
Abstract
Background
Astragalus mongholicus Bunge is an important medicinal plant used in traditional Chinese medicine. It is rich in isoflavonoids and triterpenoid saponins. Although these active constituents of A. mongholicus have been discovered for a long time, the genetic basis of isoflavonoid and triterpenoid saponin biosynthesis in this plant is virtually unknown because of the lack of a reference genome. Here, we used a combination of next-generation sequencing (NGS) and single-molecule real-time (SMRT) sequencing to identify genes involved in the biosynthetic pathway of secondary metabolites in A. mongholicus.
Results
In this study, NGS, SMRT sequencing, and targeted compound analysis were combined to investigate the association between isoflavonoid and triterpenoid saponin content, and specific gene expression in the root, stem, and leaves of A. mongholicus. Overall, 643,812 CCS reads were generated, yielding 121,107 non-redundant transcript isoforms with an N50 value of 2124 bp. Based on these highly accurate transcripts, 104,756 (86.50%) transcripts were successfully annotated by any of the seven databases (NR, NT, Swissprot, KEGG, KOG, Pfam and GO). Levels of four isoflavonoids and four astragalosides (triterpenoid saponins) were determined. Forty-four differentially expressed genes (DEGs) involved in isoflavonoid biosynthesis and 44 DEGs from 16 gene families that encode enzymes involved in triterpenoid saponin biosynthesis were identified. Transcription factors (TFs) associated with isoflavonoid and triterpenoid saponin biosynthesis, including 72 MYBs, 53 bHLHs, 64 AP2-EREBPs, and 11 bZIPs, were also identified. The above transcripts showed different expression trends in different plant organs.
Conclusions
This study provides important genetic information on the A. mongholicus genes that are essential for isoflavonoid and triterpenoid saponin biosynthesis, and provides a basis for developing the medicinal value of this plant.
Similar content being viewed by others
Background
The genus Astragalus mongholicus Bunge, family Fabaceae (Leguminosae), contains perennial herbaceous plants, many of which are conventional therapeutic plants with a long history in traditional popular medicine in China [1, 2]. A. mongholicus has been broadly utilized as a principal herbal medicine in China, including Northeast, North, and Northwest China, as well as in Mongolia and Korea [3]. Based on pharmacological studies and clinical practice, the dried roots of A. mongholicus have good potential therapeutic applications, including vital-energy tonification, abscess drainage, diuretic properties, skin reinforcement, and tissue regeneration [4,5,6]. Recent studies revealed that these bioactivities are mainly associated with isoflavonoids and triterpenoid saponins [7,8,9]. Isoflavonoids and triterpenoid saponins are widely distributed in plants, and their biosynthetic pathways are well understood, especially in some model plants [10, 11]. However, A. mongholicus is not a model organism and little genomic information is available for this plant. This profoundly limits the research into the regulatory mechanisms of development, metabolite biosynthesis, and other physiological processes in this plant.
The isoflavonoid biosynthetic pathway is a branch of the phenylpropanoid pathway [12, 13]. Isoflavonoids are a class of phenolic secondary metabolites that regulate the complex plant–microbe interactions and are mainly synthesized by legumes [14, 15]. Phenylalanine ammonia-lyase (PAL) is the first key enzyme in the phenylpropanoid pathway; cinnamate 4-hydroxylase (C4H) is the second enzyme in the pathway; and 4-coumarate CoA-ligase (4CL) produces p-coumaroyl-CoA as a precursor for the synthesis of chalcone. Chalcone synthase (CHS) catalyzes the conversion of p-coumaroyl-CoA to naringenin chalcone in the first committed step of flavonoid biosynthesis. Next, chalcone isomerase (CHI) catalyzes the cyclization of the chalcones, producing liquiritigenin or naringenin, which act as substrates for many downstream enzymatic reactions for ultimate flavonoid formation in leguminous plants. As the key step at the very beginning of the isoflavonoid metabolic pathway, isoflavone synthase (IFS) converts flavanones to their corresponding isoflavonoids.
Current research on the biosynthesis of triterpenoid saponins can be divided into three stages: synthesis of upstream precursors, synthesis of the intermediate carbocyclic skeleton, and downstream formation of different types of triterpenoid saponins by various complex functionalization reactions [16]. The upstream precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are mainly formed via the mevalonic acid pathway (MVA) and the methylerythritol pathway (MEP) [17,18,19]. The carbocyclic skeleton of triterpenoid saponins is synthesized from DMAPP in reactions catalyzed by prenyltransferase and oxidosqualene cyclases (OSCs) [20, 21]. In addition, different types of triterpenoid saponin products are generated by chemically modified cytochrome P450 monooxygenases (CYP450s), UDP-glycosyltransferases (UGTs), and glycosidases.
Transcriptome studies provide a useful perspective on the molecular biology of medicinal plants, including molecular mechanisms of gene function, cellular response, and different biological processes [22,23,24,25,26]. In the current study, we combined next-generation sequencing (NGS) and single-molecule real-time (SMRT) sequencing to obtain a complete A. mongholicus transcriptome, as a valuable resource for studying the regulatory mechanisms of development, metabolite biosynthesis, and other physiological processes of A. mongholicus in the absence of the available reference genome. We then used ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) to determine the levels of four primary isoflavonoids and four main astragalosides (triterpenoid saponins) in the root, stem, and leaves of this plant, to investigate the isoflavonoid and triterpenoid saponin biosynthesis in A. mongholicus. Candidate key genes involved in isoflavonoid and triterpenoid saponin biosynthesis in A. mongholicus were then identified. This study provides novel insights into the isoflavonoid and triterpenoid saponin biosynthesis in A. mongholicus.
Results
Accumulation of isoflavonoids and triterpenoid saponins in different organs of A. mongholicus
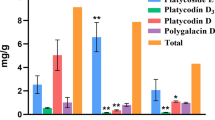
Four isoflavonoids and four triterpenoid saponins were identified by UPLC-MS/MS in the root (AR), stem (AS), and leaves (AL) of A. mongholicus, based on comparison of their retention times and MS fragmentation patterns with those of standard compounds. As shown in Fig. 1, calycosin-7-glucoside (0.244 ± 0.085 mg/g, DW), calycosin (0.086 ± 0.036 mg/g, DW), ononin (0.0483 ± 0.026 mg/g, DW), and formononetin (0.008 ± 0.005 mg/g, DW) were abundant in AR. For triterpenoid saponins, astragaloside I (0.993 ± 0.447 mg/g, DW), astragaloside II (0.078 ± 0.023 mg/g, DW), astragaloside III (0.018 ± 0.007 mg/g, DW), and astragaloside IV (0.146 ± 0.137 mg/g, DW) were also abundant in AR.

Distribution and accumulation of four isoflavonoids and four triterpenoid saponins in root, stem, and leaves of A. mongholicus. a Heatmap of four isoflavonoids and four triterpenoid saponins. b The eight components were mainly accumulated underground. AR root, AS stem, AL leaves
Transcriptome analysis of A. mongholicus using a combined sequencing approach
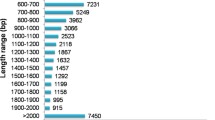
To obtain as many high-quality unigenes as possible, and differentiate the stems, leaves transcriptomes, a hybrid sequencing strategy that combined SMRT and NGS technology was used. First, nine mRNA samples from three different organs (the root, stem, and leaves) were sequenced using the DNBSEQ platform. The raw data contained low-quality reads, sequencing adapters, and unknown bases (“N” bases). These reads were removed before data analysis to ensure the reliability of the results. After quality filtering, 132.46, 133.18, and 129.68 M of clean reads were obtained from the root, stem, and leaves of A. mongholicus, respectively (Table 1). Then, full-length transcripts were reconstructed by using the PacBio Sequel platform. Overall, 14.21 Gb subreads were assembled, with a mean length of 1826 bp and an N50 length of 2191 bp (Additional file 1: Figure S1a). The ensuing analysis generated 643,812 reads of insert (ROIs), 305,998 of which were identified as full-length non-chimeric (FLNC) reads with a mean length of 1619 bp (Additional file 1: Figure S1b). We next applied Interactive Clustering and Error Correction (ICE) algorithm, combined with the Quiver program, for sequence clustering. After removal of redundant sequences using the CD-Hit program, 121,107 non-redundant transcripts with N50 value of 2124 bp were obtained (Table 2; Additional file 1: Figure S1c).
Function annotation of full-length A. mongholicus transcriptome
To obtain a putative functional annotation of A. mongholicus transcriptome, 121,107 transcripts were annotated against seven protein databases: NR (non-redundant protein sequences), NT (non-redundant nucleotide sequence), SwissProt (a manually annotated and reviewed protein sequence database), KEGG (Kyoto Encyclopedia of Genes and Genomes), KOG (Clusters of Eukaryotic Orthologous Groups), Pfam (Protein families), and GO (Gene Ontology) (Table 3). After the analysis, 36,812 (30.40%) transcripts were annotated based on the information in all these databases and 104,756 (86.50%) transcripts were annotated based on information in any of these databases. Sequences of species homologous to A. mongholicus were analyzed by aligning the obtained sequences with those in the NR database (Fig. 2a). In this manner, 30.31, 16.13, 8.67, and 8.01% sequences were mapped to the genes of Cicer arietinum (Leguminosae), Medicago truncatula (Leguminosae), Trifolium subterraneum (Leguminosae), and Glycine max (Leguminosae), respectively.

Functional annotation of transcripts of A. mongholicus among different samples. a NR homologous species distribution analysis. b GO classification of A. mongholicus transcripts. c KOG classification of A. mongholicus transcripts. d KEGG pathway annotation of A. mongholicus transcripts
Further, 74,953 transcripts were annotated using GO terms, in three major categories: molecular function, cellular component, and biological process (Fig. 2b; Additional file 2: Table S1). Most transcripts fell into the “cellular process” (30,306 transcripts), “cell” (29,746 transcripts), and “catalytic activity” (40,896 transcripts) categories. In addition, 77,196 transcripts were assigned to KOG and classified into 25 functional clusters (Fig. 2c; Additional file 3: Table S2). Among these, “general function prediction” (17,511 transcripts) was the most common annotation, followed by “signal transduction mechanisms” (12,029 transcripts), and “posttranslational modification, protein turnover, chaperones” (9,169 transcripts). After KEGG annotation, 78,294 transcripts were mapped to KEGG pathways in five functional categories (Cellular Processes, Environmental Information Processing, Genetic Information Processing, Metabolism, and Organismal Systems) (Fig. 2d; Additional file 4: Table S3). The “global and overview maps” (19,427 transcripts), “carbohydrate metabolism” (8934 transcripts), and “translation” (5904 transcripts) were the most representative pathways. Among them, 1156 transcripts were enriched in the “phenylpropanoid biosynthesis” pathway, 398 transcripts were annotated to “flavonoid biosynthesis”, and 170 transcripts were assigned to “isoflavonoid biosynthesis”. Importantly, 479 transcripts were annotated as related to “terpenoid backbone biosynthesis” and 173 transcripts were assigned to “sesquiterpenoid and triterpenoid biosynthesis”.
Identification of transcription factors (TFs)
TFs play a transient, spatially regulated role in plant development and response to stress. For TF prediction, we annotated 2455 TFs belonging to 53 different TF families in the A. mongholicus transcriptome dataset. The C3H (219 gens), GRAS (189 gens), AP2-EREBP (178 gens), MYB (177 gens), and bHLH (176 gens) TF families contained the most members (Table 4). TFs, such as those from the MYB, bHLH, AP2-EREBP, and bZIP families, play vital roles in the regulation of isoflavonoid and terpene biosynthesis in many plant species [27,28,29,30,31]. After removing from consideration the TFs, whose expression levels in the three organs analyzed were extremely low [fragments per kb per million fragments (FPKM) < 1], we identified genes for 72 MYBs, 53 bHLHs, 64 AP2-EREBPs, and 11 bZIPs. Among the 72 members of the MYB family, more than half were mainly expressed in the root. For 53 bHLH genes, 18 bHLHs showed the highest expression in the root, 16 bHLHs in the stem, and 19 bHLHs in leaves. Almost all identified members of the AP2-EREBP family showed a high expression level in the root. Finally, the expression of 5 from 11 bZIP genes was the highest in the root.
Identification of organ-specific transcripts and differentially expressed genes (DEGs)
Overall, 58,896 transcripts were simultaneously expressed in the root, stem, and leaves (Additional file 5: Figure S2a). In addition, 6087 transcripts exhibited organ-specific expression, with 2124, 1897, and 2066 transcripts specifically expressed in the root, stem, and leaves, respectively.
DEGs in different organs (AL vs. AS, AR vs. AL, and AR vs. AS comparisons) were investigated, and the results are displayed in Fig. 3a. The AR vs. AL comparison revealed 25,970 DEGs, with 12,799 genes up-regulated and 13,171 genes down-regulated (Table 5). The highest number of specific DEGs (4765) was identified in that comparison (Fig. 3), suggesting a large biological difference between the root and leaves. For the AL vs. AS comparison, 19,923 DEGs were identified, with 12,313 genes up-regulated and 7610 genes down-regulated. Further, 18,951 DEGs were identified in the AR vs. AS comparison. Among them, 12,829 genes were up-regulated and 6122 genes were down-regulated. In all comparison groups, 6035 genes were identified as DEGs, revealing that they play an important role in the metabolism in different A. mongholicus organs.

Venn diagram of DEGs in different comparisons. AR root, AS stem, AL leaves
KEGG (Fig. 4) and GO (Additional file 5: Figure S2b–d) enrichment analyses of the DEGs from different-organ comparisons were next performed to investigate the identified transcripts in detail. In the different comparisons, the widest metabolism classes were “carbohydrate metabolism”, “amino acid metabolism”, “energy metabolism”, “lipid metabolism”, and “biosynthesis of other secondary metabolites”.

Statistic of differentially expressed genes (DEGs) in different comparisons. a Root-specific gens enriched in the KEGG pathway. b Scatterplot of the top 20 KEGG pathways enrichment of DEGs in AR vs. AL. c Scatterplot of the top 20 KEGG pathways enrichment of DEGs in AR vs. AS. d Scatterplot of the top 20 KEGG pathways enrichment of DEGs in AL vs. AS. AR root, AS stem, AL leaves
Identification of DEGs involved in isoflavonoid biosynthesis
Isoflavonoids are identified almost exclusively in leguminous plants. Here, the isoflavonoid biosynthetic pathway in A. mongholicus was characterized based on the information for other legumes (Fig. 5) [32,33,34,35]. We identified 44 DEGs (FPKM > 1) encoding nine enzymes involved in isoflavonoid biosynthesis (Additional file 6: Table S4).

Heatmap of differentially expressed genes (DEGs) related to isoflavonoid biosynthesis. a Main phenylpropanoid biosynthesis and flavonoid biosynthesis of A. mongholicus. b Main isoflavonoid biosynthesis of A. mongholicus. AR root, AS stem, AL leaves
Isoflavonoids are synthesized from l-phenylalanine via a central phenylpropanoid pathway. The upstream portion of the isoflavonoid synthesis pathway is the same as that for flavonoids. For the upstream metabolism portion, 34 A. mongholicus DEGs were identified, including 12 AmPALs, two AmC4Hs, three Am4CLs, five AmCHSs, and 12 AmCHIs (Fig. 5). The expression of close to half of AmPALs in the root and stem was higher than that in the leaves. AmPAL6 showed the highest expression in the root, while AmPAL10, AmPAL3, and AmPAL11 showed the higher expression in the stem. The expression of AmPAL1, AmPAL4, and AmPAL7 was highest in the leaves. AmC4H1 showed the highest expression in the stem. AmC4H2 was predominantly expressed in the stem and rarely expressed in the leaves. Am4CL1 and Am4CL2 showed the highest expression in the stem. Am4CL3 showed a high expression in the leaves, unlike other Am4CLs. The expression of Am4CL1 in the root was higher than that of Am4CL2 and Am4CL3. AmCHS2 and AmCHS3 were mainly expressed in the root. Notably, the expression of AmCHS2 and AmCHS3 in the root was higher than that of other CHSs. AmCHS4 and AmCHS5 showed the highest expression in the stem, followed by the leaves and root. Among the 12 AmCHIs transcripts, more than half were most abundant in the root. AmCHI1, AmCHI6, AmCHI9, and AmCHI12 were expressed predominantly in the stem. In general, genes from the phenylpropanoid and flavonoid biosynthesis pathways showed a high relative expression in the root and leaves.
Isoflavonoid biosynthetic pathway is a branch of the flavonoid pathway. The following enzymes involved in the isoflavonoid pathway in A. mongholicus were identified: one AmIFSs, five AmHIDs, one Am4′-OMTs, and three AmIF7GTs. The levels of candidate transcripts were investigated in detail (Fig. 5). The genes displayed differential expression in the root, stem, and leaves of A. mongholicus. Of note, the vast majority of candidate transcripts related to isoflavonoid biosynthesis were more highly expressed in the root than in the stems and leaves, which was consistent with the isoflavonoid content of A. mongholicus. The relationship between these candidate transcript levels and the determined isoflavonoid accumulation implies that these genes play major roles in the isoflavonoid biosynthesis of A. mongholicus.
Analysis of DEGs in pathways related to triterpenoid saponin biosynthesis
Fifty-eight candidate genes encoding 16 enzymes related to triterpenoid saponin biosynthesis were identified by removing from consideration genes with extremely low expression (FPKM < 1). Among them, 44 were considered as DEGs (Additional file 7: Table S5) and were the focus of the ensuing detailed investigation.
The MVA pathway is the earliest and more traditional pathway for the synthetic biology of terpenoids. Twenty-three DEGs were identified in the MVA pathway (Fig. 6a). Half of the candidate transcripts involved in the MVA pathway were the most abundant in the root. Only a single transcript each for AmHMGCSs, AmMVKs, and AmPMKs was identified in the generated transcriptomic dataset as DEG involved in the MVA pathway. Further, these transcripts showed the same expression trends in the three plant organs analyzed, with the highest expression in the stem, followed by that in the root or leaves. Of the six AmAACTs, AmAACT4 and AmAACT6 showed a higher expression in the root than in the other two organs. However, the expression of AmAACT1 and AmAACT3 in the root was higher than that of the other AmAACTs, especially AmAACT3. These observations suggest that AmAACT3 also plays an important role in the MVA pathway of A. mongholicus. HMGR is a key enzyme in the MVA pathway; accordingly, 11 AmHMGRs were identified as DEGs in A. mongholicus. Notably, the expression levels of six AmHMGRs in the root were higher than those in the stem and leaves. Compared with other AmHMGRs, AmHMGR5 was most highly expressed in the root, which implied that the transcript plays a vital role in the MVA pathway in the A. mongholicus root. Three AmMVDs were expressed in the different organs, with the highest expression in the root, followed by that in the stem and leaves.

Heatmap of differentially expressed genes (DEGs) related to triterpenoid saponins biosynthesis. a MVA pathway. b MEP pathway. c Main sesquiterpenoid and triterpenoid biosynthesis of A. mongholicus. AR root, AS stem, AL leaves

RT-qPCR validation of 8 randomly selected genes in different organs. The actin gene was Am18S
In the transcriptomic dataset generated in the current study, 13 DEGs were identified as involved in the MEP pathway of A. mongholicus, including three AmDXSs, two AmDXRs, two AmispDs, one AmispE, one AmgcpE, and four AmispHs. As shown in Fig. 6b, almost all DEGs related to the MEP pathway showed the highest expression in the leaves and lowest expression in the root. Both the MEP and MVA pathways generate IPP and its isomer DMAPP, which are precursors for terpenoid production. Triterpenoids and sesquiterpenoids are synthesized via the MVA pathway, whereas monoterpenoids, diterpenoid, and tetraterpenoids are biosynthesized via the MEP pathway. In the current study, the A. mongholicus MVA pathway was identified as the major pathway involved in the biosynthesis of triterpenoid saponins.
The biosynthesis of triterpenoid saponins in A. mongholicus mainly involves “terpenoid backbone biosynthesis” and “sesquiterpenoid and triterpenoid biosynthesis” pathways. Six DEGs encoding three enzymes were identified in the “sesquiterpenoid and triterpenoid biosynthesis” pathway (Fig. 6c). Two candidate transcripts were identified for AmFDPSs, but only one gene was differentially expressed. AmFDPS showed similar expression in the three organs analyzed, with the highest expression in the stem. SQS and SQE play a pivotal role in the biosynthetic pathway of the carbon ring skeleton of triterpenoid saponins. Three AmSQEs showed higher expression in the root than in the stem and leaves. The two AmSQSs, AmSQS1, and AmSQS2, showed the highest expression in the stems.
Reverse transcription quantitative real-time PCR (RT-qPCR) validation of DEGs involved in flavonoid and triterpenoid saponin biosynthesis
To verify the detected gene expression levels, 10 genes were randomly selected for a real-time qPCR (RT-qPCR) experiment (Fig. 7; Additional file 8: Figure S3). The RT-qPCR data were in good agreement with the expression trends of these eight gene transcripts in the transcriptome (Fig. 7). The relative expression levels of AmCHS2, AmIF7GT3, AmIFS, AmCHI3, AmHGCMR5 and AmHGCMR11 were higher in root than those in stem or leaves. The relative expression of AmPMK and AmIDI1 was greater in stem than that in leaves or root, which was consistent with transcription data. This result proves the accuracy of the transcriptome data.
Discussion
Technological changes and advances in sequencing technology have a profound impact on transcriptome research of medicinal plants, enabling a quick, accurate, and efficient interpretation of gene-level information. Because of high-throughput, sensitivity, accuracy, and low cost, NGS is widely used as a research tool to reveal detailed information about the genes of medicinal plants [36,37,38,39]. Astragali Radix, a commonly used traditional Chinese medicine, is derived from the dried root of A. mongholicus Bunge or A. membranaceus (Fisch.) Bunge. Currently, transcriptome studies of A. mongholicus are mainly based on NGS [32, 34, 40]. Previous RNA-Seq investigations reported unigenes with an average length < 1000 bp. In another study, untargeted expressed sequence tags (EST) sequencing of A. membranaceus material yielded 9893 partial sequences [41]. Short-read sequencing often impacts the assembly of full-length transcripts. The reference genome of A. mongholicus or A. membranaceus has not yet been published, resulting in a lack of genetic information for this plant. SMRT provides a down-to-earth direct for the analysis of full-length transcripts in species without an assembled genome [42]. Further, SMRT has the advantages of single-molecule real-time sequencing and super-long reading length, which makes up for the shortcomings of NGS [43, 44]. Nonetheless, the high error rate of SMRT sequencing cannot be ignored. Therefore, a combination of NGS and SMRT is a widely recognized approach for the analysis of genes involved in the biosynthesis of secondary metabolites in plants [45,46,47]. Accordingly, we here combined NGS and SMRT sequencing to generate high-quality A. mongholicus transcriptome that is more complete/full-length of A. mongholicus transcriptome. In the current study, 643,812 CCS reads were generated, yielding 121,107 non-redundant transcript isoforms with an average length of 1619 bp and an N50 value of 2124 bp. Based on these highly accurate transcripts, 104,756 (86.50%) transcripts were successfully annotated using any of the seven databases. Of these, 16,351 transcripts might represent species-specific genes of A. mongholicus, whose function requires further exploration. Further, 2455 TFs representing 53 different TF families were identified. Comparisons of the gene expression in different tissues yielded 6035 shared DEGs.
Isoflavonoids, such as formononetin, ononin, calycosin, and calycosin-7-glucoside, are important bioactive compounds of A. mongholicus. Using UPLC-MS/MS analysis, the contents of these four isoflavonoids in the root, stem, and leaves were compared. The analysis revealed their differential accumulation in the different tissues. Nonetheless, there was a common trend of the highest isoflavonoid accumulation in the root. The tendency of active secondary compound accumulation in A. mongholicus is consistent with what has been reported previously [48, 49].
To reveal the relationship between isoflavonoid biosynthesis and accumulation in different plant organs, candidate genes responsible for isoflavonoid biosynthesis must be identified and the biosynthetic pathway characterized. Accordingly, KEGG pathway annotation revealed 1156 transcripts in the “phenylpropanoid biosynthesis” pathway, 398 transcripts in the “flavonoid biosynthesis” pathway, and 170 transcripts in the “isoflavonoid biosynthesis” pathway.
In the current study, we observed that transcripts from the same gene families exhibited different expression trends. The expression patterns of some key genes were consistent with isoflavonoid accumulation patterns in different organs. These included three PALs (AmPAL2, AmPAL6, and AmPAL12), two CHSs (AmCHS2 and AmCHS3), seven CHIs (AmCHI2, AmCHI3, AmCHI4, AmCHI5, AmCHI7, AmCHI10, and AmCHI11), one IFS (AmIFS), four HIDs (AmHID1, AmHID2, AmHID3, AmHID5), one 4′-OMT (Am4′-OMT1), and three IF7GTs (AmIF7GT1, AmIF7GT2, and AmIF7GT3). This implies that these genes play important roles in isoflavonoid biosynthesis in A. mongholicus. Of note, more genes upstream of the isoflavonoid synthesis pathway were expressed at high levels in the stem or leaves rather than in the root. By contrast, most genes in the middle and downstream pathways were expressed at high levels in the root. We further analyzed that the aboveground portion of A. mongholicus is an important biosynthesis site for phenylpropanoids and flavonoids, while the underground portion is an important biosynthesis site for isoflavonoids.
Triterpenoid saponins are important plant secondary metabolites. They have various biological activities and are widely distributed in dicotyledonous plants [50]. In the transcriptome dataset generated in the current study, 479 transcripts were annotated as related to “terpenoid backbone biosynthesis”, and 173 transcripts were assigned to “sesquiterpenoid and triterpenoid biosynthesis”. The distribution and accumulation of four triterpenoid saponins (astragaloside I, astragaloside II, astragaloside III, and astragaloside IV) in the different organs of A. mongholicus is shown in Fig. 1. The roots contained the highest levels of the analyzed triterpenoid saponins. Forty-four differentially expressed transcripts encoded for 16 enzymes involved in triterpenoid saponin biosynthesis. Some genes related to triterpenoid saponin biosynthesis were more highly expressed in the root than in the stem and leaves. Notably, the expression of HMGCRs, the key enzymes in the MVA pathway [51] (AmHMGCR1, AmHMGCR2, AmHMGCR3, AmHMGCR5, AmHMGCR6, and AmHMGCR11), was the highest in the root, in agreement with the pattern of triterpenoid saponin abundance in the different organs analyzed. Triterpenoid saponins are mainly synthesized by the MVA pathway [52]. Further, many members of the SQS and SQE families are found in plants, and some of them are key enzymes that regulate plant saponin synthesis [53]. In the current study, AmSQE1, AmSQE2, and AmSQE3 exhibited the highest expression in the root among the three organs analyzed. Hence, the above transcripts may play important roles in triterpenoid saponin biosynthesis of A. mongholicus, and can potentially be used as target markers in A. mongholicus breeding programs aimed at increasing astragaloside production.
It has been reported that MYB, bHLH, AP2-EREBP, and bZIP families of TFs regulate isoflavonoid and triterpenoid saponin biosynthesis. We identified 72 MYB unigenes, 53 bHLH unigenes, 64 AP2-EREBP unigenes, and 11 bZIP unigenes in the current study. Interestingly, more than half of MYBs and almost all AP2-EREBPs were mainly expressed in the root, the organ with the highest accumulation of isoflavones and triterpenoid saponins in A. mongholicus. This suggests that MYBs and AP2-EREBPs may play an important role in the regulation of organ-specific biosynthesis of isoflavonoids and triterpenoid saponins in A. mongholicus. In plants, TFs mainly positively regulate secondary metabolic pathways; however, the secondary metabolic network is complex, with more branches of metabolic. Inhibition of by-product branch metabolism is used as a means to efficiently increase the metabolic flow toward specific synthetic target compounds [54, 55]. Therefore, future research should focus on A. mongholicus MYBs and AP2-EREBPs, to regulate the synthesis of isoflavonoids and astragalosides in that plant.
Conclusions
In this study, the content of isoflavonoids and triterpenoid saponins in the root, stem, and leaves of A. mongholicus was determined. Further, A. mongholicus transcriptome was analyzed by a combination of short-read NGS and long-read SMRT sequencing. The analysis revealed transcripts from the two important biosynthesis pathways of A. mongholicus that were more accurate and complete than those reported previously. We also identified putative key transcripts and TFs involved in the isoflavonoid and triterpenoid saponin biosynthesis in this plant. Comprehensive transcriptome and metabolite analysis revealed gene expression and metabolite profiles in different plant organs, suggesting organ-specific biosynthesis, accumulation, and modification of isoflavonoid and triterpenoid saponin in A. mongholicus. The current study is a valuable basis for future research on gene discovery, molecular breeding, and metabolic engineering in A. mongholicus.
Materials and methods
Plant materials
Fresh roots, stems, and leaves of A. mongholicus (2-years old) were collected from Anhui University of Chinese Medicine (Anhui Province, China). Three independent biological replicates (i.e., derived from three plants) of each organ were washed with water, surface-dried, flash-frozen in liquid nitrogen, and then stored at – 80 °C until RNA and metabolite extraction.
RNA extraction, quantification, and sequencing
Total RNA was extracted from fresh samples using an improved CTAB method, according to the manufacturer’s operating manual. The RNA quality was evaluated using NanoDrop and Agilent 2100 Bioanalyzer (Thermo Fisher Scientific, MA, USA). DNBSEQ platform was used for NGS sequencing. Magnetic beads with attached oligo (dT)s were used for mRNA purification. Purified mRNA was fragmented into small pieces in a fragmentation buffer, at the appropriate temperature. Then, first-strand cDNA was generated using random hexamer-primed reverse transcription, followed by second-strand cDNA synthesis. Afterward, A-tailing mix and RNA index adapters were added and the samples were incubated to allow end-repair. The obtained cDNA fragments were then PCR-amplified. The double-stranded PCR products were heat-denatured and circularized by using a splint oligo sequence to obtain the final library. Then, raw reads were filtered to obtain clean reads by removing reads containing adaptors, poly-N stretched, or low quality reads.
For SMRT sequencing, the first-strand cDNA was synthesized from 800–1000 ng total RNA by using SMARTer PCR cDNA Synthesis Kit (Clontech, Japan). Second-strand cDNA synthesis was performed using SMARTScribe™ Reverse Transcriptase (Clontech). Double-stranded cDNA was produced by large-scale PCR using PrimeSTAR GXL DNA Polymerase (Clontech). Double-stranded cDNA fragments 0–5 kb in length were selected. The sequencing was done using PacBio Sequel sequencer. Library preparation and sequencing were done at BGI (Shenzhen, Guangdong Province, China).
Analysis of Iso-Seq data
Raw sequencing data generated by the Pacific Bioscience Sequel sequencer were processed using the SMRT analysis package version 2.3.0 (Pacific Biosciences, https://www.pacb.com/products-and-services/analytical-software/smrtanalysis). Raw polymerase reads with full pass > 0 and predicted consensus accuracy > 0.75 were selected to produce ROI. ROIs with a minimum length of 300 bp were classified into full-length non-chimeric and non-full-length transcript sequences based on whether the 5′-primer, 3′-primer, and poly-A tail were detected. The full-length sequences were processed to consensus isoforms by using the ICE algorithm and then polished using Quiver quality-aware algorithm. To improve the accuracy of full-length transcript calling, the high error rates of SMRT subreads were corrected using NGS reads by using Long-Read De Bruijn Graph Error Correction (LoRDEC) [56]. High-quality consensus isoforms (expected Quiver accuracy ≥ 0.95) from each library were merged and redundant isoforms were removed using CD-HIT [57] (parameters: -c 0.98 -T 6 -G 0 -aL 0.90 -AL 100 -aS 0.98 -AS 30) based on sequence similarity to obtain final unique full-length isoforms.
Final full-length isoforms were mapped to the NR, NT, SwissProt, KEGG (version 59), KOG, and Pfam databases by using Blast software (version 2.2.23) [58] with default parameters (E-value threshold ≤ 10–5) to obtain isoform annotations. GO annotations and functional classifications were obtained using Blast2GO program (version 2.5.0, E-value ≤ 10–5) [59] based on NR annotations. TF were predicted by mapping to PlntfDB.
DEG analysis
Clean reads from all samples were mapped to the full-length transcriptome using Bowtie2 (version 2.2.5) [60]. The expression of unigenes was then calculated by using RSEM (version 1.2.12) [61]. FPKM values were used to quantify the expression of each unigene. DEGs were identified using DEseq2 [62], at Q-value (adjust P-value) < 0.001 and fold change ≥ 2 or ≤ –2. The identified DEGs were annotated using GO terms and KEGG database with Phyper in the R package, at Q-value ≤ 0.05 [63]. The DEG FPKM values were greater than or equal to 1.
Reverse transcription quantitative real-time PCR (RT‑qPCR) analysis
The expression of 10 randomly selected genes from the transcriptome database was validated by RT-qPCR. The expression of the actin gene was used a normalization control. Primer sequences are listed in Additional file 9: Table S6. Total RNA from the root, stem, and leaves of A. mongholicus was reverse-transcribed into the first-strand cDNA using PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan). RT-qPCR analysis of gene expression was performed using three biological replicates and three technical replicates, with TB Green® Premix Ex Taq™ II (Takara) and Agilent StrataGene Mx3000P qPCR System (Agilent, USA). The relative gene expression was analyzed using the 2−ΔΔCT method.
Determination of isoflavonoid and triterpenoid saponin content
Dried and homogenized powder (0.2 g) obtained from the root, stem, or leaves of A. mongholicus was accurately weighted and suspended in 4 mL of 75% methanol. The samples were ultrasonicated at room temperature for 60 min and the supernatant collected. The solution was filtered through a 0.22 μm membrane filter, and then injected 1μL into the UPLC-MS/MS system for analysis. Each sample was analyzed in triplicate. Chromatographic separation was performed using an ExionLC AD (SCIEX, USA), an Acquity UPLC BEH C18 column (100 mm × 2.1 mm × 1.7 mm), and a C18 Pre-column (2.1 mm × 100 mm × 1.7 μm; Waters). Gradient elution (flow rate of 0.2 mL/min) with acetonitrile as solvent A and water with 0.1% formic acid as solvent B was used. The elution gradient was set as follows: 0–2 min (8–40% A), 2–5 min (40–55% B), 5–7 min (55–60% B), 7–8 min (60–80% B), and 8–10 min (80–8% B). The column temperature was maintained at 28 °C, and the column was equilibrated for 2 min between individual runs.
An AB SCIEX Triple Quad 4500 (AB SCIEX, USA) equipped with an electrospray ionization source was used to detect the MS signals. The MS detection was performed in positive ion and multiple reaction monitoring (MRM) modes, with the following parameters: capillary voltage operated at 5.5 kV; source temperature of 550 °C; nebulizer gas at 55 psi; and curtain gas at 35 psi. The cone voltage (CV) and collision energy (CE) were set to match the MRM of each marker (Additional file 10: Table S7). Data were acquired with MultiQuant software.
Availability of data and materials
The sequencing data of all sample-sequencing results have been submitted to the NCBI SRA (PRJNA681346) (https://dataview.ncbi.nlm.nih.gov/object/PRJNA681346?reviewer=cf9fmfe1blgqlaej16ksehqjrt).
Abbreviations
- CCS:
-
Circular consensus sequencing
- NR:
-
Non-redundant protein
- NT:
-
Non-redundant nucleotide sequence
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
Clusters of Eukaryotic Orthologous Groups
- Pfam:
-
Protein family
- PAL:
-
Phenylalanine ammonialyase
- C4H:
-
Trans-cinnamate 4-monooxygenase
- 4CL:
-
4-Coumarate CoA ligase
- CHS:
-
Chalcone synthase
- CHI:
-
Chalcone isomerase
- IFS:
-
2-Hydroxyisoflavanone synthase
- HID:
-
2-Hydroxyisoflavanone dehydratase
- 4’-OMT:
-
2,7,4'-Trihydroxyisoflavanone
- IF7GT:
-
Isoflavone 7-O-glucosyltransferase
- I3’H:
-
Isoflavone 3'-hydroxylase
- I2’H:
-
Isoflavone 2'-hydroxylase
- AACT:
-
Acetyl-CoA C-acetyltransferase
- HMGCS:
-
Hydroxymethylglutaryl-CoA synthase
- HMGCR:
-
Hydroxymethylglutaryl-CoA reductase
- MVK:
-
Mevalonate kinase
- PMK:
-
Phosphomevalonate kinase
- MVD:
-
Diphosphomevalonate decarboxylase
- DXS:
-
1-deoxy-D-xylulose-5-phosphate synthase
- DXR:
-
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- ispD:
-
2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase
- ispE:
-
4-diphosphocytidyl-2-C-methyl-D-erythritol kinase
- ispF:
-
2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase
- gcpE:
-
(E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase
- ispH:
-
4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase
- IDI:
-
Isopentenyl-diphosphate Delta-isomerase
- GPS:
-
Geranylgeranyl diphosphate synthase
- FDPS:
-
Farnesyl diphosphate synthase
- SQS:
-
Squalene synthase
- SQE:
-
Squalene epoxidase
- CAS:
-
Cycloartenol synthase
- UGT:
-
UDP-glycosyltransferase
References
Peng HS, Zhang HT, Peng DY, Cheng ME, Zha LP, Huang LQ. Evolution and characteristics of system, assessing quality by distinguishing features of traditional Chinese medicinal materials, of Dao-di herbs of Astragali Radix. China J Chin Mater Med. 2017;42(9):1646–51.
Li RQ, Yin MZ, Yang M, Chu SS, Han XJ, Wang MJ, Peng HS. Developmental anatomy of anomalous structure and classification of commercial specifications and grades of the Astragalus membranaceus var. mongholicus. Microsc Res Tech. 2018;81(10):1165–72.
Fu J, Wang ZH, Huang LF, Zheng SH, Wang DM, Chen SL, Zhang HT, Yang SH. Review of the botanical characteristics, phytochemistry, and pharmacology of Astragalus membranaceus (Huangqi). Phytother Res. 2014;28(9):1275–83.
Normile D. The new face of traditional Chinese medicine. Science. 2003;299(5604):188–90.
Auyeung KK, Han QB, Ko JK. Astragalus membranaceus: a review of its protection against inflammation and gastrointestinal cancers. Am J Chin Med. 2016;44(1):1–22.
Tang L, Liu Y, Wang YL, Long CL. Phytochemical analysis of an antiviral fraction of Radix astragali using HPLC-DAD-ESI-MS/MS. J Nat Med. 2010;64:182–6.
Yin MZ, Yang M, Chu SS, Li RQ, Zhao YJ, Peng HS, Zhan ZL, Sun HF. Quality analysis of different specifcation grades of Astragalus membranaceus var. mongholicus (Huangqi) from Hunyuan. Shanxi J AOAC Int. 2019;102(3):734–40.
Lee SM, Jeong JS, Kwon HJ, Hong SP. Quantification of isoflavonoids and triterpene saponins in Astragali Radix, the root of Astragalus membranaceus, via reverse-phase high-performance liquid chromatography coupled with integrated pulsed amperometric detection. J Chromatogr B. 2017;1070:76–81.
Wang SG, Li JY, Huang H, Gao W, Zhuang CL, Li B, Zhou P, Kong DY. Antihepatitis B virus activities of astragaloside IV isolated from Radix Astragali. Biol Pharm Bull. 2009;32(1):132–5.
Brenda WS. Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Bio. 2002;5(3):218–23.
Haralampidis K, Trojanowska M, Osbourn AE. Biosynthesis of triterpenoid saponins in plants. Adv Biochem Eng/Biotechnol. 2002;75(5):33–49.
Vogt T. Phenylpropanoid biosynthesis. Mol Plant. 2010;3(1):2–20.
Davies KM, Jibran R, Zhou YF, Albert NW, Brummell DA, Jordan BR, Bowman JL, Schwinn KE. The evolution of flavonoid biosynthesis: a bryophyte perspective. Front Plant Sci. 2020;11:7–28.
Keiko YS, Yasuhiro H, Ryo N. The origin and evolution of plant flavonoid metabolism. Front Plant Sci. 2020;10:943–64.
Petrussa E, Braidot E, Zancani M, Peresson C, Bertolini A, Patui S, Vianello A. Plant flavonoids—biosynthesis, transport and involvement in stress responses. Int J Mol Sci. 2013;14:14950–73.
Augustin JM, Kuzina V, Andersen SB, Bak S. Molecular activities, biosynthesis and evolution of triterpenoid saponins. Phytochemistry. 2011;72(6):435–57.
Newman JD, Chappll J. Isoprenoid biosynthesis in plant: carbon partitioning within the cytopl-asmic pathway. Crit Rev Biochem Mol Biol. 1999;34(2):95–106.
Choi D, Ward L, Bostock RM. Differential in duction and suppression of potato 3-hydroxy-3-methylglutaryl coenzyme A reduetase genes in response to Phytophthora infestans and to its elieitor arechidonic acid. Plant Cell. 1992;4(10):1333–44.
Zhao S, Wang L, Liu L, Liang Y, Sun Y, Wu J. Both the mevalonate and the non-mevalonate pathways are involved in ginsenoside biosynthesis. Plant Cell Rep. 2014;33(3):393–400.
Phillips DR, Rasbery JM, Bartel B, Matsuda SP. Biosynthetic diversity in plant triterpene cyclization. Curr Opin Plant Biol. 2006;9(3):305–14.
Tansakul P, Shibuya M, Kushiro T, Ebizuka Y. Dammarenedio-II synthase, the first dedicated enzyme for ginsenoside biosynthesis, in Panax ginseng. FEBS Lett. 2006;580(22):5143–9.
Guan S, Lu YQ. Dissecting organ-specific transcriptomes through RNA-sequencing. Plant Methods. 2013;9:42–59.
Shan CM, Wang CK, Zhang SX, Shi YY, Ma KL, Yang QS, Wu JW. Transcriptome analysis of Clinopodium gracile (Benth.) Matsum and identification of genes related to Triterpenoid Saponin biosynthesis. BMC Genomics. 2020;21(1):49–65.
Zhu FY, Chen MXI, Ye NH, Qiao WM, Gao B, Law WK, Tian Y, Zhang D, Zhang D, Liu TY, et al. Comparative performance of the BGISEQ-500 and Illumina HiSeq4000 sequencing platforms for transcriptome analysis in plants. Plant Methods. 2018;18:69–83.
Xu ZC, Luo HM, Ji AJ, Zhang X, Song JY, Chen SL. Global identification of the full-length transcripts and alternative splicing related to phenolic acid biosynthetic genes in Salvia miltiorrhiza. Front Plant Sci. 2016;7:1–10.
Xu ZC, Peters RJ, Weirather J, Luo HM, Liao BS, Zhang X, Zhu YJ, Ji AJ, Zhang B, et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015;82(6):951–61.
Zhang WW, Xu F, Cheng SY, Liao YL. Characterization and functional analysis of a MYB gene (GbMYBFL) related to flavonoid accumulation in Ginkgo biloba. Genes Genom. 2018;40:49–61.
Yang CQ, Fang X, Wu XM, Mao YB, Wang LJ, Chen XY. Transcriptional regulation of plant secondary metabolism. J Integr Plant Biol. 2012;54(10):703–12.
Keita T, Koki Y, Yasuko H, Daiki S, Ayaka C, Keiichi M, Mareshige K, Nobutaka M, Kazuki S, Toshiya M, Hikaru S. The basic helix-loop-helix transcription factor GubHLH3 positively regulates soyasaponin biosynthetic genes in Glycyrrhiza uralensis. Plant Cell Physiol. 2018;59(4):783–96.
Ji YP, Xiao JW, Shen YL, Ma DM, Li ZQ, Pu GB, Li X, Huang LL, Liu BY, Ye HC, Wang H. Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol. 2014;55(9):1592–604.
Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: an overview. Physiol Mol Biol Plants. 2013;19(3):307–21.
Chen J, Wu XT, Xu YQ, Zhong Y, Xue YL, Chen JK, Li X, Nan P. Global transcriptome analysis profiles metabolic pathways in traditional herb Astragalus membranaceus Bge. Var.. mongolicus (Bge.) Hsiao. BMC Genomics. 2015;16(S15):1–20.
Kim YB, Thwe AA, Li XH, Tuan PA, Zhao SC, Park CG, Lee JW, Park SU. Accumulation of flavonoids and related gene expressions in different organs of Astragalus mongholicus Bge. Appl Biochem Biotechnol. 2014;173(8):2076–85.
Park YJ, Thwe AA, Li XH, Kim YJ, Kim JK, Mariadhas VA, AlDhabi NA, Park SU. Triterpene and flavonoid biosynthesis and metabolic profiling of hairy roots, adventitious roots, and seedling roots of Astragalus mongholicus. J Agric Food Chem. 2015;63(40):8862–9.
Liu JP, Lan X, Lv S, Bao R, Yuan Y, Wu SQ, Quan XL. Salicylic acid involved in chilling-induced accumulation of calycosin-7-O-β-d-glucoside in Astragalus membranaceus adventitious roots. Acta Physiol Plant. 2019;41(7):120–9.
Wang CK, Peng DY, Zhu JH, Zhao DR, Shi YY, Zhang SX, Ma KL, Wu JW, Huang L. Transcriptome analysis of Polygonatum cyrtonema Hua: identifcation of genes involved in polysaccharide biosynthesis. Plant Methods. 2019;15:65–79.
Zhang HP, Chen MJ, Wen H, Wang ZH, Chen JJ, Fang L, Zhang HY, Xie ZZ, Jiang D, Cheng YJ, Xu J. Transcriptomic and metabolomic analyses provide insight into the volatile compounds of citrus leaves and flowers. BMC Plant Biol. 2020;20:7–21.
Jiang T, Zhang MD, Wen CX, Xie XL, Tian W, Wen SQ, Lu RK, Liu LD. Integrated metabolomic and transcriptomic analysis of the anthocyanin regulatory networks in Salvia miltiorrhiza Bge. flowers. BMC Plant Biol. 2020;20:349–62.
Zhou GL, Zhu P. De novo transcriptome sequencing of Rhododendron molle and identification of genes involved in the biosynthesis of secondary metabolites. BMC Plant Biol. 2020;20:414–33.
Liang JP, Li WQ, Jia XY, Zhang Y, Zhao JP. Transcriptome sequencing and characterization of Astragalus membranaceus var. mongholicus root reveals key genes involved in favonoids biosynthesis. Genes Genomics. 2020;42:901–14.
Liu XB, Ma L, Zhang AH, Zhang YH, Jiang J, Ma W, Zhang LM, Ren WC, Kong XJ. High-throughput analysis and characterization of Astragalus membranaceus transcriptome using 454 GS FLX. PLoS ONE. 2014;9(5):e95831.
Li J, Lee YH, Denton MD, Feng QJ, Rathjen JR, Qu ZP, Adelson DL. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017;3(1):17031–44.
Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, Sebra RP, Landweber LF. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell. 2014;158(5):1187–98.
Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2014;517:608–11.
Zhong FR, Huang L, Qi LM, Ma YT, Yan ZY. Full-length transcriptome analysis of Coptis deltoidea and identifcation of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms. Plant Mol Biol. 2020;102:477–99.
Rao S, Yu T, Cong X, Xu F, Cheng SY. Integration analysis of PacBio SMRT- and Illumina RNA-seq reveals candidate genes and pathway involved in selenium metabolism in hyper accumulator Cardamine violifolia. BMC Plant Biol. 2020;20:492–512.
Chen XZ, Li JR, Wang XB, Zhong LT, Tang Y, Zhou XX, Liu YT, Zhan RT, Zheng H, Chen WW, Chen LK. Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol. 2019;19:266–84.
Liu Y, Liu J, Wu KX, Guo XR, Tang ZH. A rapid method for sensitive profiling of bioactive triterpene and flavonoid from Astragalus mongholicus and Astragalus membranaceus by ultra-pressure liquid chromatography with tandem mass spectrometry. J Chromatogr B. 2018;1085:110–8.
Li Y, Guo S, Zhu Y, Yan H, Qian DW, Wang HQ, Yu JQ, Duan JA. Comparative analysis of twenty-five compounds in different parts of Astragalus membranaceus var. mongholicus and Astragalus membranaceus by UPLC-MS/MS. J Pharm Anal. 2019;9(6):392–9.
Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7(5):357–69.
Sawai S, Saito K. Triterpenoid biosynthesis and engineering in plants. Front Plant Sci. 2011;2(25):25–33.
Huang ZW, Lin JC, Cheng ZX, Xu M, Huang XY, Yang ZJ, Zheng JG. Production of dammarane-type sapogenins in rice by expressing the dammarenediol-II synthase gene from Panax ginseng C.A. Mey. Plant Sci. 2015;239:106–14.
Sun YZ, Niu YY, Xu J, Li Y, Luo HM, Zhu YJ, Liu MZ, Wu Q, Song JY, Sun C, Chen SL. Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from Panax quinquefolius. Plant Cell Tiss Org. 2013;114(2):269–77.
Lu X, Zhang L, Zhang FY, Jiang WM, Shen Q, Zhang LD, Lv ZY, Wang GF, Tang KX. AaORA, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 2013;198(4):1191–203.
Deng B, Zhang P, Ge F, et al. Enhancement of triterpenoid saponins biosynthesis in Panax notoginseng cells by co-overexpressions of 3-hydroxy-3-methylglutaryl CoA reductase and squalene synthase genes. Biochem Eng J. 2017;122(15):38–46.
Salmela L, Rivals E. LoRDEC: accurate and efficient long read error correction. Bioinformatics. 2014;30(24):3506–14.
Fu LM, Niu BF, Zhu ZW, Wu ST, Li WZ. CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics. 2012;28(23):3150–2.
George PR. Blast (basic local alignment search tool). Encyclopedia of genetics genomics proteomics & informatics. 2008.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323–39.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNAseq data with DESeq2. Genome Biol. 2014;15(12):550–71.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14.
Acknowledgements
We would like to thank the BGI Institute for assistance with experiments.
Funding
This research was supported by the National Natural Science Foundation of China (81773853, 81573543, 82073957, 81703633), and CAMS Innovation Fund for Medical Sciences (2019-I2M-5-065).
Author information
Authors and Affiliations
Contributions
MZY, LPZ and HSP conceived and designed the experiments. MZY, LPZ, SSC, TYS performed the experiments. MYZ wrote the manuscript and analyzed the data and prepared the figures and tables. All the authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Figure S1. Sequencing results of Pacbio sequel platform. a Length distribution of subreads. b The number and length distributions of FLNC reads. c The number and length distributions of non-redundant transcript isoforms.
Additional file 2
: Table S1. GO annotation of the A. mongholicus transcripts.
Additional file 3
: Table S2. KOG annotation of the A. mongholicus transcripts.
Additional file 4
: Table S3. KEGG pathway annotation of A. mongholicus.
Additional file 5
: Figure S2. Statistics of transcripts and differentially expressed genes. a Venn diagram of transcripts in different organs. b, c, d Scatterplot of the GO functional enrichment of DEGs (AR vs. AL, AR vs. AS and AL vs. AS, respectively).
Additional file 6
: Table S4. Differentially expressed genes involved in isoflavonoid biosynthesis.
Additional file 7
: Table S5. Differentially expressed genes involved in triterpenoid saponins biosynthesis.
Additional file 8
: Figure S3. RT-qPCR validation of AmAACT3, AmDXS1 in different organs. The actin gene was Am18S.
Additional file 9
: Table S6. Primers used in validation experiment of gene expression by RT-qPCR. (AR vs. AL, AR vs. AS and AL vs. AS, respectively).
Additional file 10
: Table S7. Optimized mass spectra conditions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yin, M., Chu, S., Shan, T. et al. Full-length transcriptome sequences by a combination of sequencing platforms applied to isoflavonoid and triterpenoid saponin biosynthesis of Astragalus mongholicus Bunge. Plant Methods 17, 61 (2021). https://doi.org/10.1186/s13007-021-00762-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-021-00762-1