
Abstract
Multi-pathway approaches for the treatment of complex polygenic disorders are emerging as alternatives to classical monotarget therapies and microRNAs are of particular interest in that regard. MicroRNA research has come a long way from their initial discovery to the cumulative appreciation of their regulatory potential in healthy and diseased brain. However, systematic interrogation of putative therapeutic or toxic effects of microRNAs in (models of) Alzheimer’s disease is currently missing and fundamental research findings are yet to be translated into clinical applications. Here, we review the literature to summarize the knowledge on microRNA regulation in Alzheimer’s pathophysiology and to critically discuss whether and to what extent these increasing insights can be exploited for the development of microRNA-based therapeutics in the clinic.
Similar content being viewed by others

Alzheimer’s disease complexity: on a quest for network-based approaches to therapy
Alzheimer’s disease (AD) was until recently perceived as a neuron-centric disorder with linearly evolving pathology, initiated by the deposition of β amyloid (Aβ) peptides followed by the accumulation of hyperphosphorylated TAU (pTAU) into neurofibrillary tangles and ultimately leading to a full-spectrum neurodegenerative condition with prominent dementia after a long period of 10-20 years [1,2,3,4]. Novel molecular and genetic insights have challenged the unidirectional linearity of the pathogenic cascade in AD and underscored the significance of intertwined complex cellular pathways, gene networks and feed-forward regulatory loops that may differentially impact distinct pathogenic endophenotypes and cellular phases of the disease [4,5,6,7]. Multi-omics data suggest that the genetic risk of AD functionally translates into molecular networks involved in neuroinflammation, synaptic, lysosomal and phagocytic dysfunction, vascular and metabolic alterations and white matter changes [7,8,9,10,11]. Mapping the mechanistic heterogeneity and multifactorial nature of AD is a key challenge, given the current scarcity of effective disease-modifying monotherapies [12, 13]. Just as treating multiple pathways and molecular networks in other diseases, such as cancer and human immunodeficiency virus-1 has improved outcome [13,14,15], a similar network medicine approach can be envisaged for AD [10, 13, 16, 17]. The rationale for advancing combination therapy in AD was clearly laid out recently during the Alzheimer’s Association Research Roundtable meeting [18]. Combination therapies that can exert multiple effects on disease biology (e.g. anti-amyloid and/or anti-TAU and/or anti-inflammatory agents) have been recently considered and are currently in the drug development pipeline as stand-alone or add-on treatments to those already in the clinic (cholinesterase inhibitors and NMDA receptor antagonists) for AD (for a systematic review of ongoing AD clinical trials, see [13, 19, 20]). These combination schemes can either involve the multi-modal, combinatorial administration of more than one therapeutic agents (e.g. ALZT-OPT1: anti-amyloid & anti-inflammatory treatment, NCT02547818; ANAVEX2-73: anti-amyloid & anti-TAU & anti-inflammatory, NCT03790709) or the use of multifunctional molecules (e.g. Rasagiline: neuroprotective & anti-amyloid, NCT02359552).
MicroRNA-targeted therapeutics are particularly suited for such multi-targeting purposes, as they can typically ‘hit’ multiple sensitive nodes of various molecular cascades deregulated in disease conditions. MicroRNAs (miRNAs) are small, ~22nt-long, non-protein coding RNAs that induce posttranscriptional gene silencing by binding their complementary messenger RNA (mRNA) targets and inhibiting translation and/or inducing mRNA degradation [21, 22]. Under physiological conditions, miRNA-dependent gene regulation acts to ensure precise protein output and minimal protein expression noise [23, 24]. miRNAs are also able to sense and rapidly respond to the presence of stressors in their microenvironment, providing molecular robustness to cellular stress and restoring tissue homeostasis [25,26,27]. While miRNAs may only modestly repress individual targets, their regulatory power relies on the context-specific, synergistic cross-talk between subsets of miRNAs over prioritized sets of transcripts [28,29,30,31,32]. Under pathological conditions, as it is the case in cancer tissue or in neurodegeneration, this network-based mode of gene regulation converges onto distinct molecular pathways, which potently drive disease phenotypes [30, 33, 34]. Yet, profiling the impact of miRNA-based multi-targeting therapeutic strategies in AD with respect to efficacy and toxicity remains a daunting task, which is reflected by the current sparsity of miRNA therapeutics undergoing clinical trials in AD, as we discuss below.
Functional pleiotropy of microRNAs in the central nervous system: one molecule, several pathways
miRNAs in the central nervous system (CNS) control gene expression in various cell types and in a highly regulated time-, space-, and neuronal activity- dependent manner [35,36,37,38,39]. Emerging evidence points towards a complex multicellular gene silencing repertoire for several of the studied miRNAs in the brain (for a comprehensive review refer to [40]). The miRNA transport across different brain-resident cell types along with the periphery-CNS miRNA interchange further contribute to the intercellular signaling crosstalk [41, 42].
A prototype of the intricate multicellular miRNA functions in CNS are ‘NeurimmiRs’, a term collectively referring to miRNAs acting at the interface between the neuronal and the immune systems [43]. miR-124, miR-132 and miR-146 exemplify this sort of miRNA regulatory pleiotropy with direct implications in neurodegeneration [30, 43]. miR-124 is a brain-specific miRNA regulating neurogenesis [44, 45], synaptic plasticity [46] and behavior [47]. In addition, its immunomodulatory role is exerted via the targeting of the transcription factor CCAAT/enhancer-binding protein-α, which converts microglia from an activated and inflammatory state to a quiescent phenotype [48]. Microglia endogenously express miRNA-124 and can also receive neuronally-derived exosomal miR-124 [49]. miR-124-mediated paracrine signaling between SH-SY5Y neuroblastoma cells overexpressing an AD mutation and microglia in culture induces a shift of microglial phenotype from an initially proinflammatory to a more regenerative state, re-establishing homeostasis [50].
The brain-enriched miR-132 is a key regulator of neuronal morphogenesis [51,52,53,54,55], synaptic plasticity [56,57,58,59], neuronal survival [60, 61] and cognition [62,63,64,65,66]. We recently reported a novel role for miR-132 in restoring hippocampal neurogenesis in the adult AD mouse brain by regulating several stages of the neurogenic process, including proliferation, differentiation, maturation and providing neurotrophic and neuroprotective support [67]. Emerging evidence suggests additional roles for miR-132 in immunomodulation, although extensive in vivo documentation in CNS is currently missing. More specifically, miR-132 exerts significant anti-inflammatory effects in monocytes and macrophages in vitro [63, 68]. In addition, miR-132 overexpression in the U251 human astrocytic cell line targets interleukin-1 receptor-associated kinase, resulting in decreased secretion of pro-inflammatory cytokines IL-1β and IL-6 [69]. In vivo, miR-132 targets acetylcholinesterase, thereby increasing the levels of acetylcholine, a key suppressor of pro-inflammatory cytokines [70]. Moreover, astrocytic and microglial miR-132 levels increase in epileptic rat and human brain as a protective response, while miR-132 transfection in human primary astrocytes represses expression of pro-inflammatory and pro-epileptogenic genes [71]. Although these observations support a role of miR-132 in the fine-tuning of inflammation, evidence of direct regulation of the innate immune response by miR-132 in the brain is currently lacking. Attempts to profile the baseline miR-132 levels in microglia and astrocytes in vivo and their response to AD onset and progression can provide an initial basis for further mechanistic assessment.
miR-146 is abundantly expressed in microglia and to a lesser extent in neurons and astrocytes. miR-146 represses the nuclear factor kappa-B (NF-κB) signaling pathway in several cell lines in vitro by directly targeting IRAK1 and TRAF6 [72,73,74,75]. miR-146 knockout mice fail to induce effective microglia-mediated phagocytosis in response to lipopolysaccharide pro-inflammatory stimulation, suggesting that miR-146 is essential for the microglial response to inflammation [76]. In addition, miR-146 induces pro-neurogenic effects, promoting neuronal differentiation and neuronal lineage commitment of human neural stem cells in vitro [77], while miR-146 knockout in mouse hippocampus impairs radial glia-like cell differentiation and causes severe memory impairment [78]. Interestingly, miR-146 was also shown to exert inflammation-mediated synaptic alterations in a non-cell autonomous manner via the functional intercellular crosstalk between neurons and microglia. More specifically, inflammatory microglia in culture shuttle miR-146 in extracellular vesicles to neurons, where it induces synaptic loss through targeting of Nlg and Syt1 [79]. Whether the immunomodulatory effects of miR-146 also occur in the brain under physiological or pathological conditions and if they may additionally mediate its impact on memory formation has not been addressed yet.
Evidently, considering NeurimmiRs to tackle both neuronal and immune aspects of AD pathogenesis is an attractive hypothesis. However, most of the studies discussed here do not concomitantly explore miRNA effects in both immune and neuronal functions in the brain. Moreover, current evidence is primarily based on ‘one miRNA-one target’ experimental design, which precludes the unbiased systematic mapping of both disease- and tolerability-relevant effects. In addition, miRNA effect sizes are often relatively small (20-30%) when assaying single targets. However, the network-based regulatory potential of miRNAs can be addressed in unbiased multi-target or genome-wide studies, where the global impact on multiple targets acting within one pathway or endophenotype is considered as a whole [32, 80, 81]. This novel approach to studying miRNAs elucidates the functional significance of miRNAs as network regulators, however, has not yet been widely applied in CNS systems.
MicroRNAs in key AD pathways: targeting endophenotype complexity
miRNA profiles in the brain of AD patients are altered compared to healthy controls, often in a stage- and/or region- specific manner [82,83,84,85,86,87]. How these alterations impact disease onset and progression and whether they act as cause or effect along the disease trajectory remains unclear. Nevertheless, specific early miRNA aberrations along AD progression in human brain, indicate that disruption of miRNA homeostasis may act as a (co-)driver of certain pathological cascades [84]. Indeed, miRNAs have been shown to be responsive to a wide range of neuropathological processes, including oxidative stress, neuroinflammation, protein aggregation, and alterations in neuronal connectivity and plasticity, suggesting that miRNA-regulated molecular pathways may be interfering with pathology early on in the progression of neurological disorders [54, 68, 69, 88,89,90,91,92,93,94].
Of note, evidence of genetic association between miRNAs and AD is scarce and most of the pertinent studies are underpowered. Only a handful of single nucleotide polymorphisms have been identified to date in precursor miRNA sequences in genome-wide association studies of AD patient cohorts, of which rs2291418 in the miR-1229 precursor is one of the best studied examples [95,96,97]. miR-1229 targets SORL1, an AD risk gene involved in amyloid precursor protein (APP) processing. Although potentially very interesting, this association has not yet been functionally validated. Nevertheless, strong correlative evidence links miRNAs to several key AD endophenotypes in human brain. One of the best studied miRNAs in this series is miR-132, which is among the most consistently downregulated miRNAs in AD [84, 86, 98]. We and others have previously shown that miR-132 levels are anti-correlated with deposition of both intraneuronal hyperphosphorylated TAU and extracellular amyloid aggregation in the prefrontal cortex of human AD brain [33, 84, 98]. Interestingly, miR-132 expression variation explains 6.7% of the observed variance in histopathological AD endophenotypes in the Religious Order Study and the Rush Memory Aging Project. This actually outperforms the 6.1% of variance explained in the same patient cohorts by Apolipoprotein E4 (APOE4), the largest risk factor for AD [98]. In addition, miR-132 is among the core set of variables that explain the contribution of an individual’s polygenic risk score to cognitive impairment in AD [99]. This evidence suggests a putative functional link between miR-132 and amyloidosis/TAU pathology/dementia in human AD brain and offers a direct proof-of-principle as to how one miRNA may concomitantly regulate multiple pathways associated with AD pathophysiology.
miR-203 provides another interesting example of miRNAs potentially playing multilayered roles in AD [32]. miR-203 was identified as a hub regulator of the neuronal/synaptic- and microglial/inflammatory modules in the TauP301S tauopathy mouse model. Similar modules were also mapped in other mouse models of frontotemporal dementia (FTD) and in human postmortem FTD brain. While a set of targets involved in apoptotic cell death were shown to be regulated by miR-203, miR-203 per se has not been previously reported among the deregulated miRNAs in neurodegeneration. Hence, even though the notion of network regulation is a pivotal one, the conditions under which miR-203 may actively and potently regulate neuronal death still remain to be addressed.
Recently, a meta-analysis study identified a signature of 10 commonly deregulated miRNAs with predicted or previously validated neuroimmune functions, including miR-9-5p, miR-21-5p, the miR-29 family, miR-132-3p, miR-124-3p, miR-146a-5p, miR-155-5p, and miR-223-3p, across several neurodegenerative disorders ranging from AD to multiple sclerosis and prionopathies [30]. These correlative observations strongly suggest that miRNAs may represent prime candidates for intervention as common downstream regulators of functionally diverse molecular pathways in AD. Yet, while some of these miRNAs have been previously studied and are discussed here, experimental testing of newly identified correlations in the appropriate model systems is critical for mechanistic assessment prior to any consideration in drug development pipelines.
One of the main arguments used in favor of a putatively central role for miRNAs in AD, is that many of the molecules with central roles in disease pathogenesis, such as APP [100,101,102,103,104,105,106,107,108,109,110,111,112], β-secretase (BACE1) [104, 111, 113,114,115,116,117,118,119,120,121,122,123,124], APOE [125,126,127] and TAU [33, 61, 64, 128,129,130], are direct or indirect miRNA targets. We have previously demonstrated that miR-132 can bimodally regulate both amyloidosis and Tau phosphorylation via regulating one single target, ITPKB, acting upstream of both cascades [33]. The functional convergence of miR-132 regulation on repressing Tau pathology has been demonstrated via multiple additional targets acting on Tau phosphorylation, splicing or even Tau mRNA itself [64, 84, 131, 132]. Along with a role in suppressing neuronal apoptosis via direct regulation of PTEN, FOXO3a and P300, the multi-pathway regulatory repertoire of miR-132 in AD is among the most extensively studied and validated [60].
Of note, functional evidence for beneficial roles of miRNAs in AD suggests that modifying their expression might counteract pathology. Intracerebral infusion of lentiviral constructs expressing miR-188-5p in 5xFAD mice restores dendritic spine density and memory deficits in contextual fear conditioning and T-maze tests [93], while restoring miR-132 levels through direct delivery of miR-132 synthetic mimics into the brain of different AD mouse models (APP/PS1, 3xTg AD and APPNL-G-F mice), ameliorates Aβ40-42 levels, Tau phosphorylation, deficits in adult hippocampal neurogenesis and cognition [33, 64, 67]. Additionally, stereotactic injections of lentiviral constructs expressing miR-338-5p in the dentate gyrus of 5xFAD mice, decreased BACE1 protein levels, Aβ42 and neuroinflammation, and rescued spatial memory deficits in the Morris water maze test [133]. More recently, non-invasive, nose-to-brain delivery of miR-146 synthetic antisense oligonucleotides in APP/PS1 mice, was shown to ameliorate amyloid and Tau pathologies, neuroinflammation and memory deficiency [134].
Taken together, these observations provide proof-of-concept that modulating miRNA levels in AD brain may be considered therapeutically, as it could concomitantly modify multiple aspects of the pathology (Figure 1) and potentially lead to amelioration of memory deficits. Context-specificity is a significant aspect of miRNA biology, hence, results from studies using only in vitro systems or a single transgenic mouse line may not be easily extrapolated to describe widespread miRNA-mediated effects in AD. Along the same lines, increasing the levels of miRNA in a tissue or cell population, where it was previously absent or lowly expressed, may elicit ‘de novo’ target repression with unpredictable effects. In addition, in order to enable a therapeutic effect, careful dose titration to determine the required therapeutic window must be considered. We and others have shown that ‘supraphysiological’ (>3-fold) miR-132 levels following overexpression, may exert negative effects on memory [66, 67]. This narrow dose window is in line with the role of miRNAs as fine-tuners of their target’s protein levels around a physiological set-point, indicating that both too high and too low levels can be detrimental [94]. Yet, this aspect of manipulating miRNA levels in preclinical mouse models is often overlooked in the literature.

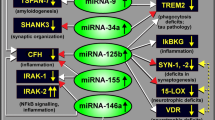
microRNAs regulate multiple AD-related cellular and molecular pathways. In healthy brain, miRNAs can maintain homeostasis through negative regulation of targets in neuronal and immune pathways. In Alzheimer’s brain, miRNAs are dysregulated impacting cellular and molecular cascades involved in AD endophenotypes.
MicroRNAs as biomarkers of Alzheimer’s disease
The remarkable stability of miRNAs in the extracellular environment and hence, in bodily fluids, together with the availability of sensitive methods for their detection and quantitation, has led to circulating miRNAs being widely employed as biomarkers for various human disorders [135]. Several miRNA-based diagnostic tests are already used in the clinic, mostly assessing panels of miRNAs, in different types of biological samples and conditions ranging from liver injury to cardiovascular disease and cancer [136,137,138].
In the AD field, currently available biomarkers include Aβ40-42, total TAU and phosphorylated TAU levels in the cerebrospinal fluid (CSF), along with brain imaging, such as positron emission tomography and structural magnetic resonance imaging scans to visualize Aβ/TAU deposits and atrophy, respectively [139,140,141]. More recently, also blood and plasma biomarkers have emerged as promising diagnostic tools, such as Aβ40/42 levels, plasma TAU phosphorylation at residue 181 (pTAU181) and plasma neurofilament light chain levels [142,143,144,145,146,147,148]. The use of peripheral miRNA levels as AD diagnostics is currently at relatively advanced stages of clinical development. miRNA profiles in CSF, blood, plasma and serum have been measured and compared between AD patients and healthy controls [83, 149,150,151,152,153,154,155]. Leidinger and colleagues have proposed a panel of 12 miRNAs in blood that discriminates AD from other CNS diseases and allows to distinguish between AD and MCI patients with an accuracy of 76%, which comes close to the 80% accuracy of blood pTau181 [144, 156]. The 12-miRNA signature could differentiate healthy from AD individuals with an accuracy of 93%, a specificity of 95% and a sensitivity of 92% [156]. These data were thereafter confirmed in independent patient cohorts and validated using machine learning approaches [157, 158], however, they have not been implemented in the clinical testing pipeline yet. Recently, a combination of 3 miRNAs and 3 Piwi-interacting RNAs (piRNA) isolated from CSF-circulating exosomes were found significantly altered in AD patients compared to controls. By adding the information of this miRNA/piRNA signature to CSF pTAU and Aβ40/42 ratio values, the classification accuracy for AD patients increased from 83% to 98% [159], suggesting that, possibly in combination with other existing biomarkers, the implementation of such signatures could contribute to stable, high-performance diagnostic tests for AD. Capitalizing on these observations, multiple products are currently in the pipeline by several companies [136]. CognimiR, a diagnostic panel of 24 brain-enriched and inflammation-associated miRNAs in blood, for early detection of mild cognitive impaired (MCI) and asymptomatic AD patients with 90% discrimination accuracy from controls (which compares with and outperforms the specificity levels achieved by plasma Aβ40-42 assessment [160]), was recently branded by DiamiR and is currently in phase I of clinical development [136]. Another miRNA-based diagnostic test for AD is being developed by Hummingbird Diagnostics, however, clinical validation has not been initiated yet [136]. Given that miRNAs in blood are easier to detect than those in CSF [161, 162], they have been found to change more consistently across independent cohorts of AD patients than CSF miRNAs [154], and there is a scarcity of standardized miRNA isolation protocols from CSF samples [161], it becomes obvious that the choice of biological fluid can be critical for the clinical development of miRNA diagnostics. Along with their minimal invasiveness, plasma- or serum-based assays may therefore be of advantage. In addition, elevated levels of miR-206 have been measured in the nasal mucosa of AD patients compared to healthy controls [163], while increased miR-200b-5p abundance was detected in tear fluid from AD patients [164]. Once validated in larger populations, these latter findings could represent novel promising non-invasive strategies for early diagnosis and further suggest that miRNAs could be harnessed for biomarker-guided drug development in AD. Future high-powered cohort studies implementing diverse patient cohorts will be required to further confirm these observations.
Bringing microRNA therapeutics into the clinical practice: hurdles and challenges
Federal Drug Agency (FDA)-approved drugs for AD include cholinesterase inhibitors, memantine and just very recently, also the first disease-modifying drug, the monoclonal antibody aducanumab, which has been shown to lower Aβ levels [20, 165, 166]. Along with a long list of other agents currently undergoing clinical trials, these approaches all exhibit distinct differences to miRNA-based strategies (Table 1). The current gap in miRNA-related clinical studies as compared to other therapeutic strategies in AD, is indicative of a set of distinct limitations, which the development of miRNA-based therapeutics is currently facing.
Even though miRNAs mechanistically differ from small-interfering RNAs (siRNAs) with respect to their multi-targeting potential (Table 2), both classes of small noncoding RNAs are short duplex RNA molecules exerting gene silencing effects at the post-transcriptional level and facing a similar set of barriers for clinical development: delivery issues, poor in vivo stability, off-target effects and safety [167].
miRNA-based therapeutics mainly comprise synthetic miRNAs used to restore endogenous miRNA levels (e.g. miRNA mimics) or antisense inhibitor oligonucleotides aimed at reducing the functionally available endogenous miRNA (e.g. antamiRs or antagomiRs). While several miRNA-based therapeutics have undergone preclinical testing and have now entered clinical trials for the treatment of a wide variety of pathologies, ranging from cardiac failure to several types of cancer [80, 81, 168,169,170,171,172,173,174,175], none of them is yet targeted towards AD.
Of note, the multi-targeting mode of miRNA action asks for extensive preclinical assessment. A single miRNA can bind several transcripts [23, 176, 177], while one mRNA can be targeted by multiple miRNAs simultaneously. In addition, a miRNA can target mRNAs that exert opposing effects within the same molecular pathway [178]. The relative endogenous abundance of a given miRNA, of other ‘competing’ miRNAs and of their common targets in a particular cell also impacts the strength of the effect of a particular miRNA on its cognate mRNA [21, 179, 180]. Yet, this complex regulatory repertoire may not merely involve ‘random’ transcripts: miRNA targets are often identified within the same molecular pathway(s) and, hence, need to be regulated in a highly coordinated manner [61, 181,182,183,184,185]. Reliable miRNA target identification is a laborious task, further complicated by the high false positive rates of most of the target prediction algorithms [186, 187]. While each miRNA target validation technology bares its own strengths and limitations (systematically reviewed in [188]), the optimization of genome editing tools, such as CRISPR/Cas9 systems, to edit miRNA binding sites in vivo holds great promise for direct and more precise functional target validation in the future [189, 190]. Systematic mapping of the genome-wide and cell (type)-specific miRNA targetomes and the affected biological cascades is an absolute requirement for clinical application, and, hence, proteomics and single-cell transcriptomics in vivo or high-throughput targetome profiling in human cell lines in vitro, are key prior to launching miRNA-based approaches in the clinical development pipeline [191, 192]. Recent studies performing extensive preclinical evaluation of miRNAs, have deployed a so-called ‘miRNA pharmacodynamic signature’ to assess dose-dependent target engagement in vivo [80, 81]. Using a panel of previously identified, robustly affected miRNA targets was shown to be an effective measure of miRNA activity in animal models, which considers the functional significance of network regulation by miRNAs. For human-validated targets, such approaches could also be considered in the clinic, if the target panel can be reliably measured in clinically relevant biological fluids, such as blood or CSF. Nevertheless, while this strategy would assess engagement of relevant targets, it would still not address genome-wide disease-relevant and non-disease-relevant effects.
In a recent small-cohort, phase I clinical trial with CDR132L, an anti-miR-132 oligonucleotide for the treatment of heart failure, besides the primary outcomes of safety, target engagement was also considered as secondary outcome [193]. Even though due to ethical constraints this information was derived from prior preclinical studies in large animals [80, 194], it still enabled the prediction of the effective human dose based on plasma miR-132 levels after administration of CDR132L [193].
On- and off-target side effects are currently a major hurdle for miRNA-therapeutics to overcome in order to transit to the clinic. The usage of miRNA therapeutics in combination with other drugs or miRNAs that target the same gene networks can reduce dose and eliminate on-target toxicity [195, 196]. Conversely, off-target toxicity can occur through activation of the immune system by administered double-stranded RNA molecules and the often highly positively charged delivery vehicles [197, 198]. In 2016, a phase I clinical trial testing a liposomal miR-34a mimic (MRX34) for the treatment of advanced solid tumors was halted due to the adverse immune reactions reported in a subset of patients, even though anti-tumor effects were observed [199]. While it is unclear whether the toxicity was caused by the vehicle or the oligonucleotide per se, these results underscore the importance of careful dosage titration and preclinical safety assessment with well-designed, standardized toxicity studies in multiple preclinical species.
Efficient and targeted delivery and cellular uptake of RNA therapeutics will be key in eliminating unwanted side effects, as it can significantly limit systemic exposure and high dose requirements. Of note, the intrinsic properties of miRNAs, including their hydrophilic nature, negative charge and relatively high molecular weight, render them poorly permeable across biological membranes [167]. Recent technological advances and insights from the cancer field have paved the way for the development of several distinct DNA and RNA delivery vehicles for transport to the brain, either by crossing the blood-brain-barrier (BBB) or by direct delivery into the CNS. Viral approaches, and in particular recombinant AAV-based systems are a potent CNS delivery platform, which can target specific tissues or cell types (making use of different capsids and promotors) and are safe in the clinic demonstrating durable transgene expression [200]. Intrathecal delivery of AAVs containing an artificial miRNA suppressing superoxide dismutase 1 (SOD1) (a genetic cause of familial amyotrophic lateral sclerosis (ALS)), was shown to be efficient in SOD1 gene silencing in nonhuman primates, yet it did induce peripheral immune responses [201]. Recently, a single-dose gene replacement therapy (Zolgensma) for spinal muscular atrophy (SMA) based on an AAV9 delivery system that can cross the BBB, was approved by the FDA [202]. However, long-term safety studies and improvement of the efficacy of AAV-mediated gene delivery are required, as high doses (1014 viral genomes/kg) of AAV9 are necessary to transduce around 20% of the target cell population in the brain, leading to numerous adverse side-effects in animal models and in humans, such as neutralizing antibodies, elevated serum aminotransferase levels and liver toxicity [202,203,204,205,206,207].
Besides viral gene delivery, non-viral strategies are also emerging for brain targeting of small RNA molecules. Most of the existing knowledge here stems again from siRNA applications, although some preliminary evidence on miRNAs has been reported. miRNA encapsulation into nanoparticles could offer novel opportunities for controlled and putatively cell type-specific miRNA delivery into the brain for therapeutic purposes, either by systemic or direct CNS administration [208, 209]. Advantages of nanoparticle-mediated delivery include increased molecular stability of the RNA payload, protection against endogenous nucleases and the possibility for cell type-specific targeting. Lipid nanoparticles are the most intensively investigated and were the ones approved by the FDA in 2018 for siRNA delivery to the liver [210]. We recently reported the feasibility of nanoparticle-encapsulated miR-132 mimic delivery via intranasal administration into the brain of an AD mouse model [211]. However, assessment of the expression levels of a limited set of predicted targets did not yield consistent results. Systematic functional validation to address broad target engagement in vivo is still pending. Key remaining challenges include the widely reported nanoparticle immunogenicity, nonspecific uptake, rapid clearance by macrophages and peripheral toxicity as a result of inefficient targeted delivery [208, 212].
The use of administration routes, such as intracerebroventricular, intrathecal or intranasal infusion, for direct delivery into the CNS, can enable the functional delivery of ‘naked’ oligonucleotides (in the absence of any delivery vehicle) both in mice and humans [134, 213,214,215,216]. Chemical modification of the RNA backbone is often required in this case, to augment stability and half-life. A very recent report by the group of Don Cleveland, demonstrated a high efficiency versus toxicity ratio for a novel modified, naked antisense oligonucleotide (ASO) intracerebroventricularly administered in mice, targeting polypyrimidine tract binding protein 1 and inducing the generation of new neurons in the aged mouse dentate gyrus [217]. Evidently, targeted delivery of oligonucleotides is key in order to improve efficacy and safety. For CNS applications, antibody- or peptide- oligonucleotide conjugates may offer a promising strategy for targeted delivery of RNA-based therapeutics across BBB and to specific brain cell types [218,219,220,221,222]. While similar strategies are chemically applicable to a wide range of oligonucleotides, this knowledge emerges once again from nucleotides other than miRNAs, like siRNAs.
Poor stability once inside the cellular environment is another major obstacle towards successful clinical development [191, 192, 223]. Naked, unmodified RNAs are degraded rapidly after administration by the abundant cellular and serum nucleases, resulting in a short half-life in vivo [167]. RNA molecules are additionally highly reactive, and hence unstable, due to the presence of the 2’—OH chemical group in the ribose sugar [136]. Chemically modified miRNA mimics or inhibitor oligonucleotides are routinely manufactured commercially and employed to improve stability and binding affinity. In particular, the addition of phosphorothioate nucleotides and methyl groups to the RNA backbone shows a significant increase in protection against nucleases and binding affinity in vivo [192, 223, 224]. Moreover, introducing locked-nucleic acid (LNA) modified nucleotides into anti-miRNA oligonucleotides greatly improves their miRNA-targeting efficacy and has been proven safe in non-human primates this far [225,226,227,228].
The knowledge gained over the last ten years on miRNA biology and synthetic oligonucleotide technologies only emphasizes the need for systematic preclinical efficacy and safety assessment of miRNA-based AD therapeutics in disease-relevant models. While miRNA-based strategies may offer certain advantages over other therapeutic approaches in AD (Table 1), the existing limitations of this type of therapeutics remain to be critically addressed.
RNA-based therapeutics in the clinic
RNA-based therapeutics are emerging as a potent new class of drugs in various clinical fields, including neurodegeneration. While extremely promising, these efforts do not yet include miRNA-based strategies targeted against AD pathology.
The first siRNA drug (Patisiran) was approved by the FDA in 2018 for the treatment of hereditary transthyretin-mediated amyloidosis. The siRNA is encapsulated in lipid nanoparticles directing it to the liver after systemic administration, where it binds the mRNA of transthyretin and prevents the production of the mutant protein [229,230,231]. Groundbreaking advances recently led to the first FDA-approved drug for SMA, a rare neuromuscular disorder. Treatment for SMA was approved at the end of 2016 using a 2′-O-2-methoxyethyl phosphorothioate-modified ASO that is administered through intrathecal infusion. The ASO interferes with the splicing of SMN2 mRNA, thereby increasing the amount of functional SMN2 protein, which can compensate for the loss of SMN1 [213, 214]. More recently, another RNA-based therapy (Zolgensma) was approved by the FDA for SMA, wherein a functional copy of SMN1 gene is delivered using an AAV9 delivery system, enabling durable SMN1 expression following a single intravenous injection [202]. In AD, the most advanced RNA-based therapeutic is based on translational inhibition of TAU mRNA also using an ASO-based approach (NCT03186989, BIIB080, IONIS-MAPTRX), and is currently at clinical trial phase I/II. Most notably, over the last year the world has witnessed the first mRNA vaccines to enter the clinic, developed my Moderna and Pfizer against the SARS-CoV-2 virus [232, 233].
The miRNA-targeted pharmaceutical market is less advanced with numerous clinical trials currently underway (CDR132L, Cardior Pharmaceuticals GmbH; RG-012, Genzyme/Sanofi/Regulus Therapeutics; MRG-106, MRG-110, MRG-201, miRagen/Viridian Therapeutics; TargomiRs [172, 234,235,236]), yet none of them in AD. Miravirsen, an LNA-modified inhibitor of miR-122 with modified phosphorothioate backbone, to treat hepatitis C infection in the liver, was the first anti-miRNA drug to enter the clinic [225, 228]. Miravirsen naturally accumulates in the liver (as a modified small RNA molecule) and therefore does not require a special delivery strategy. This facilitates its application in hepatitis patients but presents a major issue for brain delivery of similar formulations. Following successful initial clinical trials [237, 238], additional phase II clinical trials were performed by Roche. Even though the viral load did decrease in patients, further clinical development was ended due to undisclosed reasons [239]. Similarly, the clinical development of another anti-miR-122 therapy (RG-101) for the treatment of hepatitis C infection, developed by Regulus Therapeutics, was halted in 2017 due to high levels of bilirubin found in the blood of some participants of the phase II clinical trial [240]. Yet, steps towards the right direction are made also for brain diseases. Regulus Therapeutics announced in 2019 the successful termination of preclinical development of RGLS5579, an ASO to inhibit miR-10b, in the treatment of a highly aggressive brain cancer, glioblastoma multiforme. In combination with temozolomide, the inhibition of miR-10b had a synergistic effect and increased the median survival of a glioblastoma chimeric mouse model from 27% to 159% [241]. Similarly, MRG-107, a miR-155 inhibitor has been preclinically validated by miRagen Therapeutics against ALS [242]. Preparations for a phase I clinical trial are ongoing for both RGLS5579 and MRG-107, however, no information on the delivery strategy has been disclosed to date. This far, the preliminary outcomes of the clinical testing of miRNA therapeutics largely suggest that the delivered oligonucleotides can reach their target sites and can also exert functional effects. However, the suspension or discontinuation of some of these clinical trials (e.g. MRX34, Miravirsen, RG-101), calls attention to putative miRNA-specific risks, which may, at least partially, be explained by issues related to multi-targeting-related toxicity. Once again, these observations emphasize the unquestionable necessity for systematic preclinical targetome profiling and for deep understanding of the mechanistic action of miRNA-based drugs, akin to the process followed in the case of any other modality with polypharmacological potential. Interestingly, multitargeted therapeutics modulating gene expression, such as drugs targeting nuclear receptors/transcription factors or epigenetic enzymes, are already used in the clinic and can provide valuable knowledge in that regard [243, 244]. Notably, while the drop-out rate of miRNA therapeutics in clinical trials does not dramatically differ from that of siRNA-based drugs (50% versus 35,38%), there is a significant difference in the number of miRNA and siRNA formulations that enter the clinical pipeline, with over six times more siRNA target drugs [245]. Even though it is not clear whether this is attributed to negative or inadequate evaluation, there is an evident need for intensification of preclinical research on miRNAs.
Although several clinical trials are ongoing to test miRNA-based therapeutics against several peripheral diseases, no such formulations have reached clinical trials so far for the treatment of AD. Nevertheless, gemfibrozil, a previously FDA-approved drug to decrease cholesterol and lipids, has undergone a phase I trial to assess its ability to increase miR-107 levels for prevention of AD in cognitive healthy and MCI individuals (NCT02045056). 48 control and 24 MCI individuals were treated with gemfibrozil or placebo and gemfibrozil appeared safe, inducing a change in miR-107 plasma levels. The secondary outcomes, including CSF Aβ42, pTAU, Aβ42/pTau ratio, brain atrophy and plasma TNF-α levels, did show trends for a decrease in the treatment group, however, these measures did not reach statistical significance [246]. While potentially promising, such studies do not directly address efficacy and toxicity of miRNA-based oligonucleotides in AD. Future clinical studies of adequate power may lay the groundwork for further miRNA-relevant drug repurposing or development in AD.
Conclusions and future perspectives
The paradigm shift from the reductionist view ‘one target – one disease’ to the endophenotype network-informed strategy ‘multiple targets – multiple disease pathways’ has started influencing pharmacological approaches against complex multigenic disorders. Network biology and multi-modal therapies begin to attract attention also in AD research [10, 13, 17, 18]. miRNA-targeted therapeutics are particularly suited for such purposes, as they regulate multiple components of several molecular cascades converging on disease-relevant patho-phenotypes. However, as emphasized in this review, the clinical application of miRNAs in brain diseases faces distinct challenges, reflected in the current scarcity of miRNA-based clinical trials in neurodegeneration in general and in AD in particular.
The field of miRNA-based therapeutics is developing in the slipstream of other oligonucleotide-based therapeutics (siRNA, ASO). Further basic research first, to better characterize how miRNAs target pathways of interest, and second, to systematically map on- and off-target toxic effects, is a prerequisite for effective clinical application in AD and other neurodegenerative disorders. Targeted brain delivery and additional investigation of the tolerability of miRNA supplementation or inhibition strategies are key issues to address. The acquisition of miRNA-based companies by major pharmaceutical companies could signal that prime time is approaching for this novel category of drugs [136]. However, despite their promise, as discussed here, application of miRNA therapeutics in AD is lagging behind other disease areas, like cancer, which represents a more generic discrepancy between the two fields, with approximately 30 times more new molecular entities in clinical trials in cancer than in AD [247]. Technology maturation, but also more aggressive investment in the AD field, are needed to bridge the valley between promising initial miRNA research and clinical application [248], very much similar to the path previously followed with other therapeutic approaches, such as monoclonal antibodies. With several companies focusing on miRNA preclinical, clinical and large screening studies, the next years will put these newly emerging approaches to the test and will define how far miRNA biologics are from clinical practice in AD, and whether the many miRNA targets could turn out to be ‘too many’ for clinical application.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
β amyloid
- pTAU:
-
Phosphorylated TAU protein
- miRNA:
-
microRNA
- mRNA:
-
Messenger RNA
- CNS:
-
Central nervous system
- NF-κB:
-
Nuclear factor kappa-B
- APP:
-
Amyloid precursor protein
- APOE4:
-
Apolipoprotein E4
- FTD:
-
Frontotemporal dementia
- BACE1:
-
β-secretase
- AAV:
-
Adeno-associated virus
- CSF:
-
Cerebrospinal fluid
- piRNA:
-
Piwi-interacting RNA
- MCI:
-
Mild cognitive impairment
- FDA:
-
Federal drug agency
- siRNA:
-
Small-interfering RNA
- BBB:
-
Blood-brain-barrier
- SOD1:
-
Superoxide dismutase 1
- ALS:
-
Amyotrophic lateral sclerosis
- SMA:
-
Spinal muscular atrophy
- ASO:
-
Antisense oligonucleotide
- LNA:
-
Locked-nucleic acid
References
Beyreuther K, Masters CL. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol Zurich Switz. 1991 Jul;1(4):241–51.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991 Oct;12(10):383–8.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353–6.
Karran E, De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. 2016;139(Suppl 2):237–52.
Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener [Internet]. 2017 Dec [cited 2020 Apr 9];12(1). Available from: http://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-017-0184-x
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
Sierksma A, Escott-Price V, De Strooper B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science. 2020 Oct 2;370(6512):61–6.
Chen W-T, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 2020 Aug;182(4):976–991.e19.
De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016 Feb;164(4):603–15.
Fang J, Pieper AA, Nussinov R, Lee G, Bekris L, Leverenz JB, et al. Harnessing endophenotypes and network medicine for Alzheimer’s drug repurposing. Med Res Rev. 2020 Nov;40(6):2386–426.
Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med [Internet]. 2020 Mar 6 [cited 2020 Nov 18];12(3). Available from: https://onlinelibrary.wiley.com/doi/abs/10.15252/emmm.201910606
Fessel J. Alzheimer’s disease combination treatment. Neurobiol Aging. 2018 Mar;63:165.
Cummings JL, Tong G, Ballard C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J Alzheimers Dis. 2019 Feb 12;67(3):779–94.
Frei E, Karon M, Levin RH, Freireich EJ, Taylor RJ, Hananian J, et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood. 1965 Nov;26(5):642–56.
Mokhtari RB, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget. 2017 Jun 6;8(23):38022–43.
Maramai S, Benchekroun M, Gabr MT, Yahiaoui S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. BioMed Res Int. 2020;2020 Jul 3:1–27.
Hampel H, Vergallo A, Aguilar LF, Benda N, Broich K, Cuello AC, et al. Precision pharmacology for Alzheimer’s disease. Pharmacol Res. 2018;130:331–65.
Salloway SP, Sevingy J, Budur K, Pederson JT, DeMattos RB, Von Rosenstiel P, et al. Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement Transl Res Clin Interv [Internet]. 2020 Jan [cited 2021 May 19];6(1). Available from: https://onlinelibrary.wiley.com/doi/10.1002/trc2.12073
Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement Transl Res Clin Interv [Internet]. 2021 Jan [cited 2021 May 25];7(1). Available from: https://onlinelibrary.wiley.com/doi/10.1002/trc2.12179
Cummings J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol Neurodegener [Internet]. 2021 Dec [cited 2021 May 25];16(1). Available from: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-021-00424-9
Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009 Jan;136(2):215–33.
Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015 Jul;16(7):421–33.
Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008 Sep;455(7209):64–71.
Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Blüthgen N, Marks DS, et al. MicroRNA control of protein expression noise. Science. 2015 Apr 3;348(6230):128–32.
Isik M, Blackwell TK, Berezikov E. MicroRNA mir-34 provides robustness to environmental stress response via the DAF-16 network in C. elegans. Sci Rep [Internet]. 2016 Dec [cited 2020 Apr 14];6(1). Available from: http://www.nature.com/articles/srep36766
Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A MicroRNA Imparts Robustness against Environmental Fluctuation during Development. Cell. 2009 Apr;137(2):273–82.
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science. 2007 Apr 27;316(5824):575–9.
Erhard F, Haas J, Lieber D, Malterer G, Jaskiewicz L, Zavolan M, et al. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014 Jun 1;24(6):906–19.
Hsin J-P, Lu Y, Loeb GB, Leslie CS, Rudensky AY. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat Immunol. 2018 Oct;19(10):1137–45.
Juźwik CA, Drake S, Zhang Y, Paradis-Isler N, Sylvester A, Amar-Zifkin A, et al. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog Neurobiol. 2019;182:101664.
Nam J-W, Rissland OS, Koppstein D, Abreu-Goodger C, Jan CH, Agarwal V, et al. Global Analyses of the Effect of Different Cellular Contexts on MicroRNA Targeting. Mol Cell. 2014 Mar;53(6):1031–43.
Swarup V, Hinz FI, Rexach JE, Noguchi K-I, Toyoshiba H, Oda A, et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat Med. 2019;25(1):152–64.
Salta E, Sierksma A, Vanden Eynden E, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med. 2016 Sep;8(9):1005–18.
Wilk G, Braun R. Integrative analysis reveals disrupted pathways regulated by microRNAs in cancer. Nucleic Acids Res. 2018 Feb 16;46(3):1089–101.
Bak M, Silahtaroglu A, Møller M, Christensen M, Rath MF, Skryabin B, et al. MicroRNA expression in the adult mouse central nervous system. RNA N Y N. 2008 Mar;14(3):432–44.
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007 Jun 29;129(7):1401–14.
Olsen L, Klausen M, Helboe L, Nielsen FC, Werge T. MicroRNAs show mutually exclusive expression patterns in the brain of adult male rats. PloS One. 2009 Oct 6;4(10):e7225.
Barca-Mayo O, De Pietri Tonelli D. Convergent microRNA actions coordinate neocortical development. Cell Mol Life Sci CMLS. 2014 Aug;71(16):2975–95.
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA N Y N. 2003 Oct;9(10):1274–81.
Christensen M, Schratt GM. microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neurosci Lett. 2009 Dec;466(2):55–62.
Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016 Mar;17(3):160–72.
Scott H. Extracellular microRNAs as messengers in the central and peripheral nervous system. Neuronal Signal. 2017 Dec;1(4):NS20170112.
Soreq H, Wolf Y. NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol Med. 2011 Oct;17(10):548–55.
Cheng L-C, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 2009 Apr;12(4):399–408.
Sanuki R, Onishi A, Koike C, Muramatsu R, Watanabe S, Muranishi Y, et al. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci. 2011 Sep;14(9):1125–34.
Franke K, Otto W, Johannes S, Baumgart J, Nitsch R, Schumacher S. miR-124-regulated RhoG reduces neuronal process complexity via ELMO/Dock180/Rac1 and Cdc42 signalling: RhoG reduces neuronal process complexity. EMBO J. 2012 Jun 29;31(13):2908–21.
Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med. 2014 Dec;20(12):1444–51.
Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α–PU.1 pathway. Nat Med. 2011 Jan;17(1):64–70.
Veremeyko T, Kuznetsova IS, Dukhinova M. W. Y. Yung A, Kopeikina E, Barteneva NS, et al. Neuronal extracellular microRNAs miR-124 and miR-9 mediate cell–cell communication between neurons and microglia. J Neurosci Res. 2019 Feb;97(2):162–84.
Fernandes A, Ribeiro AR, Monteiro M, Garcia G, Vaz AR, Brites D. Secretome from SH-SY5Y APPSwe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie. 2018 Dec;155:67–82.
Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007 Dec;10(12):1513–4.
Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci. 2010 Nov 23;107(47):20382–7.
Wayman GA, Davare M, Ando H, Fortin D, Varlamova O, Cheng H-YM, et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci. 2008 Jul 1;105(26):9093–8.
Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005 Nov 8;102(45):16426–31.
Luikart BW, Bensen AL, Washburn EK, Perederiy JV, Su KG, Li Y, et al. miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PloS One. 2011;6(5):e19077.
Kawashima H, Numakawa T, Kumamaru E, Adachi N, Mizuno H, Ninomiya M, et al. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience. 2010 Feb;165(4):1301–11.
Mellios N, Sugihara H, Castro J, Banerjee A, Le C, Kumar A, et al. miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat Neurosci. 2011 Oct;14(10):1240–2.
Remenyi J, van den Bosch MWM, Palygin O, Mistry RB, McKenzie C, Macdonald A, et al. miR-132/212 Knockout Mice Reveal Roles for These miRNAs in Regulating Cortical Synaptic Transmission and Plasticity. Preiss T, editor. PLoS ONE. 2013 Apr 26;8(4):e62509.
Wei Z, Meng X, El Fatimy R, Sun B, Mai D, Zhang J, et al. Environmental enrichment prevents Aβ oligomer-induced synaptic dysfunction through mirna-132 and hdac3 signaling pathways. Neurobiol Dis. 2020 Feb;134:104617.
Wong H-KA, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C, et al. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet. 2013 Aug 1;22(15):3077–92.
El Fatimy R, Li S, Chen Z, Mushannen T, Gongala S, Wei Z, et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol (Berl). 2018 Oct;136(4):537–55.
Hernandez-Rapp J, Smith PY, Filali M, Goupil C, Planel E, Magill ST, et al. Memory formation and retention are affected in adult miR-132/212 knockout mice. Behav Brain Res. 2015 Jul;287:15–26.
Salta E, De Strooper B. microRNA-132: a key noncoding RNA operating in the cellular phase of Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol. 2017;31(2):424–33.
Smith PY, Hernandez-Rapp J, Jolivette F, Lecours C, Bisht K, Goupil C, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015 Dec 1;24(23):6721–35.
Hansen KF, Sakamoto K, Aten S, Snider KH, Loeser J, Hesse AM, et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn Mem. 2016 Feb;23(2):61–71.
Hansen KF, Karelina K, Sakamoto K, Wayman GA, Impey S, Obrietan K. miRNA-132: a dynamic regulator of cognitive capacity. Brain Struct Funct. 2013 May;218(3):817–31.
Walgrave H, Balusu S, Snoeck S, Vanden Eynden E, Craessaerts K, Thrupp N, et al. Restoring miR-132 expression rescues adult hippocampal neurogenesis and memory deficits in Alzheimer’s disease. Cell Stem Cell [Internet]. 2021 May [cited 2021 May 25]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1934590921002198
Nahid MA, Yao B, Dominguez-Gutierrez PR, Kesavalu L, Satoh M, Chan EKL. Regulation of TLR2-mediated tolerance and cross-tolerance through IRAK4 modulation by miR-132 and miR-212. J Immunol Baltim Md 1950. 2013 Feb 1;190(3):1250–1263.
Kong H, Yin F, He F, Omran A, Li L, Wu T, et al. The Effect of miR-132, miR-146a, and miR-155 on MRP8/TLR4-Induced Astrocyte-Related Inflammation. J Mol Neurosci. 2015 Sep;57(1):28–37.
Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 Potentiates Cholinergic Anti-Inflammatory Signaling by Targeting Acetylcholinesterase. Immunity. 2009 Dec;31(6):965–73.
Korotkov A, Broekaart DWM, Banchaewa L, Pustjens B, Scheppingen J, Anink JJ, et al. microRNA-132 is overexpressed in glia in temporal lobe epilepsy and reduces the expression of pro-epileptogenic factors in human cultured astrocytes. Glia. 2020 Jan;68(1):60–75.
Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential Regulation of Interleukin-1 Receptor-associated Kinase-1 (IRAK-1) and IRAK-2 by MicroRNA-146a and NF-κB in Stressed Human Astroglial Cells and in Alzheimer Disease. J Biol Chem. 2010 Dec;285(50):38951–60.
Fan W, Liang C, Ou M, Zou T, Sun F, Zhou H, et al. MicroRNA-146a Is a Wide-Reaching Neuroinflammatory Regulator and Potential Treatment Target in Neurological Diseases. Front Mol Neurosci [Internet]. 2020 Jun 5 [cited 2020 Dec 2];13. Available from: https://www.frontiersin.org/article/10.3389/fnmol.2020.00090/full
Saba R, Sorensen DL, Booth SA. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front Immunol [Internet]. 2014 Nov 21 [cited 2020 Dec 2];5. Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2014.00578/abstract
Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF- B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci. 2006 Aug 15;103(33):12481–6.
Martin NA, Hyrlov KH, Elkjaer ML, Thygesen EK, Wlodarczyk A, Elbaek KJ, et al. Absence of miRNA-146a Differentially Alters Microglia Function and Proteome. Front Immunol [Internet]. 2020 Jun 5 [cited 2020 Dec 2];11. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2020.01110/full
Nguyen LS, Fregeac J, Bole-Feysot C, Cagnard N, Iyer A, Anink J, et al. Role of miR-146a in neural stem cell differentiation and neural lineage determination: relevance for neurodevelopmental disorders. Mol Autism [Internet]. 2018 Dec [cited 2020 Dec 2];9(1). Available from: https://molecularautism.biomedcentral.com/articles/10.1186/s13229-018-0219-3
Fregeac J, Moriceau S, Poli A, Nguyen LS, Oury F, Colleaux L. Loss of the neurodevelopmental disease-associated gene miR-146a impairs neural progenitor differentiation and causes learning and memory deficits. Mol Autism [Internet]. 2020 Dec [cited 2020 Dec 2];11(1). Available from: https://molecularautism.biomedcentral.com/articles/10.1186/s13229-020-00328-3
Prada I, Gabrielli M, Turola E, Iorio A, D’Arrigo G, Parolisi R, et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol (Berl). 2018 Apr;135(4):529–50.
Foinquinos A, Batkai S, Genschel C, Viereck J, Rump S, Gyöngyösi M, et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat Commun [Internet]. 2020 Dec [cited 2020 Aug 4];11(1). Available from: http://www.nature.com/articles/s41467-020-14349-2
Lee EC, Valencia T, Allerson C, Schairer A, Flaten A, Yheskel M, et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat Commun [Internet]. 2019 Dec [cited 2020 Aug 4];10(1). Available from: http://www.nature.com/articles/s41467-019-11918-y
Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA Changes in Alzheimer’s Disease Brain and CSF Yields Putative Biomarkers and Insights into Disease Pathways. J Alzheimers Dis. 2008 May 9;14(1):27–41.
Herrera-Espejo S, Santos-Zorrozua B, Álvarez-González P, Lopez-Lopez E, Garcia-Orad Á. A Systematic Review of MicroRNA Expression as Biomarker of Late-Onset Alzheimer’s Disease. Mol Neurobiol. 2019 Dec;56(12):8376–91.
Lau P, Bossers K, Janky R, Salta E, Frigerio CS, Barbash S, et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med. 2013;5(10):1613–34.
Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007 Feb 12;18(3):297–300.
Pichler S, Gu W, Hartl D, Gasparoni G, Leidinger P, Keller A, et al. The miRNome of Alzheimer’s disease: consistent downregulation of the miR-132/212 cluster. Neurobiol Aging. 2017 Feb;50:167.e1-167.e10.
Li QS, Cai D. Integrated miRNA-Seq and mRNA-Seq Study to Identify miRNAs Associated With Alzheimer’s Disease Using Post-mortem Brain Tissue Samples. Front Neurosci. 2021;15:620899.
Konovalova, Gerasymchuk, Parkkinen, Chmielarz, Domanskyi. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int J Mol Sci. 2019 Nov 30;20(23):6055.
Zhang R, Zhang Q, Niu J, Lu K, Xie B, Cui D, et al. Screening of microRNAs associated with Alzheimer’s disease using oxidative stress cell model and different strains of senescence accelerated mice. J Neurol Sci. 2014 Mar;338(1–2):57–64.
Engedal N, Žerovnik E, Rudov A, Galli F, Olivieri F, Procopio AD, et al. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid Med Cell Longev. 2018;2018:1–16.
Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006 Jan;439(7074):283–9.
Cardoso AL, Guedes JR. Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production: miR-155 role during microglia activation. Immunology. 2012 Jan;135(1):73–88.
Lee K, Kim H, An K, Kwon O-B, Park S, Cha JH, et al. Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD Mouse Model of Alzheimer’s Disease. Sci Rep. 2016 06;6:34433.
Aksoy-Aksel A, Zampa F, Schratt G. MicroRNAs and synaptic plasticity—a mutual relationship. Philos Trans R Soc B Biol Sci. 2014 Sep 26;369(1652):20130515.
Ghanbari M, Ikram MA, de Looper HWJ, Hofman A, Erkeland SJ, Franco OH, et al. Genome-wide identification of microRNA-related variants associated with risk of Alzheimer’s disease. Sci Rep [Internet]. 2016 Sep [cited 2021 Jan 19];6(1). Available from: http://www.nature.com/articles/srep28387
Herrera-Espejo S, Santos-Zorrozua B, Alvarez-Gonzalez P, Martin-Guerrero I. M. de Pancorbo M, Garcia-Orad A, et al. A Genome-Wide Study of Single-Nucleotide Polymorphisms in MicroRNAs and Further In Silico Analysis Reveals Their Putative Role in Susceptibility to Late-Onset Alzheimer’s Disease. Mol Neurobiol. 2021 Jan;58(1):55–64.
Imperatore JA, Then ML, McDougal KB, Mihailescu MR. Characterization of a G-Quadruplex Structure in Pre-miRNA-1229 and in Its Alzheimer’s Disease-Associated Variant rs2291418: Implications for miRNA-1229 Maturation. Int J Mol Sci. 2020 Jan 24;21(3):767.
Patrick E, Rajagopal S, Wong H-KA, McCabe C, Xu J, Tang A, et al. Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol Neurodegener. 2017 01;12(1):51.
Tasaki S, Gaiteri C, Mostafavi S, De Jager PL, Bennett DA. The Molecular and Neuropathological Consequences of Genetic Risk for Alzheimer’s Dementia. Front Neurosci. 2018;12:699.
Delay C, Calon F, Mathews P, Hébert SS. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Mol Neurodegener. 2011 Oct 7;6:70.
Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging. 2012 Mar;33(3):522–34.
Patel N, Hoang D, Miller N, Ansaloni S, Huang Q, Rogers JT, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008 Aug 6;3:10.
Vilardo E, Barbato C, Ciotti M, Cogoni C, Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. 2010 Jun 11;285(24):18344–51.
Parsi S, Smith PY, Goupil C, Dorval V, Hébert SS. Preclinical Evaluation of miR-15/107 Family Members as Multifactorial Drug Targets for Alzheimer’s Disease. Mol Ther - Nucleic Acids. 2015;4:e256.
Long JM, Lahiri DK. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem Biophys Res Commun. 2011 Jan 28;404(4):889–95.
Hébert SS, Horré K, Nicolaï L, Bergmans B, Papadopoulou AS, Delacourte A, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis. 2009 Mar;33(3):422–8.
Liang C, Zhu H, Xu Y, Huang L, Ma C, Deng W, et al. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012 May;1455:103–13.
Long JM, Ray B, Lahiri DK. MicroRNA-153 Physiologically Inhibits Expression of Amyloid-β Precursor Protein in Cultured Human Fetal Brain Cells and Is Dysregulated in a Subset of Alzheimer Disease Patients. J Biol Chem. 2012 Sep;287(37):31298–310.
Barros-Viegas AT, Carmona V, Ferreiro E, Guedes J, Cardoso AM, Cunha P, et al. miRNA-31 Improves Cognition and Abolishes Amyloid-β Pathology by Targeting APP and BACE1 in an Animal Model of Alzheimer’s Disease. Mol Ther - Nucleic Acids. 2020 Mar;19:1219–36.
Chen X, Jiang X-M, Zhao L-J, Sun L-L, Yan M-L, Tian Y, et al. MicroRNA-195 prevents dendritic degeneration and neuron death in rats following chronic brain hypoperfusion. Cell Death Dis. 2017 Jun;8(6):e2850.
Ai J, Sun L-H, Che H, Zhang R, Zhang T-Z, Wu W-C, et al. MicroRNA-195 Protects Against Dementia Induced by Chronic Brain Hypoperfusion via Its Anti-Amyloidogenic Effect in Rats. J Neurosci. 2013 Feb 27;33(9):3989–4001.
Su D, Chai Y, Yang J, Wang X, Liu Y, Ma J, et al. Lentivirus-Carried microRNA-195 Rescues Memory Deficits of Alzheimer’s Disease Transgenic Mouse by Attenuating the Generation of Amyloid Plaques. Front Pharmacol [Internet]. 2021 Apr 26 [cited 2021 Sep 13];12. Available from: https://www.frontiersin.org/articles/10.3389/fphar.2021.633805/full
An F, Gong G, Wang Y, Bian M, Yu L, Wei C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget. 2017 Dec 26;8(69):114065–71.
Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008 Apr 29;105(17):6415–20.
Yang G, Song Y, Zhou X, Deng Y, Liu T, Weng G, et al. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol Med Rep. 2015 Aug;12(2):3081–8.
Zong Y, Wang H, Dong W, Quan X, Zhu H, Xu Y, et al. miR-29c regulates BACE1 protein expression. Brain Res. 2011 Jun 13;1395:108–15.
Zhu H-C, Wang L-M, Wang M, Song B, Tan S, Teng J-F, et al. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res Bull. 2012 Sep;88(6):596–601.
Zhang X, Huang X, Fang C, Li Q, Cui J, Sun J, et al. miR-124 Regulates the Expression of BACE1 in the Hippocampus Under Chronic Cerebral Hypoperfusion. Mol Neurobiol. 2017 May;54(4):2498–506.
Chopra N, Wang R, Maloney B, Nho K, Beck JS, Pourshafie N, et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol Psychiatry [Internet]. 2020 Jan 15 [cited 2021 Aug 11]; Available from: http://www.nature.com/articles/s41380-019-0610-2
Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and MicroRNA-328 Regulate Expression of Mouse β-Amyloid Precursor Protein-converting Enzyme 1. J Biol Chem. 2009 Jan;284(4):1971–81.
Oliverio M, Schmidt E, Mauer J, Baitzel C, Hansmeier N, Khani S, et al. Dicer1–miR-328–Bace1 signalling controls brown adipose tissue differentiation and function. Nat Cell Biol. 2016 Mar;18(3):328–36.
Kim J, Yoon H, Chung D, Brown JL, Belmonte KC, Kim J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem. 2016 May;137(3):436–45.
Zhang N, Li W-W, Lv C-M, Gao Y-W, Liu X-L, Zhao L. miR-16-5p and miR-19b-3p prevent amyloid β-induced injury by targeting BACE1 in SH-SY5Y cells. NeuroReport. 2020 Feb 5;31(3):205–12.
Zhong Z, Yuan K, Tong X, Hu J, Song Z, Zhang G, et al. MiR-16 attenuates β-amyloid-induced neurotoxicity through targeting β-site amyloid precursor protein-cleaving enzyme 1 in an Alzheimer’s disease cell model. NeuroReport. 2018 Nov 7;29(16):1365–72.
Kim J, Yoon H, Horie T, Burchett JM, Restivo JL, Rotllan N, et al. microRNA-33 Regulates ApoE Lipidation and Amyloid- Metabolism in the Brain. J Neurosci. 2015 Nov 4;35(44):14717–26.
Hsu H-W, Rodriguez-Ortiz CJ, Zumkehr J, Kitazawa M. Inflammatory Cytokine IL-1β Downregulates Endothelial LRP1 via MicroRNA-mediated Gene Silencing. Neuroscience. 2021 Jan;453:69–80.
Wang Z, Qin W, Wei CB, Tang Y, Zhao LN, Jin HM, et al. The microRNA-1908 up-regulation in the peripheral blood cells impairs amyloid clearance by targeting ApoE. Int J Geriatr Psychiatry. 2018 Jul;33(7):980–6.
Absalon S, Kochanek DM, Raghavan V, Krichevsky AM. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J Neurosci Off J Soc Neurosci. 2013 Sep 11;33(37):14645–59.
Dickson JR, Kruse C, Montagna DR, Finsen B, Wolfe MS. Alternative polyadenylation and miR-34 family members regulate tau expression. J Neurochem. 2013 Dec;127(6):739–49.
Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, et al. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014 Aug 1;33(15):1667–80.
Smith PY, Delay C, Girard J, Papon M-A, Planel E, Sergeant N, et al. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum Mol Genet. 2011 Oct 15;20(20):4016–24.
Hernandez-Rapp J, Rainone S, Goupil C, Dorval V, Smith PY, Saint-Pierre M, et al. microRNA-132/212 deficiency enhances Aβ production and senile plaque deposition in Alzheimer’s disease triple transgenic mice. Sci Rep [Internet]. 2016 Nov [cited 2021 Jun 4];6(1). Available from: http://www.nature.com/articles/srep30953
Qian Q, Zhang J, He F-P, Bao W-X, Zheng T-T, Zhou D-M, et al. Down-regulated expression of microRNA-338-5p contributes to neuropathology in Alzheimer’s disease. FASEB J Off Publ Fed Am Soc Exp Biol. 2019;33(3):4404–17.
Mai H, Fan W, Wang Y, Cai Y, Li X, Chen F, et al. Intranasal Administration of miR-146a Agomir Rescued the Pathological Process and Cognitive Impairment in an AD Mouse Model. Mol Ther - Nucleic Acids. 2019 Dec;18:681–95.
Martinez B, Peplow P. MicroRNAs as diagnostic and therapeutic tools for Alzheimer’s disease: advances and limitations. Neural Regen Res. 2019;14(2):242.
Bonneau E, Neveu B, Kostantin E, Tsongalis GJ, De Guire V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. EJIFCC. 2019 Jun;30(2):114–27.
Banizs AB, Silverman JF. The utility of combined mutation analysis and microRNA classification in reclassifying cancer risk of cytologically indeterminate thyroid nodules. Diagn Cytopathol. 2019 Apr;47(4):268–74.
Labourier E, Shifrin A, Busseniers AE, Lupo MA, Manganelli ML, Andruss B, et al. Molecular Testing for miRNA, mRNA, and DNA on Fine-Needle Aspiration Improves the Preoperative Diagnosis of Thyroid Nodules With Indeterminate Cytology. J Clin Endocrinol Metab. 2015 Jul;100(7):2743–50.
Cohen AD, Landau SM, Snitz BE, Klunk WE, Blennow K, Zetterberg H. Fluid and PET biomarkers for amyloid pathology in Alzheimer’s disease. Mol Cell Neurosci. 2019;97:3–17.
Jack CR, Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013 Dec 18;80(6):1347–58.
Schöll M, Maass A, Mattsson N, Ashton NJ, Blennow K, Zetterberg H, et al. Biomarkers for tau pathology. Mol Cell Neurosci. 2019;97:18–33.
Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) investigators, Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med. 2020 Mar;26(3):387–397.
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020 Mar;26(3):379–86.
Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020 May;19(5):422–33.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018 Feb;554(7691):249–54.
Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019 Aug 1;10.1212/WNL.0000000000008081.
Alawode DOT, Heslegrave AJ, Ashton NJ, Karikari TK, Simrén J, Montoliu-Gaya L, et al. Transitioning from cerebrospinal fluid to blood tests to facilitate diagnosis and disease monitoring in Alzheimer’s disease. J Intern Med [Internet]. 2021 May 22 [cited 2021 May 25]; Available from: https://onlinelibrary.wiley.com/doi/10.1111/joim.13332
Mattsson N, Andreasson U, Zetterberg H, Blennow K. Alzheimer’s Disease Neuroimaging Initiative. Association of Plasma Neurofilament Light With Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2017 May 1;74(5):557–66.
Denk J, Boelmans K, Siegismund C, Lassner D, Arlt S, Jahn H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer`s Disease. PloS One. 2015;10(5):e0126423.
McKeever PM, Schneider R, Taghdiri F, Weichert A, Multani N, Brown RA, et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol Neurobiol. 2018 Dec;55(12):8826–41.
Sala Frigerio C, Lau P, Salta E, Tournoy J, Bossers K, Vandenberghe R, et al. Reduced expression of hsa-miR-27a-3p in CSF of patients with Alzheimer disease. Neurology. 2013 Dec 10;81(24):2103–6.
Satoh J-I, Kino Y, Niida S. MicroRNA-Seq Data Analysis Pipeline to Identify Blood Biomarkers for Alzheimer’s Disease from Public Data. Biomark Insights. 2015;10:21–31.
Swarbrick S, Wragg N, Ghosh S, Stolzing A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol Neurobiol. 2019 Sep;56(9):6156–67.
Takousis P, Sadlon A, Schulz J, Wohlers I, Dobricic V, Middleton L, et al. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimers Dement J Alzheimers Assoc. 2019;15(11):1468–77.
Hu Y-B, Li C-B, Song N, Zou Y, Chen S-D, Ren R-J, et al. Diagnostic Value of microRNA for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front Aging Neurosci [Internet]. 2016 Feb 9 [cited 2021 Sep 9];8. Available from: http://journal.frontiersin.org/Article/10.3389/fnagi.2016.00013/abstract
Leidinger P, Backes C, Deutscher S, Schmitt K, Mueller SC, Frese K, et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013 Jul 29;14(7):R78.
Ludwig N, Fehlmann T, Kern F, Gogol M, Maetzler W, Deutscher S, et al. Machine Learning to Detect Alzheimer’s Disease from Circulating Non-coding RNAs. Genomics Proteomics Bioinformatics. 2019 Aug;17(4):430–40.
Keller A, Backes C, Haas J, Leidinger P, Maetzler W, Deuschle C, et al. Validating Alzheimer’s disease micro RNAs using next-generation sequencing. Alzheimers Dement. 2016 May;12(5):565–76.
Jain G, Stuendl A, Rao P, Berulava T, Pena Centeno T, Kaurani L, et al. A combined miRNA–piRNA signature to detect Alzheimer’s disease. Transl Psychiatry [Internet]. 2019 Dec [cited 2020 Apr 26];9(1). Available from: http://www.nature.com/articles/s41398-019-0579-2
Doecke JD, Pérez-Grijalba V, Fandos N, Fowler C, Villemagne VL, Masters CL, et al. Total Aβ 42 /Aβ 40 ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology. 2020 Apr 14;94(15):e1580–91.
Kopkova A, Sana J, Fadrus P, Slaby O. Cerebrospinal fluid microRNAs as diagnostic biomarkers in brain tumors. Clin Chem Lab Med CCLM. 2018 May 24;56(6):869–79.
Müller M, Kuiperij HB, Claassen JA, Küsters B, Verbeek MM. MicroRNAs in Alzheimer’s disease: differential expression in hippocampus and cell-free cerebrospinal fluid. Neurobiol Aging. 2014 Jan;35(1):152–8.
Moon J, Lee S-T, Kong IG, Byun J-I, Sunwoo J-S, Shin J-W, et al. Early diagnosis of Alzheimer’s disease from elevated olfactory mucosal miR-206 level. Sci Rep. 2016 Feb 4;6:20364.
Kenny A, Jiménez-Mateos EM, Zea-Sevilla MA, Rábano A, Gili-Manzanaro P, Prehn JHM, et al. Proteins and microRNAs are differentially expressed in tear fluid from patients with Alzheimer’s disease. Sci Rep [Internet]. 2019 Dec [cited 2020 Dec 2];9(1). Available from: http://www.nature.com/articles/s41598-019-51837-y
Cummings J, Aisen P, Lemere C, Atri A, Sabbagh M, Salloway S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res Ther [Internet]. 2021 Dec [cited 2021 Jun 8];13(1). Available from: https://alzres.biomedcentral.com/articles/10.1186/s13195-021-00838-z
Fillit H, Green A. Aducanumab and the FDA — where are we now? Nat Rev Neurol. 2021 Mar;17(3):129–30.
Lam JKW, Chow MYT. Zhang Y. Leung SWS. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids. 2015 Sep 15;4:e252.
Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci. 2012 Jun 26;109(26):E1695–704.
Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015 Jan 2;125(1):141–56.
Guo J, Song W, Boulanger J, Xu EY, Wang F, Zhang Y, et al. Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome. Hum Gene Ther. 2019 Jul;30(7):865–81.
Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, et al. Mir-34: A New Weapon Against Cancer? Mol Ther - Nucleic Acids. 2014 Jan;3:e195.
Witten L, Slack FJ. miR-155 as a novel clinical target for hematological malignancies. Carcinogenesis. 2020 Mar 13;41(1):2–7.
Zhang L, Liao Y, Tang L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res [Internet]. 2019 Dec [cited 2020 Aug 5];38(1). Available from: https://jeccr.biomedcentral.com/articles/10.1186/s13046-019-1059-5
Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, et al. LNA-mediated anti–miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012 Aug 23;120(8):1678–86.
Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res Off J Am Assoc Cancer Res. 2012 Nov 15;18(22):6260–70.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005 Feb;433(7027):769–73.
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008 Sep;455(7209):58–63.
Liufu Z, Zhao Y, Guo L, Miao G, Xiao J, Lyu Y, et al. Redundant and incoherent regulations of multiple phenotypes suggest microRNAs’ role in stability control. Genome Res. 2017 Oct;27(10):1665–73.
Bartel DP. Metazoan MicroRNAs. Cell. 2018 Mar;173(1):20–51.
Jens M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet. 2015 Feb;16(2):113–26.
Alexander MS, Casar JC, Motohashi N, Vieira NM, Eisenberg I, Marshall JL, et al. MicroRNA-486–dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy–associated symptoms. J Clin Invest. 2014 Jun 2;124(6):2651–67.
Mestdagh P, Boström A-K, Impens F, Fredlund E, Van Peer G, De Antonellis P, et al. The miR-17-92 MicroRNA Cluster Regulates Multiple Components of the TGF-β Pathway in Neuroblastoma. Mol Cell. 2010 Dec;40(5):762–73.
Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011 Jan;469(7330):336–42.
Le MTN, Shyh-Chang N, Khaw SL, Chin L, Teh C, Tay J, et al. Conserved Regulation of p53 Network Dosage by MicroRNA–125b Occurs through Evolving miRNA–Target Gene Pairs. McManus MT, editor. PLoS Genet. 2011 Sep 15;7(9):e1002242.
Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 Regulates OCT4, SOX2, and KLF4 and Represses Pluripotency in Human Embryonic Stem Cells. Cell. 2009 May;137(4):647–58.
Lewis BP, Shih I, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of Mammalian MicroRNA Targets. Cell. 2003 Dec;115(7):787–98.
Pinzón N, Li B, Martinez L, Sergeeva A, Presumey J, Apparailly F, et al. microRNA target prediction programs predict many false positives. Genome Res. 2017 Feb;27(2):234–45.
Mockly S, Seitz H. Inconsistencies and Limitations of Current MicroRNA Target Identification Methods. In: Laganà A, editor. MicroRNA Target Identification [Internet]. New York, NY: Springer New York; 2019 [cited 2020 Apr 17]. p. 291–314. Available from: http://link.springer.com/10.1007/978-1-4939-9207-2_16
Bassett AR, Azzam G, Wheatley L, Tibbit C, Rajakumar T, McGowan S, et al. Understanding functional miRNA-target interactions in vivo by site-specific genome engineering. Nat Commun. 2014 Aug 19;5:4640.
Ecsedi M, Rausch M, Großhans H. The let-7 microRNA directs vulval development through a single target. Dev Cell. 2015 Feb 9;32(3):335–44.
Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nat Rev Neurosci. 2009 Dec;10(12):837–41.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017 Mar;16(3):203–22.
Täubel J, Hauke W, Rump S, Viereck J, Batkai S, Poetzsch J, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021 Jan 7;42(2):178–88.
Batkai S, Genschel C, Viereck J, Rump S, Bär C, Borchert T, et al. CDR132L improves systolic and diastolic function in a large animal model of chronic heart failure. Eur Heart J. 2021 Jan 7;42(2):192–201.
Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, et al. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene. 2015 Jul;34(27):3547–55.
Rupaimoole R, Yoon B, Zhang WC, Adams BD, Slack FJ. A High-Throughput Small Molecule Screen Identifies Ouabain as Synergistic with miR-34a in Killing Lung Cancer Cells. iScience. 2020 Feb;23(2):100878.
Meng Z, Lu M. RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Front Immunol [Internet]. 2017 Mar 23 [cited 2020 Aug 3];8. Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2017.00331/full
Segal M, Slack FJ. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin Drug Discov. 2020 May;18:1–5.
Hong DS, Kang Y-K, Borad M, Sachdev J, Ejadi S, Lim HY, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020 May;122(11):1630–7.
Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. 2018 Sep;17(9):641–59.
Borel F, Gernoux G, Sun H, Stock R, Blackwood M, Brown RH, et al. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci Transl Med. 2018 Oct 31;10(465):eaau6414.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1713–22.
Pardridge WM. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci [Internet]. 2020 Jan 10 [cited 2021 May 26];11. Available from: https://www.frontiersin.org/article/10.3389/fnagi.2019.00373/full
Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med. 2020 Jul 9;383(2):151–8.
Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther. 2018 Mar;29(3):285–98.
Wang D, Zhong L, Li M, Li J, Tran K, Ren L, et al. Adeno-Associated Virus Neutralizing Antibodies in Large Animals and Their Impact on Brain Intraparenchymal Gene Transfer. Mol Ther - Methods Clin Dev. 2018 Dec;11:65–72.
Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015 Feb 2;125(2):870–80.
Lee SWL, Paoletti C, Campisi M, Osaki T, Adriani G, Kamm RD, et al. MicroRNA delivery through nanoparticles. J Control Release Off J Control Release Soc. 2019 Nov 10;313:80–95.
Zhou Y, Peng Z, Seven ES, Leblanc RM. Crossing the blood-brain barrier with nanoparticles. J Controlled Release. 2018 Jan;270:290–303.
Kulkarni JA, Witzigmann D, Chen S, Cullis PR, van der Meel R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc Chem Res. 2019 Sep 17;52(9):2435–44.
Samaridou E, Walgrave H, Salta E, Álvarez DM, Castro-López V, Loza M, et al. Nose-to-brain delivery of enveloped RNA - cell permeating peptide nanocomplexes for the treatment of neurodegenerative diseases. Biomaterials. 2020 Feb;230:119657.
Boraschi D, Italiani P, Palomba R, Decuzzi P, Duschl A, Fadeel B, et al. Nanoparticles and innate immunity: new perspectives on host defence. Semin Immunol. 2017 Dec;34:33–51.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet Lond Engl. 2016 17;388(10063):3017–3026.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med. 2018 15;378(7):625–635.
Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011 Mar 2;3(72):72ra18.
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med. 2019 Jun 13;380(24):2307–16.
Maimon R, Chillon-Marinas C, Snethlage CE, Singhal SM, McAlonis-Downes M, Ling K, et al. Therapeutically viable generation of neurons with antisense oligonucleotide suppression of PTB. Nat Neurosci [Internet]. 2021 Jun 3 [cited 2021 Jun 8]; Available from: http://www.nature.com/articles/s41593-021-00864-y
Dovgan I, Koniev O, Kolodych S, Wagner A. Antibody–Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug Chem. 2019 Oct 16;30(10):2483–501.
Dugal-Tessier J, Thirumalairajan S, Jain N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J Clin Med. 2021 Feb 18;10(4):838.
Xia C-F, Zhang Y, Zhang Y, Boado RJ, Pardridge WM. Intravenous siRNA of Brain Cancer with Receptor Targeting and Avidin–Biotin Technology. Pharm Res. 2007 Nov 5;24(12):2309–16.
Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci. 2016 Sep 27;113(39):10962–7.
Du L, Kayali R, Bertoni C, Fike F, Hu H, Iversen PL, et al. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum Mol Genet. 2011 Aug 15;20(16):3151–60.
Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017 Mar;35(3):222–9.
Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014 Dec;24(6):374–87.
Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008 Apr;452(7189):896–9.
Lima JF, Cerqueira L, Figueiredo C, Oliveira C, Azevedo NF. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018 04;15(3):338–352.
Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011 Apr;43(4):371–8.
Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010 Jan 8;327(5962):198–201.
Kristen AV, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019 Feb;9(1):5–23.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N Engl J Med. 2013 Aug 29;369(9):819–29.
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 Jul 5;379(1):11–21.
Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med [Internet]. 2020 Dec 30 [cited 2021 Jan 20]; Available from: http://www.nejm.org/doi/10.1056/NEJMoa2035389
Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020 Dec 31;383(27):2603–15.
Abplanalp WT, Fischer A, John D, Zeiher AM, Gosgnach W, Darville H, et al. Efficiency and Target Derepression of Anti-miR-92a: Results of a First in Human Study. Nucleic Acid Ther [Internet]. 2020 Jul 20 [cited 2020 Dec 2]; Available from: https://www.liebertpub.com/doi/10.1089/nat.2020.0871
Reid G, Kao SC, Pavlakis N, Brahmbhatt H, MacDiarmid J, Clarke S, et al. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics. 2016 Aug;8(8):1079–85.
van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: a first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017 Oct;18(10):1386–96.
Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013 May 2;368(18):1685–94.
van der Ree MH, van der Meer AJ, van Nuenen AC, de Bruijne J, Ottosen S, Janssen HL, et al. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther. 2016 Jan;43(1):102–13.
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020 Oct;19(10):673–94.
van der Ree MH, de Vree JM, Stelma F, Willemse S, van der Valk M, Rietdijk S, et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. The Lancet. 2017 Feb;389(10070):709–17.
Wang D, Liu K, Cattatossi G, Nelson M, Wright TM. Preclinical development of miR-10b antagomist for the treatment of glioblastoma. In New Orleans; 2018. Available from: http://regulusrx.com/wp-content/uploads/2018/12/SNO2018-miR10b-GBM.pdf
Dasgupta I, Chatterjee A. Recent Advances in miRNA Delivery Systems. Methods Protoc. 2021 Jan 20;4(1):10.
Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017 Jan;16(1):19–34.
de Lera AR, Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin Epigenetics. 2016 Dec;8(1):105.
Zhang S, Cheng Z, Wang Y, Han T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des Devel Ther. 2021 Feb;15:721–33.
Jicha GA, Kryscio RJ, Beech BF, Wang W-X, Lynn BC, Schmitt FA, et al. O3-10-05: MODULATION OF MICRORNA PATHWAYS BY GEMFIBROZIL IN PREDEMENTIA ALZHEIMER DISEASE: A RANDOMIZED, PLACEBO-CONTROLLED. DOUBLE-BLIND CLINICAL TRIAL. Alzheimers Dement. 2019 Jul;15:P910.
Cummings J, Feldman HH, Scheltens P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther. 2019 Dec;11(1):76.
Beierlein JM, McNamee LM, Ledley FD. As Technologies for Nucleotide Therapeutics Mature, Products Emerge. Mol Ther - Nucleic Acids. 2017 Dec;9:379–86.
Acknowledgements
We would like to thank the anonymous reviewers for their valuable and insightful comments that considerably contributed to the improvement of our manuscript.
Funding
H.W. is a Fonds voor Wetenschappelijk Onderzoek (FWO) fellow (1111319N) and receives funding from European Union’s Horizon 2020 research (B-SMART). L.Z. is an employee of Janssen Pharmaceutica. Work in the lab of B.D.S. is supported by the European Union (grant no. ERC-834682 337 CELLPHASE_AD), FWO, KU Leuven, VIB, a Methusalem grant from KU Leuven and the Flemish Government, the MRC, the Alzheimer Society, and Alzheimer Research UK. B.D.S. is further supported by the Opening the Future campaign of the Leuven Universiteits Fonds (LUF) and holder of the Bax-Vanluffelen Chair for Alzheimer's disease. E.S. receives funding from Alzheimer’s Association, Alzheimer Nederland and Health Holland.
Author information
Authors and Affiliations
Contributions
H.W. and E.S. wrote the manuscript. L.Z. and B.D.S. provided critical feedback and input.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Walgrave, H., Zhou, L., De Strooper, B. et al. The promise of microRNA-based therapies in Alzheimer’s disease: challenges and perspectives. Mol Neurodegeneration 16, 76 (2021). https://doi.org/10.1186/s13024-021-00496-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00496-7