
Abstract
Background
The translocation t(8;21)(q22;q22) is one of the most frequent chromosomal abnormalities associated with acute myeloid leukemia (AML) sub type M2. About 3–5 % of cases with additional chromosomal abnormalities, including structural and numerical ones, are reported to include a complex translocation t(8;21;N).
Case presentation
Here we report a chromosome rearrangement observed in a 19 years-old female diagnosed with AML-M2. When subjected to (molecular) cytogenetic analyses a complex three-way translocation involving chromosomes 8, 17 and 21 was detected, forming not a t(8;21;17) as one would expect. Real time-polymerase chain reaction analysis using 6 AML specific markers showed the presence of RUNX1/RUNX1T1 fusion gene transcripts identical to those found in classical translocation t(8;21) coupled with presence of FLT3-ITD mutation identified by fragment analysis.
Conclusions
The present case highlights importance of complex rearrangements rarely encountered in AML, suggesting that all involved regions harbor critical candidate genes regulating the pathogenesis of AML, leading to novel as well as well-known leukemia associated chromosomal aberrations.
Similar content being viewed by others


Background
It is well known that karyotyping and molecular cytogenetics play very important roles in establishing diagnosis and prognosis in acute myeloid leukemia (AML). Even though banding cytogenetics is limited to detect chromosomal rearrangements at a low resolution, it remains the gold standard for basic genetic analyses in AML. Among cytogenetic rearrangements predictive of most favorable outcomes in AML is the translocation t(8;21)(q22;q22), being detectable in approximately 7–8 % of all AML cases. This translocation results in a fusion gene located on derivative chromosome 8, composed of RUNX1T1 (also known as ETO) on 8q22 and RUNX1 gene (also known as AML1) on 21q22 [1]. RUNX1 gene has also been reported to fuse with different other genes in both de novo and therapy related acute leukemia [2]. The 2016 revision of World Health Organization (WHO) classification for hematolymphoid neoplasms underscores that AML associated with translocation t(8;21) constitutes a distinct AML subtype and is associated with favorable prognosis [3]. However, approximately 3–5 % of AML cases with translocation t(8;21) have variant chromosome partners involved [1]. The clinicopathological features of variant t(8;21) is contradictory discussed in literature and some studies have even reported a favorable outcome for patients with variant translocation t(8;21) [4, 5].
In this study, we elucidate a case with novel variant translocation t(8;21) being observed in context of a complex chromosomal rearrangement. To our knowledge, this is the first case reported here for variant three-way t(8;21) involving short arm of chromosome 17. The study suggests that the detailed characterization of RUNX1T1 via cumulative approaches, like cytogenetics, molecular cytogenetics and real time PCR is necessary to unveil complex chromosome rearrangements.
Case presentation
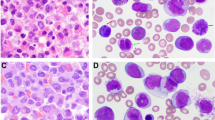
In the year 2018, a 19-years-old female of Asian ethnicity belonging to Jammu and Kashmir, India was presented with bleeding gums for the past 8 months and with high fever for 15 days. On physical examination, spleen was palpable with lymphadenopathy in the cervical and inguinal regions. At admission, blood analyses revealed white blood cell (WBC) count of 64.30 × 109/l with 48 % blasts, and platelet count of 95 × 109/l. A bone marrow (BM) examination characterized hypercellular marrow with 65 % leukemic blasts being positive for CD3, CD 15, CD19, CD34, CD-45 and HLA-DR, a clinical statues being compatible diagnosis of AML (Fig. 1a). The distinct morphologic features of marrow blasts, suggested that the patient has CBF-AML. The patient was initially treated with standard induction chemotherapy, including 7 days of infusion with cytarabine (100 mg/m2) and 3 days of daunorubicin (60 mg/m2). However, complete remission was not achieved after two cycles of chemotherapy. Afterwards she received 12 cycles of low-dose cytarabine and blood transfusions. Following bone marrow biopsy revealed a normal karyotype with ongoing morphologic remission. The patient then received another cycle of cytarabine followed by 2 cycles of 5-azacitidine (75 mg/m2 for 7 doses) before discontinuing any further therapy. As the situation stands with over 2 years of diagnosis, the patient remains in complete remission with no evidence of cytogenetic anomalies.

Morphologic and cytogenetic profile. a Bone marrow smear aspirate show blast. b Partial metaphase spread of G-banded chromosomes. A three way translocation t(8;21;17) at bands 8q22, 21q22 and 17p13 which would be consistent with AML. c FISH with the AML1/ETO dual color, dual-fusion probe. The figures depicts the RUNX1/RUNX1T1 fusion on chromosome 17. d FISH with whole-chromosome painting (WCP) for chromosomes 8, 17 and 21. A multi-color banding specific for chromosome 8 and 17, showing the origin and the breakpoints of each rearrangement
Methods
Banding cytogenetics and fluorescence in situ hybridization (FISH)
A BM aspirate collected during initial diagnosis was studied by GTG (Giemsa using trypsin) banding and molecular cytogenetic analyses. Karyotyping was performed at cytogenetics laboratory, LPL (New Delhi, India) according to ISCN 2016 [6]. Fluorescence in situ hybridization (FISH) using the RUNX1/RUNX1T1 probe (dual-color dual-fusion translocation probe) and subtelomeric probes (ST) 17pter, 17qter and 21qter (Vysis/Abbott Molecular, Des Plaines, IL, USA) were also conducted. In-house whole chromosome painting probes (WCP) for chromosomes 8, 17 and 21 and a multicolor banding probe set for chromosome 8 were used to characterize the nature of the complex rearrangement [7].
DNA extraction
Genomic DNA was extracted from bone marrow samples using QIAsymphony DNA mini kit, Qiagen according to manufacturer’s instruction.
RNA preparation and cDNA synthesis
Total RNA from the leukemic cells was isolated using QIAamp RNA blood Mini Kit (Qiagen). Total RNA (1–5 µg) was converted to cDNA using High Capacity cDNA reverse transcription kit using random primers as per the established protocols [8].
Real time detection of AML ETO fusion product
AML-ETO (or RUNX1-RUNX1T1) was detected by Real Time PCR method. The negative and positive controls were analysed in duplicate using ABI 7500 (Applied Biosystems, USA). AML-ETO transcripts were amplified in 20 µl reaction containing 300 nM of each primer, 100 nM of probe and 5 µl of cDNA. PCR master mix was prepared with Premix Ex TaqTM (Probe qPCR) and custom reaction mix containing 0.5 µl of ROX (ROX Reference Dye II; Takara). The PCR program consisted of an initial denaturation at 95 °C for 10 min followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min and extension. Similarly, endogenous control corresponding to ABL transcripts were amplified in order to compensate for differences in RNA integrity and cDNA synthesis efficiency. The AML-ETO transcripts were amplified using previously published primer probe sequences according to EAC protocol [8]. Standard TaqManTM PCR parameters (ABI 7500) were applied to all AML-ETO and ABL amplifications.
Fragment analysis
FLT3 gene exon 11 was amplified using the Veriti™ 96 well thermal cycler (Applied Biosystems™) in a 20 µl PCR mixture containing 50 ng of extracted DNA, 0.5 µmol/l each of forward primer with FAM (5′ end-labeled with carboxyfluorescein) labelled and unlabeled reverse primer [8]. The PCR program consisted of an initial denaturation at 95 °C for 10 min followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min and extension at 72 °C for 10 min. Afterwards, 2 µl of PCR products was mixed with 9 µl HIDI formamide and 0.25 µl ROX500 internal size standard. The mixtures were denatured at 94 °C for 5 min, chilled at 4 °C and then subjected to ABI PRISM 3500Dx Genetic Analyzer. The results were analyzed using the Gene Mapper software (Applied Biosystems).
Results
GTG banding technique revealed an abnormal putative karyotype of 46,XX,t(8;21;17)(q22;q22;p13) in 20 metaphases analyzed (Fig. 1b). FISH analysis for the RUNX1/RUNX1T1 probe showed 2 orange, 2 green and 1 yellow fusion signals. The latter localized on chromosome 17 indicated a cryptic fusion, RUNX1/RUNX1T1 (co-localization of ETO and AML signals) on the rearranged chromosome 17 and involvement of third chromosome (Fig. 1c). Whole chromosome paint probes for chromosome 8, 17 and 21 confirmed the presence of a complex chromosomal rearrangement, which was further characterized by FISH-probes shown in Fig. 1d as:
46,XX,del(8)(q21q22),der(17)(21qter->21q22::8q22->8q21::17p13->17qter),der(21)t(17;21)(p13;q22).
Further, molecular studies by Real Time PCR confirmed fusion of RUNX1/RUNX1T1 and fragment analysis characterized a FLT3-ITD mutation in 13q12.2 (Fig. 2a, b).

Molecular analysis of leukemia markers. a Confirmation of the AML-ETO fusion was done by Taq Man real-time PCR (Amplification plot). (i) The presented case carried AML-ETO fusion translocation t(8:21), thereby confirming the AML/ETO fusion positivity. (ii) The graph for positive and negative controls are shown along with endogenous ABL gene amplification. The x-axis shows the number of PCR cycles and the y-axis shows the normalized fluorescence intensity. The threshold cycle values are calculated automatically by determining the point at which the fluorescence exceeds a fixed threshold line. b Fragment analysis method based on capillary electrophoresis for FLT3 INDEL detection. (i) Blue peaks correspond to the FLT3 fragment detection and red peaks correspond to the marker size standards. A wild type patient showing the amplicon size expected. (ii) A mutated patient showed an insertion with an additional peak to the wild type
Discussion
The translocation t(8;21)(q22;q22) is one of the most common structural chromosomal aberrations in patients with AML, involving fusion of part of the RUNX1-gene on chromosome 21 to the almost complete open reading frame of the MTG8/ETO gene on chromosome 8 [2]. The patients with AML have favorable prognosis with achievement of complete morphological remission, and promising respond to high dose Ara-C containing regime [9]. Numerical and structural oddities leading to complex chromosomal anomalies (CCA) in t(8;21) cases involve the variant chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or X [10, 11]. But the prognostic impact of patients with complex variant involvement in addition to t(8;21)(q22;q22) is still unexplored. The CCA in the present study is a novel form of complex translocation involving chromosome 17. We attempt to reinforce a potential relevant role of a der(17) chromosome in the pathogenesis of AML M2, which contains in our patient the RUNX1/RUNX1T1 fusion gene: karyotype 46,XX,del(8)(q21q22),der(17)(21qter->21q22::8q22->8q21::17p13 > 17qter),der(21)t(17;21)(p13;q22). FISH analysis using TP53 specific probe showed that there is no loss or involvement of TP53 gene in the patient (Additional file 1: Fig. S1).
In addition, Real Time-PCR analysis revealed the presence of RUNX1/RUNX1T1 fusion transcripts coupled with presence of FLT3-ITD mutation. The FLT3 gene mutations are the most frequent genetic abnormality associated with AML. The rapid identification of these mutations is therefore significant to determine the prognostic impact against the overall genetic background of the leukemic cells. The insertion of tandem duplication into exon 11 and exon 12 in the wild-type FLT-3 produces internal tandem duplication (ITD), which results in constitutive activation of the negative regulation of the juxtamembrane domain [12]. Activating the mutation of D835 within the activation loop of FLT-3 in human hematologic malignancies leads to persistent activation of the FLT-3 receptor. In pediatric patients, FLT3 mutations have been associated with poor prognosis [13]. It has been reported that in adults with CBF (core binding factor) AML the substantial prognostic markers for the outcome are minimal residual disease levels, rather than the FLT3-ITD mutations [14].
Conclusions
The translocation t(8;21)(q22;q22) is a frequent chromosome abnormality seen in AML-M2 which is easily identified by standard chromosome analysis. The present study illustrates a unique complex translocation t(8;21;17), which reinforces that the region on chromosome 17 may harbor a critical candidate gene that could play an important role in the pathogenesis of AML. Additional cases are needed to elucidate its clinical features and prognosis. This finding supports the concept of genetic screening of complex variant translocations in AML patients, thereby envisaging not only the monitoring treatment outcome, but also for clinical manifestation and identification of additional clones that may emerge after the course of treatment.
References
Groupe Francais de Cytogenetique Hematologique. Acute myelogenous leukemia with an 8;21 translocation; a report on 148 cases from the Groupe Francais de Cytogenetique Hematologique. Cancer Genet Cytogenet. 1990;44(22):169–79.
Braekeleer ED, Guilbert ND, Le Bris MJ, Morel F, Férec C, De Braekeleer M. RUNX1-MTG16 fusion gene in acute myeloblastic leukemia with t(16;21)(q24;q22): case report and review of the literature. Cancer Genet Cytogenet. 2008;185(1):47–50.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Hagemeijer A, Garson OM, Kondo K. Fourth international workshop on chromosomes in leukemia, 1982: translocation (8;21)(q22;q22) in acute nonlymphocytic leukemia. Cancer Genet Cytogenet. 1984;11(3):284–7.
Ishii Y, Sashida G, Takaku TI, Sumi M, Nakajima A, Ohyashiki K. Cryptic chromosomal anomaly in a patient with acute myeloid leukemia leading to AML1/ETO fusion with unfavorable prognostic factors. Cancer Genet Cytogenet. 2005;160(1):94–5.
McGowan-Jordan J, Simons A, Schmid M. ISCN 2016. In: An international system for human cytogenomic nomenclature, 1st ed. Basel, Freiburg: Karger; 2016.
Othman MA, Melo JB, Carreira IM, Rincic M, Glaser A, Grygalewicz B, et al. High rates of submicroscopic aberrations in karyotypically normal acute lymphoblastic leukemia. Mol Cytogenet. 2015;8(45):45.
Van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13(1):1901–28.
Shih LY, Huang CF, Wu JH, Wang PN, Lin TL, Dunn PD, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100(7):2387–92.
Chen Z, Wang Y, Wang W, Gong J, Xue Y. All-trans retinoic acid as a single agent induces complete remission in a patient with acute leukemia of M subtype. Chin Med J. 2002;115(1):58–61.
Gmidene A, Sennana H, Frikha R, Elloumi M, Belaaj H, Saad A. An unusual three-way translocation t(21;8;1)(q22;q22;q32) in a case of acute myeloid leukemia (M2). Ann Biol Clin (Paris). 2012;70(2):213–6.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108(12):3654–61.
Jourdan E, Boissel N, Chevret S. Delabesse E, Renneville A, Cornillet P, et al. French AML Intergroup French AML Intergroup, Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121(12):2213–23.
Niparuck P, Limsuwanachot N, Pukiat S, Chantrathammachart P, Rerkamnuaychoke B, Magmuang S, et al. Cytogenetics and FLT3-ITD mutation predict clinical outcomes in non transplant patients with acute myeloid leukemia. Exp Hematol Oncol. 2019;8:3.
Acknowledgements
Authors are grateful to the patient and their family to support the case study. We would also like to thank the management of Dr. Lal Path Labs for providing excellent facilities.
Author information
Authors and Affiliations
Contributions
SRH and LR performed the cytogenetics studies and contributed to data collection. LR and SKB interpreted the data and drafted the manuscript. TL and MAKO performed the FISH with whole-chromosome painting. AT and AS conducted the Real time PCR and fragment analysis. TL, VL and SKB revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Competing interests
The author(s) declare they have no conflict of interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
FISH analysis using TP53 specific probe: a G-banded chromosomes and b FISH with TP53 specific probe indicating no loss or involvement of TP53 gene present on chromosome 17 in the patient (shown with arrows).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Mishra, S.R., Rawal, L., Othman, M.A.K. et al. Complex rearrangement in acute myeloid leukemia M2 with RUNX1/RUNX1T1 fusion involving chromosomes 8, 17 and 21. Mol Cytogenet 14, 28 (2021). https://doi.org/10.1186/s13039-021-00541-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-021-00541-6