
Abstract
The tumor microenvironment (TME) has been extensively investigated; however, it is complex and remains unclear, especially in elderly patients. Senescence is a cellular response to a variety of stress signals, which is characterized by stable arrest of the cell cycle and major changes in cell morphology and physiology. To the best of our knowledge, senescence leads to consistent arrest of tumor cells and remodeling of the tumor-immune microenvironment (TIME) by activating a set of pleiotropic cytokines, chemokines, growth factors, and proteinases, which constitute the senescence-associated secretory phenotype (SASP). On the one hand, the SASP promotes antitumor immunity, which enhances treatment efficacy; on the other hand, the SASP increases immunosuppressive cell infiltration, including myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), M2 macrophages, and N2 neutrophils, contributing to TIME suppression. Therefore, a deeper understanding of the regulation of the SASP and components contributing to robust antitumor immunity in elderly individuals with different cancer types and the available therapies is necessary to control tumor cell senescence and provide greater clinical benefits to patients. In this review, we summarize the key biological functions mediated by cytokines and intercellular interactions and significant components of the TME landscape, which influence the immunotherapy response in geriatric oncology. Furthermore, we summarize recent advances in clinical practices targeting TME components and discuss potential senescent TME targets.
Similar content being viewed by others

Introduction
In the traditional view, tumors cause a disease that is closely associated with age. However, from the perspective of cellular function, senescent cells and tumor cells have diametrically opposite behavior. Senescent cells manifest a loss of function or cessation of proliferation, while tumors manifest hyperproliferation and increased metabolism rates [1]. Recently, many studies have shown that aging involves multiple mechanisms, which both prevent cancer and promote tumorigenesis [2]. Therefore, the association between aging and tumors needs to be further explored. In addition, in elderly individuals, the cellular microenvironment is changing, which in turn allows tumors to survive in a unique cytokine, extracellular matrix (ECM), and vascular environment. This specific microenvironment contributes to tumor growth and cancer cell invasion and immune escape [3]. Senescence is a damage-induced cellular response to cancer treatment. Leonard Hayflick and colleagues first observed that human diploid fibroblasts undergo a finite number of doublings before irreversibly arresting, the process named senescence [4,5,6]. Senescence indicates a conserved response to many different types of external and internal cellular stress, including telomere shortening and oncogenic, genotoxic, metabolic, and oxidative damage, and the instances of all these responses can increase during cancer therapy [7,8,9]. Senescence has been revealed to be a broad physiological response to tissue damage that plays a pleiotropic role in aging, embryonic development, wound healing, tissue regeneration, and, importantly, responses to oncogenesis and cancer therapy [10,11,12,13,14,15].
The mechanisms of both cancer and aging are based on a time-dependent accumulation of cell damage. Previous studies have shown that many of the hallmarks of aging, including epigenetic alterations, intracellular interaction changes, changes in protein homeostasis (proteostasis), mitochondrial dysfunction and molecular senescence, are common to cancer [16]. In 2020, cancer contributed to 18% of all deaths and remained the second leading cause of death after heart diseases in the USA. However, it is the leading cause of death among women aged 40–79 years and men aged 60–79 years. In the USA, from 2017 to 2019, the probability of developing invasive cancer is 34% in 70 years and older male populations and 27.2% in female populations. In 2019, there were approximately 140,690 new cancer cases diagnosed and 103,250 cancer deaths among the “oldest old” (≥ 85 yr), also the cancer incidence rates peaked in the oldest men and women in 1990 although the rates have subsequently declined. Based on these statistics, a considerable number of cancer cells acquire aging phenotypes [17,18,19]. Many studies have highlighted that aging can markedly affect normal cells in the tumor microenvironment (TME) and thus promote tumor progression and metastasis. Fibroblasts and immune cells are thought to play necessary roles in this age-related impact [20, 21]. Tumor progression often requires genetic mutations in growth pathway genes to drive hyperproliferation, and distinct mutations trigger senescence biological processes. Aging is associated with many factors that are involved in the aforementioned processes, such as enhanced genomic damage (point mutations, deletions, and translocations), telomere attrition, epigenetic alternation, impaired proteostasis, and deregulation of nutrient sensing [22, 23]. Many environmental factors, such as ultraviolet radiation exposure, alcohol, smoking, and pollution, contribute to the chronic accumulation of DNA damage and other events associated with cellular aging. Previous studies suggested that the cellular aging process of somatic selection is nonautonomous and is, in fact, defined by TME-imposed increases in positive selection for previously accumulated genetic and/or phenotypic diverse senescent tissues, which is leveraged to ensure that senescent models can induce cancer across tissues and species [24]. Many factors involved in senescent tissue evolution result in the final transformation to malignancy and hyperplastic growth in self-renewing tissues, which contribute to growth arrest, apoptosis, and the degradation of other cells and structural tissue components. With increasing age, the cancer risk and many degradative features within tissues and cells exponentially increase [23]. An increasing number of studies have focused on the complex interrelationship between an aged local and systematic TME and its fundamental role in tumor development and progression (Fig. 1). In addition, age-induced reprogramming of stromal components in an established TME seems to contribute to tumor metastasis and progression. Interestingly, clear impact of senescent stromal cells on cancer outcomes is not determined yet or even contradictory, which suggests that different stromal tissue environments in the body may be reprogrammed differently during the aging process. This mechanism ultimately influences tumor growth and progression with respect to the original tissue [25,26,27]. In this review, we will (1) discuss the interactions between tumors and an aged TME, identifying how TME changes during aging facilitate the reprogramming of stromal cell populations, the ECM, and immune cell infiltration to initiate cancer and progression; (2) then investigate how an aging TME regulates the potential responses of cancer cells to chemotherapy, radiotherapy, targeted therapy, and immunotherapies; and (3) summarize recent clinical progress in geriatric oncology and aging TME. With these foci, we hope to identify additional tumor therapy methods for this special population of aging individuals.

The hallmarks of cancer and new additions. Besides the original hallmarks of cancer, four new hallmarks of “senescent cells,” “unlocking phenotypic plasticity,” “nonmutational epigenetic reprogramming,” and “polymorphic microbiomes” have been added to these parameters. The four new hallmarks are believed to stimulate debate, discussion and experimental elaboration, also give us multiple facets of understanding on human cancer. Cancer senescence can be regarded as a distinct TME feature as it leads to further research interest. TME, tumor microenvironment
Intracellular changes in senescent cells
Increased levels of transcription and protein synthesis in senescent cells converge to promote a distinct aspect of senescence, the acquisition of the senescence-associated secretory phenotype (SASP), which promotes senescent cells to communicate damage signals with neighboring cells, including immune cells, fibroblasts, endothelial cells, and adjacent nontumor epithelial cells in the TME in a nonautonomous manner in cells [28, 29] (Table 1). Enhancer regions of hundreds of SASP factors are made accessible, transcribed by NF-κB and other factors, and translated in an mTOR-dependent manner, contributing to the robust secretion of inflammatory cytokines and chemokines and angiogenic, growth, and ECM-degrading signals [21]. The SASP is also associated with the upregulation of cell surface molecules that modulate the interactions between senescent cells and the immune system [30, 31]. Notably, hallmarks of senescence are neither specific nor universal to all types of senescent cells and that multiple markers are necessary to distinguish senescence from other biological outcomes of cancer therapy [7].
The dynamic nature of the SASP and its ability to modulate the surrounding tissue TME and immune responses in different ways is thought to be associated with many contrasting physiological characteristics of senescence (Fig. 2 and Table 2). In fact, senescence can lead to anti- or protumorigenic outcomes depending on the senescence inducer, the duration of the senescent period, the SASP factors produced, and the tissue and disease context [32, 33]. Biomarkers of cellular senescence have been thoroughly investigated in precancerous tissues in different solid organs in humans, including the lungs, prostate, pancreas, and skin, and have been found to be lost during neoplastic progression [15, 34]. Therefore, it has been postulated that senescence may suppress tumors and thus block tumor development by preventing the proliferation of potentially malignant cells. Supporting this hypothesis, oncogene-induced senescence following aberrant RAS activation led to the arrest of premalignant cells and secretion of proinflammatory SASP factors that promoted innate and adaptive immunity cell clearance of incipient cancer cells and blocked tumor formation [35]. Similarly, therapy-induced senescence (TIS) has been shown to inhibit tumor growth and lead to an influx of cytotoxic CD8+ T cells and natural killer (NK) cells that promote tumor regression [36, 37]. In contrast, some evidence has demonstrated that the SASP after chemotherapy can promote tumors through the secretion of immune suppressive factors and attraction of immune suppressive cells, as well as the production of angiogenic and other growth factors, which enhance the invasion and metastasis of adjacent non-senescent tumor cells [38, 39]. Senescence is generally presumed to be neither a permanent nor irreversible state and tumor cells that bypass senescence through the acquisition of genomic instability (e.g., polyploidy) likely achieve enhanced stemness and tumorigenic potential that contributes to drug resistance and tumor relapse [40,41,42,43].

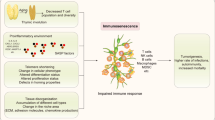
Differences between young and aged TME and senescence-induced factors. One of the critical factors involved in age-related pathologies is immunosenescence defined as a significant decline in overall immune function. The subpopulations of effector immune cells including T cells, NK cells, macrophages, and DCs exhibit a dramatic decrease in cytotoxic activity during the senescence. Age-related immunosenescence plays a key role in promoting tumor formation and accumulation of SASP-secreting cells; the SASP-related decreases in effector immune cell function can also induce tissue-specific switching toward more immunosuppressive cell populations. In the elderly, immunosuppressive MDSCs and Tregs are significantly increased in aged tissue and blood; besides, neutrophils and macrophages appear to switch phenotypically toward immunosuppressive N2 and M2 states, both of which have been shown to promote tumorigenesis of various cancer types, while more direct evidence on the involvement in age-related tumorigenesis is warranted. The accumulation of SASP stromal components results in inflammaging that disrupts acute inflammatory response toward malignant tissue, induces infiltration of immunosuppressive MDSCs and Tregs, and improves secretion of anti-inflammatory components such as cytokines, chemokines, and inflamma-microRNAs. Inflammaging seems to downregulate antitumor activity in aged tissues. NK cells, natural killer cells; DCs, dendritic cells; SASP, senescence-associated secretory phenotype; MDSCs, myeloid-derived suppressor cells; Tregs, regulatory T cells; ARG1, arginase 1; CRP, C-reactive protein; ECM, extracellular matrix; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFN-γ, interferon-γ; IL, interleukin; ROS, reactive oxygen species; TGF-β, transforming growth factor-β; TNF, tumor necrosis factor (Mainly from https://doi.org/10.1038/s41568-019-0222-9 [3])
During aging, cells undergo a series of intracellular changes. In the nucleus, during cell division and senescence, telomeres at the ends of DNA sequences are shortened, resulting in their reduced binding to protective protein complexes, which protect DNA from DNA damage response (DDR) factors. Shortened telomeres and DDR activation may establish connections between senescent cells and tumor cells [44]. Telomeres act a necessary role in protecting chromosome ends, preventing DDR, and maintaining genomic stability. There are two telomere maintenance mechanisms (TMMs) in human cancer to keep the infinite capacity for tumor proliferation: one is telomerase-mediated maintenance (observed in 85%) and the other is alternative lengthening of telomeres (ALT) (observed in 15%). Unique characteristics of ALT include very long telomeres, telomere length heterogeneity, abundant extrachromosomal linear and circular telomere DNA, increased telomere-sister chromatid exchange (T-SCE) events, and the formation of ALT-associated promyelocytic leukemia (PML) bodies. It is crucial to understand the molecular mechanism underlying ALT and its impact on cancer prognosis as ALT can be therapeutic target [44, 45]. Similarly, with aging, genetic mutations gradually accumulate, especially when cells are exposed to tobacco and other chemicals, ultraviolet rays, ionizing radiation, or exogenous mutagens. Mutation ultimately increases the probability of cancer occurrence. This theory has been verified by cancer driver mutations detected in many middle-aged and older individuals [46]. Moreover, p16, a tumor suppressor protein that regulates Rb protein phosphorylation, accumulates with aging in most mammals and ultimately participates in tumorigenesis [47]. Furthermore, another classic tumor suppressor, p53, has been shown to induce senescence by affecting the downstream factor p51 [48]. In addition to changes in DNA and gene expression, the epigenetic inheritance of senescent cells regulates tumor formation and progression. The rate of methylation, a common gene repressive modification, increases with age. Therefore, methylation of tumor suppressor gene promoters, such as the VHL promoter, has been suggested to contribute to angiogenesis, thereby promoting tumors, and hypermethylated cells prefer to undergo oncogenic transformation [49, 50].
In addition to changes at the genetic level, cytoplasmic alterations are involved in aging and tumors. Reactive oxygen species (ROS) are byproducts of mitochondrial electron transfer in aerobic cells. High levels of ROS lead to cell damage and increase genomic instability to induce oncogenic functions [51]. Studies have shown that during aging, dysfunctional mitochondria gradually accumulate, and deleterious events, including membrane potential reduction and proton leakage, eventually lead to increased ROS levels [7]. Moreover, MAPK, PI3K, and STAT3 can be regulated by ROS, which promotes cell proliferation and survival, as has been discovered in breast cancer, lung cancer, pancreatic cancer, and other malignant tumors [52]. In addition, changes in mitochondria cause corresponding changes in AMP/ATP, AMP/ATP, and NAD+/NADH ratios, leading to cell cycle arrest, NF-κB activation, and other changes that are considered to be important to tumor formation [7]. In addition, the endoplasmic reticulum is an important subcellular organelle in lipid synthesis and protein synthesis, and the unfolded protein response (UPR) is triggered when excessive ROS levels cause protein misfolding. In normal senescent cells, the UPR drives cell death, but in breast cancer, the UPR prevents cell death and promotes cancer cell immortality [53]. In addition, centrosome dysfunction may regulate aging, tumorigenesis, and tumor immunity [54].
In addition, the role of the cGAS-STING signaling pathway, constituted by cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING), has been extensively studied. cGAS recognizes DNA in the cytoplasm and activates IFN expression and NF-κB through multiple cascade reactions. The effects of cGAS-STING on tumors are diverse. On the one hand, the interferon produced by short-term cGAS-STING activation can recruit dendritic cells and CD8+ T cells and promote their maturation. After tumor cells are killed, the tumor-associated antigens that are released are by surrounding DCs, which will further enhance the immune response, thus forming a positive feedback mechanism [55]. A positive correlation between cGAS expression and survival has been reported in human lung adenocarcinoma patients [56]. On the other hand, long-term cGAS-STING activation may promote tumorigenesis. cGAS-STING can assist in the formation of the SASP, ultimately promoting the epithelial–mesenchymal transition (EMT), tumor progression, and invasion [57]. Another study demonstrated that activation of STING might disrupt calcium homeostasis in T cells, leading to cell death [58]. De Cecco found that senescent cells, loss of the nuclear lamin protein Lamin B1 and chromatin fragments located in the cytoplasm can activate the cGAS-STING pathway. In the same study, high activity of long-interspersed element-1 (LINE-1) reverse-transcribes mRNA into cDNA and activates cGAS-STING [59]. Additionally, infection with various DNA viruses, such as human cytomegalovirus and hepatitis B virus, which are common infections in elderly individuals, can activate the cGAS/STING pathway [60]. This process, in turn, modulates the tumor microenvironment in elderly individuals. The aforementioned evidence suggests that cells engage in unique mechanisms to inhibit tumor formation during the early stages of aging and that when inactivated or mutated, these mechanisms may in turn promote tumor formation and progression.
The SASP can reinforce senescent growth arrest and/or promote immune surveillance to suppress cancer [21]. Oncogene-induced and therapy-induced senescent cells secrete the inflammatory cytokine IL-1α, which is a crucial SASP initiator and regulator [61]. IL-1α facilitates an autocrine inflammatory response through the activation of NF-κB, which leads to the transcription of IL-6 and IL-8 [61]. Subsequently, these inflammatory cytokines reinforce senescence-related proliferation arrest through the increased production of reactive oxygen species and a sustained DNA damage response, particularly in oncogene-induced senescent cells [61, 62]. In addition, IL-1α mediates paracrine senescence in neighboring cells to suppress tumor progression [63], and IL-1α, IL-6, and IL-8 mediate the recruitment of M1-like macrophages, T helper 1 cells, and NKs to the TME. Infiltrative immune cells drive the elimination of senescent tumor cells and may also eliminate non-senescent cancer cells via a bystander effect [35, 64]. Some immune cells, such as T helper 1 cells, can also trigger senescence in cancer cells through the secretion of inflammatory cytokines [65]. The SASP of senescent cancer cells is thought to initially suppress tumorigenesis but to be mostly detrimental in the long term [38, 66]. In an in vivo study, proliferation and tumorigenesis of both premalignant and malignant epithelial cells were increased when they were coinjected with human senescent fibroblasts into mice [67]. Another study showed that MMPs secreted by senescent human fibroblasts were critical for promoting tumorigenesis [68]. Prominent SASP factors are involved in ECM processing and degradation, which can promote tumor cell proliferation and invasion [69]. Additionally, MMPs promote the release of many other cytokines and growth factors supporting tumorigenesis, such as vascular endothelial growth factor (VEGF), which promotes tumor-driven angiogenesis [70], and the chemokine CXCL1, which promotes tumor growth [71].
IL-6 and IL-8, known SASP-associated factors, mediate the protumorigenic effects of senescent cells because they establish a chronic inflammatory TME that triggers tumor growth [28, 72]. In addition, IL-6 and IL-8 drive the transcription of genes encoding MMPs and drive the epithelial-to-mesenchymal transition, thereby promoting tumor invasiveness [73,74,75,76]. IL-6 also recruits myeloid-derived suppressor cells (MDSCs) to the TME to mediate the protumorigenic effects of senescent cells, which block IL-1α signaling and antagonize senescence in cancer cells [77, 78]. In addition, MDSCs block immune surveillance by inhibiting CD8+ T cells and NK cells through the actions of IL-6 and CCL2, respectively [64, 78, 79]. However, the protumorigenic and antitumorigenic effects of senescent cancer cells are likely mediated by a comprehensive interaction between multiple SASP factors and the immune TME. Furthermore, the effects of SASP factors are likely impacted by tissue type, residual immune cells, inflammatory networks, and senescence inducers. Therefore, it is difficult to precisely identify whether the effects of senescent cancer cells are protumorigenic or antitumorigenic.
Several studies have demonstrated that senescence-induced therapies are associated with complex reprogramming that ultimately drives stemness in both tumor and normal cells [40, 41]. Moreover, senescent cancer cells that are not eliminated by the immune system can spontaneously resist proliferation arrest under certain circumstances and reenter the cell cycle [40, 80]. A study confirmed that oncogene-induced senescent cells entered the cell cycle, particularly by restoring telomerase activity through the derepression of the telomerase reverse transcriptase (TERT) gene [81]. Senescent cells show WNT-dependent enhanced growth and tumor-initiating potential to resume growth [40]. This senescence-associated stemness results in a highly aggressive nature driven by WNT pathway activation independent of WNT ligand binding via the SASP and is enriched in relapsed tumors [40]. Additionally, the expression of β-catenin in pituitary stem cells provokes the acquisition of a senescence signature and the SASP and induces craniopharyngioma tumors in a paracrine fashion. Importantly, mice with a decreased senescent cell burden and an attenuated SASP response exhibited decreased tumorigenic potential, indicating that the SASP may promote tumor induction [82]. Extracellular vesicles and exosomes, components of the SASP, have also garnered considerable interest in the field of senescence, and small vesicles from senescent cells can promote tumors [83,84,85]. The complex and often unpredictable role of senescence-inducing therapies is derived from the dual role of the SASP. The effect of the SASP is highly dependent on context and cell type and varies during different stages of cancer progression [21, 38, 86]. Specifically, SASP-mediated immunosuppression promotes tumor growth in later stages of tumor progression, while the SASP is a tumor suppressor in the early stages of tumorigenesis [64]. In addition, TP53 may be involved in determining whether the senescence-induced inflammatory response suppresses or promotes tumorigenesis [87]. In the long term, the SASP of senescent tumors is suggested to be primarily detrimental to neoplastic growth, therapy resistance, immunosuppression, metastasis and angiogenesis [67, 78, 87, 88]. However, senescent cancer cells potentially remain dormant for a long time, evading therapy and posing a risk for tumor relapse [15, 89, 90]. It has also been revealed that many genotoxic chemotherapies lead to debilitating side effects caused by senescence induced in normal tissues. Normal senescent cells remain present in the long term and promote local and systematic inflammation caused by the SASP, which results in or exacerbates chemotherapy side effects [88]. Accordingly, it may be helpful to combine senescence-promoting therapy with senolytic therapy in the context of cancer. In addition to direct targeting of cancer cells by delivering a one-two punch and decreasing the side effects of chemoradiotherapies on normal tissues, senolytics may eliminate incipient preneoplastic senescent cells or other senescent cells in the TME to suppress the detrimental effects of the SASP [66, 72].
Changes in cytokines and their receptors
The tumor microenvironment consists of a variety of cytokines that can affect tumor progression, metastasis, and the formation of an immunosuppressive microenvironment. In aging patients, changes in the crosstalk between cytokines and immune cells characterize a unique aging tumor microenvironment [91] (Fig. 3).

The formation of SASP in senescent cell. The formation of SASP undergoes multiple mechanism and regulator; many time-dependent damage will accelerate the formation of SASP. Actually, transformation of SASP involves many signaling pathways. The deletion of p53 and the upregulated expression of RAS aggravate the paracrine activity of SASP. Besides, the three-dimensional structure of the genome in senescent cells can enhance the SAE activity through the transcription factor C/EBPα/β, thereby promoting the secretion of SASP. DDR-dependent SASP activation accompanies by chromatin remodeling frequently, in which HDAC might be involved. SAE SASP of senescent cells also damages the DNA of adjacent cells and induces senescence, thereby forming senescence-induced senescence. The cytoplasm alternation is also involved in aging and tumors. ROS is a by-product of mitochondrial electron transfer in aerobic cells. High levels of ROS will lead to cell damage and increase genomic instability to exert oncogenic functions. In the process of aging, dysfunction mitochondria will gradually accumulate, and events including membrane potential reduction and proton leakage will occur, eventually leading to increased ROS levels. Not only that, changes in mitochondria will cause corresponding changes in AMP/ATP, AMP/ATP, and NAD + /NADH ratios, thereby leading to cell cycle arrest, NF-κB activation, and other changes that are considered important to tumor formation. Transcribed by NF-κB and other factors, and translated in an mTOR-dependent manner contributing to the robust secretion of SASP-related inflammatory cytokines, chemokines, angiogenic growth, and ECM-degrading signals. SASP, senescence-associated secretory phenotype; DDR, DNA damage response; SAE, senescence activation enhancer; C/EBPα/β, CCAAT/enhancer-binding protein α/β; HDAC, histone deacetylase; ATP, adenosine triphosphate; AMP, adenosine monophosphate
Formation of senescence and SASP
Cellular senescence serves as a powerful protective mechanism against tumorigenesis [15]. The activation of oncogenes such as HRASV12 triggers growth arrest, referred to as oncogene-induced senescence (OIS), which was first reported in 1997 [10, 13, 92, 93]. In 2005, the concept of OIS was extended to multiple carcinogenesis models, including lymphomas, prostate cancer, lung adenomas, hyperplastic pituitary gland, and melanocytic nevi [93,94,95,96,97]. Melanocytic nevi induced by BRAF mutations gradually remain senescent for decades, preventing their progression into melanoma [95]. Similarly, the lack of tumor suppressor genes, such as PTEN, can also induce senescence in the primary prostate epithelium, referred to as PTEN loss-induced cellular senescence (PICS) [94]. In older adults, the composition of cytokines and immune cells changes because of SASP acquisition. The acquisition of the SASP involves multiple mechanisms and regulators. Time-dependent damage accelerates SASP acquisition. Several studies have shown that + transformation of the SASP involves many signaling pathways [3]. The deletion of P53 and the upregulated expression of RAS aggravate the paracrine activity of the SASP [28]. Moreover, the three-dimensional structure of the genome in senescent cells enhances senescence activation enhancer (SAE) activity through the action of the transcription factor CCAAT/enhancer-binding protein α (C/EBPα), thereby promoting the secretion of SASP factors [98]. DDR-dependent SASP acquisition is frequently accompanied by chromatin remodeling, in which histone deacetylase (HDAC) might be involved [99]. The SASP in senescent cells also damages the DNA of adjacent cells and induces their senescence, causing senescence-induced senescence [100, 101]. In addition, OIS is mediated by activation of the INK4A-RB pathway, independent of p53 activation and DNA damage signaling [10, 92, 102]. Moreover, senescence can be triggered by other oncogenic pathways, such as the activated MYC pathway, which increases the levels of the ARF-encoding transcript at the CDKN2A locus, resulting in stabilized p53 [103] and hyperactivated WNT-β-catenin signaling, leading to the DNA damage response via the p53-p21 pathway [104,105,106,107,108].
Malignant cells can be forced to enter a senescent state via therapy-induced senescence, and conventional therapeutics such as chemotherapy or radiotherapy show the ability to induce senescence in cancer cells [9, 36, 37, 109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124] (Table 3). In a chemotherapy-induced senescent state, apoptosis is induced when higher doses of drugs are applied [125,126,127]. Mechanistically, many chemotherapies cause DNA damage in cancer cells, which triggers senescence through ATM-CHK2 and ATR-CHK1 kinase-mediated activation of the interconnected p53-RB pathways [128, 129]. Topoisomerase I and II inhibitors, such as doxorubicin, have been shown to dysregulate the re-ligation of DNA strands after supercoil unwinding, leading to large-scale DNA damage and increasing expression of p53 and its downstream targets CDKN1A and SERPINE1, thereby inducing senescence [130,131,132]. Platinum-based therapies, including cisplatin, carboplatin, and oxaliplatin, induce DNA damage through DNA cross-linking, leading to senescence induction [133, 134]. Alkylating agents such as temozolomide, dacarbazine, and busulfan cross-link with DNA by reacting with atoms in DNA, triggering a DNA damage-mediated senescence response [135]. Cell cycle dysfunction caused by microtubule inhibitors (paclitaxel and docetaxel) may cause extensive DNA damage and trigger a p53-p21 pathway-facilitated senescence response [136, 137]. Methotrexate and gemcitabine induce genotoxic stress by blocking DNA synthesis, thereby inducing cellular senescence [138, 139]. Radiotherapy is widely used for the treatment of multiple cancer types and can induce irreparable DNA damage response that activates ATM or ATR and p53-p21 pathway-mediated apoptosis and cellular senescence [129, 140, 141]. Since radiotherapy is applied locally, the tissue surrounding a tumor shows an increase in senescent cell burden that results in immunosuppressive effects [38, 78, 142].
Upregulation of cyclin-dependent kinase (CDK) inhibitor proteins such as INK4A and p21 to induce cell cycle arrest is a hallmark of senescent cells [143]. CDK4/6 are important for the progression from the G1 phase to the S phase of the cell cycle and are overexpressed in a number of human cancers. CDK4/6 mimic the function of INK4A and induce senescence in various cancer cells [144,145,146,147,148,149,150,151]. A triple CDK2/4/6 inhibitor (PF-06873600) that is still being investigated for the treatment of breast cancer has been shown to be a potential senescence inducer in various cancer models [152, 153]. Inhibition of DNA replication through small-molecule inhibition of the kinase CDC7, for example, by XL413 or TAK-931, leads to senescence induction in liver cancer cells. This senescence response has been observed only in TP53-mutant tumors, presumably because TP53-mutant tumors retain the ability to be arrested in the cell cycle upon CDC7 inhibition [124].
Numerous compounds inhibiting telomerase complex action have been identified as candidates for anticancer therapy [154], among which BIBR15 and GRN163L are potent telomerase inhibitors that greatly promote senescence and suppress cancer cell proliferation [155,156,157]. However, the use of GRN16 for senescence-promoting therapy should be examined further, as it also induces apoptosis in pancreatic cancer cells [157]. Vorinostat, a histone deacetylase inhibitor, upregulates the expression of multiple tumor suppressor genes, such as CDKN2A and TP53, and induces senescence via these two major pathways in various cancer cell lines [158, 159]. In mouse models, genetic restoration of Trp53 resulted in the regression of sarcomas and liver carcinomas by inducing a senescence response, and an apoptotic response was observed in lymphoma regression [160,161,162]. Additionally, senescence induction has been shown to be accompanied by the acquisition of the SASP and recruitment of immune cells into tumors, suggesting efficient clearance of senescent cancer cells [160]. The MDM2 inhibitors nutlin-3 and RG7112 interact with p53-MDM2 and show promising results for inducing senescence in tumors retaining wild-type TP53 in human cancer cell models [163,164,165]. Inactivation of PTEN shows the potential for use in senescence-promoting cancer therapy in vitro and in mice [166]. The PTEN status in vitro has been shown to be a crucial determinant of glioma cell fate after ionizing radiation exposure; PTEN-mutant cells underwent premature senescence, while cancer cells expressing PTEN underwent apoptosis [167]. In addition, the inactivation of PTEN resulted in p53-mediated senescence and suppression of tumorigenesis in mice [94]. While these genes are mediators in the senescence response of cancer cells, they are not essential for senescence induction in cancer [86].
Changes in association with immune molecules and cells
The SASP can affect the tumor microenvironment in many ways (Fig. 4). SASP components include immunoregulatory factors, including IL-6, IL-8, and MCP-2; growth factors, including HGF and IGFBP; and exfoliated cell survival factors, including ICAMs and UPAR [28]. As an autocrine proinflammatory factor, Il-1α binds to cell receptors to form a positive feedback mechanism mediated through the NF-κB pathway, which might maintain the SASP and the secretion of IL-1β, IL-6, and IL-8 [168, 169]. A previous study showed that IL-6 functioned through complex mechanisms. Under oncogenic stress, IL-6 is an autocrine factor that inhibits cell proliferation via cell cycle arrest. However, when acting as a paracrine factor, IL-6 promotes angiogenesis, which contributes to tumor progression [29]. Other SASP components, namely, chemokines, function in the tumor microenvironment. Multiple studies have confirmed that CCL5 inhibits the activation of Th1 cells and cytotoxic T cells and recruits MDSCs, T-regulatory cells (Tregs), and mesenchymal stem cells (MSCs), which reduce the killing ability of T cells and NK cells [170]. Moreover, CXCL1 induces adjacent cell senescence and immune escape through paracrine signaling [171]. These secreted immunoregulatory factors might lead to chronic inflammation and contribute to the transformation of an immunosuppressive microenvironment by regulating immune system cell infiltration [160].

Components and potential effects of senescent cell in tumor. NF-κB signaling is activated in senescent cancer cells and elevates multiple production of IL-1α, IL-6, IL-8, CCL5, and growth factors like VEGF, FGF, PDGF, HMGB1, and MMP. To be detailed, IL-6, IL-8, CCL5, CXCL1, etc., help to recruit MDSCs to the TME to mediate protumorigenic effect of senescent cells, which block IL-1α signaling and antagonize the establishment of senescence in cancer cells. Simultaneously, MDSCs block immune surveillance by inhibiting CD8+ T cells and NK cells through IL-6 and CCL2, respectively, while IL-6 and IL-8 can recruit NK cells and T cells to reinforce immune surveillance. The ILs will spread senescence to surrounding cancer cells in a paracrine fashion, which further mediates tumor growth. The prominent SASP factors are involved in ECM processing and degradation, which can promote tumor cell proliferation and invasion. IL-6 secreted by senescent cancer cells or released from the ECM by MMPs recruits MDSCs, leading to an immunosuppressive TME. Moreover, the cleaved ECM components release growth factors such as VEGF, FGF, PDGF, and ILs that can promote tumor growth and EMT, promoting tumor metastasis. Additionally, MMPs also promote the release of many other cytokines and growth factors such as VEGF supporting tumorigenesis and chemokine CXCL1 to promote tumor growth. The SASP can stimulate blood vessel formation and vascular remodeling that contributes to tumor metastasis. Activated TLR4 promotes tumor progression in breast, prostate, and colon cancers and is associated with poor prognosis, but the antitumor activity is increased in skin cancers. TLR4 also recognizes HMGB1 and facilitates SASP phenotype formation. NF-κB, nuclear factor-κB; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; PDGF, platelet-derived growth factor; MMP, matrix metalloproteinases; IL, interleukin; TME, tumor microenvironment; MDSCs, myeloid-derived suppressor cells; NK cell, natural killer cell; SASP, senescence-associated secretory phenotype; ECM, extracellular matrix
From an overall perspective, aged individuals are often in a state of chronic inflammation. Obesity, changes in intestinal microbes, and tissue degradation exacerbate this chronic inflammation. Compared with those in young people, the serum levels of IL-1, IL-6, IL-8, and TNF-α in aged people are significantly increased [172]. This chronic inflammatory condition and the interrelated pathways usually exert an immunosuppressive effect and can increase the risk of cancer [22, 173]. Increased IL-6 and IL-8 levels can promote the EMT and tumor cell invasion [174]. The specific inflammatory environment also leads to the accumulation of Treg cells, Th2 cells, and activated B cells, increasing the secretion of IL-4, IL-6, IL-10, IL-33, and TGF-β, which are important growth-promoting factors [175].
In addition to molecular changes, changes in immune cell receptors, especially T-cell receptors, are important in the elderly tumor microenvironment. In contrast to highly expressed suppressing molecules in mice, the levels of PD-1 and TIM-3 in human T cells were not significantly changed, but CTLA-4 and LAG-3 were slightly elevated during senescence [176]. During aging, the proportion of CD8+ T cells that do not express CD28, a co-stimulatory molecule, increases, and this increase is accompanied by the upregulation of CD57 and killer inhibitor receptor (KIR) [177]. The immunoreceptor tyrosine‐based inhibitory motif (ITIM) domain protein (TIGIT), which is highly expressed in elderly CD8+ T cells, can exert an immunosuppressive effect on CD226 by competing with the ligand CD155 [176]. In addition, the expression and activity of CD38, a key molecule in NAD + depletion, are upregulated with advancing age. Activated CD38 may play an immunomodulatory role and has been considered a new immune checkpoint [178]. In addition to the aforementioned CD molecules, TLRs are involved in aging. The role played by TLR4 is tissue-specific. Activated TLR4 promotes tumor progression in breast, prostate, and colon cancers and is associated with poor prognosis but shows antitumor activity in skin cancers [179]. TLR4 recognizes HMGB1 and facilitates SASP acquisition [180]. Experiments have shown that the expression of TLR4 is elevated in aging mice [181]. This may indicate that TLRs are also involved in changes in immune cells in the aged tumor microenvironment.
Changes associated with cell growth
During the aging process, molecules and receptors of growth factors exhibit significant functions and tissue specificity in various ways. GDF 15, an important growth factor in the SASP, promotes epithelial cell proliferation, migration, and invasion through the MAPK and PI3K signaling pathways, thereby promoting tumor progression [182]. In addition, in a family of receptor protein tyrosine kinases, ErbB receptors directly or indirectly interact with classical downstream pathways such as MAPK, PI3K, and JAK [183]. The regulatory effects of ErbB on tumor cell proliferation, progression, and invasion have been widely studied in colorectal, breast, and lung cancers [184,185,186]. Among the ErbB family members, ErbB-1 (EGFR) binds to EFG, another SASP component, and promotes cell division [187, 188]. Moreover, clinical trials have revealed that aging is a risk factor for advanced lung cancer with EGFR mutant subtypes [189]. In vitro studies have also shown that p53 induces senescence by downregulating EGFR in multiple cell lines [190]. This finding is consistent with the opinion that aging prevents the growth of cells that are at risk of tumor transformation and thus inhibits tumorigenesis [191]. In addition, FGFR inhibitors have been reported to inhibit breast, gastric, and clonal cancers [192,193,194]. Ota et al. showed that FGFR promotes DNA-associated senescence, but loss of p53 and dysregulation of c-Myc reversed this effect and promote tumor transformation. However, in the same study, activation of FGFR can downregulate the expression of c-Myc, which may have suppressed tumor formation [195]. Additionally, FGFR may also act as a negative regulator of mesenchymal stem cell senescence. Reduced expression of FGFR has been revealed in a variety of aging tissues [196]. By interacting with RACK1, FGFR promotes the degradation of p53 in lung squamous cell carcinoma, ultimately inhibiting tumor cell senescence [197]. By binding to FGFR, sulfated heparin prevents premature replicative senescence and ultimately prohibits p53 expression. The indirect depletion of surface sulfated heparin leads to cellular senescence in tumors. Therefore, Jung et al. speculated that FGFR initiates new tumor defense mechanisms by regulating premature senescence [198]. The evidence illustrates a regulatory role and increased complexity of growth factors in aging and tumorigenesis.
As an upstream and downstream molecule regulated in vivo, GH regulates the secretion of IGF-1, which constitutes the GH/IGF-1 axis. As the corresponding hormone receptors, GHR and IGFR play important roles in cell senescence and tumor formation. Most tumor cells express GH, which may indicate that the autocrine function of GH on tumor cells activates GHR more than GH secreted by the pituitary gland, thereby driving cancer progression [199]. Correspondingly, in patients lacking GHR (Laron syndrome), the concentration of IGF in the patient's serum is significantly reduced, but these patients are free from aging-related disorders and rarely develop tumors [200]. Similarly, overexpression of IGF-1R has been observed in various tumors, such as those in thyroid cancer and breast cancer [201]. Additionally, these receptors may all be engaged in aging. Strous found that GH knockout significantly extended the lifespan of mice and delayed immune system-related aging. In addition, they observed the downregulation of the GH/IGF-1 axis activity in elderly individuals [199]. Similarly, it has been confirmed that the lifespan of mice was significantly prolonged after heterozygous IGF-1R knockout [202]. A similar conclusion suggested that in individuals older than 100 years, the activity of IGF-1 is reduced, which may be mediated by IGF-1R mutation, and these individuals exhibit profound anti-inflammatory characteristics. These two changes both depend on p53, which reduces the risk of tumor development [203, 204]. The activation of IGF-1R leads to the activation of PI3K, Ras-Raf, JAK/STAT3, and other pathways, ultimately upregulating p21, which promotes cell proliferation, survival, migration, and adaptation to hypoxia and inhibits autophagy, apoptosis, and anoikis [205]. Therefore, we speculate that IGFR may be involved in the forkhead pathway in cellular senescence and tumor formation. The ultimate fate of a cell may depend on which of the two states prevails. In formal theory, in the early phase, aging inhibits tumor formation by preventing the growth of cells [191]. In this process, cell surface receptors activate a variety of signal transduction pathways and engage in crosstalk with each other, ultimately affecting the activity of p53 and other key proteins. Intracellular receptors can also sense various changes in senescent cells and ultimately participate in their regulation. All of these factors might reflect constant competition between aging and tumor cells.
Impact of senescence on the TME
The effects of the ECM
The tumor microenvironment matrix constitutes endothelial cells, fibroblasts, pericytes, adipocytes, immune cells, and the ECM, which is composed of collagen, fibronectin, laminin, and elastin [3]. Matrix fibroblasts inhibit tumor cell proliferation [206]. The different cross-linking and arrangements of ECM proteins change the physical features of the ECM, such as its stiffness and mechanical force [207]. Moreover, through focally assembling integrin adhesion complexes, cells can sense changes in the ECM and regulate the cell cycle and energy metabolism via the FAK/Src, ILK-PINCH-parvin-kindlin, and α-actinin-zyxin-VASP signaling pathways [208]. This molecular and supramolecular heterogeneity affects the infiltration and migration of cancer cells and angiogenesis [207]. Moreover, the ECM contains growth factors, including IGFS, FGFs, TGF-β, and HGF [209]. These stimulating factors can induce adipocytes, mesenchymal stem cells, pericytes, and other cells to transform cancer-associated fibroblasts (CAFs). Similarly, miRNA-21 induces CAF formation via its inhibition of the Smad7 pathway. These CAFs induce ECM remodeling by secreting matrix metalloproteinase (MMP) [210, 211]. On the basis of the product levels of unique collagen and other ECM molecule genes, such as COL10A1 and COL4A1, a previous study showed that CAFs can be classified into multiple subtypes. These multiple CAF subtypes increase the complexity and heterogeneity of the tumor microenvironment [212].
Many time-related mutations accelerate normal stromal cells, which are transformed into CAFs, contributing to immune cell regulation, angiogenesis, and ECM remodeling. The promotion of CAF activation may be a result of the impaired function of p53/p21 and CLS/RBP-Jκ, a transcription factor in dermal fibroblasts, which functionally and physically interacts with p53 in the Notch signaling pathway [213]. Activated CAFs upregulate the expression of VCAM-1 in adjacent tumor cells and promote the adhesion of monocytes. Several studies have shown that activated M2 macrophages might, in turn, promote the formation of CAFs, thus participating in crosstalk with malignancy factors in pancreatic cancer and neuroblastoma [214, 215]. CAFs also inhibit NK cells activate receptors on the surface of cells and killer particles [216] and recruit normal DCs to form IDO-producing regulatory dendritic cells [217]. Moreover, CAF cells stimulate miR21/Toll-like receptors through lactate to promote CD4+ T-cell polarization from Th2 to Th1 cells and maintain Treg cells [218,219,220]. The number of CD8+ T cells is reduced by CAF-upregulated immune checkpoint molecules such as PD-1, which facilitates tumor cell immune escape [221]. Senescent fibroblasts express the nonclassical MHC molecule HLA-E, which inhibits the immune response by interacting with NKG2A on the surface of NK cells and CD8+ cells [222].
In the aging tumor microenvironment, change in the ECM is an important factor. As other secreted SASP factors, MMP-1, MMP-3, MMP-10, and other MMP levels are elevated in the aging matrix, which contributes to ECM remodeling [3]. Through this increased MMP expression, senescent cells show reduced contact inhibition, facilitating cancer growth. Under the action of MMP, the collagen in the ECM undergoes fibrosis, accompanied by the destruction and reorganization of the elastin structure, composition alteration of laminin, and decreased hydration capacity of hyaluronic acid, eventually resulting in the loss in ECM mass and moisture, increased fibrosis, and tissue dysfunction [223]. A study demonstrated that fibronectin inhibits tumor cell proliferation in the tumor microenvironment but plays the opposite role in the normal stroma. In the aging ECM, the expression of fibronectin is upregulated; however, due to hypoxia, mutations, nutritional deficiency, viral infections, and other adverse factors, structures often undergo misalignment [224]. The decline in heart function in old age enhances the chances of ECM [225]. In addition, the decline in NAD+ levels in senescent cells leads to increased stability of HIF-1α, which induces a pseudohypoxic state [226]. Activation of HIF-1α can mediate cancer cell invasion through the action of fibronectin [227]. Hypoxia can also change the stiffness of the ECM mediated through LOX, leading to immune cell infiltration and tumor cell migration [228,229,230].
Angiogenesis
In young individuals, angiogenesis vitally contributes to tumor progression. Previous studies have shown that solid tumors need enough blood to grow. When solid tumors are larger than 2 mm in diameter, new blood vessels must be formed to maintain the blood supply; without new vasculature, the tumor undergoes necrosis due to hypoxia [231]. In addition, angiogenesis is related to tumor cell invasion, immune cell infiltration, and chronic systemic inflammation.
In normal tumors, angiogenesis can be initiated in many ways, including vasculogenesis, sprouting angiogenesis, and vasculogenic mimicry (VM) [232]. Vasculogenesis refers to endothelial progenitor cells (EPCs) differentiating into endothelial cells and de novo formation of a primary blood vessel network. Sprouting angiogenesis refers to the proliferation and migration of existing vascular endothelial cells, which generate new capillaries, which is the process in embryonic development, after birth and during tumor progression [233]. Moreover, in the tumor microenvironment, a variety of angiogenic factors, including VEGF, FGF, and PDGF, promote angiogenesis [234]. In contrast to the two aforementioned methods, VM does not depend on endothelial cells [235]. Many studies have reported that VM is involved in a variety of tumors [236]. In the VM process, tumor cells are arranged into tubes and covered by glycoproteins to form blood vessels [232]. There is evidence to suggest that VM may be the blood supply source in early tumor progression and that these vessels are gradually replaced via vasculogenesis and sprouting angiogenesis in a later stage [237]. Regardless of the mechanism, angiogenesis is closely related to hypoxia. Under hypoxic conditions, the content of HIF-1α increases. Activated HIF upregulates the expression of VEGF, TGF, and other angiogenic factors; IGF, c-Myc, and other proteins associated with cell survival and proliferation; and GLUT1, GLUT3, and other proteins that change cell metabolism and enhance adaptability [238]. Moreover, hypoxic conditions can accelerate the EMT of tumor cells and the secretion of MMP, which leads to an incomplete vascular matrix that enables cells to migrate, thereby increasing the aggressiveness of tumor cells [239]. Circulating tumor cells (CTCs) originating from a primary focal point may die rapidly after entering the blood due to the shear force of the blood flow and anoikis [240, 241]. However, CTCs can interact with blood platelets, macrophages, lymphocytes, and other cells to prolong survival and immune escape [242]. In addition, a study demonstrated that the microvasculature may be a premetastatic niche formed by tumor cells before they enter the circulatory system [243].
In the aging tumor microenvironment, the change in angiogenesis mechanisms is diverse. A study showed that SRPX expression is increased in senescent cells and decreased in tumor cells, and SRPX itself promotes angiogenesis through the FAK pathway [244]. Thus, cells acquire the SASP during senescence. Among SASP factors, VEGF, PDGFA/B, FGF, and other secreted angiogenic factors and CCL5, CXCL-1, Il-6, and other secreted proinflammatory factors may promote vascular remodeling [37]. Changes in miRNA expression during cell senescence may promote tumor metastasis. Mir-21 can inhibit the regeneration of endothelial cells and promote angiogenesis in vitro and in vivo, and it is secreted by CAFs in exosomes [210, 245]. In addition, previous reports have noted that exosomes carrying miRNA can regulate the microenvironment of a metastatic site, thereby promoting tumor colonization [211]. In addition, HDAC, which functions as part of the SASP, also contributes to vascular endothelial formation. Many studies have shown that HDAC6, HDAC7, and HDAC9 promote angiogenesis by inducing endothelial cell migration, while HDAC5 exerts an antiangiogenic effect in endothelial cells [99, 246,247,248].
Moreover, angiogenesis in the tumor microenvironment can be affected by changes in the aged physiological state. Previous studies have shown that with advancing age, the number of small arteries in certain tissues decreases, which decreases blood flow and in turn leads to the downregulation of Notch in endothelial cells [249]. As mentioned above, MMP9 is highly expressed in the heart during aging, causing collagen deposition and cross-linking [250]. ECM deposition can cause cardiac insufficiency, reduce angiogenesis and further reduce the blood supply to the whole body [225]. These mechanisms together reduce blood flow into the tumor microenvironment, causing hypoxic and nutrient-deficient conditions, which activate HIF and other signaling pathways. Moreover, studies have shown that molecules including Il-4 and CD163 secreted by M2-polarized macrophages can induce pathological angiogenesis, which may increase the blood supply to the tumor microenvironment [251]. In addition, CTCs can be cloaked with platelets, reducing the effect of the shearing force of the blood. Platelets can also support CTC adhesion to the vasculature and facilitate CTC immune escape [242, 252, 253]. In aging bone marrow, macrophages promote an increase in platelet hematopoietic stem cells through the action of Il-1, which ultimately increases the number of circulating platelets in mice [254]. These mechanisms together maintain the material supply to an aged tumor microenvironment.
Stromal population
The stromal TME within tissue is made up of various components, including fibroblasts, endothelial cells, pericytes, adipocytes, the ECM, and immune cells, and plays a major role in TME homeostasis. Fibroblasts are the most common stromal component; these cells are required for the synthesis of collagen and for the structural integrity of connective tissue and play key roles in wound healing and inflammation [255]. Their predominant mode in regulating many of these processes is through the secretion of soluble factors, including cytokines, chemokines, growth factors, enzymes, and structural components of the ECM [23, 38]. Given the complexity of different tissues, the TME plays a specific role in the regulation of the soluble factors secreted by fibroblasts along with their migratory and proliferative characteristics. The fibroblast renewal rate, defined as the sum of the total number and proliferative capacity of fibroblasts, greatly varies among different tissues, with factors such as local temperature, vascularization, mechanical stress, and hormonal responses contributing to the renewal rate [256]. Changes in fibroblasts during aging are likely to be different between organ sites and often involve senescence. Senescence is a classic example of antagonistic pleiotropy, and the accumulation of senescent cells is a key pathological feature associated with aging [257,258,259,260]. Cellular senescence is linked to many of the cellular processes of aging and can be a direct result of responses to intrinsic or extrinsic oncogenic stimuli; notably, many forms of senescence are not aging-related (e.g., oncogene-, replication-, stress- and therapy-induced) [261,262,263] (Fig. 5).

Potential risk factors for stromal cell senescence. Senescence can be induced in stromal cells with different signals from different avenues. Cancer cells can release cytokines and growth factors (IL-1α, TGF-β, CXCL1) that induce stromal cell senescence directly. Cancer cells can alternate TME via regulating metabolism, environmental stress, physical forces, and matrix disruption to indirectly induce stromal cell senescence. Different types of cancer therapy (i.e., chemotherapy, radiotherapy, immunotherapy, and personalized therapy) may have the possibility to induce paracrine senescence of cancer cell senescence and induce stromal cell senescence via DNA damage signal. Immune cell may emit diverse immune inflammatory factors to promote stromal cell senescence. Microbiota in the gut or TME can also cause stromal senescence by generating toxins and metabolites. The common cancer risk factors including alcohol, smoking, radiation, and genetic disease are associated with DNA damage signal to induce stromal senescence. Furthermore, other age-associated damage signals may arouse stromal senescence. TME, tumor microenvironment; CAFs, cancer-associated fibroblast (Mainly from https://doi.org/10.1016/j.trecan.2022.09.002)
There are still some disputes on how the accumulation of senescent cells occurs in elderly individuals. It should be hypothesized that, as we age, a reduction in immune function decreases the recognition and clearance of these growth-arrested cells, which eventually results in their accumulation [264]. Typically, the SASP is believed to be made up of approximately 75 secreted factors, including granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-6, IL-8, and IL-10 [38, 260]. However, many of these secreted factors were identified in studies using oncogene-induced senescence models and may not necessarily reflect true age-induced senescence. While the mechanisms underlying age-related SASP transformation are still under investigation, many genetically engineered mouse models (GEMMs) have been key in determining their pathological and homeostatic role. p16INK4A activation seems to be one of the important contributors toward senescence induction in cells [265], yet the contributions to age-related accumulation of senescent cells need more description. To be consistent, when modeled in vivo, its contribution toward senescent cell accumulation is described as more of a “molecular” form of aging as opposed to a “chronological” form [265]. The GEMM demonstrates that a dramatic accumulation of p16INK4A-expressing cells occurs across various tissues throughout the aging process and characterizes the pathological changes associated with the age-induced SASP in peritoneal macrophages, illustrating the potential for other stromal components. Fibroblasts contribute to many SASP-related pathologies, and studies have shown that tumor-associated fibroblasts undergo chromatin remodeling via histone deacetylase (HDAC) modulation to achieve a SASP irrespective of DNA damage [99, 266]. Recently, it was shown that LINE-1 retrotransposable elements are derepressed at the transcriptional level to elicit a type I IFN response, which contributes to the maintenance of a SASP [59]. These findings further support the conclusion that dynamic changes within an aged TME play a key role in reprogramming cells toward a SASP.
Other TME components
Other senescent cell populations, such as endothelial cells, epithelial cells, immune cells, stem cells, and even certain tumor cells, play clear roles in modulating the TME by acquiring the SASP [21, 267]. Many examples have shown that senescent cell populations can contextually produce protumorigenic or antitumorigenic effects; however, direct age-related evidence for these effects within these cell types remains limited. A recent study based on a xenograft model with human BPLER triple-negative breast cancer cells in nude mice found that tumors showed delayed onset, slower growth kinetics, and reduced metastasis in aged mice (> 10 months old) than in young mice (8–10 weeks old). Furthermore, a subset of tumor-infiltrating hematopoietic cells in young mice showed upregulated CSF1 receptor (CSF1R) expression and secreted the growth factor granulin to induce robust tumor growth and metastasis. Importantly, bone marrow-derived cells from young mice and transplanted into aged mice were sufficient to activate the tumor-supportive TME and induce tumor progression.
Cellular senescence has been confirmed to be a key contributor to inflammaging in many age-related malignancies [268, 269]. SASP acquisition in stromal cell populations results in the persistently increased secretion of multiple inflammatory cytokines that maintain low adaptive immune response levels. Other age-related changes to the gut microbiota, obesity, and tissue degradation also appear to drive the inflammaging response [22]. In total, these age-related processes appear to drive chronic inflammation by increasing systemic levels of IL-1, IL-6, IL-1α, IL-1β, IL-33, GM-CSF, IFN-γ, TNF, and C-reactive protein (CRP). All these factors contribute to multiple morbidities and mortalities in elderly individuals [22, 270]. Similar effects have also been observed with increased infiltration of immunosuppressive Treg cell populations in chronic inflammatory mouse models, mimicking the chronic inflammation that often precedes and may lead to certain malignancies such as melanoma and colorectal cancer [271]. Treg cells play a key role in maintaining tolerance to self-antigens and suppressing the induction and proliferation of effector T cells (such as CD4+ and CD8+ T cells) via the secretion of cytokines and enzymes [272]. Another study induced chronic, tumor-promoting allergic contact dermatitis (ACD) in 6- to 8-week-old mice by treating them with 1-fluoro-2,4-dinitrobenzene (DNFB) and found that IL-33 expression was key in inducing the transition from acute, tumor-suppressing inflammation to chronic inflammation [271]. The number of Treg cells was significantly reduced in DNFB-treated IL-33-knockout mice, and knocking out IL-33R in Treg cells significantly reduced ACD-induced skin carcinogenesis. Interestingly, in colitis-induced colorectal cancer, the IL-33-Treg cell axis was identified as a key driver of carcinogenesis. However, more evidence on the age-related effects of the IL-33-Treg cell axis in tumorigenesis is needed [270, 273, 274].
Several microRNAs (miRNAs) termed identified in association with many human malignancies are called “inflamma-miRs” [275]. Age-related increases in miR-19b, miR-21, miR-126, and miR-146a appear to drive the progression of many types of cancer, and possibly other diseases, through inflammaging [276]. miR-21 has been shown to be overexpressed in many malignancies and to reduce the expression of the potent anti-inflammatory factors IL-10 and TGF-β [277], but when binding to Toll-like receptor 8 (TLR8), it induces the secretion of the inflammaging cytokines IL-6 and TNF [278]. Immunosenescence is another contributor to many age-related pathologies, including cancer. Immunosenescence is defined as the age-related dysregulation of the immune system, whereby subpopulations of effector immune cells and overall immune function decline. This process is a result of multiple factors, including thymic atrophy [279], a decrease in the number of naive T cells [280], a reduction in memory T-cell function [281], and decreased recognition of diverse antigens by T cells [282, 283]. Along with T-cell dysfunction, NK cells, macrophages, and dendritic cells, all of which play early roles in tumor-immune recognition and suppression, appear to undergo phenotypic decreases in cytotoxic activity as humans age [284,285,286,287]. The key involvement of these processes in cancer progression has been identified in a squamous cell carcinoma (SCC) model of aging [288]. In GEMM models of conditionally expressed mutant HRAS in keratinocytes, aged mice (18–22 months) developed SCC more quickly than young mice (2–4 months). Molecular analyses of the immune system of the aged mice revealed a shift toward a protumorigenic T helper 2 cell anti-inflammatory response, as well as increased expression of inhibitory programmed cell death ligand 1 (PD-L1) and senescence-associated β-galactosidase on effector immune cells in the dermis.
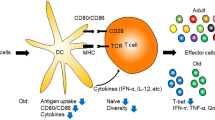
Age-induced immunosenescence is largely mediated by effector T cells and other immune cell types required for antitumor immunity (Fig. 6). Changes in these cells have been hypothesized to induce a shift toward the activation and infiltration of more immunosuppressive cell populations in elderly individuals, which is important to their increased predisposition to cancer cell invasion and metastasis [289]. The number of M2 tumor-associated macrophages (TAMs), which are negatively associated with tumor immunity, is significantly higher in the spleen and bone marrow of aged mice (> 24–28 months old) [290]. Additionally, stimulation of macrophages isolated and cultured from aged mice in mesothelioma or lung carcinoma cell-derived culture supernatants increased the levels of the M2-derived immunosuppressive cytokine IL-4. Stimulation of M2 TAMs toward switch to an M1 proinflammatory phenotype by treating aged mice with a combination consisting of an IL-2 agonist and anti-CD40 therapy reduced immunosuppressive IL-4 and IL-10 expression and inhibited tumor growth [291]. While the binary M1/M2 classification of macrophages has been hotly debated, a large amount of evidence suggests that M2-like immunosuppressive macrophages promote tumor progression in an aging context [291]. TAMs have also been demonstrated to play key roles in establishing a premetastatic niche in the liver by secreting CXCL1 and inducing the recruitment of MDSCs, which are necessary for the efficient formation of colorectal cancer liver metastases [292]. p16Ink4a and p21Waf1/Cip1 could suppress tumor progression by inducing cellular senescence, the deletion of p16Ink4 and p21Waf1/Cip1 reduces CX3CR1 expression and inhibits monocytic-MDSCs (Mo-MDSCs) accumulation in tumors expressing CX3CR1, hence suppresses the tumor proliferation in mice model. The regulation of Mo-MDSCs is a valuable strategy to inhibit tumor progression [293]. More direct evidence on the systematic effects of M2 TAMs is warranted in the future.

Biological function and effect of senescent immune cells. A T-cell senescence may be activated by soluble factors secreted by cancer cells, Tregs, and occur during the aging process. The senescence markers such as p16, SA-βGal are elevated, co-stimulatory receptors (CD27, CD28) are decreased and CD57 is increased. Senescent T cells may have poor immune function and reveal immune suppressive TME. B p16 and SA-βGal highly expressed, senescent macrophages may enhance phagocytic activity or increase macrophage polarization by transferring M2 into M1. C In senescent MDSCs, p16 and p21 are highly expressed and upregulate the expression of the chemokine receptor CX3CR1, which mediates the recruitment of MDSCs to tumor site. Tregs, regulatory T cells, MDSCs, myeloid-derived suppressor cells (Mainly from https://doi.org/10.1016/j.trecan.2022.09.002)
Neutrophils undergo immunosenescence throughout aging, and they have been confirmed to infiltrate injured tissue in elderly people [294]. Studies have shown that neutrophils in aged patients and mice produce more anti-inflammatory cytokines than their younger counterparts [295]. Neutrophils are well-characterized regulators that mediate tumor progression through proinflammatory effects in tumor models of young mice; however, subpopulations of anti-inflammatory “N2 tumor-associated neutrophils (TANs)” have recently been implicated in many kinds of cancers [296, 297]. Studies conducted with young mice revealed that the number of immunosuppressive N2 TANs is systematically increased in aged patients and that they exhibit a function similar to that of MDSCs [298]. Nevertheless, direct age-related investigations into N2 TAN involvement in the TME should be performed.
Many studies suggest a dramatic increase in Treg cell numbers and function in age-related pathologies and in organs such as the lymph nodes and spleen [299,300,301,302]; however, other reports showed no change in Treg cell numbers or function or their reduced contribution to other aged tissues and cancers [303,304,305], suggesting that Treg cells may play a context-specific role in different TMEs and cancer environments. Furthermore, Treg cell recruitment also appears to be a significant factor in the establishment of the premetastatic niche in many cancer types [306]. Nevertheless, whether the age-related increases in the number of Treg cells that is observed systematically in certain mouse models are directly linked with increases in age-related metastasis remains unclear. MDSCs exhibit a consistent increase in human blood during aging [307] and in the bone marrow and lymphoid organs of 17- to 19-month-old mice [308, 309]. Regression within young mice harboring breast cancer correlated with significant effector T-cell infiltration, whereas aged mice showed significantly increased numbers of MDSCs in the TME. Importantly, MDSCs are among the immune cell types most closely associated with the formation of the premetastatic niche in cancer [310]. Stromal senescence significantly increased the numbers of immunosuppressive MDSCs and Treg cells adjacent to senescent populations in healthy mice, primarily via the secretion of IL-6 [78]. These studies additionally suggest that the accumulation of senescent stromal cells is sufficient to establish a tumor-permissive chronic inflammatory TME that allows tumors to grow and progress unabated by the immune system. Age-related increases in the numbers of systemic MDSCs and acceleration of other age-related processes, such as inflammaging and ECM modulation, may directly link MDSCs, Treg cells, and other immunosuppressive cell subpopulations with age-related cancer predisposition and premetastatic niche formation. Details on the roles played by immunosuppressive immune cell types in these processes are needed. In prostate cancer, effector T cells and proinflammatory cytokines appear to contribute to increases in tumor growth. Prostate fibroblasts cultured from young (< 55 years old) and aged (> 65 years old) healthy individuals showed that the aged fibroblasts secrete a greater number of cytokines and interleukins, which promote the growth of epithelial cells and may affect the function of immune cells [311]. Another study on prostate cancer showed that CD3+, CD4+, and CD8+ T-cell infiltration exerts a protumorigenic effect and is associated with tumor growth [312]. These findings suggest that effector versus immunosuppressive cell infiltration in the TME with advancing age depends on the context and is related to tumor growth and premetastatic niche formation.
Aging and potential response to therapy
The treatment for cancer in elderly patients is still challenging because age-related health conditions often leave clinicians in a dilemma, as it is frequently unclear whether potentially beneficial therapies can be safely administered at standard dosages and will improve the prognosis or whether potential side effects will likely affect the patient’s quality of life (QoL). Approximately 50% of all cancers are diagnosed in patients over 65 years old, and this percentage may increase to 70% as life expectancy continues to increase, but survival data from clinical trials for patients above this age are relatively rare [313]. In fact, 40% of patients enrolled in cancer trials are over 65 and fewer than 10% are older than 75 years; therefore, more evidence is required to facilitate clinical management for elderly cancer patients. Recent advances in understanding the mechanisms of senescence, the aged TME, and responses to therapy will yield crucial knowledge to allow more efficient targeting and less-intensive treatment of different cancer types in elderly patients. In the following section, we provide descriptions of clinical therapy for patients presenting with both cellular senescence and cancer.
Chemotherapy
Chemotherapy is a nonspecific, aggressive treatment that results in the targeting of fast-growing malignant cells; however, chemotherapy causes many side effects associated that can often be life-threatening in elderly individuals. A recent study showed that chemotherapy-induced senescent fibroblasts within mice caused a consistent inflammaging response, and the elimination of these fibroblasts significantly decreased both short-term and long-term side effects of chemotherapy-induced cytotoxicity, decreased the possibility of cancer recurrence and reduced the extent of cancer metastasis [88]. Many similar studies have suggested that chemotherapy may be initially beneficial; however, in many cases, it may later contribute to accelerated aging of the TME and increase residual disease in patients [314]. Chemotherapy can also induce off-target effects that include stem cell pluripotency decline and bone marrow exhaustion. Studies showed that MSCs in 16-month-old mice were much more sensitive to doxorubicin treatment than those in 1-month-old and 8-month-old young mice [315]. Patients can undergo stem cell transplantation while receiving high-dose chemotherapy regimens to prevent off-target effects; however, stem cell transplantation may induce toxicity that exceeds the safety threshold in many elderly individuals, which limits the broad use of this strategy [315]. In a cohort of patients with different types of cancers, including myeloma, lymphoma, and leukemia and who underwent hematopoietic stem cell transplantation, the expression of p16INK4A was significantly increased in effector T cells. Further analysis of gene expression in effector T cells from these cancer patients showed clear signs of immunosenescence and T-cell aging [316]. The off-target effect associated with stem cell transplantation and chemotherapy can cause damage to the thymus by accelerating thymic aging [316]. Overall, there may be a potential clinical benefit in targeting chemotherapy-induced acceleration of age-related tumorigenic events to decrease cytotoxicity and increase survival. There are many cases where chemotherapy in elderly patients is tolerated well and prolongs survival irrespective of the location of the cancer [317]. In any case, the key consideration to determine proper chemotherapeutic regimens in the elderly should be whether the benefits of treatment outweigh the side effects. Further research is warranted to provide more insights into the cytotoxic effects at the molecular level and the degree of organ function decline in older patients.
Targeted therapy
Targeted therapy has been regarded as one of the standard personalized approaches to target cancer cells or the TME for specifically inhibiting tumor development or progression. However, drug resistance and adverse events are always major concerns for this type of treatment [317, 318]. Targeted therapy often induces fewer off-target effects than chemotherapy or radiotherapy, as cancer cells targeted for therapy appear to undergo more intrinsic changes based on genetic mutations, epigenetic alterations, and genomic instability to induce off-target effects. A recent study illustrated that healthy aged dermal fibroblasts facilitated increased resistance to targeted BRAF therapy in allogeneic mouse models of melanoma by secreting SFRP2 into the TME [319]. Furthermore, using recombinant SFRP2 to treat young mice led to increased resistance in a formerly sensitive mouse model [319]. Studies focusing on melanoma have shown that B-cell infiltration in the TME results in the secretion of insulin-like growth factor 1 (IGF1), which promotes drug resistance to BRAF and MEK inhibitors [320]. Another recent study reported that immunosuppressive age-associated B-cell counts are significantly increased in aged mice (> 24 months old) but that the levels of other B-cell subtypes were reduced [320, 321]. The number of MDSCs, as previously described, increases with age, and these cells appear to induce resistance to antiangiogenic therapies as well as other targeted therapies used in the treatment of multiple myeloma [322], prostate cancer [323], liver cancer [324] and melanoma [325]; however, a direct link between MDSC drug resistance and age has not been investigated in these models. Notably, combination therapy with drugs targeting treatment resistance-promoting components in the stroma and ECM of the TME may increase the persistence of targeted therapy effects [326,327,328]. Given the complex mechanism by which protumorigenic effects are induced with age, research on the aging TME, potential premetastatic niches, and their contributions to targeted therapy resistance is definitely needed.
Immunotherapy
Immunotherapy has been established to modulate the immune TME and thus target and eliminate tumor cells. The efficacy of immunotherapy is considerable; however, not all cancer patients receive clinical benefits from immunotherapy [329]. Therefore, investigators need to identify more-robust biomarkers for this kind of treatment. Marked changes in immune profiles and function are found in aging humans, and therapy targeting the immune system within the elderly has shown considerable clinical implications. However, few studies have incorporated elderly individuals into trials to evaluate the efficacy of immunotherapies in older cancer models. The most widely used immunotherapies that have proven clinical efficacy across a wide range of cancers are immune checkpoint inhibitors (ICIs) targeting programmed cell death 1 (PD-1), PD-L1, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and lymphocyte activation gene-3 (LAG-3). Many cancer cells and other stromal components upregulate cell signaling through immune checkpoint pathways to evade antitumor immune responses. Interestingly, PD-1 surface expression has been suggested to contribute to the age-dependent functional decline of effector memory T cells [330]. PD-1 levels are increased on T cells with age, and anti-PD-1 therapy increases T-cell function, especially in aged mouse models [331, 332]. Rapamycin, an mTOR inhibitor, has been shown to reduce age-related increases in PD-1 levels, suggesting a role of these inhibitors in increasing tumor immunity in aged tissues [332]. A marked increase in PD-L1 expression in CD8+ effector T cells of aged mice compared with young mice has been observed, and anti-PD-L1 immunotherapy reduced cell proliferation in vitro and antitumor immunity in aged hosts compared with the effect in young mouse lymphoma models [333]. In another study, PD-L1 and indoleamine 2,3-dioxygenase 1 (IDO1) levels were increased during aging in the brains of healthy human adults, while the number of circulating Treg cells increased and that of CD8+ T cells decreased during aging [334]. These findings suggest that older patients with cancers such as lymphoma, glioblastoma and leukemia may be less responsive to immunotherapy.
Additionally, within the aged mouse model TME, CD8+ T cells displayed a tendency for exhaustion, and IFN-γ levels were significantly decreased; similarly, in aged patients with triple-negative breast cancer, IFN-γ gene expression levels were found to be decreased. Inflammation of the TME mediated by IFN-γ in aged tumor-bearing mice significantly increased the sensitivity of the mouse responses to ICIs [335]. Considerable high-quality evidence and substantial published datasets have confirmed that currently available ICIs show high efficacy in older adults [336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362,363] (Table 4); however, some of these findings are controversial and because of a lack of enrolled aged patients (maybe ≥ 65 years) in clinical trials, to determine whether the investigated treatments show clinical benefits or whether toxicity is increased in elderly patients, more study is needed [305, 364]. Patients older than 60 years are likely to respond more efficiently to anti-PD-1 immunotherapy, and the likelihood of eliciting a response to anti-PD-1 increases with age. These findings have been recapitulated in young and aged melanoma mouse models.
Moreover, research on TME immune cell subtypes revealed that aged mice showed significantly increased CD8+ T-cell-to-Treg cell ratios, indicating that they carried more immunogenic tumors [305]. The depletion of Treg cells via anti-CD25 therapy significantly increased the anti-PD-1 response in young mice [305], and Treg cell depletion in aged melanoma mouse models was ineffective in inducing antitumor immunity but completely decreased tumor growth in young mice [308]. However, other studies have shown contradictory findings. For example, in one study, anti-PD-L1 treatment of B16 melanomas exhibited substantial efficacy only in young mice, but combination therapy with anti-PD-L1 and anti-CTLA4 antibodies showed partial efficacy in aged mice [365]. Some retrospective studies on melanoma [364] and non-small cell lung cancer (NSCLC) [366] in which either anti-PD-1 or anti-PD-L1 was administered showed little difference in overall survival (OS), progression-free survival (PFS) or toxicity between age groups. Because the diversity in immune cell profiles, infiltration status, and activity across various tumor models as well as in the TME at different tissue sites is large, the immunotherapeutic response likely differs marked in young versus aged patients. Genetic and environmental diversity in different populations also limits collective analyses across various racial and ethnic backgrounds. Given the discrepancy found when targeting one immune checkpoint compared with another in aged models, tailoring specific immunotherapeutic treatments to aged patients may be warranted. Importantly, some groups are focusing on seeking strategies for recruiting older patients with cancer for clinical trials [367]. These strategies, along with a focus on furthering the understanding of age-related changes at the molecular level in TMEs and premetastatic niches, may be critical in efficiently predicting patient responses across a wide range of cancers. Considering these possibilities, we may also identify other avenues for effective cotargeting of tumor-promoting immune cell subpopulations.
Conclusions and further perspectives
Recent studies have suggested that the aging TME may exert dramatic effects on tumor progression. Normal age-related changes in stromal and immune populations may function together to drive the progression of tumor cells from an initial or slow-growing state to a highly aggressive and metastatic disease state. The outcomes of these changes involve variations in secreted factors, changes in the biophysical architecture of the TME, and even changes on a macroscopic level, such as the breakdown in vasculature integrity [368, 369]. Several challenges remain that need to be resolved in the future. (1) There is a lack of drugs that are highly effective in inducing senescence in a high proportion of cancer cells. In addition, drugs need to show a preferential affinity for cancer cells over normal cells, as inducing senescence in normal tissues can cause detrimental side effects [88]. (2) We need to establish more gold standard signatures and biomarkers for identifying the senescent state. No obvious biomarker can be measured to unambiguously discriminate between senescence and other growth-arresting states [86]. There have been several multi-gene signatures to identify senescence in primary cells. A panel of cancer cells have been selected to identify the SENCAN classifier for cancer senescence [86]. In vivo, investigators should use galacto-conjugated fluorescent nanoparticles to detect senescent cells. This method has been tested in models of chemotherapy-induced senescence [370, 371]. The noninvasive imaging system could be ideal for measuring the efficacy of senescence induction in cancer cells. Radioactive β-gal positron emission computed tomography (PET) tracer seems to be feasible [372]; however, β-gal-based screening strategy might be unpowered to detect all senescent cells accurately. On the one hand, cells from some tissue types do not induce SA-β-gal activity when they turn to senescent [8], on the other hand, macrophages may also be able to exhibit increased SA-β-gal activity [373, 374], consequently, false positives may result from macrophages inside inflamed tumor tissues. Other noninvasive methods, for example, oxylipin biosynthesis may also help to detect clearance of senescent cells [375]. Furthermore, other biomarkers that can be potentially used in noninvasive approaches to detect senescent cancer cells should be similarly established [376, 377]. Researchers need to explore whether these markers can be used to detect senescent cancer cells in different contexts, such as cells with different genetic backgrounds, derived from different tissue types or with senescence induced by different agents [378]. (3) The next challenge is that there is still no unique senescence-based therapy due to tumor heterogeneity. Intratumoral heterogeneity leads to varying drug responses that may limit the effectiveness of senescence induction within tumors. Senescent cells can spread the senescent phenotype through the SASP to the surrounding non-senescent cells within tumors [101], which will sensitize non-senescent cancer cells to senolytic treatments. Additionally, such bystander effects could be fortified by the local TME shaped by SASP of the senescent cells. Further efforts to understand senescence-based therapy outcomes may overcome tumor heterogeneity and guide the timing of personalized treatments [101, 379]. Most senolytic drugs were developed with the aim to reverse the effects of aging and were consequently tested mostly on primary cells. In a study, the commonly used senolytic (navitoclax) on a panel of senescent cancer cells showed that it has widely variable activity as a senolytic [86]. Beyond identifying absolute biomarkers for the senescent state, the field is also in need of a druggable and broadly present vulnerability of senescent cancer cells. Novel CRISPR-Cas9-based genetic screening platform allows for the performance of drop-out screens to identify new senolytic targets on the genome scale. If universal vulnerabilities of senescent cancer cells exist, unbiased genetic screens should allow the identification. (4) There is still no abundant knowledge on how the SASP acquired by senescent cancer cells impacts the interaction between senescent cancer cells and immune system cells. Several early studies have pinpointed a synergy between senescence-promoting therapy and checkpoint immunotherapy [36, 37]. It is possible that not all pro-senescence therapies and not all cancer types will benefit from combination checkpoint immunotherapy. This possibility is supported by the substantial heterogeneity in SASP factors produced by different types of cells undergoing senescence [86]. It is important to ascertain when a SASP provokes an immune response that can be enhanced by checkpoint immunotherapy and when it does not. It is possible to use the so-called senomorphic drugs as the NF-κB-inhibiting drugs apigenin and kaempferol or the mTOR inhibitor rapamycin [380, 381], which can modify the SASP of senescent cells to become more responsive to checkpoint therapy clearance [382]. (5) We need to be significantly cautious in ablating senescent normal cells via anti-senescence therapies in aged individuals. In elderly individuals, senescent cells can constitute a high percentage of the net number of cells in some tissues and this may jeopardize tissue structural integrity or affect vascular endothelial cells, leading to blood–tissue barrier disorder that potentially leads to liver and perivascular tissue fibrosis and health collapse [383, 384]. This issue highlights the need for the development of cancer-selective senolytics.
In this article, we discuss the impact of aging on the TME from multiple perspectives and review treatments as well as recent clinical trials with data on elderly individuals. The effects of aging on tumors are two-sided. In the early stage of tumor formation, aging is often associated with tumor suppression. However, once a tumor progresses past a certain threshold, the tumor-suppressive mechanisms are exploited by tumors, which increases their malignancy. To some extent, these mechanisms exhibit a screening function for tumors. In addition, in special elderly groups, in addition to age-associated alterations at the local cellular and molecular levels, changes in organs may play an important role in the tumor microenvironment. Therefore, for tumor prevention and treatment in elderly people, in addition to focusing on existing treatment methods, the influence of various organs and biological systems needs to be comprehensively considered.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- ACD:
-
Allergic contact dermatitis
- ALT:
-
Alternative lengthening of telomeres
- CDK:
-
Cyclin-dependent kinase
- cGAS:
-
Cyclic GMP-AMP synthase
- CRP:
-
C-reactive protein
- CSF1R:
-
CSF1 receptor
- CTC:
-
Circulating tumor cells
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen 4
- C/EBPα:
-
CCAAT/enhancer-binding protein α
- DDR:
-
DNA damage response
- DNFB:
-
Dinitrobenzene
- miRNAs:
-
MicroRNAs
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial–mesenchymal transition
- EPC:
-
Endothelial progenitor cells
- GEMMs:
-
Genetically engineered mouse models
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HDAC:
-
Histone deacetylase
- ICIs:
-
Immune checkpoint inhibitors
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IGF1:
-
Insulin-like growth factor 1
- LAG-3:
-
Lymphocyte activation gene 3
- LINE-1:
-
Long-interspersed element-1
- MDSCs:
-
Myeloid-derived suppressor cells
- MSCs:
-
Mesenchymal stem cells
- Mo-MDSCs:
-
Monocytic-MDSCs
- NK cells:
-
Natural killer cells
- OIS:
-
Oncogene-induced senescence
- OS:
-
Overall survival
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand 1
- PET:
-
Positron emission computed tomography
- PFS:
-
Progression-free survival
- PICS:
-
PTEN-loss-induced cellular senescence
- PML:
-
Promyelocytic leukemia
- QoL:
-
Quality of life
- ROS:
-
Reactive oxygen species
- SAE:
-
Senescence activation enhancer
- SASP:
-
Senescence-associated secretory phenotype
- SCC:
-
Squamous cell carcinoma
- STING:
-
Stimulator of interferon genes
- TAMs:
-
Tumor-associated macrophages
- TANs:
-
Tumor-associated neutrophils
- TERT:
-
Telomerase reverse transcriptase
- TIME:
-
Tumor-immune microenvironment
- TIS:
-
Therapy-induced senescence
- TLR8:
-
Toll-like receptor 8
- TME:
-
Tumor microenvironment
- TMMs:
-
Telomere maintenance mechanisms
- Treg:
-
T-regulatory cells
- T-SCE:
-
Telomere sister chromatid exchange
- UPR:
-
Unfolded protein response
- VEGF:
-
Endothelial growth factor
- VM:
-
Vasculogenic mimicry
References
Selman M, Pardo A. Fibroageing: An ageing pathological feature driven by dysregulated extracellular matrix-cell mechanobiology. Ageing Res Rev. 2021;70: 101393.
Tomaselli D, Steegborn C, Mai A, Rotili D. Sirt4: A Multifaceted Enzyme at the Crossroads of Mitochondrial Metabolism and Cancer. Front Oncol. 2020;10:474.
Fane M, Weeraratna AT. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 2020;20:89–106.
Pazolli E, Stewart SA. Senescence: the good the bad and the dysfunctional. Curr Opin Genet Dev. 2008;18:42–7.
Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621.
Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1965;37:614–36.
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell. 2019;179:813–27.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79.
Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536–46.
Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96.
Rhinn M, Ritschka B, Keyes WM. Cellular senescence in development, regeneration and disease. Development. 2019;146:151837.
Prieto LI, Graves SI, Baker DJ. Insights from in vivo studies of cellular senescence. Cells. 2020;9:954.
Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408.
Wang B, Kohli J, Demaria M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. 2020;6:838–57.
Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7.
Aunan JR, Cho WC, Søreide K. The Biology of Aging and Cancer: A Brief Overview of Shared and Divergent Molecular Hallmarks. Aging Dis. 2017;8:628–42.
Hsu T. Educational initiatives in geriatric oncology - Who, why, and how? J Geriatr Oncol. 2016;7:390–6.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48.
DeSantis CE, Miller KD, Dale W, Mohile SG, Cohen HJ, Leach CR, et al. Cancer statistics for adults aged 85 years and older, 2019. CA Cancer J Clin. 2019;69:452–67.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186:243–78.
Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439–53.
Zinger A, Cho WC, Ben-Yehuda A. Cancer and Aging - the Inflammatory Connection. Aging Dis. 2017;8:611–27.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705.
Rozhok AI, Salstrom JL, DeGregori J. Stochastic modeling indicates that aging and somatic evolution in the hematopoetic system are driven by non-cell-autonomous processes. Aging. 2014;6:1033–48.
Galluzzi L, Zitvogel L, Kroemer G. Immunological Mechanisms Underneath the Efficacy of Cancer Therapy. Cancer Immunol Res. 2016;4:895–902.
Swanton C. Cell-cycle targeted therapies. Lancet Oncol. 2004;5:27–36.
Rixe O, Fojo T. Is cell death a critical end point for anticancer therapies or is cytostasis sufficient? Clin Cancer Res. 2007;13:7280–7.
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31.
Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol. 2018;40: 101275.
Kale A, Sharma A, Stolzing A, Desprez PY, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing. 2020;17:16.
Rao SG, Jackson JG. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer. 2016;2:676–87.
Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. 2017;7:278.
Braig M, Schmitt CA. Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res. 2006;66:2881–4.
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51.
Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362:1416–22.
Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020;181:424-441.e21.
Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Gonzalez-Meljem JM, Apps JR, Fraser HC, Martinez-Barbera JP. Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer. 2018;118:1283–8.
Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553:96–100.
Triana-Martínez F, Loza MI, Domínguez E. Beyond tumor suppression: senescence in cancer stemness and tumor dormancy. Cells. 2020;9.
Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017;31:172–83.
Sikora E, Czarnecka-Herok J, Bojko A, Sunderland P. Therapy-induced polyploidization and senescence: coincidence or interconnection? Semin Cancer Biol. 2022;81:83–95.
BernardesdeJesus B, Blasco MA. Telomerase at the intersection of cancer and aging. Trends Genet. 2013;29:513–20.
Sung J-Y, Lim H-W, Joung J-G, Park W-Y. Pan-cancer analysis of alternative lengthening of telomere activity. Cancers. 2020;12:2207.
Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–9.
Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–25.
Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–43.
Klutstein M, Nejman D, Greenfield R, Cedar H. DNA Methylation in Cancer and Aging. Cancer Res. 2016;76:3446–50.
Saul D, Kosinsky RL. Epigenetics of aging and aging-associated diseases. Int J Mol Sci. 2021;22.
Park SH, Ozden O, Jiang H, Cha YI, Pennington JD, Aykin-Burns N, et al. Sirt3, mitochondrial ROS, ageing, and carcinogenesis. Int J Mol Sci. 2011;12:6226–39.
Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. 2020;77:4459–83.
Minakshi R, Rahman S, Jan AT, Archana A, Kim J. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp Mol Med. 2017;49: e389.
Wu Q, Li B, Liu L, Sun S, Sun S. Centrosome dysfunction: a link between senescence and tumor immunity. Signal Transduct Target Ther. 2020;5:107.
Zheng J, Mo J, Zhu T, Zhuo W, Yi Y, Hu S, et al. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol Cancer. 2020;19:133.
Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A. 2017;114:E4612-e4620.
Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–70.
Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216:867–83.
De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566:73–8.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7.
Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–6.
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90.
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–47.
Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–5.
Sieben CJ, Sturmlechner I, van de Sluis B, van Deursen JM. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018;28:723–37.
Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–7.
Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–26.
Khalilgharibi N, Mao Y. To form and function: on the role of basement membrane mechanics in tissue development, homeostasis and disease. Open Biol. 2021;11: 200360.
Donnini S, Monti M, Castagnini C, Solito R, Botta M, Schenone S, et al. Pyrazolo-pyrimidine-derived c-Src inhibitor reduces angiogenesis and survival of squamous carcinoma cells by suppressing vascular endothelial growth factor production and signaling. Int J Cancer. 2007;120:995–1004.
Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:16472–7.
Lasry A, Ben-Neriah Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. 2015;36:217–28.
Wang L, Tang C, Cao H, Li K, Pang X, Zhong L, et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol Ther. 2015;16:1220–30.
Goulet CR, Champagne A, Bernard G, Vandal D, Chabaud S, Pouliot F, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer. 2019;19:137.
Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26:38–47.
Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–41.
Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515:134–7.
Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762.
Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol. 2020;13:151.
Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65:2795–803.
Patel PL, Suram A, Mirani N, Bischof O, Herbig U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc Natl Acad Sci U S A. 2016;113:E5024-5033.
Gonzalez-Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017;8:1819.
Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. 2017;8:15729.
Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, Wang R, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864–71.
Jakhar R, Crasta K. Exosomes as emerging pro-tumorigenic mediators of the senescence-associated secretory phenotype. Int J Mol Sci. 2019;20:2547.
Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K, et al. The Cancer SENESCopedia: a delineation of cancer cell senescence. Cell Rep. 2021;36: 109441.
Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–56.
Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–76.
Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594:100–5.
Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. 2019;21:94–101.
Cai Y, Song W, Li J, Jing Y, Liang C, Zhang L, et al. The landscape of aging. Sci China Life Sci. 2022;65:2354–454.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602.
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5.
Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30.
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4.
Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642.
Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol. 2005;25:2660–72.
Guan Y, Zhang C, Lyu G, Huang X, Zhang X, Zhuang T, et al. Senescence-activated enhancer landscape orchestrates the senescence-associated secretory phenotype in murine fibroblasts. Nucleic Acids Res. 2020;48:10909–23.
Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, Stewart SA. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012;72:2251–61.
de Magalhaes JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018;170:2–9.
Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–9.
Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403.
Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028–33.
Zhang DY, Wang HJ, Tan YZ. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS ONE. 2011;6: e21397.
Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505.
Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–85.
Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2:862–73.
Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51.
Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–7.
Gewirtz DA, Holt SE, Elmore LW. Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation. Biochem Pharmacol. 2008;76:947–57.
Kansara M, Leong HS, Lin DM, Popkiss S, Pang P, Garsed DW, et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J Clin Invest. 2013;123:5351–60.
Tesei A, Arienti C, Bossi G, Santi S, De Santis I, Bevilacqua A, et al. TP53 drives abscopal effect by secretion of senescence-associated molecular signals in non-small cell lung cancer. J Exp Clin Cancer Res. 2021;40:89.
Meng Y, Efimova EV, Hamzeh KW, Darga TE, Mauceri HJ, Fu YX, et al. Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther. 2012;20:1046–55.
Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–36.
Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009;113:3503–11.
Hao X, Zhao B, Zhou W, Liu H, Fukumoto T, Gabrilovich D, et al. Sensitization of ovarian tumor to immune checkpoint blockade by boosting senescence-associated secretory phenotype. iScience. 2021;24:102016.
Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89.
Xu Q, Long Q, Zhu D, Fu D, Zhang B, Han L, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell. 2019;18: e13027.
Vilgelm AE, Johnson CA, Prasad N, Yang J, Chen SC, Ayers GD, et al. Connecting the dots: therapy-induced senescence and a tumor-suppressive immune microenvironment. J Natl Cancer Inst. 2016;108:djv406.
Punt S, Malu S, McKenzie JA, Manrique SZ, Doorduijn EM, Mbofung RM, et al. Aurora kinase inhibition sensitizes melanoma cells to T-cell-mediated cytotoxicity. Cancer Immunol Immunother. 2021;70:1101–13.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–5.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984-997.e924.
Guan X, LaPak KM, Hennessey RC, Yu CY, Shakya R, Zhang J, et al. Stromal senescence by prolonged CDK4/6 inhibition potentiates tumor growth. Mol Cancer Res. 2017;15:237–49.
Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature. 2019;574:268–72.
Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–15.
Lee M, Lee JS. Exploiting tumor cell senescence in anticancer therapy. BMB Rep. 2014;47:51–9.
Petrova NV, Velichko AK, Razin SV, Kantidze OL. Small molecule compounds that induce cellular senescence. Aging Cell. 2016;15:999–1017.
Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5.
Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18:7644–55.
Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9:338–50.
Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802.
Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–84.
Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer. 2021;21:37–50.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Hall AG, Tilby MJ. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992;6:163–73.
Perez EA. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8:2086–95.
Klein LE, Freeze BS, Smith AB 3rd, Horwitz SB. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle. 2005;4:501–7.
Dabrowska M, Skoneczny M, Uram L, Rode W. Methotrexate-induced senescence of human colon cancer cells depends on p53 acetylation, but not genomic aberrations. Anticancer Drugs. 2019;30:374–82.
Song Y, Baba T, Mukaida N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem Biophys Res Commun. 2016;477:515–9.
Sabin RJ, Anderson RM. Cellular senescence - its role in cancer and the response to ionizing radiation. Genome Integr. 2011;2:7.
Fei P, El-Deiry WS. P53 and radiation responses. Oncogene. 2003;22:5774–83.
Li M, You L, Xue J, Lu Y. Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front Pharmacol. 2018;9:522.
Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther. 2012;13:451–7.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Yoshida A, Lee EK, Diehl JA. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 2016;76:2990–3002.
Klein ME, Dickson MA, Antonescu C, Qin LX, Dooley SJ, Barlas A, et al. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene. 2018;37:5066–78.
Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19:6173–82.
Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597–607.
Guha M. Blockbuster dreams for Pfizer’s CDK inhibitor. Nat Biotechnol. 2013;31:187.
Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013;31:2024–8.
Kumarasamy V, Vail P, Nambiar R, Witkiewicz AK, Knudsen ES. Functional determinants of cell cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res. 2021;81:1347–60.
Freeman-Cook KD, Hoffman RL, Behenna DC, Boras B, Carelli J, Diehl W, et al. Discovery of PF-06873600, a CDK2/4/6 inhibitor for the treatment of cancer. J Med Chem. 2021;64:9056–77.
Gomez DL, Armando RG, Cerrudo CS, Ghiringhelli PD, Gomez DE. Telomerase as a cancer target. Development of new molecules. Curr Top Med Chem. 2016;16:2432–2440.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem. 2002;277:15566–72.
Burchett KM, Yan Y, Ouellette MM. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE. 2014;9: e85155.
Elknerova K, Myslivcova D, Lacinova Z, Marinov I, Uherkova L, Stöckbauer P. Epigenetic modulation of gene expression of human leukemia cell lines - induction of cell death and senescence. Neoplasma. 2011;58:35–44.
Kaletsch A, Pinkerneil M, Hoffmann MJ, Jaguva Vasudevan AA, Wang C, Hansen FK, et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin Epigenetics. 2018;10:100.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60.
Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34.
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5.
Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett. 2013;4:466–9.
Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007;67:7350–7.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8.
Augello G, Puleio R, Emma MR, Cusimano A, Loria GR, McCubrey JA, et al. A PTEN inhibitor displays preclinical activity against hepatocarcinoma cells. Cell Cycle. 2016;15:573–83.
Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, et al. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011;18:666–77.
Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102.
Lau L, Porciuncula A, Yu A, Iwakura Y, David G. Uncoupling the senescence-associated secretory phenotype from cell cycle exit via interleukin-1 inactivation unveils its protumorigenic role. Mol Cell Biol. 2019;39(12):e00586-e618.
Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014;2014: 292376.
Acosta JC, Banito A, Wuestefeld T, Wuestefeld T, Georgilis A, Georgilis A, Janich P, Janich P, Morton JP, Morton J, Athineos D, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990.
Sarkar D, Fisher PB. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006;236:13–23.
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2017;8:1960.
Coppé JP, Patil CK, Rodier F, Rodier F, Sun Y, Sun Y, Muñoz DP, Muñoz DP, Goldstein J, Goldstein J, Nelson PS, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868.
DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:212.
Song Y, Wang B, Song R, Hao Y, Wang D, Li Y, et al. T-cell immunoglobulin and ITIM domain contributes to CD8(+) T-cell immunosenescence. Aging cell. 2018;17.
Mou D, Espinosa J, Lo DJ, Kirk AD. CD28 negative T cells: is their loss our gain? Am J Transplant. 2014;14:2460–6.
Hogan KA, Chini CCS, Chini EN. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front Immunol. 2019;10:1187.
Chen CY, Kao CL, Liu CM. The cancer prevention, anti-inflammatory and anti-oxidation of bioactive phytochemicals targeting the TLR4 signaling pathway. Int J Mol Sci. 2018;19.
Loo TM, Miyata K, Tanaka Y, Takahashi A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020;111:304–11.
Dimitrijević M, Stanojević S, Vujić V, Aleksić I, Pilipović I, Leposavić G. Aging oppositely affects TNF-α and IL-10 production by macrophages from different rat strains. Biogerontology. 2014;15:475–86.
Guo YA-O, Ayers JA-O, Carter KA-O, Wang TA-O, Maden SA-O, Edmond DA-O, et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell. 2019;18:e13013.
Wang Z. ErbB receptors and cancer. Methods Mol Biol. 2017;1652:3–35.
Messa C, Russo F, Caruso MG, Di Leo A. EGF, TGF-alpha, and EGF-R in human colorectal adenocarcinoma. Acta Oncol. 1998;37:285–9.
Balanis N, Carlin CR. Stress-induced EGF receptor signaling through STAT3 and tumor progression in triple-negative breast cancer. Mol Cell Endocrinol. 2017;451:24–30.
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80.
Xiao ZQ, Majumdar AP. Increased in vitro activation of EGFR by membrane-bound TGF-alpha from gastric and colonic mucosa of aged rats. Am J Physiol Gastrointest Liver Physiol. 2001;281:G111-116.
Zeng Q, Du S, Xu Y, Yang F, Wu L, Wang N, et al. Assessing the potential value and mechanism of Kaji-Ichigoside F1 on arsenite-induced skin cell senescence. Oxid Med Cell Longev. 2022;2022:9574473.
Choi WI, Jeong J, Lee CW. Association between EGFR mutation and ageing, history of pneumonia and gastroesophageal reflux disease among patients with advanced lung cancer. Eur J Cancer. 2019;122:101–8.
Xu X, Liu Q, Zhang C, Ren S, Xu L, Zhao Z, et al. Inhibition of DYRK1A-EGFR axis by p53-MDM2 cascade mediates the induction of cellular senescence. Cell Death Dis. 2019;10:282.
Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56.
Campbell J, Ryan CJ, Brough R, Bajrami I, Pemberton HN, Chong IY, et al. Large-scale profiling of kinase dependencies in cancer cell lines. Cell Rep. 2016;14:2490–501.
Verstraete M, Debucquoy A, Gonnissen A, Dok R, Isebaert S, Devos E, et al. In vitro and in vivo evaluation of the radiosensitizing effect of a selective FGFR inhibitor (JNJ-42756493) for rectal cancer. BMC Cancer. 2015;15:946.
Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 2016;6:838–51.
Ota S, Zhou ZQ, Link JM, Hurlin PJ. The role of senescence and prosurvival signaling in controlling the oncogenic activity of FGFR2 mutants associated with cancer and birth defects. Hum Mol Genet. 2009;18:2609–21.
Coutu DL, Galipeau J. Roles of FGF signaling in stem cell self-renewal, senescence and aging. Aging. 2011;3:920–33.
Chen T, Wang F, Wei S, Nie Y, Zheng X, Deng Y, et al. FGFR/RACK1 interacts with MDM2, promotes P53 degradation, and inhibits cell senescence in lung squamous cell carcinoma. Cancer Biol Med. 2021;18:665–74.
Jung SH, Lee HC, Yu DM, Kim BC, Park SM, Lee YS, et al. Heparan sulfation is essential for the prevention of cellular senescence. Cell Death Differ. 2016;23:417–29.
Strous GJ, Almeida ADS, Putters J, Schantl J, Sedek M, Slotman JA, et al. Growth hormone receptor regulation in cancer and chronic diseases. Front Endocrinol. 2020;11: 597573.
Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13.
Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31:805.
Xu J, Gontier G, Chaker Z, Lacube P, Dupont J, Holzenberger M. Longevity effect of IGF-1R(+/−) mutation depends on genetic background-specific receptor activation. Aging Cell. 2014;13:19–28.
Salvioli S, Capri M, Bucci L, Lanni C, Racchi M, Uberti D, et al. Why do centenarians escape or postpone cancer? The role of IGF-1, inflammation and p53. Cancer Immunol Immunother. 2009;58:1909–17.
Bonafè M, Barbieri M, Marchegiani F, Olivieri F, Ragno E, Giampieri C, et al. Polymorphic variants of insulin-like growth factor I (IGF-I) receptor and phosphoinositide 3-kinase genes affect IGF-I plasma levels and human longevity: cues for an evolutionarily conserved mechanism of life span control. J Clin Endocrinol Metab. 2003;88:3299–304.
Hua H, Kong Q, Yin J, Zhang J, Jiang Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J Hematol Oncol. 2020;13:64.
Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N, et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. 2011;128:2793–802.
Eble JA, Niland S. The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis. 2019;36:171–98.
Humphries JD, Chastney MR, Askari JA, Humphries MJ. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol. 2019;56:14–21.
Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. FASEB J. 1997;11:51–9.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131.
Sandiford OA, Moore CA, Du J, Boulad M, Gergues M, Eltouky H, et al. Human aging and cancer: role of miRNA in tumor microenvironment. Exosomes, stem cells and MicroRNA: aging, cancer and age related disorders. Springer, Cham 2018. p. 137–152.
Lambrechts DA-OX, Wauters E, Boeckx B, Aibar SA-O, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–1289.
Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. 2015;17:1193–204.
Hashimoto O, Yoshida M, Koma Y, Yanai T, Hasegawa D, Kosaka Y, et al. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J Pathol. 2016;240:211–23.
Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423–31.
Ziani L, Safta-Saadoun TB, Gourbeix J, Cavalcanti A, Robert C, Favre G, et al. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget. 2017;8:19780–94.
Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5:e198–e198.
Bourhis M, Palle J, Galy-Fauroux I, Terme M. Direct and indirect modulation of T cells by VEGF-A counteracted by anti-angiogenic treatment. Front Immunol. 2021;12: 616837.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463-479.e410.
Comito G, Iscaro A, Bacci M, Morandi AA-O, Ippolito L, Parri M, et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene. 2019;38:3681–3695.
Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. 2018;9:948.
Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers EA-O, Subramanian P, Patel N, et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun. 2019;10:1–13.
Jo YA-O, Hwang SA-O, Jang JA-O. Employing extracellular matrix-based tissue engineering strategies for age-dependent tissue degenerations. Int J Mol Sci. 2021;22:9367.
Vranken NPA, Lindelauf A, Simons AP, Ariës MJH, Maessen JG, Weerwind PW. Cerebral and limb tissue oxygenation during peripheral venoarterial extracorporeal life support. J Intensive Care Med. 2020;35:179–86.
Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. Geroscience. 2017;39:7–18.
Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–38.
Ryu MH, Park H, Chung J, Chung J, Lee CH, Lee C, Park HR, Park HR. Hypoxia-inducible factor-1alpha mediates oral squamous cell carcinoma invasion via upregulation of alpha5 integrin and fibronectin. Biochem Biophys Res Commun. 2010;393:11–15.
Mohammadi H, Sahai EA-O. Mechanisms and impact of altered tumour mechanics. Nat Cell Biol. 2018;20:766–774.
Baluk P, Hashizume H, McDonald DM, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15:102–111.
Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. 2015;7:1120–1134.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6.
Luo Q, Wang J, Zhao W, Peng Z, Liu X, Li B, et al. Vasculogenic mimicry in carcinogenesis and clinical applications. J Hematol Oncol. 2020;13:19–19.
Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4.
Jiang X, Wang J, Deng X, Xiong F, Zhang S, Gong Z, et al. The role of microenvironment in tumor angiogenesis. J Exp Clin Cancer Res. 2020;39:204–204.
Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–752.
Li S, Meng W, Guan Z, Guo Y, Han X. The hypoxia-related signaling pathways of vasculogenic mimicry in tumor treatment. Biomed Pharmacother. 2016;80:127–35.
Chen Y-S, Chen Z-P. Vasculogenic mimicry: a novel target for glioma therapy. Chin J Cancer. 2014;33:74–9.
Tirpe AA, Gulei D, Ciortea SA-O, Crivii C, Berindan-Neagoe I. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci. 2019;20(24):6140.
Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782–94.
Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–7.
Strilic B, Offermanns S. Intravascular survival and extravasation of tumor cells. Cancer Cell. 2017;32(3):282–93.
Heeke S, Mograbi B, Alix-Panabières C, Hofman P. Never travel alone: the crosstalk of circulating tumor cells and the blood microenvironment. Cells. 2019;8:714.
Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17(5):302–17.
Casella G, Munk R, Kim KM, Piao Y, De S, Abdelmohsen K, et al. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019;47:7294–305.
Zhu S, Deng S, Ma Q, Zhang T, Jia C, Zhuo D, Yang F, et al. MicroRNA-10A* and MicroRNA-21 modulate endothelial progenitor cell senescence via suppressing high-mobility group A2. Circ Res. 2013;112(1):152–164.
Kaluza D, Kroll J, Gesierich S, Manavski Y, Boeckel J-N, Doebele C, Zelent A, et al. Histone deacetylase 9 promotes angiogenesis by targeting the antiangiogenic microRNA-17–92 cluster in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(3):533–543.
Mottet D, Bellahcène A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101(12):1237–46.
Urbich C, Rössig L, Kaluza D, Potente M, Boeckel J-N, Knau A, Diehl F, et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113(22):5669–79.
Ramasamy SK, Kusumbe AP, Schiller M, Zeuschner D, Bixel MG, Milia C, et al. Blood flow controls bone vascular function and osteogenesis. Nat Commun. 2016;7:13601.
Yabluchanskiy A, Ma Y, Chiao YA, Lopez EF, Voorhees AP, Toba H, et al. Cardiac aging is initiated by matrix metalloproteinase-9-mediated endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2014;306:H1398–407.
Li PH, Zhang R, Cheng LQ, Liu JJ, Chen HZ. Metabolic regulation of immune cells in proinflammatory microenvironments and diseases during ageing. Ageing Res Rev. 2020;64: 101165.
Stegner D, Dütting S, Nieswandt B. Mechanistic explanation for platelet contribution to cancer metastasis. Thromb Res. 2014;133(Suppl 2):S149–57.
Lim M, Park S, Jeong H-O, Park SH, Kumar S, Jang A, et al. Circulating tumor cell clusters are cloaked with platelets and correlate with poor prognosis in unresectable pancreatic cancer. Cancers. 2021;13:5272.
Forte D, Krause DS, Andreeff M, Bonnet D, Méndez-Ferrer S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica. 2019;104:1928–34.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Ruchti C, Haller D, Nuber M, Cottier H. Regional differences in renewal rates of fibroblasts in young adult female mice. Cell Tissue Res. 1983;232:625–36.
Faragher RG, McArdle A, Willows A, Ostler EL. Senescence in the aging process. F1000Res. 2017;6:1219.
McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77.
Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. 2019;99:1047–78.
Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol. 2014;39:45–61.
Özcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging. 2016;8:1316–29.
Nelson DM, McBryan T, Jeyapalan JC, Sedivy JM, Adams PD. A comparison of oncogene-induced senescence and replicative senescence: implications for tumor suppression and aging. Age. 2014;36:9637.
Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114.
Burton DGA, Stolzing A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17–25.
He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017;169:1000–11.
Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, et al. Cells exhibiting strong p16(INK4a) promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A. 2019;116:2603–11.
Liedtke C, Rody A, Gluz O, Baumann K, Beyer D, Kohls EB, et al. The prognostic impact of age in different molecular subtypes of breast cancer. Breast Cancer Res Treat. 2015;152:667–73.
Leonardi GC, Accardi G, Monastero R, Nicoletti F, Libra M. Ageing: from inflammation to cancer. Immun Ageing. 2018;15:1.
Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018;2018:9076485.
Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23:1966–71.
Ameri AH, Moradi Tuchayi S, Zaalberg A, Park JH, Ngo KH, Li T, et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc Natl Acad Sci U S A. 2019;116:2646–51.
Shevach EM. Foxp3(+) T regulatory cells: still many unanswered questions-a perspective after 20 years of study. Front Immunol. 2018;9:1048.
Garbern JC, Williams J, Kristl AC, Malick A, Rachmin I, Gaeta B, et al. Dysregulation of IL-33/ST2 signaling and myocardial periarteriolar fibrosis. J Mol Cell Cardiol. 2019;128:179–86.
Kim DW, Kim DK, Kong IG, Eun KM, Cho SH. Age-Related Increase Of IL-33 In Non-Eosinophilic Nasal Polyps. J Allergy Clin Immunol. 2018;141:AB67.
Olivieri F, Rippo MR, Monsurrò V, Salvioli S, Capri M, Procopio AD, et al. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev. 2013;12:1056–68.
Olivieri F, Spazzafumo L, Santini G, Lazzarini R, Albertini MC, Rippo MR, et al. Age-related differences in the expression of circulating microRNAs: miR-21 as a new circulating marker of inflammaging. Mech Ageing Dev. 2012;133:675–85.
Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhão JG, et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011;4:ra75.
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109:E2110-2116.
Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316.
Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc Natl Acad Sci U S A. 2003;100:15053–8.
Saule P, Trauet J, Dutriez V, Lekeux V, Dessaint JP, Labalette M. Accumulation of memory T cells from childhood to old age: central and effector memory cells in CD4(+) versus effector memory and terminally differentiated memory cells in CD8(+) compartment. Mech Ageing Dev. 2006;127:274–81.
Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J Exp Med. 2008;205:711–23.
Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol. 2019;10:2247.
Gayoso I, Sanchez-Correa B, Campos C, Alonso C, Pera A, Casado JG, et al. Immunosenescence of human natural killer cells. J Innate Immun. 2011;3:337–43.
Colonna-Romano G, Aquino A, Bulati M, Lio D, Candore G, Oddo G, et al. Impairment of gamma/delta T lymphocytes in elderly: implications for immunosenescence. Exp Gerontol. 2004;39:1439–46.
Linton PJ, Thoman ML. Immunosenescence in monocytes, macrophages, and dendritic cells: lessons learned from the lung and heart. Immunol Lett. 2014;162:290–7.
Linehan E, Fitzgerald DC. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol. 2015;5:14–24.
Golomb L, Sagiv A, Pateras IS, Maly A, Krizhanovsky V, Gorgoulis VG, et al. Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ. 2015;22:1764–74.
Hurez V, Padrón Á, Svatek RS, Curiel TJ. Considerations for successful cancer immunotherapy in aged hosts. Exp Gerontol. 2018;107:27–36.
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ. Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell. 2013;12:345–57.
Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–91.
Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77:3655–65.
Okuma A, Hanyu A, Watanabe S, Hara E. p16Ink4a and p21Cip1/Waf1 promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat Commun. 2017;8:2050.
Gomez CR, Nomellini V, Faunce DE, Kovacs EJ. Innate immunity and aging. Exp Gerontol. 2008;43:718–28.
Schröder AK, Rink L. Neutrophil immunity of the elderly. Mech Ageing Dev. 2003;124:419–25.
Grecian R, Whyte MKB, Walmsley SR. The role of neutrophils in cancer. Br Med Bull. 2018;128:5–14.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612:141–7.
Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33:949–55.
Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y. Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. J Leukoc Biol. 2007;81:1386–94.
Kryczek I, Liu R, Wang G, Wu K, Shu X, Szeliga W, et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res. 2009;69:3995–4000.
Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol. 2007;188:117–27.
Sharma S, Dominguez AL, Lustgarten J. High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol. 2006;177:8348–55.
Thomas DC, Mellanby RJ, Phillips JM, Cooke A. An early age-related increase in the frequency of CD4+ Foxp3+ cells in BDC2.5NOD mice. Immunology. 2007;121:565–576.
Kozlowska E, Biernacka M, Ciechomska M, Drela N. Age-related changes in the occurrence and characteristics of thymic CD4(+) CD25(+) T cells in mice. Immunology. 2007;122:445–53.
Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age correlates with response to Anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin Cancer Res. 2018;24:5347–56.
Aguado BA, Bushnell GG, Rao SS, Jeruss JS, Shea LD. Engineering the pre-metastatic niche. Nat Biomed Eng. 2017;1.
Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, et al. Blood CD33(+)HLA-DR(-) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol. 2013;93:633–7.
Hurez V, Daniel BJ, Sun L, Liu AJ, Ludwig SM, Kious MJ, et al. Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res. 2012;72:2089–99.
Enioutina EY, Bareyan D, Daynes RA. A role for immature myeloid cells in immune senescence. J Immunol. 2011;186:697–707.
Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. 2019;10:172.
Begley LA, Kasina S, MacDonald J, Macoska JA. The inflammatory microenvironment of the aging prostate facilitates cellular proliferation and hypertrophy. Cytokine. 2008;43:194–9.
Strasner A, Karin M. Immune infiltration and prostate cancer. Front Oncol. 2015;5:128.
Daste A, Chakiba C, Domblides C, Gross-Goupil M, Quivy A, Ravaud A, et al. Targeted therapy and elderly people: a review. Eur J Cancer. 2016;69:199–215.
Morre M, Beq S. Interleukin-7 and immune reconstitution in cancer patients: a new paradigm for dramatically increasing overall survival. Target Oncol. 2012;7:55–68.
Bashiri Dezfouli A, Pourfathollah AA, Salar-Amoli J, Khosravi M, Nikogoftar-Zarif M, Yazdi M, et al. Evaluation of age effects on doxorubicin-induced toxicity in mesenchymal stem cells. Med J Islam Repub Iran. 2017;31:98.
Wood WA, Krishnamurthy J, Mitin N, Torrice C, Parker JS, Snavely AC, et al. Chemotherapy and stem cell transplantation increase p16(INK4a) expression, a biomarker of T-cell aging. EBioMedicine. 2016;11:227–38.
Grigorescu AC. Chemotherapy for elderly patients with advanced cancer: a pilot study in Institute of Oncology Bucharest. J Transl Int Med. 2015;3:24–8.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26.
Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH 3rd, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016;532:250–4.
Somasundaram R, Zhang G, Fukunaga-Kalabis M, Perego M, Krepler C, Xu X, et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun. 2017;8:607.
Riley RL, Khomtchouk K, Blomberg BB. Age-associated B cells (ABC) inhibit B lymphopoiesis and alter antibody repertoires in old age. Cell Immunol. 2017;321:61–7.
Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev Mol Diagn. 2018;18:675–83.
Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559:363–9.
Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435–47.
Steinberg SM, Shabaneh TB, Zhang P, Martyanov V, Li Z, Malik BT, et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017;77:1599–610.
Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20.
Almeida FV, Douglass SM, Fane ME, Weeraratna AT. Bad company: Microenvironmentally mediated resistance to targeted therapy in melanoma. Pigment Cell Melanoma Res. 2019;32:237–47.
Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–77.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Shimada Y, Hayashi M, Nagasaka Y, Ohno-Iwashita Y, Inomata M. Age-associated up-regulation of a negative co-stimulatory receptor PD-1 in mouse CD4+ T cells. Exp Gerontol. 2009;44:517–22.
Lages CS, Lewkowich I, Sproles A, Wills-Karp M, Chougnet C. Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/ PD-L1 pathway. Aging Cell. 2010;9:785–98.
Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell. 2015;14:945–56.
Mirza N, Duque MA, Dominguez AL, Schrum AG, Dong H, Lustgarten J. B7–H1 expression on old CD8+ T cells negatively regulates the activation of immune responses in aged animals. J Immunol. 2010;184:5466–74.
Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front Pharmacol. 2019;10:200.
Sceneay J, Goreczny GJ, Wilson K, Morrow S, DeCristo MJ, Ubellacker JM, et al. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative breast cancer. Cancer Discov. 2019;9:1208–27.
Elias R, Hartshorn K, Rahma O, Lin N, Snyder-Cappione JE. Aging, immune senescence, and immunotherapy: a comprehensive review. Semin Oncol. 2018;45:187–200.
Elias R, Karantanos T, Sira E, Hartshorn KL. Immunotherapy comes of age: Immune aging & checkpoint inhibitors. J Geriatr Oncol. 2017;8:229–35.
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26.
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–32.
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–56.
Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–35.
Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378:1789–801.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–33.
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393:1819–30.
Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–39.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377:1919–29.
Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–92.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379:2040–51.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381:2020–31.
Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301.
Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet. 2021;398:1344–57.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13.
Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–90.
Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1116–27.
Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380:1103–15.
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–26.
Galsky MD, Arija JÁA, Bamias A, Davis ID, De Santis M, Kikuchi E, et al. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395:1547–57.
Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383:1218–30.
Betof AS, Nipp RD, Giobbie-Hurder A, Johnpulle RAN, Rubin K, Rubinstein SM, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963–71.
Figueiredo ASP, Hurez V, Liu A, Curiel TJ. Age and sex affect αCTLA-4 efficacy alone and combined with αB7-H1 or regulatory T cell depletion in a melanoma model. J Immunol. 2016;196:213.214–213.214.
Lichtenstein MRL, Nipp RD, Muzikansky A, Goodwin K, Anderson D, Newcomb RA, et al. Impact of age on outcomes with immunotherapy in patients with non-small cell lung cancer. J Thorac Oncol. 2019;14:547–52.
Freedman RA, Dockter TJ, Lafky JM, Hurria A, Muss HJ, Cohen HJ, et al. Promoting accrual of older patients with cancer to clinical trials: an alliance for clinical trials in oncology member survey (A171602). Oncologist. 2018;23:1016–23.
Xu X, Wang B, Ren C, Hu J, Greenberg DA, Chen T, et al. Age-related impairment of vascular structure and functions. Aging Dis. 2017;8:590–610.
Wang Y, Zhao B, Chen W, Liu L, Chen W, Zhou L, et al. Pretreatment geriatric assessments of elderly patients with glioma: development and implications. Aging Dis. 2020;11:448–61.
Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, Paez-Ribes M, et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10(9): e9355.
Lozano-Torres B, Galiana I, Rovira M, Garrido E, Chaib S, Bernardos A, et al. An OFF-ON two-photon fluorescent probe for tracking cell senescence in vivo. J Am Chem Soc. 2017;139:8808–11.
Dolgin E. Send in the senolytics. Nat Biotechnol. 2020;38:1371–7.
Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging. 2017;9:1867–84.
de Mera-Rodríguez JA, Álvarez-Hernán G, Gañán Y, Martín-Partido G, Rodríguez-León J, Francisco-Morcillo J. Is senescence-associated β-galactosidase a reliable in vivo marker of cellular senescence during embryonic development? Front Cell Dev Biol. 2021;9: 623175.
Wiley CD, Sharma R, Davis SS, Lopez-Dominguez JA, Mitchell KP, Wiley S, et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 2021;33:1124-1136.e1125.
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583:127–32.
Schafer MJ, Zhang X, Kumar A, Atkinson EJ, Zhu Y, Jachim S, et al. The senescence-associated secretome as an indicator of age and medical risk. JCI Insight. 2020;5.
Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020;18: e3000599.
Zhao B, Xia Y, Yang F, Wang Y, Wang Y, Wang Y, et al. Molecular landscape of IDH-mutant astrocytoma and oligodendroglioma grade 2 indicate tumor purity as an underlying genomic factor. Mol Med. 2022;28:34.
Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17:1205–17.
Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012;122:2601–12.
Niedernhofer LJ, Robbins PD. Senotherapeutics for healthy ageing. Nat Rev Drug Discov. 2018;17:377–377.
Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, et al. Defined p16(High) senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32:87-99.e86.
Omori S, Wang TW, Johmura Y, Kanai T, Nakano Y, Kido T, et al. Generation of a p16 reporter mouse and its use to characterize and target p16(high) cells in vivo. Cell Metab. 2020;32:814-828.e816.
Acknowledgements
The authors thank professor Jianjun Xu, MD (Department of Thoracic Surgery, The Second Affiliated Hospital of Nanchang University) for his advice. The authors also thank the support of the National Natural Science Foundation of China and Natural Science Foundation of Jiangxi Province.
Funding
This study was supported by the National Natural Science Foundation of China (NSFC, Grant Number: 81560345) and Natural Science Foundation of Jiangxi Province (Grant Number: 20212BAB206050). Role of the Funding: The funding had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
WZ had full access to all of the data in the manuscript and took responsibility for the integrity of the data and the accuracy of the data analyses. All authors contributed to concept, design, acquisition, analysis, or interpretation of the data. BZ, BW, Xiang Zhang, Xin Zhang, and ZX drafted the manuscript. BZ, BW, NF, YW, and WZ contributed to critical revision of the manuscript for important intellectual content. BZ and WZ supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent participate
The current study investigated the publicly available data, and no ethical approval was required.
Consent for publication
Not applicable.
Informed consent
For this type of study, formal consent is not required.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, B., Wu, B., Feng, N. et al. Aging microenvironment and antitumor immunity for geriatric oncology: the landscape and future implications. J Hematol Oncol 16, 28 (2023). https://doi.org/10.1186/s13045-023-01426-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01426-4