
Abstract
Background
Pogostemon cablin (Blanco) Benth. (Patchouli) is an important aromatic and medicinal plant and widely used in traditional Chinese medicine as well as in the perfume industry. Patchoulol is the primary bioactive component in P. cablin, its biosynthesis has attracted widespread interests. Previous studies have surveyed the putative genes involved in patchoulol biosynthesis using next-generation sequencing method; however, technical limitations generated by short-read sequencing restrict the yield of full-length genes. Additionally, little is known about the expression pattern of genes especially patchoulol biosynthesis related genes in response to methyl jasmonate (MeJA). Our understanding of patchoulol biosynthetic pathway still remained largely incomplete to date.
Results
In this study, we analyzed the morphological character and volatile chemical compounds of P. cablin cv. ‘Zhanxiang’, and 39 volatile chemical components were detected in the patchouli leaf using GC-MS, most of which were sesquiterpenes. Furthermore, high-quality RNA isolated from leaves and stems of P. cablin were used to generate the first full-length transcriptome of P. cablin using PacBio isoform sequencing (Iso-Seq). In total, 9.7 Gb clean data and 82,335 full-length UniTransModels were captured. 102 transcripts were annotated as 16 encoding enzymes involved in patchouli alcohol biosynthesis. Accorded with the uptrend of patchoulol content, the vast majority of genes related to the patchoulol biosynthesis were up-regulated after MeJA treatment, indicating that MeJA led to an increasing synthesis of patchoulol through activating the expression level of genes involved in biosynthesis pathway of patchoulol. Moreover, expression pattern analysis also revealed that transcription factors participated in JA regulation of patchoulol biosynthesis were differentially expressed.
Conclusions
The current study comprehensively reported the morphological specificity, volatile chemical compositions and transcriptome characterization of the Chinese-cultivated P. cablin cv. ‘Zhanxiang’, these results contribute to our better understanding of the physiological and molecular features of patchouli, especially the molecular mechanism of biosynthesis of patchoulol. Our full-length transcriptome data also provides a valuable genetic resource for further studies in patchouli.
Similar content being viewed by others

Background
Pogostemon cablin (Blanco) Benth. (Patchouli), a widely cultivated tropical and subtropical herb of Asia, is an important medicinal plant in Lamiaceae family due to its various clinical properties, including antimicrobial [1], antioxidant [2], analgesic, anti-inflammatory [3], antimutagenic [4] and antiemetic [5]. Patchouli plant have been extensively used in several traditional medicinal systems worldwide. In the traditional Chinese medicine (TCM) system, the dried aerial parts of patchouli known as ‘Guang-Huo-Xiang’ and Pogostemonis Herba, are frequently used to remove moisture, provide relief from summer heat and exterior syndrome, prevent vomiting and stimulate the appetite [6] which are documented in Chinese Pharmacopoeia. The raw herb is also widely used in numerous Chinese herbal compound prescriptions and Chinese patent medicine owing to its significant clinical efficacy. In addition, patchouli is also of great commercial importance due to its essential oil, which can be obtained through steam distillation of the shade dried leaves and is widely applied in fragrance industries to manufacture perfumes products, soaps and cosmetic products [7]. Therefore, due to the clinical and industrial need, there is a vastly increasing demand of P. cablin.
Modern phytochemical and pharmacological studies have indicated that patchouli oil is the major constituent obtained from the leaves of P. cablin. Of which, sesquiterpenes is the most abundant, mainly the patchouli alcohol (patchoulol, PA), a tricyclic sesquiterpene. Many studies have confirmed that patchoulol is the key constituent in regulating and controlling patchouli oil quality and largely contributes to the various biological activities of patchouli oil [8,9,10,11]. However, although with great medicinal value of patchoulol in patchouli, the functional genes related to patchoulol biosynthesis have not yet been fully identified. Recently, the high throughput, high accuracy and cost-effective NGS (Next-Generation RNA Sequencing) technology was widely utilized to identify and characterize genes in many non-model plants without the aid of a reference genome [12], and had been utilized for gene characterization at the transcriptome and genome level in P. cablin [13,14,15,16]. Using Illumina HiSeq 2000 platform, He et al. reported the leaf and stem transcriptome characteristic of P. cablin in which 108,996 unigenes were identified and many genes related to secondary metabolic production were characterized, and particularly 17 candidate genes were found involved in PA biosynthesis pathway [13]. Furthermore, the draft genome and complete chloroplast genome sequences of P. cablin were also revealed based on Illumina HiSeq 2500 platform, which would promote the phylogenic, population and genetic engineering research investigation of P. cablin [15, 16]. NGS has promoted the study of patchouli, however, the relatively short length of the reads generated from NGS restrict the yield of full-length genes [17], which brings problems in gene cloning and metabolic analysis. Moreover, in some cases, the low-quality transcripts generated by short-read RNA-seq sequencing can also lead to incorrect annotation. Thus, the global terpenoid biosynthesis pathway in P. cablin remains to be fully characterized.
Fortunately, with the development of sequence technology, the third-generation long-read sequencing has overcome these limitations, as it can construct full-length transcripts without an assembly step by providing amplification-free, single-molecule sequencing. SMRT (Single molecular real-time) sequencing performed with the PacBio Iso-Seq protocol is the most popular third-generation sequencing platform in most publications to date [18]. Because of its long-read sequencing feature, Iso-Seq has been widely used in full-length transcriptome analyses in numerous non-model organisms, including medical plants like Astragalus membranaceus [19], Salvia miltiorrhiza [20] and safflower [21],etc., and has become a powerful strategy for obtaining full-length gene sequences, reconstructing biosynthesis pathways, investigating alternative splicing, and validating and improving the existing genomes. In addition, combining NGS and SMRT sequencing can provide high-quality and more complete assemblies in genome and transcriptome studies [20, 22].
Herein, we combined morphological analysis, metabolic profile and transcriptome sequencing to acquire comprehensive information of the Chinese-cultivated P. cablin cv. ‘Zhanxiang’. To achieve a more complete view of P. cablin at transcriptional level, we generated full-length transcriptome of P. cablin from leaf and stem tissues via PacBio Iso-Seq. Functional annotations of the obtained full-length transcripts were systematically carried out, and the complexity of the biological processes and metabolic pathways in P. cablin were also revealed. Additionally, jasmonate (JA) and its derivative methyl jasmonate (MeJA) were reported to be general inducers of biosynthesis of plant secondary metabolite [23], but till now little is known about the expression pattern of genes especially patchoulol biosynthesis related genes in response to methyl jasmonate (MeJA). Therefore, with the full-length transcript models as a reference data, we established the transcriptome profiling of P. cablin induced by MeJA to analyze expression pattern of genes related to patchouli alcohol biosynthesis. The present work provided a better understanding of patchouli especially the molecular mechanism of patchoulol biosynthesis. Moreover, this transcriptome data also provide a valuable genetic resource for further study in patchouli.
Results
Morphology and volatile chemical components survey of the Chinese-cultivated P. cablin
Pogostemon cablin (Blanco) Benth. (Lamiaceae) is an herbaceous plant native to southern and southeast Asia [24], it is cultivated mainly in the Philippines, Indonesia, Malaysia, India, Brazil and China [25]. Here, we identified the Chinese P. cablin cultivar ‘Zhanxiang’, which is originally from Zhanjiang city in southern China and has been cultivated there for a long time (Additional file 1: Figure S1). With respect to morphology, the plant height of this species ranges from 87.05 cm to 109.30 cm, and length/width of leaf ranges from 1.73 to 1.88. Distinct stumpy and rhombic square stems were observed for P. cablin cv. ‘Zhanxiang’; compared with another Chinese P. cablin landrace [13] in Yangjiang city and the Indonesia germplasm, this cultivar also exhibits swelling at the nodes and a rather small ramification angle (Additional file 1: Figure S1).
Patchouli oil mainly consists of volatile components, which are the important and unique perfume bases in the fragrance industry and the active ingredients in drugs. To investigate volatile constituents in P. cablin cv. ‘Zhanxiang’, volatile chemical components in leaves were adequately examined by GC-MS with a variety of solvents, including chloroform (Additional file 2: Figure S2c), ethyl alcohol, ethyl acetate and n-Hexane (Table 1). In total, 39 of the characteristic components were identified, which may maximum number isolated so far, and most of them were sesquiterpenes. Important compounds including β-patchoulene, caryophyllene, α-guaiene, seychellene, β-guaiene, δ-guaiene, spathulenol and patchoulol were identified, in accordance with previous reports. We found that trans-caryophyllene, α-guaiene, seychellene, α-patchoulene, α-bulnesene, patchoulol, and squalene were found in all extracts. Among them, patchoulol (Additional file 2: Figure S2a) was the main sesquiterpene alcohol (C15H26O) medicinal constituent in P. cablin. We also detected another important medicinal constituent pogostone (4-hydroxy-6-methyl-3-(4-methylpentanoyl)-2H-pyran-2-one) in chloroform (Additional file 2: Figure S2b). These results provided informative insight into the medicinal utilization of patchouli and revealed a foundation for the biosynthesis of natural active constituent.
Reconstruction of unique full-length transcript models of P. cablin via PacBio Iso-Seq
Full-length cDNAs from pooled poly (A) RNA of two tissues (leaf and stem) were normalized and subjected for SMRT sequencing via the PacBio Sequel system to generate a full-length transcriptome of P. cablin. In total, 8,211,525 subreads (9.7 Gb) were generated from two SMRT cells, with an average read length of 1182 bp and N50 of 2205 bp. A total of 502,101 reads of insert (ROIs) were generated, of which, 387,463 sequences with two primers and poly-A tail were identified as full-length ROIs. The full-length ROIs were subsequently classified into chimeric and non-chimeric reads using the Iso-Seq pipeline, and 379,146 were identified as full-length non-chimeric (FLNC) reads with an average read length of 1885 bp (Fig. 1a).

PacBio single-molecule long-read sequencing of P. cablin. a Summary of reads from PacBio Iso-Seq. b Comparison of quality from cluster consensus transcripts and corrected transcripts. c Length distribution of Iso-Seq consensus Transcripts and full-length UniTransModels. d Length distribution of CDS
By applying Iterative Clustering for Error Correction (ICE) algorithm and the final Arrow polishing algorithm to the above nonchimeric transcripts, we produced 251,015 full-length consensus transcripts for patchouli, including 26,155 polished high-quality (HQ, >99% accuracy) and 224,860 low-quality (LQ) transcripts. Additionally, the total length of these full-length consensus transcripts was 439,224,625 bp and was 3.6 times greater than that of the assembled unigenes via Illumina NGS in a previous study [13]. Compared with that of Illumina sequencing assembled unigenes, there were more sequences with >2000 bp length generated by Iso-Seq (Additional file 3: Table S1 and Fig. 1c). These results have demonstrated that PacBio Iso-Seq provides a pragmatic approach for producing full-length transcripts without the step of assembly, and it is an important improvement for transcriptome studies on species without a reference genome.
Performing error correction to acquire more accurate transcripts from Iso-Seq is necessary, since SMRT sequencing generates reads of several thousand bases but with a high error rate. While generating consensus transcripts via the pipeline of standard Iso-Seq bioinformatics, we applied self-correction by iterative clustering of circular-consensus reads. Afterwards, we used Proovread software to correct the errors of both HQ and LQ consensus transcripts, and the quality of the corrected bases is shown in Fig. 1b. Subsequently, CD-HIT-EST (c = 0.99) was used for further clustering to obtain the non-redundant transcripts [26], we successfully reconstructed a total of 82,335 full-length UniTransModels for pooled samples of patchouli leaves and stems. In this pipeline, we observed that the numbers of transcripts were significantly reduced, while the length distributions of the full-length UniTransModels were similar to the consensus transcripts.
Functional annotation
Transcripts in P. cablin were functionally annotated and classified via BLASTx or tBLASTx according to sequence similarities (E-value ≤1e-5) against seven different protein and nucleotide databases. A total of 65,826 (79.95%) UniTransModels were successfully matched to known proteins in at least one out of the seven databases, and the successful rates in each single database ranged from 18.27 to 71.89% (Table 2). The homologous species of P. cablin were predicted by aligning sequences to the Nr database, and the largest number of sequences was distributed in Sesamum indicum (34,662), followed by Erythranthe guttata (9827) and Dorcoceras hygrometricum (1910) (Additional file 4: Figure S3).
In functional classification, GO terms were assigned to each UniTransModels via BLAST2GO program base on annotation of Nr database. Overall, there are 15,036 transcripts were assigned into the three major categories (biological process, molecular function and cellular component; Fig. 2a). Major representatives in classification of biological processes were “cellular process” (GO:0009987) and “metabolic process” (GO:0008152). The major subgroups of cellular component were “cell” (GO:0005623) and “cell part” (GO:0004464). For molecular function category, “binding” (GO:0005488) and “catalytic activity” (GO:0003824) were represented most.

Functional annotation and classification of P. cablin UniTransModels. a GO classification of P. cablin UniTransModels. b KOG functional classification of P. cablin UniTransModels. c KEGG pathway classification of P. cablin UniTransModels
A total of 33,793 UniTransModels were assigned to 24 functional clusters in KOG. The largest three categories were “general function prediction only” (18.04%, 6095), “posttranslational modification, protein turnover, chaperones” (12.12%, 4095), “signal transduction mechanisms” (11.21%, 3787). Secondary metabolites contribute a lot to the pharmacological activity and quality of Patchouli, and 1280 (3.79%) UniTransModels were clustered into the “Secondary metabolites biosynthesis, transport and catabolism” category (Fig. 2b).
For exploration of biological functions and interactions, Patchouli UniTransModels were queried against the KEGG. Totally, 57,164 transcripts were annotated and assigned to 363 KEGG pathway annotations (KOs) which involved six functional categories (Fig. 2c). Among these pathways, the “Metabolism” pathway included the most isoforms, and represented 24.9% of our PacBio data sets; in which “Carbohydrate metabolism” and “energy metabolism” were the most represented pathways observed (both representing 3.7%). “Metabolism of terpenoids and polyketides” was assigned to 395 of these transcripts. Together, the above results from the functional annotation and classification of the UniTransModels, not only reveal a comprehensive functional characterization for the full-length P. cablin transcriptome to us, but also provide a great help in further research on gene function.
Coding sequence prediction and annotation
Protein-coding sequences (CDSs) were determined from the UniTransModels via the ANGEL pipeline. We identified a total of 32,232 CDSs, among which, 12,323 (38%) were longer than 900 nt (Fig. 1d). The CDSs were subsequently compared to the protein sequences of Viridiplantae, Arabidopsis and rice curated in UniProtKB to acquire a wide-ranging functional annotation of the P. cablin full-length transcriptome. Approximately 73.7% of the CDSs were annotated via aligning sequences from these well-curated databases (Table 3, Additional file 5: Table S2), and the remaining 1272 unannotated CDSs might represent novel P. cablin species-specific genes.
Identification of transcription factor and lncRNA
Transcription factors (TFs) and Transcription regulators (TRs), vital elements in gene regulatory networks, are prevalent in all living organisms and many different kinds of biological processes including growth, development and environmental responses [27]. By applying our sequencing data to the software iTAK, we annotated 3176 TFs belonging to 90 different families totally. Compared with those in A. thaliana and O. sativa, isoforms of the C3H, MYB-related, GRAS, B3-ARF and SBP families were more abundant in P. cablin. TRs such as SNF2, SET, AUX/IAA and Jumonji families also had a particularly higher frequency of isoforms in P. cablin (Fig. 3c).

Identification of lncRNAs and Transcription Factors (TFs) in P. cablin transcriptome. a Venn diagram of the number lncRNAs predicted by CPC, CNCI and Pfam protein structure analysis. b Length distribution of lncRNAs of P. cablin. c Comparison of the primary transcription factor families identified in P. cablin transcriptome to which in Arabidopsis thaliana and Oryza sativa
Aside from protein-coding RNAs, noncoding RNAs are a main consitiuent of the transcriptome. As key functional regulators in a wide spectrum of biological processes, lncRNAs have become an emerging important area of research in molecular biology. In this study, a total of 15,803 candidate lncRNAs were identified by three methods from 82,335 UniTransModels in P. cablin (Fig. 3a). Most (91.5%) of these lncRNAs were shorter than 1000 bp, and only 62 (0.4%) were longer than 4000 bp (Fig. 3b). Only seven of these lncRNAs have high homology with known pre-miRNAs by querying against the miRBase; among them, three lncRNAs served as precursors for miR159, two for miR166a and the rest two respectively for miR172 and miR2916 (Additional file 6: Table S3).
Identification of candidate genes involved in patchoulol biosynthesis
P. cablin is rich in terpenoids, in this study, based on the KEGG pathway assignments, we found a total of 395 transcripts were assigned to the “metabolism of terpenoids and polyketides” in our full-length sequences; among them, 152 were assigned to the terpenoid backbone (ko00900), 12 to monoterpenoid (ko00902), 47 to sesquiterpenoid and triterpenoid (ko00909) and 34 to diterpenoid biosynthesis (ko00904) (Additional file 7: Table S4).
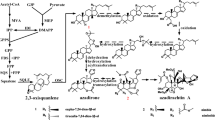
Patchoulol, the major components in patchouli oil, belongs to sesquiterpenes. Its biosynthesis includes terpenoid backbone biosynthesis and sesquiterpenoid biosynthesis pathways. All known that in plant, all terpenoids including patchouli alcohol are originated from the C5 unit isopentenyl diphosphate (IPP), which can be synthesized through the cytosolic mevalonate (MVA) pathway and plastidial methyl-erythritol phosphate (MEP) pathway. In P. cablin full-length transcriptome, 32 transcripts encoding six enzyme genes were identified to be involved in MVA pathway, including 8 AACT (acetyl-CoA acyltransferase, EC 2.3.1.9) genes, 3 HMGS (3-hydroxy-3-methylglutaryl-CoA synthase, EC 2.3.3.10) genes, 5 HMGR (3-hydroxy-3-methylglutaryl-CoA reductase, EC 1.1.1.34) genes, 8 MVK (mevalonate kinase, EC 2.7.1.36) genes, 4 PMK (phosphomevalonate kinase, EC 2.7.4.2) genes and 4 MVD (mevalonate 5-dinhophate decarboxylase, EC 4.1.1.33) genes. In addition, 39 transcripts encoding seven enzymes were also found in the MEP pathway, including 10 DXS (1-deoxy-D-xylulose 5-phosphate synthase, EC 2.2.1.7) genes, 1 DXR (1-deoxy-D-xylulose 5-phosphate reductoisomerase, EC 1.1.1.267) gene, 1 MCT (MEP cytidyltransferase, EC 2.7.7.60) gene, 3 CMK (4-(Cytidine 5-diphospho)-2-C-methylerythritol kinase, EC 2.7.1.148) genes, 4 MDS (2-C-Methy-D-erythritol 2,4-cyclodiphosphate synthase, EC 4.6.1.12) genes, 5 HDS (hydroxymethylbutenyl 4-diphosphatesynthase, EC 1.17.7.1) genes and 15 HDR (4-hydroxy-3-methylbut-2-enyldiphosphatereductase, EC 1.17.1.2) genes (Fig. 4d). In this work, with the exception of DXR and MCT, the remaining synthase-enzyme genes in the MVA and MEP pathways displayed multiple transcripts, presenting in the transcriptome as multiple paralogous genes. Additionally, we also found 8 transcripts encoding IDI (isopentenyl diphosphate isomerase, EC 5.3.3.2), which catalyzes the conversion of the common precursor IPP to DMAPP (Dimethylallyl diphosphate). Subsequently, with the catalysis of FDPS (farnesyl diphosphate synthase, EC 2.5.1.10), IPP and DMAPP are condensed to form farnesyl diphosphate (FPP), which is further catalyzed by patchoulol synthase (PatPTS) to produce patchouli alcohol. In this full-length transcriptome, 4 and 19 transcripts were identified to encode FDPS and PatPTS, respectively (Fig. 4d).

Patchoulol content and genes expression pattern in patchouli leaves induced by MeJA. a GC-MS chromatogram of leaves extracts from plant sprayed with or without MeJA. b Contents of patchoulol in leaves significantly increased after MeJA treatment. The amount of patchoulol was first calculated comparing with external standard and then dividing by the fresh weight of the samples. Error bars indicate the SD of three biological replicates. ** indicates significant difference of the means at p < 0.01 between non-treantment (CK) and MeJA-treatment leaves for each parameter mesureed (n = 3). c Abundance of DEGs in leaf tissues in response to MeJA treatment. The heat map showing genes having a fold change ratio ≥ 2, with corrected p-value<0.01. d Abundance changes of transcripts involved in the cytosolic MVA, plastidial MEP, and P. cablin patchoulol biosynthesis pathways in response to MeJA. The expressions are indicated by heat map, estimated using Log2(foldchange) value for each transcript. Magenta means high expression and green means low expression, the color gradually from green to magenta represents gene expression abundance from low to high
Expression analysis of patchoulol biosynthesis genes under MeJA treatment
Jasmonic acid (JA) is a kind of plant hormone which involving in many physiological processes of plant during growth and development, including secondary metabolism [28]. In this study, the patchoulol content of P. cablin leaves significantly increased after MeJA treatment (Fig. 4a, b). To investigate the gene expression patterns, transcriptome profiling of P. cablin leaves under MeJA treatment were further performed with a reference transcript of our P. cablin full-length transcript models, and the relative gene expression level was normalized as FPKM. Results showed that a total of 655 DEGs were identified with at least a two-fold change in response to MeJA treatment (Fig. 4c and Additional file 8: Table S5), of which 427 were up-regulated and 228 were down-regulated, indicating that more activation than suppression of genes induced by MeJA.
We further detected the expression patterns of genes related to patchoulol biosynthesis in response to MeJA treatment. As shown in Fig. 4d and Additional file 9: Table S6, most of the genes in the MVA and MEP pathways were up-regulated after MeJA treatment, in accord with the increasing tendency of patchoulol content. Particularly, transcripts encoding HMGR (HMGR_1 and HMGR_2) in the MVA pathway and encoding DXS (DXS_4, DXS_6, DXS_8) and HDR (HDR_8) in the MEP pathway were clearly up-regulated with at least two-fold change under MeJA treatment, suggesting that these genes are key regulatory factors in the biosynthesis of patchoulol in P. cablin. IDI converts the IPP to DMAPP, and then IPP and DMAPP were condensed to generate FPP, the common precursors of sesquiterpenes, by FDPS. In our gene expression profile data, the relative expression of IDI multi-transcripts were varied, and all were insensitive to MeJA. In addition, four transcripts encoding FDPS were all up-regulated in response to MeJA, although the fold change was less than two; especially, FDPS_3 had a very high expression level, which thereby becomes noteworthy. Patchoulol synthase (PatPTS) was detected as a multi-isoform that was mainly up-regulated in response to MeJA, which was also consistent with the increasing of patchoulol content. Among them, PTS_2, PTS_7, PTS_8, PTS_19 were up-regulated with a fold-change more than two; notably, PTS_3, PTS_4, PTS_5, PTS_6, PTS_9 and PTS_11 were exhibited with a very high expression level. In brief, majority of genes related to patchoulol biosynthesis were up-regulated in response to MeJA treatment, which had a positive correlation with the patchoulol content. However, experiment evidence for gene functional specialization in production of patchoulol still need further study.
Expression analysis of genes associated with the JA synthesis and signaling pathway
We also determined the transcription levels of key genes involved in the JA biosynthesis and signaling pathway as well as their expression patterns under exogenous MeJA treatment. Genes having a fold change ratio ≥ 1.5, with corrected p-value<0.05 were used for further analysis. JAR1, which converts JA into the biologically active jasmonyl-iso-leucine (JA-Ile), is the key synthases in jasmonate-amino acid synthesis. We observed that patchouli contained 21 JAR1 co-orthologs (Additional file 10: Table S7), and four of them were differently expressed under MeJA treatment (Fig. 5a). PatUTM.I-23903 (8.55) and PatUTM.I-40660 (11.97) were markly down-regulated under MeJA treatment and another two with a more than 1.5-fold up-regulated in expression were identified. CORONATINE INSENSITIVE1 (COI1), an F-box protein acting as a receptor for jasmonate, was demonstrated to directly bind to JA-Ile and coronatine (COR) [29]. We found that eight UniTransModel. Isoforms were annotated as COI1 (Additional file 11: Table S8), however, none were significantly differentially expressed. The SCFCOI1 complex recognizes JASMONATE ZIM DOMAIN (JAZ) whose degradation enables TFs (such as MYC2) release from complex. There are 14 JAZ UniTransModel. Isoforms were significantly upregulated following application of MeJA (Fig. 5a). In the present study, twelve JAZ genes were recognized (Additional file 12: Figure S4) and cloned, of which five (JAZ2, 3, 4, 6, 11) were further confirmed by qRT-PCR (Fig. 6). Our work indicated that these five JAZs may serve as transcriptional regulatory factors in the JA signaling pathway, which establishes research foundation for further dissection of the molecular mechanism clearly.

Expression profiles of genes associated with JA synthesis and signaling in patchouli induced by MeJA (foldchange > 1.5. p-value ≤0.05). a Heat map depicting abundance change of JAZ- and JAR1 co-orthologues. Blue color in the map represents low transcript abundance and red represents high level of transcript abundance. The same below. b Heat map depicting differential expression pattern of transcription factors in P. cablin induced by MeJA

Validation of expression patterns of JAZ proteins and ERFs by qRT-PCR. The relative expression levels were calculated according to the 2-ΔΔCT method using Pat18S as internal reference gene. Error bars represent standard deviations. * indicates significant difference of the means at p < 0.05 between non-treantment (CK) and MeJA-treatment leaves for each parameter mesureed (n = 3). The relative expression of qRT-PCR is indicated on the left y-axis and the normalized expression level (FPKM) of RNA sequencing is indicated on the right y-axis
TFs of the bHLH (basic/helix-loop-helix) and ERE (ethylene responsive element) families have been previously identified as key regulatory factors in plant JA signaling processes. In this study, we surveyed the bHLHs and AP2/ERFs, and results showed that 67 of them were expressed in 73 bHLH UniTransModel. Isoforms, of which including one bHLH-MYC2 homolog (PatUTM.I-27152, 2.46) was up-regulated and 8 weredown-regulated with a more than 1.5-fold change (Fig. 5b and Additional file 13 Table S9). Six of the down-regulated bHLH were PIF homologs (PatUTM.I-08185, PatUTM.I-09729, PatUTM.I-53598, PatUTM.I-61036, PatUTM.I-07628 and PatUTM.I-08186). On the other hand, we had surveyed the expression patterns of other eleven MYC homologs and found that all of them were up-regulated in response to MeJA treatment (Additional file 14: Figure S5), suggesting that these bHLH transcription factors (MYCs) might be released from JAZ-like repressors, activating transcriptional and expression levels of downstream genes which in charge of the biosynthesis of secondary metabolites. Furthermore, five AP2/ERF TFs were detected as MeJA-responsive genes: PatUTM.I-28888 was significantly upregulated while the rest four were downregulated 1.85 to 4.23 folds. (Fig. 5b and Additional file 13 Table S9). Besides, three AP2/ERF TFs were selected for qRT-PCR analysis: PatUTM.I-09997, PatUTM.I-35815 and PatUTM.I-51420 (Fig. 6). Additionally, MYB and bZIP TFs were also differentially expressed under MeJA treatment (Fig. 5b and Additional file 13 Table S9).
Discussion
Patchouli was brought into China for its great value of perfume and medicinal industries, and currently is widespread in southern China. According to the morphological characters and proteins patterns, it was classified to three cultivars as follow: Pogostemon cablin (Blanco) Benth. cv. shipaiensis, P. cablin (Blanco) Benth. cv. gaoyaoensis, and P. cablin (Blanco) Benth. cv. zhanjiangensis. P. cablin populations have evolved diverse morphological and metabolism characteristics, because of cultivation under varying environmental conditions in different localities in China. Distinct stumpy and rhombic square stem was observed in P. cablin cv. ‘Zhanxiang’, which also possessed swelling at the nodes and rather small ramification angle compared with another Chinese P. cablin landrace cultivated in yangjiang [25]. Additionally, in order to have a comprehensive understanding of chemical components in P. cablin cv. ‘Zhanxiang’, we made a survey with a variety of solvents and detected by GC-MS, results showed that thirty-nine compounds were identified in in P. cablin, which mainly were sesquiterpenes. The investigation of morphological and metabolic characteristics provides insights into its biological foundation for resource utilization.
Previous studies on P. cablin transcriptome were mainly based on NGS method, but the short reads resulting from that methodology have prohibited the accurate reconstruction of full-length reads without a reference genome sequence [13, 14]. PacBio Iso-seq, the most popular application of third-generation sequencing, has proven useful for accurately characterizing the diverse landscape of isoforms due to its advantage of obtaining full-length transcripts [30]. Utilizing this superior methodology, this study provides the first comprehensive set of full-length isoforms in P. cablin. We generated 9.7 G clean data, including 502,101 ROIs and 379,146 FLNC reads. A total of 251,015 consensus transcripts were clustered from the FLNC reads, which included 26,155 HQ isoforms. Except from self-correction, we corrected the consensus transcripts using publicly available Illumina short reads to improve the long read accuracy [31]. After removing redundant sequences, we subjected this polished consensus to a new and innovative pipeline (Cogent: Coding GENome Reconstruction Tool) to merge transcripts to partition gene families and reconstruct 82,335 full-length transcript models.
Based on these highly accurate transcripts, a total of 79.95% UniTransModels were successfully annotated to known proteins in at least one out of the seven functional databases (Nr, SwissProt, KEGG, KOG, GO, Nt and Pfam) and 73.7% of CDSs were annotated with hits in the UniProtKB. KEGG pathway annotation also revealed that 395 transcripts were assigned to the “metabolism of terpenoids and polyketides” in our full-length sequences. Additionally, 15,803 candidate lncRNA and 3176 TFs (belonging to 90 different families) were also obtained. These results in our study also indicated the high capacity of PacBio transcriptome sequencing to generate full-length transcript sequence information, which is of great use and importance for genome annotation and gene function analysis.
Extensive works as well as the chemical components survey in this study have confirmed that P. cablin is rich in sesquiterpenes, of which patchoulol is the major component and contributes largely to the clinical effects, and its synthesis pathway has attracted extensive interests. Previous studies using NGS technology have roughly mined the genes related to the synthesis pathway of patchoulol [13], however, due to the technical restriction of NGS, few of them have the full-length sequences, which may lead to the inaccurate gene annotation and restrict the gene function study. Therefore, it still needs a more accurate and comprehensively study and the full-length transcript sequence information in this present work will help a lot. It has become evident that, all terpenoids including patchoulol are derived from IPP and its allylic DMAPP using the MVA and MEP pathways [32]. In this patchouli full-length transcriptome, a total of 102 transcripts encoding enzymes related to patchoulol biosynthesis were identified, including 32 encoding six enzymes in MVA pathway, 39 encoding seven enzymes in MEP pathway, 8 encoding IDI, 4 encoding FDPS and 19 encoding PatPTS, the key enzyme directly responsible for the production of patchouli alcohol in P. cablin.
Previous studies have proved that the important plant hormone JA can remarkably improve the biosynthesis of some secondary metabolites in plant; for instance, JA up-regulated TIA biosynthesis in Catharanthus roseus [33], artemisinin biosynthesis in Artemisia annua [34, 35], and tanshinone biosynthesis in Salvia miltiorrhiza [28]. In the present work, after MeJA treatment, we also found the significant increasing of patchoulol content in P. cablin leaves. Multiple studies have manifested the enhancement of concentration of terpenoids in response to various abiotic stresses is often mediated by an increase in transcriptional activity of the specific terpenoid biosynthetic genes [36,37,38]. So we analyzed the transcriptome profiling of patchouli induced by MeJA and found that, most of genes related to the patchoulol biosynthesis were mainly up-regulated expression under MeJA treatment, which accorded with the increasing tendency of patchoulol content. Particularly, DEGs including HMGR_1, HMGR_2, DXS_ (4, 6, 8), HDR_8 and PTS_ (2, 7, 8, 19) genes were up-regulated with at least two-fold change after MeJA treatment. All know that HMGR catalyzes the conversion of HMG-CoA to mevalonate, the rate-limiting step in the MVA pathway. Previous studies have demonstrated the crucial regulatory role of HMGR in sesquiterpenoids biosynthesis, although flux control often involves additional downstream enzymes like sesquiterpene synthases [39, 40]. DXS catalyzes the first reaction in the MEP pathway, it functions as an important regulatory and rate-limiting enzyme in the biosynthesis of plastidial terpenes [32], up- or down-regulation of DXS would increase or decrease the accumulation of specific isoprenoid final products in plants such as arabidopsis [41], tomato [42] and potato [43]. HDR also plays an important role in controlling the production of MEP-derived precursors [44], the alteration of HDR gene expression level might directly influence the synthesis of IPP and DMAPP. Patchoulol synthase (PatPTS) is the key enzyme directly catalyzed the production of patchoulol, many studies have confirmed the relation between PatPTS expression level and the accumulation of patchoulol content in P. cablin [45]. In view of the important roles of the above DEGs in controlling carbon flux distribution, it indicated that they mainly participated in MeJA promoting the synthesis of patchoulol. Therefore, it was apparent that MeJA activates the expression of genes related to patchoulol biosynthesis pathway and consequently promoted the production of patchoulol in P. cablin leaves; however, the specific regulation mechanisms still need further investigation.
Furthermore, numerous studies have reported on the important roles of TFs in JA signaling pathway, TFs like the bHLH family members MYCs [35, 46] and MYBs [47, 48] as well as EFR family members AP2/ERFs [33, 49] have been identified as key regulators of JA signaling. When jasmonates accumulate and be perceived, the degradation of JAZ proteins liberates the repression of various TFs that execute JA responses. To date, some TFs regulated by the jasmonates hormone signaling cascade that activate the transcription of sesquiterpenoid biosynthesis genes have already been reported [23], however, studies on the TFs participated in JA regulation of PA biosynthesis in patchouli are scarce. In the present study, transcriptome profiling analysis of crucial genes take part in JA biosynthesis and signaling pathways revealed that, two JAR1 genes and 14 JAZ genes were up-regulated under MeJA treatment. Additionally, nine bHLHs were also markedly induced by MeJA, including one MYC which may play important regulatory roles in secondary metabolite biosynthesis. On the other hand, five AP2/ERFs, seven MYBs and 13 bZIPswere found to be MeJA-responsive TFs in P. cablin. However, additional researches on the detailed molecular mechanism still need to be carried out.
Conclusions
The current study comprehensively reported the morphological characterization, volatile chemical compositions and transcriptome characterization of the Chinese-cultivated P. cablin cv. ‘Zhanxiang’. The established full-length transcriptome provides a fully characterization of patchouli and offer a valuable genetic resource for further study in patchouli. Our MeJA-induced transcriptome profiling analysis also deepens the understanding of the MeJA regulatory mechanism on patchouli alcohol biosynthesis.
Methods
Plant material and RNA isolation
The cutting branches of Pogostemon cablin (Blanco) Benth. cv. zhangjiangensis were collected from ‘Shizhen’ Mountain, Guangzhou University of Traditional Chinese Medicine (23.03°N, 113.23°E), which are from twigs of single plant. The rooting and culturing of cutting twigs were performed in a growth chamber in the Research Center of Chinese Herbal Resource Science and Engineering, Guangzhou University of Chinese Medicine. The original plant was identified by Professor Ruoting Zhan of the Research Center of Chinese Herbal Resource Science and Engineering and a voucher specimen was deposited in the Research Center of Chinese Herbal Resource Science and Engineering.
Leaves and stems of Pogostemon cablin (Blanco) Benth. cv. zhanjiangensis were harvested, then snap-frozen via liquid nitrogen after a quick rinse by distilled water.
For hormone treatment, 300 μM MeJA (Sigma-Aldrich) was used to spray on the leaves, whereas 0.5% ethanol was used as a negative control. Leaf samples were washed and collected at 8 h after spraying with three biological replicates. All plants used in this study were at the 4-leaf stage of growth.
The GeneMark Plant Total RNA Purification Kit (GeneMarkBio, Taiwan) was used to extract RNA from patchouli tissues, and then an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) was used to measure the RNA integrity and a Qubit Fluorometer (Thermo Fisher Scientific) was used to estimate the RNA concentration. High-quality mRNA isolation was performed via the oligo d(T) magnetic beads and further processed for cDNA preparation.
cDNA construction and PacBio Iso-Seq
According to the PacBio Isoform Sequencing protocol, the Clontech SMARTer PCR cDNA Synthesis kit (Clontech, Mountain View, CA, USA) was used for cDNA synthesis and library construction. The full-length cDNA Iso-Seq template was sequenced in two SMRT cells on a PacBio Sequel System after purification, size selection, re-amplification and SMRTbell template preparation. Size selection was performed via a BluePippin (Sage Science, Beverly, MA) to construct a cDNA library of size > 4 kb. The same amount of non-selected cDNA and > 4 kb cDNA were combined to form Iso-Seq library for SMRT sequencing.
Iso-Seq data processing with standard bioinformatics pipeline
Raw sequencing data were processed using the SMRTlink4.0 software through standard Iso-Seq protocol. First, reads of insert (ROIs) were generated from subreads removing adapters and artifacts, and then classified into full-length nonchimeric (FLNC) and non-full-length (nFL) reads by detecting primer and polyA tail with ‘pbclassify.py’. Clustering of the full-length transcripts was performed via Iterative Clustering for Error Correction (ICE) algorithm. Followed by final Arrow polishing, the Quiver algorithm was used to polish and categorize the consensus sequences produced above using nFL reals. Error correction was performed via ‘proovread (2.13.12)’ [31, 50] using a Illumina RNA-seq dataset.
Full-length unique transcript model reconstruction
Redundancy isforms were removed using the program CD-HIT (v4.6.8) [26] to generate a high-quality transcripts dataset for patchouli by applying the all error-corrected full-length polished consensus transcripts. In Cogent (Coding GENome Reconstruction Tool), above non-redundant transcripts were submitted to create its k-mer profile and calculate pairwise distance, and clustered into families with their k-mer similarity. One or several unique transcript model(s) (UniTransModels) were reconstructed from each of the resultant transcript families via a De Bruijn graph method [22].
Functional annotation of UniTransModels
The obtained UniTransModels were mapped to seven databases to acquire the annotation information. We used the software BLAST and set the e-value ‘1e-5’ in Nt (Nucleotide database) database analysis and used software Hmmscan in Pfam (Protein family) database analysis. Nr (Non-Redundant Protein Database), KEGG (Kyoto Encyclopedia of Genes and Genomes), KOG (Cluster of Orthologous Group), Swiss-Prot and and GO (Gene Ontology) databases annotations were conducted using BLASTX with a cut-off e-value of ‘1e-5’.
Prediction and further annotation of coding sequences
To determine protein coding sequences (CDS) from cDNAs, UniTransModels were processed via ANGEL pipeline using this species or closely related species’ confident protein sequences for ANGEL training and prediction [51]. The predicted CDS were then used to search and confirm against three well-characterized protein databases: UniProtKB (UniProt Knowledgebase)_Viridiplantae, UniProtKB_Arabidopsis thaliana and UniProtKB_Rice (including all green plant, Arabidopsis and rice curated protein entries, respectively) via software BLASTX and set the e-value ‘1e-5′.
Identification of transcription factors and LncRNA
The iTAK software was used to identify and classify TFs (transcription factors) and TRs (transcription regulators) in P. cablin [52]. The identified transcripts of each gene family were compiled and the number was compared with those in A. thaliana and O. stativa (the data were retrieved from iTAK: Plant Transcription factor & Protein Kinase Identifier and Classifier, http://itak.feilab.net/cgi-bin/itak/index.cgi).
In this study, the transcripts were applied to the Pfam, CNCI (Coding-Non-Coding Index) and CPC (Coding Potential Calculator) databases for lncRNA prediction through screening coding potential. To predict potential miRNA precursors (pre-miRNAs), the searchable miRNA database miRBase (www.mirbase.org) was used to select hits from the candidate lncRNAs which were similar (sequence coverage > 90%) with published miRNA sequences [53, 54].
Illumina library construction and sequencing
Leaves treated with MeJA were harvested and used for total RNAs extraction. RNAs were sequenced with three biological replicates via an Illumina HiSeq 2500 platform (Illumina Inc., CA, USA), and the obtained sequences were mapped to the patchouli reference full-length UniTransModels built in this study. The amount of fragments mapping to each transcript was counted and used to determine each transcript’s relative expression level, and FPKMs (fragments per kilobase of transcript per million mapped reads) method was used to normalize the relative gene expression levels. Genes having a fold change ratio ≥ 2, with corrected p-value<0.01 were defined as differentially expressed genes (DEGs).
Determination of the volatile chemical composition using gas chromatography mass spectrometry (GC-MS)
Leaf samples of P. cablin were extracted using four different solvents, i.e. chloroform, ethyl alcohol, ethyl acetate, and n-Hexane. A HP6890/5973 GC–MS (Agilent Technologies, Palo Alto, CA, USA) was used to analyze the extracted with a split ratio of 10:1, an injection temperature of 230 °C, an interface temperature of 280 °C and ion source temperature was 230 °C. Separation of compounds was performed on DB-FFAP capillary column (30 m × 0.25 mm × 0.25 μm). The flow rate of Helium carrier gas was set at 1.0 mL∙min− 1. The oven temperature was set to 50 °C initially, then heating of 20 °C∙min− 1 to 130 °C and a final 1 min at 130 °C. Follow by ramping to 150 °C at 2 °C∙min− 1 and held for 5 min, and ramping at 20 °C∙min− 1 to 230 °C. Electron impact ionization mode of the mass spectrometric was applied to record the mass spectra at an ionizing energy of 70 eV in scanning rang of m/z 29–500. The spectra peaks were identified via WILEY7n.L (Palisade Corporation, N.Y., USA) and Nist08.L (NIST, Gaithersburg, Md., USA) databases, and data normalization were performed with fresh weight of each sample and external standard.
Patchoulol exaction and analysis
Approximately, 0.25 g of leaf samples extracted with 50 mL Ethyl acetate using an ultrasonic cleaner for 20 min, repeat two times, and then concentrated by rotary evaporation. Concentrate dissolved in hexane to a volume of 5 mL. The extraction was analyzed using Agilent 7890B Gas Chromatograph with 5977A inert Mass Selective Detector (Agilent, United States). The injection volume is 1 μl of continuous filtrate, and helium is carrier gas. Agilent HP-5MS column (30 m × 250 mm × 0.25 mm film thickness) as a column for separation. GC oven temperature was programmed at an initial temperature of 50 °C for 2 min with an increase of 20 °C/min to 130 °C, and raised to 150 °C at a rate of 2 °C / min for 5 min. And temperature was then raised to 230 °C at a rate of 20 °C / min. Quadrupole and ion source temperatures are set to 150 °C and 230 °C. NIST14/Wiley275Mass Spectral Library was used for metabolite identification. This study used the external standard method to quantify the patchouli alcohol; there were three biological replicates and two technical replicates for each tissue.
Quantitative real-time PCR
RNA extraction was performed as described above, and first-strand cDNA synthesis was used HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech Co., Ltd.). The primers in these experiments were designed via Primer Premier 5.0 and were listed in Additional file 15: Table S10. QRT-PCRs for genes were carried out with three biological replicates and two technical replicates by ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech) on CFX96 Real-Time PCR Detection System (Bio-Rad, United States). Then, the reference gene Pat18s was served as control for normalization and calculated the transcript levels of target genes via the 2-△△Ct method.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its Additional files. The PcaBio SMRT reads and the Illumina RNA-seq data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra). Accession numbers for two SMRT cells of sequencing are SRR8761881 and SRR8761882 under BioProject PRJNA528262. Accession number for Illumina cDNA libraries obtains from the control and samples of leaves treated with MeJA are SRR8756845, SRR8756846, SRR8756847, SRR8755475, SRR8755476, and SRR8755477 under BioProject PRJNA511937.
Abbreviations
- AACT:
-
Acetyl-CoA acyltransferase
- AP2/ERF:
-
APETALA2/ethylene response factor
- bHLH:
-
basic helix-loop-helix
- CDS:
-
Coding sequences
- CK:
-
Control
- CMK:
-
4-diphosphocytidyl-2-C-methyl-D-erythritol kinase
- Cogent:
-
Coding GENome Reconstruction Tool
- COR:
-
Coronatine
- DEGs:
-
Differentially expressed genes
- DMAPP:
-
Dimethylallyl pyrophosphate
- DXR:
-
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- DXS:
-
1-deoxy-D-xylulose-5-phosphate synthase
- ERF:
-
Ethylene responsive factor
- FDPS:
-
Farnesyl diphosphate synthase
- FDR:
-
False discovery rate.
- FLNC:
-
Full-length nonchimeric
- FPKMs:
-
Fragment per kilobase of exon model per million mapped reads
- FPP:
-
Farnesyl diphosphate
- HDR:
-
1-hydroxy-2-methyl-2-butenyl 4-diphosphate reductase
- HDS:
-
1-hydroxy-2-methyl-2-butenyl 4-diphosphate synthase
- HMGR:
-
3-hydroxy-3-methylglutaryl-CoA reductase
- HMGS:
-
3-hydroxy-3-methylglutaryl-CoA synthase
- ICE:
-
Iterative Clustering for Error Correction
- IDI:
-
Isopentenyl diphosphate isomerase
- JA-Ile:
-
Jasmonyl-iso-leucine
- JAZ:
-
Jasmonate ZIM domain-containing protein
- lncRNA:
-
Long non-coding RNA
- MCT:
-
MEP cytidyltransferase
- MDS:
-
2-C-Methy-D-erythritol 2,4-cyclodiphosphate synthase
- MeJA:
-
Methyl jasmonate
- MVD:
-
Mevalonate 5-dinhophate decarboxylase
- MVK:
-
Mevalonate kinase
- MYC:
-
Myelocytomatosis oncogene
- nFL:
-
non-full-length
- PatPTS:
-
Patchoulol synthase
- PMK:
-
Phosphomevalonate kinase
- ROIs:
-
Reads of insert
- TF:
-
Transcription factor
- TR:
-
Transcription regulator
- UniTransModels:
-
Unique transcript model(s)
References
Tadtong S, Puengseangdee C, Prasertthanawut S, Hongratanaworakit T. Antimicrobial constituents and effects of blended Eucalyptus, rosemary, patchouli, pine, and cajuput essential oils. Nat Prod Commun. 2016;11(2):267–70.
Wei A, Shibamoto T. Antioxidant activities and volatile constituents of various essential oils. J Agric Food Chem. 2007;55(5):1737–42.
Yoon SC, Je IG, Cui X, Park HR, Khang D, Park JS, Kim SH, Shin TY. Anti-allergic and anti-inflammatory effects of aqueous extract of Pogostemon cablin. Int J Mol Med. 2016;37(1):217–24.
Miyazawa M, Okuno Y, Nakamura S, Kosaka H. Antimutagenic activity of flavonoids from Pogostemon cablin. J Agric Food Chem. 2000;48(3):642–7.
Yang Y, Kinoshita K, Koyama K, Takahashi K, Tai T, Nunoura Y, Watanabe K. Anti-emetic principles of Pogostemon cablin (Blanco) Benth. Phytomedicine. 1999;6(2):89–93.
Committee NP: pharmacopoeia of the People's Republic of China. Beijing: Chemical Industry Press; 2010.
Swamy MK, Sinniah UR. A comprehensive review on the phytochemical constituents and pharmacological activities of Pogostemon cablin Benth.: an aromatic medicinal plant of industrial importance. Molecules. 2015;20(5):8521–47.
Hu LF, Li SP, Cao H, Liu JJ, Gao JL, Yang FQ, Wang YT. GC-MS fingerprint of Pogostemon cablin in China. J Pharm Biomed Anal. 2006;42(2):200–6.
Näf F, Decorzant R, Giersch W, Ohloff G. A Stereocontrolled access to (±)-, (−)-, and (+)-patchouli. Alcohol. 1981;64.
Donelian A, Carlson LHC, Lopes TJ, Machado RAF. Comparison of extraction of patchouli (Pogostemon cablin) essential oil with supercritical CO2 and by steam distillation. J Supercrit Fluids. 2009;48(1):15–20.
Ramya HG, Palanimuthu V, Rachna S. An introduction to patchouli (Pogostemon cablin Benth.) - a medicinal and aromatic plant: It's importance to mankind. Agricultural Engineering International : The CIGR e-journal. 2013;15(2):243–50.
Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24(3):133–41.
He Y, Deng C, Xiong L, Qin S, Peng C. Transcriptome sequencing provides insights into the metabolic pathways of patchouli alcohol and pogostone in Pogostemon cablin (Blanco) Benth. Genes and Genomics. 2016;38(11):1031–9.
Chen H, Wu Y, Yang Y, Pang Z, Zhang J, Yu J. Transcriptome data assembly and gene function annotation of young and mature Leves from Pogostemon cablin. Mol Plant Breed. 2018;16(7):2139–54.
He Y, Xiao H, Deng C, Xiong L, Yang J, Peng C. The complete chloroplast genome sequences of the medicinal plant Pogostemon cablin. Int J Mol Sci. 2016;17(6).
He Y, Xiao H, Deng C, Xiong L, Nie H, Peng C. Survey of the genome of Pogostemon cablin provides insights into its evolutionary history and sesquiterpenoid biosynthesis. Sci Rep. 2016;6:26405.
Steijger T, Abril JF, Engström PG, Kokocinski F, Abril JF, Akerman M, Alioto T, Ambrosini G, Antonarakis SE, Behr J, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10(12):1177–84.
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szczesniak MW, Gaffney DJ, Elo LL, Zhang X, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13.
Li J, Harata-Lee Y, Denton MD, Feng Q, Rathjen JR, Qu Z, Adelson DL. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017;3:17031.
Xu Z, Peters RJ, Weirather J, Luo H, Liao B, Zhang X, Zhu Y, Ji A, Zhang B, Hu S, et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015;82(6):951–61.
Chen J, Tang X, Ren C, Wei B, Wu Y, Wu Q, Pei J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genomics. 2018;19(1):548.
Zhang JJ, Su H, Zhang L, Liao BS, Xiao SM, Dong LL, Hu ZG, Wang P, Li XW, Huang ZH, et al. Comprehensive characterization for Ginsenosides biosynthesis in ginseng root by integration analysis of chemical and transcriptome. Molecules. 2017(6):22.
De Geyter N, Gholami A, Goormachtig S, Goossens A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012;17(6):349–59.
Patil SA, GBN SSB, Tembe RP, Khan RR. Impact of biofield treatment on growth and anatomical characteristics of Pogostemon cablin (Benth.). Biotechnology. 2012;11(3):154–62.
Wu L, Wu Y, Guo Q, Li S, Zhou K, Zhang J. Comparison of genetic diversity in Pogostemon cablin from China revealed by RAPD, morphological and chemical analyses. Journal of medicinal plant research. 2011;5(18):4549–59.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–2.
Saibo NJ, Lourenco T, Oliveira MM. Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann Bot. 2009;103(4):609–23.
Shi M, Zhou W, Zhang J, Huang S, Wang H, Kai G. Methyl jasmonate induction of tanshinone biosynthesis in Salvia miltiorrhiza hairy roots is mediated by JASMONATE ZIM-DOMAIN repressor proteins. Sci Rep. 2016;6:20919.
Yan J, Zhang C, Gu M, Bai Z, Zhang W, Qi T, Cheng Z, Peng W, Luo H, Nan F, et al. The Arabidopsis CORONATINE INSENSITIVE1 protein is a jasmonate receptor. Plant Cell. 2009;21(8):2220–36.
Tilgner H, Jahanbani F, Blauwkamp T, Moshrefi A, Jaeger E, Chen F, Harel I, Bustamante CD, Rasmussen M, Snyder MP. Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat Biotechnol. 2015;33:736.
Au KF, Underwood JG, Lee L, Wong WH. Improving PacBio long read accuracy by short read alignment. PLoS One. 2012;7(10):e46679.
Vranova E, Coman D, Gruissem W. Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol. 2013;64:665–700.
van der Fits L, Memelink J. ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science. 2000;289(5477):295–7.
Yu ZX, Li JX, Yang CQ, Hu WL, Wang LJ, Chen XY. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol Plant. 2012;5(2):353–65.
Shen Q, Lu X, Yan T, Fu X, Lv Z, Zhang F, Pan Q, Wang G, Sun X, Tang K. The jasmonate-responsive AaMYC2 transcription factor positively regulates artemisinin biosynthesis in Artemisia annua. New Phytol. 2016;210(4):1269–81.
Tholl D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Plant Biol. 2006;9(3):297–304.
Nagegowda DA. Plant volatile terpenoid metabolism: biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett. 2010;584(14):2965–73.
Xi Z, Bradley RK, Wurdack KJ, Wong K, Sugumarn M, Bomblies K, Rest JS, Davis CC. Horizontal transfer of expressed genes in a parasitic flowering plant. BMC Genomics. 2012;13(1):227.
Chappell J, Vonlanken C, VöGeli U. Elicitor-inducible 3-hydroxy-3-methylglutaryl coenzyme a reductase activity is required for sesquiterpene accumulation in tobacco cell suspension cultures. Plant Physiol. 1991;97(2):693–8.
Aquil S, Husaini AM, Abdin MZ, Rather GM. Overexpression of the HMG-CoA reductase gene leads to enhanced artemisinin biosynthesis in transgenic Artemisia annua plants. Planta Med. 2009;75(13):1453–8.
Estevez JM, Cantero A, Reindl A, Reichler S, Leon P. 1-deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem. 2001;276(25):22901–9.
Enfissi EM, Fraser PD, Lois LM, Boronat A, Schuch W, Bramley PM. Metabolic engineering of the mevalonate and non-mevalonate isopentenyl diphosphate-forming pathways for the production of health-promoting isoprenoids in tomato. Plant Biotechnol J. 2005;3(1):17–27.
Morris WL, Ducreux LJ, Hedden P, Millam S, Taylor MA. Overexpression of a bacterial 1-deoxy-D-xylulose 5-phosphate synthase gene in potato tubers perturbs the isoprenoid metabolic network: implications for the control of the tuber life cycle. J Exp Bot. 2006;57(12):3007–18.
Botella-Pavia P, Besumbes O, Phillips MA, Carretero-Paulet L, Boronat A, Rodriguez-Concepcion M. Regulation of carotenoid biosynthesis in plants: evidence for a key role of hydroxymethylbutenyl diphosphate reductase in controlling the supply of plastidial isoprenoid precursors. Plant J. 2004;40(2):188–99.
Yu ZX, Wang LJ, Zhao B, Shan CM, Zhang YH, Chen DF, Chen XY. Progressive regulation of sesquiterpene biosynthesis in Arabidopsis and patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol Plant. 2015;8(1):98–110.
Major IT, Yoshida Y, Campos ML, Kapali G, Xin XF, Sugimoto K, de Oliveira Ferreira D, He SY, Howe GA. Regulation of growth-defense balance by the JASMONATE ZIM-DOMAIN (JAZ)-MYC transcriptional module. New Phytol. 2017;215(4):1533–47.
Zhou M, Sun Z, Ding M, Logacheva MD, Kreft I, Wang D, Yan M, Shao J, Tang Y, Wu Y, et al. FtSAD2 and FtJAZ1 regulate activity of the FtMYB11 transcription repressor of the phenylpropanoid pathway in Fagopyrum tataricum. New Phytol. 2017;216(3):814–28.
Delgado LD, Zuniga PE, Figueroa NE, Pastene E, Escobar-Sepulveda HF, Figueroa PM, Garrido-Bigotes A, Figueroa CR. Application of a JA-Ile biosynthesis inhibitor to methyl Jasmonate-treated strawberry fruit induces upregulation of specific MBW complex-related genes and accumulation of Proanthocyanidins. Molecules. 2018;23(6).
De Boer K, Tilleman S, Pauwels L, Vanden Bossche R, De Sutter V, Vanderhaeghen R, Hilson P, Hamill JD. Goossens a: APETALA2/ETHYLENE RESPONSE FACTOR and basic helix-loop-helix tobacco transcription factors cooperatively mediate jasmonate-elicited nicotine biosynthesis. Plant J. 2011;66(6):1053–65.
Hackl T, Hedrich R, Schultz J, Förster F. Proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics. 2014;30(21):3004–11.
Shimizu K, Adachi J, Muraoka Y. ANGLE: a SEQUENCING ERRORS RESISTANT PROGRAM FOR PREDICTING PROTEIN CODING REGIONS IN UNFINISHED cDNA. J Bioinforma Comput Biol. 2006;04(03):649–64.
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, Banf M, Dai X, Martin GB, Giovannoni JJ, et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant. 2016;9(12):1667–70.
Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue:D152–7.
Joshi RK, Megha S, Basu U, Rahman MH, Kav NN. Genome wide identification and functional prediction of long non-coding RNAs responsive to Sclerotinia sclerotiorum infection in Brassica napus. PLoS One. 2016;11(7):e0158784.
Acknowledgements
We appreciate Professor Rui He (Guangzhou University of Chinese Medicine) for kind suggestions.
Funding
Financial support for this research was provided in part by a grant from the Project 81803657 supported by National Natural Science Foundation of China, the Guangdong education department key promotion platform construction project, Lingnan key laboratory of Chinese medicine resources ministry of education (2014KTSPT016), Special funds for the construction of traditional Chinese medicine in Guangdong province (No. 20181075) and the earmarked fund for Guangdong education department innovation strong school project (No. E1-KFD015181K28/2017KQNCX039). Authors declare that none of the funding bodies have any role in the design of the study and collection, analysis, and interpretation of data as well as in writing the manuscript.
Author information
Authors and Affiliations
Contributions
LKC, XZC and JRL conceived the study. HZ, XXZ, RTZ and WWC collected samples. XZC, JRL, XBW, LTZ, YT and YTL performed the experiments. XZC, JRL and LKC analyzed the data. XZC and JRL wrote the manuscript. LKC is the corresponding author and is responsible for all contact and correspondence. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Morphology characteristics of different Pogostemon cablin cultivars, including (a) P. cablin cv. ‘Zhanxiang’, (b) P. cablin cv. ‘Yangjiang’, and (c) P. cablin cv. ‘Indonesia’. (DOCX 458 kb)
Additional file 2:
Figure S2. Analyses of the volatile chemical components of P. cablin using gas chromatography mass spectrometry. a Eluted time and Mass-to-charge (m/z) of patchouli alcohol. b Eluted time and Mass-to-charge(m/z) of pogostone. c GC-MS base peak chloroform extract of P. cablin. (DOCX 433 kb)
Additional file 3:
Table S1. Comparison results between SMRT sequencing and Illumina sequencing. (XLSX 9 kb)
Additional file 4:
Figure S3. Homologous species distribution of P. cablin annotated in Nr database. (DOCX 152 kb)
Additional file 5:
Table S2. Annotation of CDS with different protein databases. (XLSX 2631 kb)
Additional file 6:
Table S3. Known RNA motifs annotated in P. cablin lncRNAs. (XLSX 8 kb)
Additional file 7:
Table S4. Summary of re-characterisation and transcription isoforms of genes annotated in pathways related to terpenoids biosynthesis. (XLSX 22 kb)
Additional file 8:
Table S5. List of DEGs in Pogostemon cablin under MeJA treatment. (XLSX 190 kb)
Additional file 9:
Table S6. MeJA-induced expression pattern of genes involved in patchoulol biosynthesis. (XLSX 33 kb)
Additional file 10:
Table S7. Annotation of JAR1 co-orthologs in Pogostemon cablin. (XLSX 9 kb)
Additional file 11:
Table S8. Eight UniTransModel. Isoforms annotated as COI1. (XLSX 9 kb)
Additional file 12:
Figure S4. Expression changes of JAZ proteins in Pogostemon cablin induced by MeJA. The heat map showing log2(FPKM+ 1) values of each protein. (DOCX 126 kb)
Additional file 13:
Table S9. Expression profiles of MeJA-responsive genes associated with JA synthesis and signaling in patchouli. (XLSX 24 kb)
Additional file 14:
Figure S5. Expression changes of PatMYCs in Pogostemon cablin induced by MeJA. (DOCX 184 kb)
Additional file 15:
Table S10. Primers used for qPCR analysis. (XLSX 9 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, X., Li, J., Wang, X. et al. Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol 19, 266 (2019). https://doi.org/10.1186/s12870-019-1884-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1884-x