, Pickering C, Aiyetan P, Xu G, Rice R, et al. Liquid-biopsy-derived glycoproteomic profiling as a novel means for noninvasive diagnosis of ovarian cancer. J Clin Oncol. 2022;40:e17604–e17604.
Lindpaintner K, Pickering C, Mitchell A, Xu G, Cong X, Serie D. Abstract 5314: A peripheral blood-based glycoproteomic predictor of checkpoint inhibitor treatment benefit in advanced non-small cell lung cancer. Cancer Res. 2023;83:5314–5314.
Lindpaintner K, Srinivasan A, Mitchell A, Dixit A, Xu G, Cong X, et al. 158 A novel, highly accurate liquid biopsy-based glycoproteomic predictor of checkpoint inhibitor treatment benefit in advanced non-small cell lung cancer. Regular and Young Investigator Award Abstracts. BMJ Publishing Group Ltd; 2022. p. A171–A171.
Lindpaintner K, Cheng M, Prendergast J, Normington K, Wong M, Xu G, et al. 30 Blood-based glycoprotein signatures in advanced non-small-cell lung carcinoma (NSCLC) receiving first-line immune checkpoint blockade. J Immunother Cancer. 2021;9:A35–A35.
Wang Y-N, Lee H-H, Hsu JL, Yu D, Hung M-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J Biomed Sci. 2020;27:77.
Lee H-H, Wang Y-N, Xia W, Chen C-H, Rau K-M, Ye L, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36:168–78.
Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632.
Li C-W, Lim S-O, Chung EM, Kim Y-S, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201.
Funding
None.
Author information
Authors and Affiliations
Contributions
KH, DWH: Conceptualization, writing—original draft, supervision; MB, HK, TC, GX: Conceptualization, writing—original draft; CL, CB: Writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
MB, TC, GX, DWH: employees of InterVenn Biosciences.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, K., Baniasad, M., Kwon, H. et al. Decoding the glycoproteome: a new frontier for biomarker discovery in cancer. J Hematol Oncol 17, 12 (2024). https://doi.org/10.1186/s13045-024-01532-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01532-x