
Abstract
Cellular senescence is a normal biological process that is initiated in response to a range of intrinsic and extrinsic factors that functions to remove irreparable damage and therefore potentially harmful cells, from the proliferative pool. Senescence can therefore be thought of in beneficial terms as a tumour suppressor. In contrast to this, there is a growing body of evidence suggesting that senescence is also associated with the disruption of the tissue microenvironment and development of a pro-oncogenic environment, principally via the secretion of senescence-associated pro-inflammatory factors. The fraction of cells in a senescent state is known to increase with cellular age and from exposure to various stressors including ionising radiation therefore, the implications of the detrimental effects of the senescent phenotype are important to understand within the context of the increasing human exposure to ionising radiation. This review will discuss what is currently understood about senescence, highlighting possible associations between senescence and cancer and, how exposure to ionising radiation may modify this.
Similar content being viewed by others


Cellular Senescence
Cellular senescence is a metabolically active form of irreversible growth arrest that halts the proliferation of ageing and/or damaged cells and as a consequence, prevents the transmission of damage to daughter cells. This complicated cellular event is initiated in response to a variety of intrinsic and extrinsic genotoxic stimuli [1–4] and mediated through tumour suppressor pathways involving p53, and p16INK4A/pRb [5, 6]. This ultimately leads to the inhibition of cyclin-dependant kinases [7, 8]. Accordingly, cellular senescence can be thought of as a tumour suppressor mechanism. Indeed the majority of cancers have mutations in p53 and/or the pRb/p16 pathways, while germ-line mutations in these pathways result in a cell-specific ability to overcome senescence-inducing signals, thereby greatly increasing their susceptibility to cellular transformation [9–11]. The importance of cellular senescence as a tumour suppressor is further demonstrated by cell fusion experiments [12] that provide evidence that growth arrest observed in senescent cells has a strong influence over the growth in proliferating cells and cellular oncogenes of tumour cells. When proliferating cells were fused with senescent cells, DNA replication was inhibited even in the presence of mitogens, and when senescent cells were fused with tumour cells, DNA replication was similarly inhibited. These fusion experiments led to the assumption that senescent cells contained control elements capable of exerting a dominant effect over proliferating pre-senescent cells. Importantly, this tumour suppressive mechanism of cellular senescence has been supported in both mice and human studies [13].
As well as possessing tumour suppressive mechanisms, senescence has been found to play an important role in wound healing and tissue repair and/or communication to surrounding tissues/cells of damage crisis to assist healing [14, 15]. For instance, senescent cells have been shown in in vivo mouse models to play a role in the resolution of fibrosis by matrix metalloproteinases (MMPs) after acute liver injury. Under normal conditions, proliferating hepatic stellate cells triggered in response to acute liver injury, produce fibrotic scars in advance of entering into a senescent state, followed by secretion of MMPs and scar dissolution. However in cells deficient in either p53/pRb pathways, liver injury results in severe, irresolvable fibrosis [16]. Similarly, the matricellular protein CCN1, which is expressed at sites of cutaneous wound repair, has been shown to initiate DNA damage response pathways and reactive-oxygen species dependent activation of p16INK4A/pRb pathway, resulting in senescence and the expression of anti-fibrotic genes in wild-type mice. Mutant mice that express a senescence-defective CCN1 protein however show increased fibrosis at sites of wound repair [17]. Cellular senescence has been also shown to be important in the prevention of epithelial-mesenchymal transition (EMT) whereby the metastatic dissemination of cancerous cells is prevented [18, 19]. Thus, the functional significance of cellular senescence includes a diverse range of roles which are essentially beneficial to the organism.
Phenotype
The major phenotype of senescence that characteristically distinguishes senescence from quiescence is irreversible growth arrest that cannot be reversed by any known physiological stimuli, associated with resistance to apoptosis and increased sensitivity to cellular injury [20–26]. Other characteristic changes include altered gene expression with increased expression of proteins including p53, p16, p19 and p21 [27, 28], an increase in senescence-associated beta galactosidase (SA-β-gal) activity at pH 6.0 [29], the presence of persistent telomere and non-telomere DNA damage foci [30, 31], senescence-associated heterochromatic foci (SAHF) [6] and a senescence-associated secretory phenotype (SASP) [32, 33]. Accordingly, identification of senescent cells can be achieved by assaying for a combination of the above characteristics. The application of proliferation cell-cycle specific markers, such as Ki-67, can also be used [34–36]. For example Kill et al (1996) showed that 56% of human dermal fibroblasts were Ki-67 positive at early passage 4 [37] compared to only 30% at passage 38 [38] with the decrease in fraction of Ki-67 positive cells reflecting an increase in senescence.
A range of morphological changes have also been documented with senescent fibroblasts showing enlarged and flattened morphology accompanied by the loss of elongated, spindle-like properties, when compared to normal proliferating fibroblasts. Specifically, the mean nuclear area of fibroblasts was shown to be 255 μm2 at early passage, compared to 293 μm2 at later passage [39]. Interestingly, the sub-nuclear organisation of chromosomes has also been shown to be different in senescent and proliferating mammalian somatic cells, whereby gene poor chromosomes such as chromosomes 13 and 18 are thought to alter their preferential nuclear position from near the nuclear periphery and relocate to the nuclear interior when induced to senesce. Thus, the interphase organisation of particular chromosome territories changes such that their position correlates according to the size of the chromosome, rather than the density [40, 41].
Replicative Senescence
Seminal work carried out by Hayflick and Moorhead (1961) demonstrated that normal cells grown in culture dishes are only able to undergo a finite number of cellular divisions before their growth is irreversibly arrested [42–44]. This 'Hayflick Limit' was the first demonstration of a senescence phenotype and described the replicative capacity of diploid cells in culture before the cells ceased to divide [45] and has since been demonstrated for many different types of cells both in vitro and in vivo [46, 47]. Importantly, diploid cells within cell populations do not all reach senescence at the same time, rather there is a progressive decrease in the fraction of proliferating cells that are capable of undergoing cellular division with each round of replication [48, 49]. Also, different cell types and lineages will vary in the rate at which they enter a senescent state [50]. For example, in vitro studies comparing the growth rates and passage number of fibroblast and keratinocyte cell types observed that the decline in cell growth rate was notably higher for keratinocytes which had senesced by P6, compared to fibroblasts that were passaged beyond P10 in all donor age groups above and below 40 years of age; suggesting that the growth rate of the two cell types is independent of donor age [51]. Indeed a 'memory' for the number of completed population doublings was observed when WI-38 fibroblasts were found to enter senescence with respect to their remaining replicative capacity, even after cryopreservation for a period of 23 years [44].
The mechanism for replicative senescence is believed to be associated with a progressive shortening of telomeres that occurs with each DNA replication cycle [52, 53]. Functional telomeres protect the ends of chromosomes however, approximately 100-300 bp of these repeat sequences are lost as a result of incomplete replication of the extending 3' overhang of nucleotides [54, 55]. Support for this comes from studies that show critically short telomeres trigger a DNA damage response which results in cellular senescence [30, 56], while immortal cancer and germ cell lines overcome the action of telomeric shortening by the expression of the telomerase enzyme [57]. Telomerase synthesizes and maintains telomeric end sequences, preventing the exposure of uncapped ends [58] thereby permitting continued cellular proliferation [59–63].
Stress-induced Premature Senescence (SIPS)
Stress-induced premature senescence (SIPS), also known as premature senescence, culture shock and STASIS (stress or aberrant signalling-induced senescence) [64–66], occurs rapidly in response to a variety of intrinsic or extrinsic stressors, including DNA damage from ionising and non-ionising radiation, cytotoxic DNA damaging agents, oxidative stress and as a consequence of oncogenic activation [3, 4, 8, 67]. Serrano et al (1997) were among the first to identify a form of SIPS that was not attributable to telomere attrition that they described as oncogene-induced senescence (OIS). The group showed that oncogenic ras expression permanently arrested primary human and rodent cells in G1 and that the cells displayed features similar to those of replicative senescence, including the accumulation of p53 and p16 [65]. Importantly the expression of the catalytic subunit of the telomerase enzyme hTERT has been shown not to abrogate SIPS, demonstrating that cellular senescence can be triggered prematurely independently of telomere attrition [68, 69]. Thus, oncogenic activation and stressors that lead to DNA damage but which are independent of loss or dysfunction of telomeres, can initiate a response that results in a cellular phenotype indistinct to that observed for replicative senescence [63, 70].
Molecular Pathways of Senescence
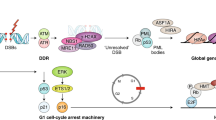
The initiating event for both replicative senescence and SIPS involves the recognition of DNA damage and the activation of the DNA damage response (DDR) pathway [3, 56, 71–74]. The key mediator in this process, ATM, phosphorylates important sensors and effectors of the DDR including H2AX, 53BP1 [31, 75–77] and p53 leading to the up-regulation of cyclin-dependant kinase inhibitor p21, which in-turn acts to inhibit the action of CDK2 kinase activity arresting the cell cycle in G1 [76] (Figure 1). In addition, p21 also activates pRb through the inhibition of cyclin E/CDK2 [1]. Where SIPS differs from replicative senescence is in the formers dependence on the P16INK4 family of tumour suppressor proteins, which are activated upstream to pRb [10, 78]. Accordingly, increased P16INK4A expression is considered as another useful marker of senescence in vitro, and indeed elevated protein levels have been detected in ageing baboon fibroblasts along with markers of telomere damage and SAHF [79]. The hypophosphorylated state of pRb results in inhibition of the transcription factor gene E2F and this acts to bring about G1 cell cycle arrest. For this reason, the p53 and p16/pRB dependent senescent pathways are not completely separable and as well as the common link through p21, pRB has been shown to regulate the activity of MDM2 which acts to control the stability of p53 [80]. Thus increased expression of p21 is important for senescence [81]. Consequently, the DNA damage response, apoptotic and senescence pathways share common molecular mediators through p53 and p21. What directs a cell to senesce or apoptose remains unclear, but cell type, the type of damaging agent and the dose administered may be important; as well as the post-translational modifications that p53 undergoes [82]. For instance in normal cells, senescence has been shown to be more favourable than apoptosis to deal with low levels of DNA damage, perhaps as the cell makes the decision to attempt to repair instead of removal from the cell pool [3, 76, 83]. By contrast, adult human dental pulp stem cells (DPSCs) were found to enter premature senescence in the G2 phase of the cell cycle after exposure to much higher doses (2-20 Gy) of ionising radiation, as detectable by phosphorylated p53 and increasing p16 expression observed over 13 days and SA-β-gal activity from day 3 after irradiation [84]. Possible mechanisms that may be involved in determining cellular fate include the status of the tumour suppressor phosphate and tensin homolog (PTEN). For instance, Lee J, et al (2010) showed that PTEN-deficient glioma cells preferentially entered senescence, while PTEN-proficient glioma cells generally apoptosed in response to ionising radiation. The authors concluded that SIPS may be a compensatory mechanism in place of apoptosis when PTEN tumour suppressor protein is absent [85].

Scheme highlighting initiating and molecular mediators of cellular senescence. The senescent phenotype includes expression of SA-β-galactosidase (SA-β-gal), increased expression of p16INK4a leading to cell cycle arrest and an increase in the secretion of pro-inflammatory factors termed as senescence-associated secretory phenotype (SASP). Senescent cells have been observed in normal ageing cells and in cells/tissues of various age-related pathologies.
What is clear is that the convergence of multiple pathways through p53 and pRb are required to establish and maintain the senescent state and removal of either of these has been shown to prevent senescence in mouse embryonic fibroblasts [86]. In humans, it is thought that both p53 and pRb pathways must be inactivated in order to prevent the onset of cellular senescence [87], consistent with the majority if findings that show ~50% of all tumours show evidence of mutated/non-functional p53 and/or pRb [88, 89]. Interestingly though, more recent experiments in humans and mice have shown that senescence can be prevented, or significantly delayed as a result of inactivation of either p53 or Rb alone while some cell types exhibit delayed onset of senescence upon p16 inactivation [90]. Therefore the relationship between these two pathways and the potential for redundancy in either pathway may provide further protection against senescence bypass in different cell types [3, 91].
Senescence and Cancer
Senescence, in addition and in contrast to the previously noted beneficial tumour suppression and tissue repair effects, has also been linked to reduced tissue functionality and is increasingly thought to play a role in age-related pathologies such as cancer, Alzheimer's disease, diabetes and obesity [54, 92–96]. For instance, senescent cells have been observed in many ageing mice tissues [20, 97], baboon skin fibroblasts [79] and human tissues [29] indicating that senescence may have a causal role in ageing in vivo as well as in vitro. Senescent endothelial cells have been shown to increase in atherosclerosis, thrombosis and at sites of inflamed vascular endothelium [18, 98], demonstrating possible links with pathology. Therefore, the accumulation of senescent haemopoietic stem cells has been suggested as a possible mediator for the decline in tissue regeneration and repair with age.
Markers of DNA damage are known to accumulate in ageing stem cells [64, 99–101] and other senescing human and mouse cell types [102–104]. For instance, studies employing the DSB marker, γ-H2AX, reveal γ-H2AX foci to accumulate in normal human fibroblasts, WI38 fibroblasts and PrEC prostate epithelial cells with increasing passage in a manner that correlates with an increasing fraction of SA-β gal positive cells. Specifically, early passage cultures show 0.2-0.3 γ-H2AX foci/cell increasing to 2.2-4.1 foci/cell in senescent cultures [31]. Further to this, radiation-induced γ-H2AX foci have both been shown to increase in both murine and human senescent cells in vitro [56, 73, 74] while, in vivo studies have shown the long-term expression of senescence markers, including an increased expression of p16INK4a to be coupled with the persistence of DNA damage foci 45 weeks post irradiation to a sub-lethal dose of radiation [105]. Interestingly, γ-H2AX foci (along with other DNA damage markers such as 53BP1) have been shown to localise both at telomeres as telomere-dysfunction-induced-foci (TIFs) in both early and late passage fibroblasts [106] and also throughout the genome as a consequence of ionising radiation exposure. Therefore γ-H2AX foci seen in senescent cells are not necessarily telomere-associated foci, representative of replicative senescence, but may represent SIPS-induced sites of DSB, highlighting that mediators of SIPS may contribute to age-related pathologies, including cancer. Accordingly, there is a relationship between cellular ageing and the accumulation of residual DNA damage both in vitro and in vivo, however as yet there is no evidence to determine whether senescence is a resultant part of ageing and age-related pathologies or whether it is a state that contributes to the development of ageing tissues and tissue pathology. Interestingly though, mouse models of accelerated ageing that are deficient for p16INK4a show delayed onset of age-related phenotypes, highlighting the role of increasing p16INK4a in maintaining the senescent state and its role in age-related decline of tissue regeneration and repair [107]. Further studies will hopefully decipher the evidential link between increasing populations of senescent cells and the contribution they have in the development of age-related pathologies such as cancer [108, 109].
Senescence-Associated Secretory Phenotype (SASP)
It is well established that senescent cells secrete factors such as interleukins, chemokines, growth factors and proteases, encompassing what is known as the senescence-associated secretory phenotype (SASP) [15, 32, 110]. The function of SASP is to mediate the characteristic growth arrest of senescence via the autocrine activities of pro-inflammatory cytokines (including IL-6 and IL-8), in addition to pro-apoptotic protein insulin growth-factor binding protein 7 (IGFBP7), epithelial growth factors (heregulin and VEGF), matrix metalloproteinases including MMP-3 and plasminogen activator inhibitor 1 (PAI-1) [111–113]. Interestingly the name 'senescence-messaging secretome' (SMS) was proposed to highlight that the associated factors of the secretory phenotype were not only essential for initiating the senescent state but also for its maintenance and communication of this state to the local microenvironment [114, 115]. A study that highlights this communication shows that re-activation of endogenous p53 in p53-deficient tumours in a mosaic mouse model of hepatocellular carcinoma led to tumour regression. This was proposed to occur through the induction of cellular senescence and up-regulation of inflammatory cytokines, triggering an innate immune response in vivo that ultimately led to tumour clearance [116]. Thus, inflammatory cytokines are necessary for both the establishment and maintenance of senescence, suggesting SASP/SMS products are important for the suppression of malignancy [111, 114]. However, SASP is also known to influence the proliferation of neighbouring cells and disrupt tissue architecture [117], principally through these pro-inflammatory influences. For instance, an increase in VEGF, as a result of senescent fibroblasts, has been seen to stimulate tumour vascularisation and invasion of basement membranes [118]. Further, inflammation is thought of as a key mediator in cancer development and inflammatory cytokines and MMPs are being increasingly implicated as a contributing factor in this multistep process.
What this suggests is that senescent cells can actually promote, in addition to preventing, the progression of malignancy; a relationship that is described as antagonistically pleiotrophic [21, 118–121]. For instance, senescent human fibroblasts have been shown to stimulate pre-malignant and malignant fibroblasts to hyperproliferate and form tumours in mouse models when senescent cells comprised ~10% of the fibroblast population [122]. Close proximity of senescent fibroblasts to pre-neoplastic cells are thought to be the trigger for this change. Additionally, after exposure to the DNA-damaging agent bleomycin, human SIPS fibroblasts co-transplanted into xenografts of immunodeficient mice were seen to stimulate nearby cancer cells to proliferate, either directly or through local tissue damage and inflammation mediated by MMPs [123]. These findings support SASP as being an important mediator of the transformation process of pre-neoplatic cells. In addition, studies carried out by Zhou et al (2011) have shown SIPS in normal airway epithelial cells to result in an impairment of repair of drug-induced damage initiating a p38 MAPK dependant increase of pro-inflammatory cytokines that was subsequently seen to exacerbate the airway injury [124]. Interestingly this cytokine secretion, which primarily involves IL-6 and IL-8, is only established as a result of persistent DNA damage response signalling (DDR) and not as a result of transient signalling [125] suggesting the presence of long-lived, irreparable DNA lesions are important in this process.
Thus alteration of the tissue microenvironment that results in the promotion of cell growth as a consequence of the senescence phenotype, through inflammation and persistent tissue damage [4, 15, 126] may provide a mechanism whereby senescent cells may also contribute to cancer promoting effects in otherwise normal tissues [127]. If demonstrated then senescence may functionally protect young animals from cancer via tumour suppression, whilst contributing to the deleterious effects in aged organisms through persistent inflammation and tissue injury [122, 126].
Ionising radiation and Senescence
Ionising radiation is known to induce SIPS in both normal and cancer cell types after exposure to relatively high doses (10Gy) of radiation [125, 128–131]. Thus, an important implication is what contribution, if any, senescence plays as a possible mediator of tumour recurrence after radiotherapy, given the effects of SASP in stimulating pre-neoplastic cells as discussed earlier. SIPS is also induced after exposure to lower doses of radiation [125, 130, 132, 133] which similarly has consequences for understanding human cancer risk to radiation exposure, but this time within the context of SIPS in normal tissue after e.g. diagnostic exposures. For instance it is well established that radiation induces damage in cells that are not directly irradiated but which are in communication with irradiated cells. This radiation-induced non-targeted bystander (NTE) phenomenon is known to dominate at low radiation doses and to mediate a range of cellular effects such as DNA damage [134, 135], cell death [136], cell proliferation, adaptive protective effects and malignant transformation [75, 137–141]. To date, such NTE have been observed in microbeam-irradiated human tissue [141, 142], in vivo animal models [143–145] and interestingly, in cells cultured in both non-irradiated tumour and senescent cell conditioned medium [14, 75].
Thus, it is reasonable to ask if there is a possible concordance between radiation-induced SIPS and SASP, and candidate mediators of NTE effects. Reactive oxygen species (ROS) are known to be important damaging agents involved in NTE [143, 146, 147], but additionally, activated macrophage, NO, IL-6, IL-8, IFN-γ and TGF-β have all also been implicated [143, 144, 148]. For instance, one study used radiation-induced AML susceptible and resistant mouse strains, CBA/ca and C57BL/6 respectively, to correlate radiation-induced up-regulation of gene expression of a M1 pro-inflamatory macrophage profile with more NTE in CBA/ca and an M2 anti-inflammatory macrophage profile, with less NTE in C57BL/6 strains [144]. Thus, candidate sources for the mediation of radiation-induced NTE include inflammation-associated cytokines and chemokines secreted from irradiated (or otherwise stressed) cells. Whether the irradiated cells that contribute to this plethora of inflammatory signals remain within the proliferative pool upon repair of damage or whether they become senescent is unknown however it is clear that even low doses of radiation induce SIPS and these cells subsequently secrete inflammatory cytokines, including IL-6 and IL-8 [27, 110, 133, 149–153]. Interestingly Tsai et al (2009) showed that stromal fibroblasts that were induced to senesce after low dose radiation exposure stimulated the proliferation of breast-carcinoma cells when co-cultured in the same medium [125, 130, 132, 133].
This potential relationship between exposure to radiation, cellular age and deleterious inflammatory (NTE) responses is further demonstrated by human and animal studies which show a correlation exists between the immunological imbalances caused as a result of exposure to radiation and, those effects which are seen in normal aged immune cells, implying ionising radiation may accelerate immunological ageing [154]. For instance, the normal age-related decrease of total CD4+ T-cells was found to be ~4% per 10 years, compared to a radiation-induced decrease of ~2% Gy-1, equivalent to a 5 year age increase per 1 Gy [155, 156]. This group also demonstrated a dose-dependent increase in CD25+/CD127- regulatory T-cells and attributed T-cell immunosenescence to a higher level of inflammatory markers in A-bomb survivors. For instance, changes in the immunological profiles of cytokines, known to be involved in the coordination of the inflammatory response (TNF-α, IFN-γ, IL-6 and IL-10) were seen in both A-bomb survivors and liquidators which may contribute to the persistent subclinical inflammatory status that is seen in these individuals [157–160]. There is the suggestion therefore that radiation-induced enhancement of inflammatory reactions might contribute to the development of radiation-induced disorders and premature ageing [155, 161]. Indeed, it is also well known that A-bomb survivors show increased cardiovascular and respiratory diseases associated with persistent inflammation [162, 163].
Taken all together it is tempting to speculate radiation-induced SIPS and SASP as important mediators and, or amplifiers of radiation-induced NTE, which in turn may perpetuate inflammatory signals that subsequently, also contribute to increasing SIPS. In elucidating the importance of any such relationship in contributing to cancer risk, particularly at low doses, future work needs to understand the relevance of radiation quality, dose and dose rate in initiating SIPS and the long term tissue damage and pathological alterations that may arise as a consequence.
Conclusion
The beneficial tumour suppressive role of senescence whereby damage is prevented from being transmitted to daughter cells is well established. What is only recently becoming apparent is that pro-inflammatory factors such as those encompassing the senescence-associated secretory phenotype (SASP) are linked to cellular proliferation, a persistent low grade inflammation, elevated DNA damage foci and transformation of pre-neoplastic cells. Human populations are increasingly being exposed to ionising radiation from a range of diagnostic, treatment and occupational sources highlighting the potential risks of SASP whereby stress-induced premature senescence (SIPS) is initiated instead of apoptosis. The potential effects of this are two-fold; accelerated cellular ageing and an amplification of any detrimental effects produced by SASP. Thus, further research is required to understand the relationship between exposures to radiation, SIPS and how, in turn, SIPS may modify the biological effect of radiation exposure.
References
Campisi J: Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11 (11): S27-S31. 10.1016/S0962-8924(01)02151-1.
Wright WE, Shay JW: Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr Opin Genet Dev. 2001, 11 (1): 98-103. 10.1016/S0959-437X(00)00163-5.
Ben-Porath I, Weinberg RA: The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005, 37 (5): 961-976. 10.1016/j.biocel.2004.10.013.
Davalos AR, Coppe JP, Campisi J, Desprez P: Senescent cells as a source of inflammatory factors for tumour progression. Cancer Metastasis Rev. 2010, 29: 273-283. 10.1007/s10555-010-9220-9.
Kulju KS, Lehman JM: Increased p53 Protein Associated with Aging in Human Diploid Fibroblasts. Exp Cell Res. 1995, 217 (2): 336-345. 10.1006/excr.1995.1095.
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW: Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell. 2003, 113 (6): 703-716. 10.1016/S0092-8674(03)00401-X.
Mehta IS, Figgit M, Clements CS, Kill IR, Bridger JM: Alterations to Nuclear Architecture and Genome Behaviour in Senescent Cells. Ann N Y Acad Sci. 2007, 1100 (1): 250-263. 10.1196/annals.1395.027.
Blagosklonny MV: Cell senescence and hypermitogenic arrest. EMBO Rep. 2003, 4 (4): 358-362. 10.1038/sj.embor.embor806.
Ohtani N, Yamakoshi K, Takahashi A, Hara E: The p16INK4a-RB pathway: molecular link between cellular senescence and tumour supression. J Med Invest. 2004, 51 (3-4): 146-153.
Gil J, Peters G: Regulation of the INK4b-ARF-INK4a tumour supressor locus: all for one and one for all. Nat Rev Mol Cell Biol. 2006, 7 (9): 667-677.
Collins CJ, Sedivy JM: Involvement of the INK4a/Arf gene locus in senescence. Aging Cell. 2003, 2 (3): 145-150. 10.1046/j.1474-9728.2003.00048.x.
Smith JR, Pereira-Smith OM: Replicative Senescence: Implications for in Vivo Aging and Tumor Suppression. Science. 1996, 273 (5271): 63-67. 10.1126/science.273.5271.63.
Serrano M, Collado M: Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010, 10: 51-57. 10.1038/nrc2772.
Coppe J-P, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J: Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the P53 tumour supressor. PLoS Biol. 2008, 6 (12): 2853-2868.
Coppe J-P, Desprez P-Y, Krtolica A, Campisi J: The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol. 2010, 5 (1): 99-118. 10.1146/annurev-pathol-121808-102144.
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW: Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell. 2008, 134 (4): 657-667. 10.1016/j.cell.2008.06.049.
Jun JI, Lau LF: The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010, 12 (7): 676-685. 10.1038/ncb2070.
Evan GI, d' Adda di Fagagna F: Cellular senescence: hot or what?. Curr Opin Genet Dev. 2009, 19 (1): 25-31. 10.1016/j.gde.2008.11.009.
Birchmeier W, Birchmeier C: Epithelial-mesenchymal transitions in development and tumour progression. EXS. 1995, 74: 1-15.
Wang E: Senescent Human Fibroblasts Resist Programmed Cell Death, and Failure to Suppress bcl2 Is Involved. Cancer Res. 1995, 55 (11): 2284-2292.
Campisi J: Cancer, aging and cellular senescence. In Vivo. 2000, 14 (1): 183-188.
Ryu SJ, Oh YS, Park SC: Failure of stress-induced down regulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14 (5): 1020-1028.
Rochette PJ, Brash DE: Progressive apoptosis resistance prior to senescence and control by the anti-apoptotic protein BCL-xL. Mech Ageing Dev. 2008, 129 (4): 207-214. 10.1016/j.mad.2007.12.007.
Chaturvedi V, Bacon P, Bodnar B, Nickoloff BJ: Proliferating cultured human keratinocytes are more susceptible to apoptosis compared with mouse keratinocytes. J Invest Dermatol. 2004, 123 (6): 1200-1203. 10.1111/j.0022-202X.2004.23514.x.
Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Durr P: Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol. 2004, 39 (11-12): 1713-1721. 10.1016/j.exger.2004.05.010.
Labinskyy N, Csiszar A, Orosz Z, Smith K, Rivera A, Buffenstein R, Ungvari Z: Comparison of endothelial function, superoxide and H2O2 production, and vascular oxidative stress resistance between the longest-living rodent, the naked mole rat, and mice. Am J Physiol Heart Circ Physiol. 2006, 291 (6): H2698-H2704. 10.1152/ajpheart.00534.2006.
Shelton DN, Chang E, Whittier PS, Choi D, Funk WD: Microarray analysis of replicative senescence. Curr Biol. 1999, 9 (17): 939-945. 10.1016/S0960-9822(99)80420-5.
Kristnamurthy J: p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006, 443: 443-457.
Dimri GP: A biomarker that identifies senescent human cells in culture and in ageing skin in vivo. Proc Natl Acad Sci USA. 1995, 9363-9367. 92
Herbig U, Jobling WA, Chen BPC, Chen DJ, Sedivy JM: Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, p53, and p21CIP1, but Not p16INK4a. Mol Cell. 2004, 14 (4): 501-513. 10.1016/S1097-2765(04)00256-4.
Sedelnikova OA: Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004, 6 (2): 168-170. 10.1038/ncb1095.
Campisi J: Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell. 2005, 120 (4): 513-522. 10.1016/j.cell.2005.02.003.
Kuilman T, Peeper TS: Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009, 9 (2): 81-34. 10.1038/nrc2560.
Bridger JM, Kill IR, Lichter P: Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Res. 1998, 6: 13-24. 10.1023/A:1009210206855.
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H: Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984, 133 (4): 1710-1715.
Gerdes J: Ki-67 and other proliferation markers useful for immunohistological diagnostic and prognostic evaluations in human malignancies. Semin Cancer Biol. 1990, 1 (3): 199-206.
Kill IR: Localisation of the Ki-67 antigen within the nucleolus. Evidence for a fibrillarin-deficient region of the dense fibrillar component. J Cell Sci. 1996, 109 (6): 1253-1263.
Bridger JM, Boyle S, Kill IR, Bickmore WA: Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol. 2000, 10 (3): 149-152. 10.1016/S0960-9822(00)00312-2.
Mehta IS, Figgitt M, Clements CS, Kill IR, Bridger JM: Alterations to nuclear architecture and genome behaviour in senescent cells. Ann N Y Acad Sci. 2007, 1100: 250-263. 10.1196/annals.1395.027.
Bridger JM, Boyle S, Kill IR, Bickmore WA: Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol. 2000, 10 (3): 149-152. 10.1016/S0960-9822(00)00312-2.
Meaburn KJ, Cabuy E, Bonne G, Levy N, Morris GE, Novelli G, Kill IR, Bridger JM: Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell. 2007, 6 (2): 139-153. 10.1111/j.1474-9726.2007.00270.x.
Hayflick L, Moorhead PS: The serial cultivation of human diploid cell strains. Exp Cell Res. 1961, 25: 585-621. 10.1016/0014-4827(61)90192-6.
Hayflick L: How and why we age. Exp Gerontol. 1998, 33 (7): 639-653. 10.1016/S0531-5565(98)00023-0.
Hayflick L: The cell biology of aging. Clin Geriatr Med. 1985, 1 (2): 15-27.
Shay JW, Wright WE: Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000, 1 (1): 72-76. 10.1038/35036093.
Hayflick L: The cell biology of aging. J Invest Dermatol. 1979, 73 (1): 8-14. 10.1111/1523-1747.ep12532752.
Itahana K, Campisi J, Dimri GP: Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007, 371: 21-31. 10.1007/978-1-59745-361-5_3.
Cristofalo VJ, Sharf BB: Cellular senescence and DNA synthesis: Thymidine incorporation as a measure of population age in human diploid cells. Exp Cell Res. 1973, 76 (2): 419-427. 10.1016/0014-4827(73)90394-7.
Merz GS, Ross JD: Viability of human diploid cells as a function of in vitro age. J Cell Physiol. 1969, 74 (3): 219-222. 10.1002/jcp.1040740302.
Thomas E, Al-Baker E, Dropcova S, Denyer S, Ostad N, Lloyd A, Kill IR, Faragher RGA: Different Kinetics of Senescence in Human Fibroblasts and Peritoneal Mesothelial Cells. Exp Cell Res. 1997, 236 (1): 355-358. 10.1006/excr.1997.3760.
Ng MH, Aminuddin BS, Hamizah S, Lynette C, Mazlyzam AL, Ruszymah BHI: Correlation of donor age and telomerase activity with in vitro cell growth and replicative potential for dermal fibroblasts and keratinocytes. J Tissue Viability. 2009, 18 (4): 109-116. 10.1016/j.jtv.2009.06.003.
Harley CB, Futcher AB, Greider CW: Telomeres shorten during ageing of human fibroblasts. Nature. 1990, 345 (6274): 458-460. 10.1038/345458a0.
Vaziri H, Benchimol S: Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998, 8 (5): 279-282. 10.1016/S0960-9822(98)70109-5.
Artandi SE, DePinho RA: Telomeres and telomerase in cancer. Carcinogenesis. 2010, 31 (1): 9-18. 10.1093/carcin/bgp268.
Makarov VL, Hirose Y, Langmore JP: Long G Tails at Both Ends of Human Chromosomes Suggest a C Strand Degradation Mechanism for Telomere Shortening. Cell. 1997, 88 (5): 657-666. 10.1016/S0092-8674(00)81908-X.
d' Adda di Fagagna F, Reaper PM, Clay-Farrence L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NPJSP: A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003, 426 (6963): 194-198. 10.1038/nature02118.
Shay JW, Bacchetti S: A survey of telomerase activity in human cancer. Eur J Cancer. 1997, 33 (5): 787-791. 10.1016/S0959-8049(97)00062-2.
Hayflick L: Mortalilty and immortality at the cellular level. A review. Biochemistry (Mosc). 1997, 62 (11): 1180-1190.
van Steensel B, Smogorzewska A, de Lange T: TRF2 Protects Human Telomeres from End-to-End Fusions. Cell. 1998, 92 (3): 401-413. 10.1016/S0092-8674(00)80932-0.
Shay JW, Wright WE: Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006, 5 (7): 577-584. 10.1038/nrd2081.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE: Extension of life-span by introduction of telomerase into normal human cells. Science. 1998, 279 (5349): 349-352. 10.1126/science.279.5349.349.
Hayflick L: The illusion of cell immortality. Br J Cancer. 2000, 83 (7): 841-846. 10.1054/bjoc.2000.1296.
Pazolli E, Stewart SA: Senescence: the good the bad and the dysfunctional. Curr Opin Genet Dev. 2008, 18 (1): 42-47. 10.1016/j.gde.2007.12.002.
Chen J: Senescence and functional failure in hematopoietic stem cells. Exp Haematol. 2004, 32 (11): 1025-1032. 10.1016/j.exphem.2004.08.001.
Serrano M: The Tumor Suppressor Protein p16INK4a. Exp Cell Res. 1997, 237 (1): 7-13. 10.1006/excr.1997.3824.
Ramirez RD, Morales CP, Herbert B-S, Rohde JM, Passons C, Shay JW, Wright WE: Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001, 15 (4): 398-403. 10.1101/gad.859201.
Serrano M, Blasco M: Putting the stress on senescence. Curr Opin Cell Biol. 2001, 13 (6): 748-753. 10.1016/S0955-0674(00)00278-7.
Robles SJ, Adami GR: Agents that cause DNA double strand breaks lead to P16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998, 16 (9): 1113-1125. 10.1038/sj.onc.1201862.
Wei S, Wei W, Sedivy JM: Expression of Catalytically Active Telomerase Does Not Prevent Premature Senescence Caused by Overexpression of Oncogenic Ha-Ras in Normal Human Fibroblasts. Cancer Res. 1999, 59 (7): 1539-1543.
Zglinicki TV, Saretzki G, Ladhoff J, Fagagna FdAd, Jackson SP: Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005, 126 (1): 111-117. 10.1016/j.mad.2004.09.034.
Rodier F, Kim S-H, Nijjar T, Yaswen P, Campisi J: Cancer and aging: the importance of telomeres in genome maintenance. In J Biochem Cell Biol. 2005, 37 (5): 977-990. 10.1016/j.biocel.2004.10.012.
Ben-Porath I, Weinberg RA: When cells get stressed: an integrative view of cellular senescence. J Clin Invest. 2004, 113: 8-13.
Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, Bonner WM: Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. 2008, 1 (1): 6-10.1186/1756-8935-1-6.
Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ: A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992, 71 (4): 587-597. 10.1016/0092-8674(92)90593-2.
Dickey JS, Baird BJ, Redon CE, Sokolov MV, Sedelnikova OA, Bonner WM: Intercellular communication of cellular stress monitored by gamma-H2AX induction. Carcinogenesis. 2009, 30 (10): 1686-1695. 10.1093/carcin/bgp192.
Abraham RT: Checkpoint signalling: focusing on 53BP1. Nat Cell Biol. 2002, 4 (12): 277-279. 10.1038/ncb1202-e277.
Lee H, Kwak H-J, Cho I-T, Park SH, Lee C-H: S1219 residue of 53BP1 is phosphorylated by ATM kinase upon DNA damage and required for proper execution of DNA damage response. Biochem Biophys Res Commun. 2009, 378 (1): 32-36. 10.1016/j.bbrc.2008.10.150.
Finkel T, Serrano M, Blasco MA: The common biology of cancer and ageing. Nature. 2007, 448 (7155): 767-774. 10.1038/nature05985.
Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U: Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007, 128 (1): 36-44. 10.1016/j.mad.2006.11.008.
Yap DB, Hsieh JK, Chan FS, Lu X: mdm2: a bridge over the two tumour suppressors, p53 and Rb. Oncogene. 1999, 18 (53): 7681-7689. 10.1038/sj.onc.1202954.
Zhang J, Liu WL, Tang DC, Chen L, Wang M, Pack SD, Zhuang Z, Rodgers GP: Identification and characterization of a novel member of olfactomedin-related protein family, hGC-1, expressed during myeloid lineage development. Gene. 2002, 283 (1-2): 83-93. 10.1016/S0378-1119(01)00763-6.
Webley K, Bond JA, Jones CJ, Blaydes JP, Craig A, Hupp T, Wynford-Thomas D: Posttranslational Modifications of p53 in Replicative Senescence Overlapping but Distinct from Those Induced by DNA Damage. Mol Cell Biol. 2000, 20 (8): 2803-2808. 10.1128/MCB.20.8.2803-2808.2000.
Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M: Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res. 2001, 155 (1 Pt 2): 248-253.
Muthna D, Soukup T, Vavrova J, Mokry J, Cmielova J, Visek B, Jiroutova A, Havelek R, Suchanek J, Filip S, English D, Rezacova M: Irradiation of adult human dental pulp stem cells provokes activation of p53, cell cycle arrest, and senescence but not apoptosis. Stem Cells Dev. 2010, 19 (12): 1855-1862. 10.1089/scd.2009.0449.
Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, Lee JS: PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionising radiation. Cell Death Differ. 2010, 18 (4): 666-677.
Dirac AMG, Bernards R: Reversal of Senescence in Mouse Fibroblasts through Lentiviral Suppression of p53. J Biol Chem. 2003, 278 (14): 11731-11734. 10.1074/jbc.C300023200.
Smogorzewska A, de Lange T: Different telomere damage signalling pathways in human and mouse cells. EMBO. 2002, 21 (16): 4338-4348. 10.1093/emboj/cdf433.
Kaul SC, Yaguchi T, Taira K, Reddel RR, Wadhwa R: Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp Cell Res. 2003, 286 (1): 96-101. 10.1016/S0014-4827(03)00101-0.
Shay JW, Pereira-Smith OM, Wright WE: A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991, 196 (1): 33-39. 10.1016/0014-4827(91)90453-2.
Ansieau S, Bastid J, Doreau A, Morel A-P, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A: Induction of EMT by Twist Proteins as a Collateral Effect of Tumor-Promoting Inactivation of Premature Senescence. Cancer Cell. 2008, 14 (1): 79-89. 10.1016/j.ccr.2008.06.005.
Wei W, Herbig U, Wei S, Dutriaux A, Sedivy JM: Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBO. 2003, 4 (11): 1061-1066. 10.1038/sj.embor.7400001.
Erusalimsky JD, Kurz DJ: Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp Gerontol. 2005, 40 (8-9): 634-642. 10.1016/j.exger.2005.04.010.
Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, Clark IM: The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002, 1 (1): 57-65. 10.1046/j.1474-9728.2002.00008.x.
DePinho RA: The age of cancer. Nature. 2000, 408 (6809): 248-254. 10.1038/35041694.
Beghe C, Balducci L: Biological basis of cancer in the older person. Cancer Treat Res. 2005, 124: 189-221. 10.1007/0-387-23962-6_10.
Wu X, Pandolfi PP: Mouse models for multistep tumorigenesis. Trends Cell Biol. 2001, 11 (11): 2-9. 10.1016/S0962-8924(01)02127-4.
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, Von Zglinicki T: DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009, 8 (3): 311-323. 10.1111/j.1474-9726.2009.00481.x.
Erusalimsky JD: Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 2009, 106 (1): 326-332.
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL: Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007, 449 (7160): 288-291. 10.1038/449288a.
Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, Enver T, Bell JI, Slijepcevic P, Goodnow CC, Jeggo PA, Cornall RJ: DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007, 447 (7145): 686-690. 10.1038/nature05875.
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT: Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006, 443 (7110): 421-426.
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM: A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000, 10 (15): 886-895. 10.1016/S0960-9822(00)00610-2.
Rothkamm K, Lobrich M: Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003, 100 (9): 5057-5062. 10.1073/pnas.0830918100.
Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM: Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 2002, 158 (4): 486-492. 10.1667/0033-7587(2002)158[0486:QDOIID]2.0.CO;2.
Le ON, Rodier F, Fontaine F, Coppe J-P, Campisi J, DeGregori J, Laverdiere C, Kokta V, Haddad E, Beausejour CM: Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell. 2010, 9 (3): 398-409. 10.1111/j.1474-9726.2010.00567.x.
Bakkenist CJ, Drissi R, Wu J, Kastan MB, Dome JS: Disappearance of the Telomere Dysfunction-Induced Stress Response in Fully Senescent Cells. Cancer Res. 2004, 64 (11): 3748-3752. 10.1158/0008-5472.CAN-04-0453.
Baker DJ, Jin F, van Deursen JM: The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle. 2008, 7 (18): 2795-2802. 10.4161/cc.7.18.6687.
Sikora E, Arendt T, Bennett M, Narita M: Impact of cellular senescence signature on ageing research. Ageing Res Rev. 2011, 10 (1): 146-152. 10.1016/j.arr.2010.10.002.
Vijg J, Campisi J: Puzzles, promises and a cure for ageing. Nature. 2008, 454 (7208): 1065-1071. 10.1038/nature07216.
Freund A, Orjalo AV, Desprez P-Y, Campisi J: Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010, 16 (5): 238-246. 10.1016/j.molmed.2010.03.003.
Acosta JC, O'Loghlen A, Banito A, Raguz S, Gil J: Control of senescence by CXCR2 and its ligands. Cell Cycle. 2008, 7 (19): 2956-2959. 10.4161/cc.7.19.6780.
Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR: Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell. 2008, 132 (3): 363-374. 10.1016/j.cell.2007.12.032.
Kortlever RM, Higgins PJ, Bernards R: Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006, 8 (8): 877-884.
Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS: Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell. 2008, 133 (6): 1019-1031. 10.1016/j.cell.2008.03.039.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS: The essence of senescence. Genes Dev. 2010, 24 (22): 2463-2479. 10.1101/gad.1971610.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW: Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007, 445 (7128): 656-660. 10.1038/nature05529.
Parrinello S, Coppe J-P, Krtolica A, Campisi J: Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005, 118 (3): 485-496. 10.1242/jcs.01635.
Coppe J-P, Kauser K, Campisi J, Beausejour CM: Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence. J Biol Chem. 2006, 281 (40): 29568-29574. 10.1074/jbc.M603307200.
Yeager TR, DeVries S, Jarrard DF, Kao C, Nakada SY, Moon TD, Bruskewitz R, Stadler WM, Meisner LF, Gilchrist KW, Newton MA, Waldman FM, Reznikoff CA: Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998, 12 (2): 163-174. 10.1101/gad.12.2.163.
Reddel RR: The role of senescence and immortalization in carcinogenesis. Carcinogenesis. 2000, 21 (3): 477-484. 10.1093/carcin/21.3.477.
Campisi J, d'Adda di Fagagna F: Cellular senescence: when bad things happen to good cells. Nature Reviews Mol Cell Biol. 2007, 8 (9): 729-740. 10.1038/nrm2233.
Krtolica A, Parrinello S, Lockett S, Desprez P-Y, Campisi J: Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci USA. 2001, 98 (21): 12072-12077. 10.1073/pnas.211053698.
Liu D, Hornsby PJ: Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67 (7): 3117-3126. 10.1158/0008-5472.CAN-06-3452.
Zhou F, Onizawa S, Nagai A, Aoshiba K: Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res. 2011, 12 (78): 1-18.
Rodier F, Coppe J-P, Paril CK, Hoeijmakers WAM, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J: Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009, 11 (8): 973-979. 10.1038/ncb1909.
Bassi P, Sacco E: Cancer and aging: The molecular pathways. Urol Oncol. 2009, 27 (6): 620-627. 10.1016/j.urolonc.2009.07.013.
Krtolica A, Ortiz de Solorzano C, Lockett S, Campisi J: Quantification of epithelial cells in coculture with fibroblasts by fluorescence image analysis. Cytometry. 2002, 49 (2): 73-82. 10.1002/cyto.10149.
Suzuki M, Boothman DA: Stress-induced premature senescence (SIPS) - influence of SIPS on radiotherapy. J Radiat Res. 2008, 48: 105-112.
Raffetto JD, Leverkus M, Park HY, Menzoian JO: Synopsis on cellular senescence and apoptosis. J Vasc Surg. 2001, 34: 173-177. 10.1067/mva.2001.115964.
Tsai KK, Stuart J, Chuang YY, Little JB, Yuan ZM: Low-dose radiation-induced senescent stromal fibroblasts render nearby breast cancer cells radioresistant. Radiat Res. 2009, 172 (3): 306-313. 10.1667/RR1764.1.
Mirzayans R, Scott A, Cameron M, Murray D: Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat Res. 2005, 163 (1): 53-62. 10.1667/RR3280.
Tsai KKC, Chuang EY-Y, Little JB, Yuan Z-M: Cellular Mechanisms for Low-Dose Ionizing Radiation-Induced Perturbation of the Breast Tissue Microenvironment. Cancer Res. 2005, 65 (15): 6734-6744. 10.1158/0008-5472.CAN-05-0703.
Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe J-P, Campeau E, Beausejour CM, Kim SH, Davalos AR, Campisi J: DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2010, 124: 68-81.
Azzam EI, de Toledo SM, Little JB: Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha-particle irradiated to nonirradiated cells. Proc Natl Acad Sci USA. 2001, 98 (2): 473-478. 10.1073/pnas.011417098.
Little JB, Nagasawa H, Li GC, Chen DJ: Involvement of the nonhomologous end joining DNA repair pathway in the bystander effect for chromosomal aberrations. Radiat Res. 2003, 159 (2): 262-267. 10.1667/0033-7587(2003)159[0262:IOTNEJ]2.0.CO;2.
Lyng F M, Seymour C B, Mothersill C: Initiation of apoptosis in cells exposed to medium from the progeny of irradiated cells: a possible mechanism for bystander-induced genomic instability?. Radiat Res. 2002, 157 (4): 365-370. 10.1667/0033-7587(2002)157[0365:IOAICE]2.0.CO;2.
Nagasawa H: Effect of dose rate on the survival of irradiated human skin fibroblasts. Radiat Res. 1992, 132 (3): 375-379. 10.2307/3578247.
Mothersill C, Seymour C: Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int J Radiat Biol. 1997, 71 (4): 421-427. 10.1080/095530097144030.
Lorimore SA, Kadhim MA, Pocock DA, Papworth D, Stevens DL, Goodhead DT, Wright EG: Chromosomal instability in the descendants of unirradiated surviving cells after alpha-particle irradiation. Proc Natl Acad Sci USA. 1998, 95 (10): 5730-5733. 10.1073/pnas.95.10.5730.
Sawant SG, Randers-Pehrson G, Metting NF, Hall EJ: Adaptive response and the bystander effect induced by radiation in C3H 10T(1/2) cells in culture. Radiat Res. 2001, 156 (2): 177-180. 10.1667/0033-7587(2001)156[0177:ARATBE]2.0.CO;2.
Belyakov OV, Mitchell SA, Parikh D, Randers-Pehrson G, Marino SA, Amundson SA, Geard CR, Brenner DJ: Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc Natl Acad Sci USA. 2005, 102 (40): 14203-14208. 10.1073/pnas.0505020102.
Sedelnikova OA, Nakamura A, Kovalchuk O, Koturbash I, Mitchell SA, Marino SA, Brenner DJ, Bonner WM: DNA Double-Strand Breaks Form in Bystander Cells after Microbeam Irradiation of Three-dimensional Human Tissue Models. Cancer Res. 2007, 67 (9): 4295-4302. 10.1158/0008-5472.CAN-06-4442.
Coates PJ, Lorimore SA, Wright EG: Damaging and protective cell signalling in the untargeted effects of ionizing radiation. Mutat Res. 2004, 568 (1): 5-20.
Coates PJ, Robinson JI, Lorimore SA, Wright EG: Ongoing activation of p53 pathway responses is a long-term consequence of radiation exposure in vivo and associates with altered macrophage activities. J Pathol. 2008, 214 (5): 610-616. 10.1002/path.2321.
Ilnytskyy Y, Koturbash I, Kovalchuk O: Radiation-induced bystander effects in vivo are epigenetically regulated in a tissue-specific manner. Environ Mol Mutagen. 2009, 50 (2): 105-113. 10.1002/em.20440.
Rzeszowska-Wolny J, Przybyszewski WM, Widel M: Ionizing radiation-induced bystander effects, potential targets for modulation of radiotherapy. Eur J Pharmacol. 2009, 625 (1-3): 156-164. 10.1016/j.ejphar.2009.07.028.
Buonanno M, de Toledo SM, Pain D, Azzam EI: Long-term consequences of radiation-induced bystander effects depend on radiation quality and dose and correlate with oxidative stress. Radiat Res. 2011, 175 (4): 405-415. 10.1667/RR2461.1.
Dieriks B, De Vos WH, Derradji H, Baatout S, Van Oostveldt P: Medium-mediated DNA repair response after ionizing radiation is correlated with the increase of specific cytokines in human fibroblasts. Mutat Res. 2010, 687 (1-2): 40-48.
Milis AJT, Hoyle M, McCue HM, Martini H: Differential expression of metalloproteinases and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp Cell Res. 1992, 201: 373-379. 10.1016/0014-4827(92)90286-H.
West MD, Pereira-Smith OM, Smith JR: Replicative senescence of human skin fibroblasts correlates with a loss of regulation and overexpression of collagenase activity. Exp Cell Res. 1989, 184 (1): 138-147. 10.1016/0014-4827(89)90372-8.
Pasi F, Facoetti A, Nano R: IL-8 and IL-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 2010, 30 (7): 2769-2772.
McBride WH, Pajonk F, Chiang CS, Sun JR: NF-kappa B, cytokines, proteasomes, and low-dose radiation exposure. Mil Med. 2002, 167: 66-67.
Schaue D, McBride WH: Links between Innate Immunity and Normal Tissue Radiobiology. Radiat Res. 2010, 173 (4): 406-417. 10.1667/RR1931.1.
Park H-R, Jo S-K: Lasting effects of an impairment of Th1-like immune response in gamma-irradiated mice: A resemblance between irradiated mice and aged mice. Cell Immunol. 2011, 267 (1): 1-8. 10.1016/j.cellimm.2010.10.004.
Kusunoki Y, Yamaoka M, Kubo Y, Hayashi T, Kasagi F, Douple EB, Nakachi K: T-cell immunosenescence and inflammatory response in atomic bomb survivors. Radiat Res. 2010, 174 (6): 870-876. 10.1667/RR1847.1.
Kusunoki Y, Kyoizumi S, Hirai Y, Suzuki T, Nakashima E, Kodama K, Seyama T: Flow cytometry measurements of subsets of T, B and NK cells in peripheral blood lymphocytes of atomic bomb survivors. Radiat Res. 1998, 150 (2): 227-236. 10.2307/3579858.
Hayashi T, Kusunoki Y, Hakoda M, Morishita Y, Kubo Y, Maki M, Kasagi F, Kodama K, Macphee DG, Kyoizumi S: Radiation dose-dependent increases in inflammatory response markers in A-bomb survivors. Int J Radiat Biol. 2003, 79 (2): 129-136.
Hayashi T, Morishita Y, Kubo Y, Kusunoki Y, Hayashi I, Kasagi F, Hakoda M, Kyoizumi S, Nakachi K: Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am J Med. 2005, 118 (1): 83-86. 10.1016/j.amjmed.2004.06.045.
Kusunoki Y, Yamaoka M, Kasagi F, Hayashi T, MacPhee DG, Kyoizumi S: Long-last changes in the T-cell receptor V beta repertoires of CD4 memory T-cell populations in the peripheral blood of radiation-exposed people. Br J Haematol. 2003, 122 (6): 975-984. 10.1046/j.1365-2141.2003.04520.x.
Nakachi K, Hayashi T, Imai K, Kusunoki Y: Perspectives on cancer immuno-epidemiology. Cancer Science. 2004, 95 (12): 921-929. 10.1111/j.1349-7006.2004.tb03178.x.
Yamada M, Wong FL, Fujiwara S, Akahoshi M, Suzuki G: Noncancer disease incidence in atomic bomb survivors, 1958-1998. Radiat Res. 2004, 161 (6): 622-632. 10.1667/RR3183.
Van der Meeren A, Tourdes F, Gremy O, Grillon G, Abram MC, Poncy JL, Griffiths N: Activation of alveolar macrophages after plutonium oxide inhalation in rats: involvement in the early inflammatory response. Radiat Res. 2008, 170 (5): 591-603. 10.1667/RR1150.1.
Neriishi EN: Persistent subclinical inflammation among A-bomb survivors. Int J Radiat Biol. 2001, 77 (4): 475-482. 10.1080/09553000010024911.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RJS drafted the manuscript. RA participated in its design and helped to draft the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Sabin, R.J., Anderson, R.M. Cellular Senescence - its role in cancer and the response to ionizing radiation. Genome Integrity 2, 7 (2011). https://doi.org/10.1186/2041-9414-2-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2041-9414-2-7