
Abstract
The unique environment of the brain and retina is tightly regulated by blood–brain barrier and the blood-retinal barrier, respectively, to ensure proper neuronal function. Endothelial cells within these tissues possess distinct properties that allow for controlled passage of solutes and fluids. Pericytes, glia cells and neurons signal to endothelial cells (ECs) to form and maintain the barriers and control blood flow, helping to create the neurovascular unit. This barrier is lost in a wide range of diseases affecting the central nervous system (CNS) and retina such as brain tumors, stroke, dementia, and in the eye, diabetic retinopathy, retinal vein occlusions and age-related macular degeneration to name prominent examples. Recent studies directly link barrier changes to promotion of disease pathology and degradation of neuronal function. Understanding how these barriers form and how to restore these barriers in disease provides an important point for therapeutic intervention. This review aims to describe the fundamentals of the blood-tissue barriers of the CNS and how the use of transgenic animal models led to our current understanding of the molecular framework of these barriers. The review also highlights examples of targeting barrier properties to protect neuronal function in disease states.
Similar content being viewed by others


Introduction
The central nervous system (CNS), including the brain and retina, has a relatively high energy requirement due to neuronal activity, but in order to maintain proper neuronal function, the neural micro-environment must be rigorously controlled. These organs are highly vascularized and in the case of the retina, possess a unique vasculature structure, that provide the necessary oxygen and glucose for proper neuronal activity. To safeguard the neuronal environment, the vessels in the CNS possess well developed junctional complexes in the vascular endothelium, contributing to the formation of the blood–brain (BBB) or inner blood-retinal barrier (iBRB) controlling paracellular flux [1, 2]. In combination with active transcellular transporters, the BBB and BRB act as selective filters that regulate the flux of fluids, signaling molecules, metabolic intermediates, and cells into and out of the CNS, regulating the neural environment and distinguishing the microvasculature of the brain and retina from that of other vascular beds. The development of BBB/BRB, or barriergenesis, occurs spatially and temporally along with angiogenesis in the CNS [3]. In a wide variety of disease affecting proper neural function in the CNS including stroke, brain tumors, dementia and in the retina, diabetes, retinal vein occlusions and age-related macular degeneration, loss of the BBB and BRB contribute to disease pathogenesis [4, 5].
This review will focus on the vascular components creating the BBB and BRB although both the brain and retina possess additional membranes necessary for proper control of fluids and solute movement to the CNS. These include the choroid plexus with an epithelial layer that filters fluid generating cerebrospinal fluid (CSF) and constitutes the blood-CSF barrier [6] and the retinal pigmented epithelium adjacent to photoreceptors creating the outer BRB [7].
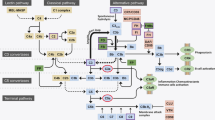
Brain and retina ECs possess certain properties that are unique and contribute to the integrity of the BBB and BRB [2]. They have continuous intercellular tight junctions (TJs) that control the passage of ions and nutrients within the paracellular space. They have limited vesicles reducing the rate of transcytosis, creating a transcellular barrier to hydrophilic molecules. Finally, and as a consequence of their limited permeability, the expression of dedicated transporters facilitates nutrient uptake such as glucose, amino acids, and metabolically relevant ions and removal of potentially neurotoxic substances and drugs. These properties of the BBB have been reviewed in detail in [2] and are depicted in Fig. 1. A recent study has shown that neural activity regulates endothelial barrier function specifically related to efflux transporters [8]. Using a transgenic approach to stimulate or inhibit glutamatergic neurons through designer receptors exclusively activated by designer drugs (DREADDs) the investigators demonstrated that BBB efflux, particularly through the transporter P-glycoprotein, is regulated by neuronal activity through endothelial circadian clock genes [8].

The Neurovascular Unit. In the CNS, ECs, pericytes, glia and neurons form an interdependency that allows for proper neuronal function and establishes a blood-tissue barrier that controls the passage of solutes and fluids. The BBB and BRB ECs possess unique characteristics that allow for this control, specifically the presence of TJs that seal the paracellular space and low rates of transcytosis. TJs are formed by long strands of transmembrane proteins including: claudins, occludin and tricellulin that are connected to the actin cytoskeleton by scaffold ZO proteins
Animal models have been indispensable in clarifying the underlying mechanisms of controlled transport, barrier permeability, cellular architecture, and related morphological changes, which have provided most of our basic knowledge about the BBB/BRB. Recent research utilizing transgenic animals has identified critical elements in the structural components of the BBB and BRB as well as elucidate signaling factors required for the developmental process that controls the formation of the BBB and BRB in the CNS. Furthermore, studies have begun to elucidate how changes to the BBB and BRB affect CNS function and contribute to disease pathogenesis and have begun to explore potential mechanisms to prevent loss of BBB and BRB or restore these barriers in order to intervene in disease processes. This review will specifically explore these transgenic mouse models used to study the development and functional significance of the BBB and BRB in normal physiology and disease focusing on what transgenic models have revealed about the protein components that make up the BBB and BRB and required pathways in barriergenesis. Finally, this review will highlight studies that elucidate the relationship of the BBB and BRB to proper neural function and the neurovascular unit.
Transport across the BBB/BRB
Tight junctions and paracellular transport
At the BBB and iBRB, signals from glial cells and pericytes induce vascular ECs to form well-developed TJs that restrict the passage of molecules through the paracellular space thus sealing the blood vessel lumen from the CNS parenchyma [9]. Additionally, TJs also have roles in numerous signaling pathways involved in their assembly, function, and polarity as well as a role in gene expression [10, 11].
The TJ complex consists of numerous interacting proteins and include transmembrane claudin family members, MARVEL (MAL and related proteins for vesicle trafficking and membrane link) proteins, and junctional adhesion molecules (JAMs), which form intercellular contacts (Fig. 1) [10]. These transmembrane proteins are linked to the actin cytoskeleton by cytoplasmic scaffold proteins including zonula occludens proteins (ZO-1, ZO-2 and ZO-3) [2]. Recent studies using gene deletion and conditional gene deletion have shed important insight into the proteins that regulate the paracellular permeability in the BBB and BRB.
Claudin family
The claudin family is considered the main structural component of the TJs, forming the intramembrane strands. They have a central role in controlling paracellular transport by forming charge specific interactions between claudins that restrict or allow particular ion diffusion and restrict solute permeability [12]. At the plasma membrane, claudins interact with each other across opposing cells (trans-interaction), with the interaction between the same claudin type being a homotypic interaction and between different claudins a heterotypic interaction [13, 14]. In addition, claudins interact within the same cell (cis-interaction) in a homomeric or heteromeric interaction [13, 14]. Structurally, the extracellular loops (ECL) of claudins determine their functionality, with the first ECL being involved in the ion selectivity of claudin strands while the second ECL seems to be important for the intercellular binding that narrows the paracellular space [14, 15]. Additionally, almost every claudin (except for claudin-12) has a PDZ binding motif at the C-terminal that allows binding to the PDZ domain of cytoplasmic scaffolding proteins such as ZO-1, determining its spatial organization [16].
The claudin family is composed of 27 members [17] and they can functionally be divided into barrier sealing or pore forming claudins. Examples of sealing claudins include claudin-1, 3 and 5 that are found in several epithelia and endothelia with a moderate to high transelectrical resistance [18, 19]; while example of pore-forming claudins include claudin-2, -10, -16, -17 and -19 [20] that allow charge and size restricted permeability. The expression and combination of specific claudin members determines size and charge-selectivity and permeability of the TJ in a tissue specific manner. During development and in response to disease stress, claudin expression patterns change modulating paracellular permeability [10, 21].
Expression and localization of claudins-1, -3, -5, and -12 at the BBB and BRB have been reported, where claudin-5 seems to be the most highly enriched [22,23,24]. However, the lack of specific claudin antibodies has led to erroneous reports regarding which claudins are expressed at the BBB and BRB. The generation of more recent knockout mice and single-cell RNA sequencing technique have allowed more direct assessment of the contribution of claudins to the BBB and BRB. Claudin-1 knockout mice die within 1 day after birth due to excessive dehydration from the skin demonstrating that claudin-1 has a key role in skin barrier formation [25]. No BBB defects were reported which has led to the suggestion that claudin-1 is not essential for BBB TJ complex function under physiological conditions. More recent data has suggested claudin-1 expression is rather associated with altered TJ function in pathology, such as ischemic stroke delaying barrier reformation [26]. Claudin-3 was thought to be enriched at the BBB; however, a recent study failed to identify claudin-3 expression in brain microvessels from wildtype animals by single cell RNA sequencing or microvessel isolation and qRT-PCR [27]. Further, claudin-3 knockout mice demonstrated that absence of claudin-3 does not impair brain barrier function [27]. A separate study showed that claudin-3 knockout mice did present with exacerbated ischemic lesion in a mouse model of stroke resulting in BBB breakdown associated with a decrease in claudin-1 expression and shorter TJ strands [28]. Claudin-12 knockout mice were recently generated and revealed that this claudin is not essential in establishing or maintaining BBB TJs [29]. Additionally, using a claudin-12 reporter mouse it was shown that this claudin has a very low expression in BBB ECs, being predominantly expressed in neurons, astrocytes and smooth muscle cells [22, 29]. Whether other claudins compensate after gene deletion of a particular claudin remains a possibility in these types of studies. But current evidence from single cell sequencing, PCR or gene marker studies have revealed under normal conditions claudins (1, 3 and 12) are either not expressed or expressed at low concentration in brain vascular endothelium and their loss by gene deletion does not alter barrier properties revealing these claudins are not necessary for the BBB.
Claudin-5 remains the one claudin clearly present and required for BBB. The importance of claudin-5 at the BBB was demonstrated by Nitta et al., revealing that claudin-5 knockout mice died shortly after birth due to loss of size-selectivity permeability of the BBB, allowing the diffusion of molecules smaller than 800 Da, but not of larger molecules [30]. Surprisingly, these mice showed no abnormal vessel development or edema, nor did they present morphologic alterations in TJ strands. Whether other claudins or TJ proteins compensate for the lack of claudin-5 in TJ formation remains unknown. Claudin-5 also seems to be the highest expressed claudin at the BRB [31]. However, since claudin-5 knockout mice die before the retinal vasculature is formed its direct role in BRB permeability has yet to be explored. Nonetheless, decreased claudin-5 expression correlates with increased BBB and BRB permeability in several models of disease [32].
MARVEL and angulin family
The TJ associated MARVEL proteins (TAMP) family includes occludin (MARVELD1), tricellulin (MARVELD2) and MARVELD3. Similar to the claudin family, they have 4 transmembrane domains with 2 ECL domains, however they cannot form TJ strands alone [33]. They are thought to enhance strength of claudin strands through cis interactions [34].
Occludin was the first transmembrane TJ protein to be described [35] and its high expression at the BBB and BRB may be a crucial determinant of the TJ permeability properties of these endothelial cell when compared to other vascular beds [36]. Surprisingly, occludin knockout mice revealed that occludin was not required for TJ strand formation as these animals presented intact BBB with morphologically normal TJs [37]. These mice exhibit complex phenotypes with hyperplasia of the gastric epithelium, brain calcification, thinning of the compact bone and male infertility [37, 38]. However, this occludin knockout mouse, thought to be a homozygous null model, still expressed a recently described occludin isoform (Ocln-ΔN) that lacks the N-terminus and the first 3 transmembrane domains [39]. Since this isoform still contains the C-terminus, which has been shown to mediate ZO interactions, localization to the TJ and essential signaling [40, 41], the definite contribution of occludin in BBB formation remains unknown. The role of occludin in regulating barrier properties and maintaining paracellular permeability has become clearer with recent research. Phosphorylation of several residues in the C-terminus region have been shown to regulate occludin interaction with ZO proteins and TJ integrity [42,43,44,45].
Tricellulin (MARVELD2) is another transmembrane protein that is present specifically in tricellular endothelial junctions in the brain and retina [46, 47]. Several studies have shown that ECs of CNS are particularly enriched with tricellulin when compared to other vascular beds [22, 23, 48]. Even though tricellulin and occludin only share 32% homology, tricellulin has been shown to compensate for the lack of occludin, relocating to bicellular TJs [46, 49], suggesting a compensatory mechanism. Additionally, mice lacking tricellulin are also viable but develop hearing loss, similar to occludin deficient mice [49,50,51]. Recently, it has been shown that tricellulin and tricellular junctions might be involved in regulating T cell entry into the CNS [48]. Furthermore, using Madine Darby Canine Kidney cells, a clear role for TAMP family members occludin and tricellulin was demonstrated by creating double knockout lines [52]. In these experiments, loss of occludin or tricellulin alone had limited effect on barrier properties. However, in the double knockout lines, electrical resistance was reduced in half and small solute flux increased fivefold associated with loss of TJ branching, observed by electron microscopy. These studies support a role for Marvel D1 and 2 having a specific role in TJ branch formation, increasing TJ complexity and strengthening barrier function. The role for occludin and tricellulin gene deletion on the BBB has yet to be established.
Lipolysis-stimulated lipoprotein receptor (LSR) (also known as angulin-1), has been shown to be necessary to recruit tricellulin to tricellular junctions [53]. However, despite limited tricellular junctions in the BBB vascular compared to epithelial monolayers, LSR has been found to be specifically expressed at the BBB and BRB [47, 54]. LSR is a type I trans-membrane proteins with an extracellular immunoglobulin-like domain. The intracellular domain is responsible for the recruitment of tricellulin to the tricellular TJ [55]. And the extracellular domain is thought to form a homophilic trimer structure at tricellular contacts [56]. LSR knockout are embryonic lethal [57] as the BBB fails to seal with a size-selective permeability defect to small molecules [54]. However, these mice do not present any ultrastructural differences in TJ strands, resembling the claudin-5 knockout [30]. These studies point to LSR as having a central role in tricellular TJ assembly and BBB barriergenesis.
MARVELD3 is the third member of the TAMP family and is also a tetraspan protein; however, it lacks the C-terminus found in occludin and tricellulin [58] and its role at the BBB/BRB remains unknown. TJs are known to regulate signaling mechanisms, and MARVELD3 was shown to be a dynamic junctional regulator of the MEKK1–c-Jun NH2-terminal kinase (JNK) pathway. Loss of MarvelD3 expression in epithelial cells resulted in increased cell migration and proliferation [59]. MARVELD3 was also identified as a regulator of ocular morphogenesis, as its depletion disrupted the normal expression pattern of eye-field transcription factors and led to the development of smaller eyes or to an eye-less phenotype [60].
JAMs
JAMs are a family of single span proteins with an extracellular immunoglobulin-like domain [61]. They interact with ZO-1 through a PDZ domain at the C-terminus and can dimerize through both cis- or trans-interaction [62, 63]. Dimerization of JAM-A is required for its interaction with the scaffolding proteins and TJs assembly [64, 65].
In brain and retinal ECs, JAM-A is the predominant JAM isoform [66, 67]. JAM family members have been shown to also regulate leukocyte adherence and transmigration by interacting with integrins expressed on leukocytes [68], endothelial cell-specific deletion of JAM-A blocked leukocyte migration through venular walls [69]. JAM-C deficient animals showed increased JAM-A expression and enhanced retinal vascularization [70, 71], suggesting that they have compensatory roles and denote a role for JAM-A in retinal angiogenesis.
Zonula occludens
Zonula occludens (ZO) are part of the MAGUK family and act as scaffold proteins that link transmembrane proteins to the actin cytoskeleton stabilizing TJ strands [72]. Three isoforms have been described, ZO-1, -2 and -3 [73,74,75]. ZO-1 and ZO-2, but not ZO-3, bind the transmembrane TJ proteins to the actin cytoskeleton at the BBB, allowing for TJ complexes to assemble [76]. At the structural levels, PDZ domains allow ZO-1 to interact with other ZO, claudins and JAMs [16, 40, 77] while the SH3-GUK domains together allow occludin interaction [41]. In addition, several cell signaling molecules and additional junction proteins and cytoskeletal proteins bind to and are organized by ZO family members [78].
The central role of ZO family in the formation of TJs has been demonstrated in cells deficient for ZO-1 and -2 which fail to form TJs with impaired claudin strand polymerization [79, 80]. ZO-1 knockout mice are embryonic lethal showing vascular defects with abnormal angiogenesis in the yolk sac [81]. ZO-2 deficient mice are also embryonic lethal, with embryos dying shortly after implantation due to increased gastrula apoptosis [82], suggesting that these proteins do not have redundant functions in early development. ZO-1 and -2 also have a role in regulating cell proliferation through binding and regulating localization of the transcription factor ZO-1 associated nucleic acid binding protein (ZONAB) [83]. ZO-1 has also been shown to regulate cell–cell tension, cell migration and angiogenesis [84].
Transcellular transport and caveolae
In ECs of the CNS small lipophilic molecules can passively diffuse across the lipid bilayer but, in many cases, are returned to the blood by efflux transporters. Larger lipophilic molecules along with hydrophilic molecules and ions are dependent on transcellular transport to cross the BBB/BRB through channels, pinocytosis, receptor and non-receptor mediated vesicular transport and excluded by efflux pumps [85, 86]. To maintain tight control of neuronal environment of the CNS, ECs have low rates of transcytosis, express low levels of transporters while expressing high levels of efflux pumps.
Some transporters are expressed early in development, such as the glucose transporter GLUT1, and its distribution pattern correlates with BBB maturation [87]. GLUT-1 haploinsufficient mice have severe defects in BBB physiology and neurovascular function, showing decreased brain glucose uptake, reduced cerebral blood flow and increased BBB permeability due to lower TJ protein expression [88, 89]. GLUT-1 deletion in ECs not only affects glucose uptake to the brain but may also alter endothelial metabolism contributing to cell dysfunction or death [90]. This suggests that loss of BBB specific transporters can ultimately influence general BBB integrity.
Transcytosis in ECs occurs primarily through caveola. Caveolae are plasma membrane invaginations consisting of lipid rafts and the proteins caveolin-1 or flotillin-1 and cavin. Caveolae vesicles undergo endocytosis and within the cytoplasm of cells can initiate signal transduction, be recycled to the plasma membrane, or transport their contents to the opposite plasma membrane [91]. Caveolin-1 knockout mice have a significant increase in BRB permeability, potentially due to compensatory flux through the paracellular pathway, specifically in branch veins, without apparent alteration in junctional proteins [92]. Plasmalemma vesicle-associated protein (PLVAP), a structural protein of caveolae in peripheral ECs is absent or has a low expression in BBB/BRB ECs [93]. However, PLVAP is up-regulated in a number of diseases characterized by increased BBB/BRB leakage and is spatially correlating with increased vascular permeability sites [94,95,96], making PLVAP a good marker for increased transcytosis specifically in CNS EC. Data suggests that during barriergenesis, transcellular and paracellular control of permeability are coordinated at the BRB with high permeability vessels possessing both high PLVAP and low claudin-5 expressing ECs [97]. In the brain, a similar mechanism is found, where circumventricular organs vessels, which physiologically lack BBB properties, have high PLVAP and low claudin-5 expressing ECs as opposed to the tight BBB vessels that have low PLVAP and high claudin-5 expressing ECs [98].
Another molecule implicated in regulating transcytosis in the CNS is the transporter major facilitator superfamily domain-containing 2a (Mfsd2a). In brain ECs, Mfsd2a transports phospholipids from the outer to the inner leaflet of the plasma membrane, including omega-3 fatty acids like docosahexaenoic acid (DHA), which is important for brain development [99]. Gene deletion studies revealed that lack of Mfsd2a leads to microcephaly, neuronal cell loss and cognitive deficits [100, 101]. These mice were also shown to have increased BBB and BRB permeability due to increased levels of transcytosis without TJ alterations [100, 102]. In contrast, other studies have failed to detect BRB breakdown in Mfsd2a deficient mice using the same molecular weight tracers [103, 104]. Besides ECs, Mfsd2a is also expressed in astrocytes which may lead to insight to resolve the contradicting studies [22]. Nonetheless, these studies did show that DHA transport by Mfsd2a was essential for long-term photoreceptor survival, denoting the importance the lipid transport by Mfsd2a in retinal homeostasis [103, 104]. Subsequent work using endothelial-specific transgenic mice with a mutation in Mfsd2a that blocks its transporter activity (Mfsd2a D96A) revealed that the increase in unsaturated phospholipids by functional Mfsd2a alters the lipid content of the plasma membrane, which inhibits the development of caveolae and hence prevents transcytosis across the BBB [105], contributing to BBB formation and maturation.
Neurovascular unit
The development and maintenance of the BBB and BRB results from the close contact of the vasculature with neurons, glial cells and pericytes and may also include microglia [106]. The term neurovascular unit (NVU) is used to describe the interdependence of these cells for proper neural signaling. The vasculature barrier and control of blood flow ensure proper neural support and maintain the required environment for neuronal function, while neurons, glia and pericytes signal to ECs to induce and maintain the unique barrier properties of the BBB and BRB and control proper blood flow (Fig. 1). Early evidence for the importance and requirement of such signaling was demonstrated through studies showing that transplantation of CNS neural tissue to the periphery can drive ectopic BBB formation [107, 108]. More recent data using transgenic mice to specifically elucidate the signaling mechanisms of BBB and BRB formation are highlighted below.
Pericytes
Pericytes are mural cells that ensheathe the abluminal surfaces of CNS capillaries and support microvessel stability and regulation of blood flow [109, 110]. In contrast with other vascular beds the ratio of pericytes to ECs is significantly higher in the CNS, with 1 pericyte:1 endothelial cell in the retina [111] and 1 pericyte: 4 endothelial cells in the brain [112], suggesting that pericytes play an important role in the BBB and BRB.
Pericytes are recruited to the CNS vasculature by release of platelet-derived growth factor-b (Pdgf-b) from nascent ECs during embryonic development, which binds to platelet-derived growth factor receptor-β (Pdgfrβ) expressed on the pericyte cell surface. This recruitment signaling was demonstrated in both Pdgfb and Pdgfrb knockout mice which completely lack CNS pericytes [113, 114]. Pericyte-deficient mice are embryonically lethal and are characterized by high vascular permeability, showing increased transcytosis as well as TJ defects, demonstrating that pericytes are required for BBB formation [115,116,117]. Additionally, decreasing pericyte recruitment without completely abolishing it revealed that adult mice present a strong correlation between pericyte density and BBB permeability [116, 117]. However, Park and colleagues recently showed that pericyte ablation in the adult mouse using conditionally expressed diphtheria toxin did not lead to BRB leakiness, suggesting that pericytes are important for initial BBB formation but not maintenance [118]. The same study also demonstrated that loss of pericytes made the retinal vessels highly susceptible to VEGF signaling, leading to increased hemorrhage and vascular permeability, suggesting that pathological pericyte loss may contribute to barrier loss and disease etiology.
In general ECs and pericytes are separated by a shared extracellular matrix (ECM); however, in certain areas, they form specialized junctions called peg and socket which allows for direct signaling between pericytes and ECs [119]. After recruitment, adhesion between ECs and pericytes is mediated by transforming growth factor-β (TGF-β) [120]. Both cell types secrete TGF-β and express its receptor TGF-βR2, which activates Smad pathway to promote transcription of target genes. TGF-β signaling promotes the production of ECM molecules by pericytes and upregulates N-cadherin, an adherens junction protein, in endothelial cells enhancing pericyte adhesion [120]. Endothelial specific deletion of Smad4 induces pericyte detachment, vascular defects, increased BBB permeability, and hemorrhage [121]. Pericytes also promote the known Ang-Tie2 angiogenic pathway by secreting Ang 1 and Ang2 that signal to Tie2 receptor in ECs. While Ang1 is known to stabilize vessels via Tie2 receptor in ECs [122], its counterpart, Ang2 can act as an antagonist of Ang1 signaling to Tie2 leading to ECs destabilization and retinal overexpression of Ang2 in mice leads to pericyte detachment and increased BRB leakage [123]. Studies using intravitreal injection of Ang2 revealed that the loss of endothelial barrier function was associated with induction of VE-Cadherin phosphorylation and degradation [124]. At the BBB, endothelial cell-specific expression of Ang2 gain-of-function was shown to increase vascular permeability via an increase in both paracellular and transcellular routes along with reduced pericyte coverage [125]. Ang2 gain-of-function mice also have exacerbated infarct sizes and vessel permeability after experimental stroke. These effects were rescued by activation of Tie2 signaling using a vascular endothelial protein tyrosine phosphatase inhibitor [125]. These studies highlight the notion that pericyte function is central in regulating BBB/BRB function. However, the factors made by pericytes that promote the BBB and BRB remain unknown.
Glia
In addition to pericytes, glial cells also contribute to the regulation of the NVU and BBB/BRB. Recent research has clarified critical signaling pathways from astrocytes that promote barrier properties. The most prevalent glial cells in the brain and retina are astrocytes as well as Müller cells in the retina. Due to their organization and spatial arrangement, glial cells are considered to be the bridge between the neurons and the vasculature, providing immunosurveillance, nutritional and regulatory support [126]. Astrocyte interaction with ECs in brain and retina may be distinct. At the BBB, astrocytes are generally thought to aid in the maintenance of BBB rather than its induction, since astrocytes initially appear at the NVU postnatally [127]. In the retina, astrocytes are present at an early stage, serving as a meshwork template for growing vessels and guiding endothelial cell migration [128, 129]. The majority of retinal astrocytes are limited to the inner vascular plexus, while Müller cells, a specialized retinal glial cell, spans the retina and interacts with all vascular plexus taking over astrocyte functions in the deeper vascular plexus [130,131,132].
Early experiments showed that transplantation of astrocytes could induce peripheral ECs to form non-leaky vessels [107] implying that glial cells express molecules that regulate barrier properties in ECs. In addition to Wnts, which will be discussed in further detail in the next section, several other astrocyte-derived signals have been identified that regulate BBB function, including sonic hedgehog, angiopoietins, angiotensin, and ApoE.
Sonic hedgehog (Shh) signaling is involved in numerous developmental processes, including neuronal differentiation, axon guidance, and angiogenesis [133]. Shh is produced by astrocytes and has also been identified as important for BBB formation. In physiological conditions, Shh binds and inhibits the EC receptor patched (Ptch) causing de-repression of the co-receptor smoothened (Smo) and consequent activation of the Gli family of transcription factors [134]. Shh knockout mice are embryonic lethal and present abnormalities in BBB formation, having decreased expression of TJ proteins occludin and claudin-5 [135]. Conditional deletion of Smo in ECs revealed that this decrease in TJs protein expression was correlated with vessel leakage, indicating a requirement for Shh signaling in BBB formation during embryonic development.
Astrocytes are also known to secrete angiotensinogen [136], a precursor of angiotensin II that signals through type 1 Ang receptor (AT1) in ECs. Activation of AT1 was shown to restrict ECs permeability through recruitment of occludin to lipid raft membrane microdomains. Accordingly, angiotensinogen deficient mice exhibit a leaky BBB with disorganized occludin strands [137].
Additionally, astrocytes are one of the main sources of ECM, critical in maintaining BBB function. Astrocyte-specific deletion of laminin induces pericyte dysfunction, loss of astrocyte end-feet coverage and BBB leakage associated with decreased expression of occludin and claudin-5 [138, 139], supporting the concept that astrocytes are important in the maintenance of BBB/NVU integrity.
Wnt/Norrin-beta-catenin signaling
The Wnt/beta-catenin signaling pathway has been identified as a unique CNS angiogenic program that also induces barrier properties in ECs [140,141,142]. Wnt molecules are secreted by neural progenitors and glial cells to induce signaling in ECs required for BBB/BRB formation.
Different Wnt signaling pathways are activated when Wnt proteins connect with high affinity to Frizzled (Fzd) receptors. The canonical Wnt signaling pathway causes beta-catenin to stabilize and accumulate in the nucleus, where it binds to transcription factors from the lymphoid enhancer factor/T cell factor (Lef/Tcf) family to induce the transcription of target genes [143].
Endothelial targeted beta-catenin gene deletion leads to specific CNS angiogenesis defects and loss of BBB properties. Besides vascular malformation, these mice show decreased expression of GLUT1, reduced border staining of claudin-5, and upregulation of PLVAP, leading to increased paracellular and transcellular BBB permeability [140,141,142]. Liebner and colleagues also showed that canonical Wnt signaling is active in ECs during brain angiogenesis and becomes progressively downregulated during vessel maturation, and that inducing beta-catenin signaling earlier is sufficient to accelerate BBB maturation [141]. Targeting astrocytic release of Wnt molecules by conditionally knocking out the Wnt secretion mediator evenness interrupted (Evi) gene leads to brain edema, increased vascular permeability and decreased coverage of brain capillaries by astrocytic end-feet, highlighting the importance of Wnt signaling from astrocytes to EC and barrier maintenance [144].
In mammals, 19 Wnt proteins have been identified and ten different Frizzled receptors have been discovered. Throughout the CNS, Wnt ligands, Frizzled receptors and co-receptors are differentially expressed, with Wnt1, Wnt3, Wnt3a, Wnt4, and Wnt7a/b having a role in BBB formation [140, 142]. Regarding Frizzled receptors, CNS ECs preferentially express Fzd4, Fzd6 and Fzd8 [140].
Wnt7a and Wnt7b have been identified as the dominant ligands in the brain cortex [140, 142] and mice lacking both Wnt7a and Wnt7b are embryonic lethal, presenting reduced angiogenesis, abnormal vascular structures and defects in BBB formation [142]. Genetic deletion of GPR124, an orphan member of the G protein-coupled receptor family expressed in ECs and pericytes revealed that these mice presented a similar phenotype to mice lacking Wnt7a and Wnt7b, displaying CNS vascular abnormalities including reduced vascular sprouting, hemorrhages, and also presenting increased Glut1 and PLVAP expression [145,146,147]. Endothelial cell specific deletion of GPR124 revealed that the effects observed in the global knockout were due to decreased signaling in ECs [147]. GPR124, together with RECK (a GPI-anchored protein) were later identified as coactivators of Wnt7a and 7b signaling through Fzd4 and LRP5/6, enhancing beta-catenin signaling dramatically [148,149,150]. Reck endothelial-specific knockout mice present a similar phenotype to EC-specific GPR124 knockout and both can be rescued by beta-catenin gain-of-function [151]. Further mechanistic insight revealed that the potentiation of Wnt signaling by GPR124 relies on dishevelled, a critical component of Wnt signaling [152]. Dishevelled recruits Gpr124 and the associated Reck-bound Wnt7 into dynamic Wnt/Reck/Frizzled/Lrp5/6 signalosomes, thus concentrating ligand availability for Fzd signaling [153]. This complex seems to contribute an essential role in brain-specific angiogenesis and BBB formation (Fig. 2).

Beta-catenin signaling in CNS ECs. In the brain Wnt7a/b binds specifically to the receptor complex Fzd4/Lrp5/Gpr124/Reck (left) and in the retina norrin binds specifically to Fzd4/Lrp5/Tspan12. Upon ligand binding the canonical Wnt signaling promotes the accumulation of beta-catenin in the cytoplasm and its translocation to the nucleus where it binds to TCF/LEF to promote the transcription of target genes that include genes promoting the induction and maintenance of the barrier
Interestingly, BRB formation requires a different, but also specific ligand/receptor complex. In the retina, Norrin, a TGF-beta family molecule, binds with high affinity to Fzd4, [154], dependent on the presence of co-receptor TSPAN12 [155] and together with LRP5/6, strongly promotes beta-catenin signaling (Fig. 2) [155,156,157]. In humans, mutations in any of these genes lead to retinal diseases including the Norrie disease and familial exudative vitreoretinopathies (FEVR), characterized by severe hypovascularization, hemorrhages and blindness [158]. These phenotypes have been recapitulated in knockout mice for Norrin (Ndp), Fzd4, LRP5 and Tspan12. These mice all lack the deep capillary beds and exhibit high vascular permeability, revealing that this signaling complex is required for both retinal angiogenesis and BRB formation [97, 155, 156, 159]. Gene deletion of either Ndp or Fzd4 showed that the increase in vascular permeability was correlated with a reduction in claudin-5 at the cell border and an increase in PLVAP, suggesting that this signaling is required to restrict both the transcellular and paracellular pathways [159]. Importantly, these phenotypes could be rescued by expressing stabilized beta-catenin, demonstrating the requirement of canonical Wnt signaling for retinal barriergenesis. Recently, it was proposed that norrin signaling also plays a role in limiting BRB transcytosis through direct transcriptional activation of Mfsd2a. LRP5 and Ndp knockout mice present high retinal vascular permeability with increased levels of caveolin-1 induced transcytosis together with low levels of Mfsd2a, a known repressor of caveolin-1 mediated transcytosis. Over expression of Mfsd2a in Lrp5 knockout retinas was able to restore transcytosis levels back to normal [160].
Wnt/beta-catenin signaling has also been shown to be necessary for BBB/BRB integrity in mature ECs. Adult mice with Fzd4 deletion or anti-Frizzled 4 blocking antibody showed similar alterations to those observed in Ndp and Fzd4 developmental knockout, with high PLVAP expression and permeability of the BBB/BRB [159, 161]. Additionally, disruption of endothelial-cell specific beta-catenin alone induced BBB breakdown and downregulation of the TJs proteins claudin-1 and -3 in adult mice [162]. Conversely, beta-catenin gain-of-function in FEVR models is sufficient to restore BRB integrity postnatally [141, 149, 159]. Therefore, beta-catenin signaling is required not only for the induction of BRB and BBB characteristics during development, but also for barrier maintenance.
Importantly, adult BBB maintenance requires a continuous and low beta-catenin signal [162]. Recently, Sox17, a known angiogenesis inducer was identified as an important signaling molecule in regulating beta-catenin activity in adult brain ECs. Sox17 is also required for BBB formation and acts in concert with Wnt/beta-catenin signaling. Beta-catenin signaling increases Sox17 that, in turn, maintains the correct beta-catenin activity after vascular development [163]. Further, additional studies have shown that prolonged high levels of beta-catenin signaling lead to alterations in vascular stability [97, 164, 165] including alpha-catenin mutations in humans leading to overactivation of beta-catenin and a FEVR phenotype [165]. Together, these data indicate that Wnt/beta-catenin signaling is required for BBB/BRB formation and maintenance and that beta-catenin must be appropriately regulated for barrier maintenance.
BBB and neural function
Multiple reviews have cited the requirement of a functional and intact BBB/BRB to maintain the microenvironment required for proper neural signaling and numerous studies correlate breakdown of BBB integrity with neurological dysfunction (Fig. 3). For example, expression of claudin-5 is decreased in the brains of depressed patients, and it positively correlates with resilience to social stress in mice [166] and in a mouse model of depression, transient reduction of claudin-5 levels with siRNA exacerbates depression-like behaviors, while chronic antidepressant treatment rescued claudin-5 loss and promoted resilience [166]. Similarly, claudin-5 protein levels are significantly decreased in surgically resected brain tissue from patients with treatment-resistant epilepsy who present widespread BBB disruption by MRI [167]. In mice, targeted disruption of claudin-5 in the hippocampus exacerbates the excitotoxin kainic acid-induced seizures and BBB disruption, while upregulating claudin-5 expression was sufficient to reduce kainic acid-evoked seizures [167]. Alzheimer’s disease has also been associated with changes in BBB permeability, but the relationship is not simple (reviewed in [168]). However, only recently has modern genetic approaches been used to elucidate precisely how loss of the BBB/BRB impacts neural function or how restoration of the BBB may prevent or restore loss of neural function. For example, claudin-5 gene deletion in the adult mouse brain was shown to be sufficient to induce a psychosis like-behavioral phenotype, with impairments in learning and memory, anxiety-like behavior and sensorimotor gating [169]. Persistent suppression of claudin-5 in these mice led to the development of seizures and 100% rate mortality within 40 days [167, 169]. These studies both emphasize the crucial role of functional TJs in normal neurological function but also demonstrate that loss of barrier properties may be sufficient to induce disease phenotype.

Neurovascular unit breakdown. In several disease states, such as stroke or diabetic retinopathy, a number of factors contribute to BBB breakdown and both paracellular and transcellular transport are increased. Loss of pericytes and gliosis further exacerbate the increase in permeability leading to infiltration of harmful substances to neurons and causing neuronal dysfunction. The process of barrier loss and neural degeneration is hypothesized to lead to a feed-forward pathology extending the disease process. Methods that target barrier breakdown may halt this process
Recent studies have also highlighted the importance of maintaining vascular endothelial TJs in preserving visual function in models of diabetic retinopathy. In retinal ECs, occludin phosphorylation at S490 [170] promotes its ubiquitination and internalization leading to TJ disassembly and vascular permeability [171,172,173]. Endothelial specific expression of the non-phosphorylatable alanine mutant (S490A) is able to prevent diabetes-induced loss of BRB integrity by preventing TJ disassembly [174]. Importantly, prevention of diabetes-induced permeability resulted in preservation of visual function, as measured by visual acuity and contrast sensitivity, highlighting the importance of early protection of the BRB in diabetes and the relationship of the barrier to visual function [174]. A second study has demonstrated the role of occludin phosphorylation at S490 as a major regulator of tissue plasminogen activator (tPA)-induced BBB permeability in the context of stroke [175]. Preventing occludin phosphorylation either genetically using vascular endothelial specific expression of the stable, S490A non-phosphorylatable form of occludin, or by inhibiting PKCbeta, blocked stroke-induced permeability while decreasing stroke volume and improving functional outcome. Importantly, blocking the increase in BBB permeability with the S490A expression in the vascular endothelium or by providing PKCbeta inhibitor 5 h after stroke, was also able to prevent intracerebral hemorrhage induced by late-thrombolytic therapy [175]. Late tPA induced hemorrhage is associated with high mortality [176] and PKCbeta treatment may provide a means to allow late tPA thrombolytic activity without barrier breakdown.
Others have demonstrated the value of preserving the BBB in stroke. Vascular endothelial conditional expression of a constitutively active mutant form of actin depolymerizing factor (ADFm) or cofilin, reducing actin polymerization leads to reduced permeability after stroke and reduced TJ disassembly at endothelial cell border [177]. Importantly, preserving the BBB significantly improved neurological outcome in the stroke mice [177]. A second study using vascular endothelial conditional expression of heat shock protein 27 (HSP27), which also prevents actin polymerization, also demonstrated reduced BBB permeability and improved neurologic outcome after stroke [178].
Analysis of poststroke human and mouse blood microvessels have identified claudin-1 as highly expressed in leaky brain microvessels [26]. This newly synthesized claudin-1 was shown to be incorporated into the TJ complex with established interaction with ZO-1 and building homophilic cis- and trans-interactions [26]. Claudin-5 strands, both homophilic cis- and trans-interactions, and the claudin-5/ZO-1 interaction were reduced when claudin-1 was introduced into the TJ complex. This altered claudin-5 incorporation into the TJ complex prevents BBB recovery causing BBB permeability. Using a specific peptide that targets claudin-1 reduced BBB permeability after stroke and consequently improved neurological recovery [26].
Recent studies have also targeted microRNAs that regulate TJs expression. Specifically, miR-15a/16-1 cluster directly binds to claudin-5 3′UTR, decreasing its expression and endothelial cell-specific deletion of this cluster in mice was able to increase claudin-5 expression, leading to improvement of BBB dysfunction and post-stroke functional outcome [179]. Additionally, miR-501-3p was shown to directly bind to the 3′UTR region of ZO-1 to decrease its expression [180]. Administration of a locked nucleic acid-modified antisense oligonucleotide targeting miR-501-3p was able to rescue ZO-1 expression and BBB disruption in a mouse model of vascular cognitive impairment, significantly improving working memory deficits [180].
Considering that barrier function also regulates the exchange of nutrients and waste with the parenchyma, a reduction in permeability may also have a detrimental impact on neuronal activity.
Inducing beta-catenin signaling in the leaky vessels of the circumventricular organs converts them into BBB-like vessels by increasing claudin5 and decreasing PLVAP expression, overall reducing vascular permeability [98]. Endothelial tightness was accompanied by an increase in neuronal activity in that region; however, this non-physiological increase presented detrimental effects in basic behaviors [98].
In Alzheimer's disease (AD), which is characterized by the accumulation of neurotoxic amyloid-β (Aβ) peptides leading to a gradual decline in cognitive function [181], the impairment of BBB ability to properly promote Aβ clearance has been shown to have pathological effects on neuronal function and contribute to AD progression. Reduced levels of glucose transporter GLUT1 were reported in brain microvessels of AD patients and murine models [89]. Transgenic mice overexpressing human amyloid β-peptide showed accelerated Aβ accumulation in the brain when Glut1 was conditionally deleted from ECs [89]. These mice presented increased progressive neuronal dysfunction, behavioral deficits, neuronal loss and neurodegeneration [89]. GLUT1 deficiency effects in reducing Aβ-clearance and accelerating AD pathology were due to consequent reduced expression of LRP1, a key Aβ clearance transporter, in the microvasculature [89]. LRP1-mediated transcytosis has been shown to contribute to Aβ-clearance [182]. Endothelial-specific deletion of Lrp1 showed reduced brain efflux of injected Aβ-peptides in wild-type mice. In a transgenic mouse model of AD, this conditional deletion of Lrp1 lead to a reduction of plasma Aβ levels and elevated soluble brain Aβ, resulting in a decline of spatial learning and memory deficits [182]. In a separate study, blocking Lrp1 expression with antisense mRNA resulted in similar accumulation of radiolabeled Aβ-peptides in the brain and impaired learning and memory [183]. Targeting restoration of LRP1 or GLUT1 expression may lead to increased LRP1-mediated Aβ clearance, representing a novel strategy for AD therapy. Indeed, additional efflux transporters such as P-glycoprotein may also be targets to promote Aβ clearance [184].
Recent studies have shown that targeting the paracellular route might also enhance Aβ clearance. In a murine model of familial AD suppression of the TJ proteins claudin-5 and occludin by systemic siRNA allowed for diffusion of soluble human Aβ monomers from the brain to the blood [185]. These animals presented increased plasma levels of Aβ monomers and reduced brains levels of neurotoxic Aβ with a concomitant improvement in cognitive function [185]. Importantly, the authors reported that siRNA to claudin-5 and occludin did not induce changes in BBB permeability to large plasma proteins such as immunoglobulins that could have a detrimental effect.
Targeting paracellular permeability has also been tested in the context of cerebral edema and brain swelling, which are associated with higher risk of permanent brain damage and mortality. Administration of siRNA against claudin-5 post induction of a cold-induced cerebral edema allowed for enhanced exchange of accumulated extra-neural water, decreasing brain-swelling, and improving the cognitive outcome after the focal injury [186]. The increase in BBB permeability was achieved in a temporary and size-selective manner [186]. A similar approach for the use of siRNA targeting claudin-5 for clearance of edema from the optic nerve head by intravitreal injection has been proposed by the same group [187] but has yet to be tested. Collectively, these studies emphasize the importance of the BRB and BBB and demonstrate that targeting endothelial vascular permeability can improve neurologic outcome.
Conclusion
Transgenic approaches have proven to be highly valuable in understanding the complex multi-cellular interaction necessary for formation of the BBB and BRB and have provided critical insight into required factors for proper barriergenesis. The use of transgenic mice has demonstrated the importance of the BBB and BRB for proper neural function in a physiological setting. These studies have also begun to elucidate how manipulation of the barrier, either by promoting tightening of the barrier or permeability, may provide important therapeutic options to address diseases of the CNS. Future studies using stem cells and organ culture systems may provide critical information on the factors sufficient to promote formation of the BBB and provide new avenues to quickly address fundamental questions regarding the NVU. However, the use of transgenic animals in the study of the BBB and BRB promises to continue to provide important physiological insight and novel understanding of the NVU.
Availability of data and materials
Not applicable.
References
Bauer H, Traweger A. Tight junctions of the blood-brain barrier—a molecular gatekeeper. CNS Neurol Disord Drug Targets. 2016;15(9):1016–29.
Diaz-Coranguez M, Ramos C, Antonetti DA. The inner blood-retinal barrier: cellular basis and development. Vision Res. 2017;139:123–37.
Umans RA, Henson HE, Mu F, Parupalli C, Ju B, Peters JL, et al. CNS angiogenesis and barriergenesis occur simultaneously. Dev Biol. 2017;425(2):101–8.
O'Leary F, Campbell M. The blood-retina barrier in health and disease. FEBS J. 2021.
Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: Important unanswered questions. J Exp Med. 2020;217(4).
Solar P, Zamani A, Kubickova L, Dubovy P, Joukal M. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17(1):35.
Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol. 2021;17(4):195–206.
Pulido RS, Munji RN, Chan TC, Quirk CR, Weiner GA, Weger BD, et al. Neuronal activity regulates blood-brain barrier efflux transport through endothelial circadian genes. Neuron. 2020;108(5):937–52.
Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79(10):707–17.
Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016;17(9):564–80.
Takano K, Kojima T, Sawada N, Himi T. Role of tight junctions in signal transduction: an update. EXCLI J. 2014;13:1145–62.
Furuse M, Tsukita S. Claudins in occluding junctions of humans and flies. Trends Cell Biol. 2006;16(4):181–8.
Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778(3):631–45.
Piontek J, Winkler L, Wolburg H, Muller SL, Zuleger N, Piehl C, et al. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 2008;22(1):146–58.
Colegio OR, Van Itallie CM, McCrea HJ, Rahner C, Anderson JM. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol. 2002;283(1):C142–7.
Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. 1999;147(6):1351–63.
Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011;585(4):606–12.
Piontek J, Fritzsche S, Cording J, Richter S, Hartwig J, Walter M, et al. Elucidating the principles of the molecular organization of heteropolymeric tight junction strands. Cell Mol Life Sci. 2011;68(23):3903–18.
Ohtsuki S, Yamaguchi H, Katsukura Y, Asashima T, Terasaki T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J Neurochem. 2008;104(1):147–54.
Gunzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol. 2012;2(3):1819–52.
Goncalves A, Ambrosio AF, Fernandes R. Regulation of claudins in blood-tissue barriers under physiological and pathological states. Tissue Barriers. 2013;1(3): e24782.
Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554(7693):475–80.
Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS ONE. 2010;5(10): e13741.
Luo Y, Xiao W, Zhu X, Mao Y, Liu X, Chen X, et al. Differential expression of claudins in retinas during normal development and the angiogenesis of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2011;52(10):7556–64.
Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156(6):1099–111.
Sladojevic N, Stamatovic SM, Johnson AM, Choi J, Hu A, Dithmer S, et al. Claudin-1-dependent destabilization of the blood-brain barrier in chronic stroke. J Neurosci. 2019;39(4):743–57.
Castro Dias M, Coisne C, Lazarevic I, Baden P, Hata M, Iwamoto N, et al. Claudin-3-deficient C57BL/6J mice display intact brain barriers. Sci Rep. 2019;9(1):203.
Winkler L, Blasig R, Breitkreuz-Korff O, Berndt P, Dithmer S, Helms HC, et al. Tight junctions in the blood-brain barrier promote edema formation and infarct size in stroke—ambivalent effects of sealing proteins. J Cereb Blood Flow Metab. 2021;41(1):132–45.
Castro Dias M, Coisne C, Baden P, Enzmann G, Garrett L, Becker L, et al. Claudin-12 is not required for blood-brain barrier tight junction function. Fluids Barriers CNS. 2019;16(1):30.
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–60.
Ohtsuki S, Sato S, Yamaguchi H, Kamoi M, Asashima T, Terasaki T. Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J Cell Physiol. 2007;210(1):81–6.
Greene C, Hanley N, Campbell M. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS. 2019;16(1):3.
Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. 1998;143(2):391–401.
Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9(7):268–73.
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6 Pt 2):1777–88.
Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110(Pt 14):1603–13.
Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11(12):4131–42.
Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669(1):34–42.
Bendriem RM, Singh S, Aleem AA, Antonetti DA, Ross ME. Tight junction protein occludin regulates progenitor Self-Renewal and survival in developing cortex. Elife. 2019;8.
Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S, et al. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127(6 Pt 1):1617–26.
Tash BR, Bewley MC, Russo M, Keil JM, Griffin KA, Sundstrom JM, et al. The occludin and ZO-1 complex, defined by small angle X-ray scattering and NMR, has implications for modulating tight junction permeability. Proc Natl Acad Sci U S A. 2012;109(27):10855–60.
Dorfel MJ, Westphal JK, Bellmann C, Krug SM, Cording J, Mittag S, et al. CK2-dependent phosphorylation of occludin regulates the interaction with ZO-proteins and tight junction integrity. Cell Commun Signal. 2013;11(1):40.
Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, et al. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A. 2009;106(1):61–6.
Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193(3):565–82.
Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, et al. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J Biol Chem. 2009;284(3):1559–69.
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171(6):939–45.
Iwamoto N, Higashi T, Furuse M. Localization of angulin-1/LSR and tricellulin at tricellular contacts of brain and retinal endothelial cells in vivo. Cell Struct Funct. 2014;39(1):1–8.
Castro Dias M, Odriozola Quesada A, Soldati S, Bosch F, Gruber I, Hildbrand T, et al. Brain endothelial tricellular junctions as novel sites for T cell diapedesis across the blood-brain barrier. J Cell Sci. 2021;134(8).
Kitajiri S, Katsuno T, Sasaki H, Ito J, Furuse M, Tsukita S. Deafness in occludin-deficient mice with dislocation of tricellulin and progressive apoptosis of the hair cells. Biol Open. 2014;3(8):759–66.
Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, et al. Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet. 2006;79(6):1040–51.
Kamitani T, Sakaguchi H, Tamura A, Miyashita T, Yamazaki Y, Tokumasu R, et al. Deletion of tricellulin causes progressive hearing loss associated with degeneration of cochlear hair cells. Sci Rep. 2015;5:18402.
Saito AC, Higashi T, Fukazawa Y, Otani T, Tauchi M, Higashi AY, et al. Occludin and tricellulin facilitate formation of anastomosing tight-junction strand network to improve barrier function. Mol Biol Cell. 2021;32(8):722–38.
Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, Akashi M, et al. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J Cell Sci. 2011;124(Pt 4):548–55.
Sohet F, Lin C, Munji RN, Lee SY, Ruderisch N, Soung A, et al. LSR/angulin-1 is a tricellular tight junction protein involved in blood-brain barrier formation. J Cell Biol. 2015;208(6):703–11.
Nakatsu D, Kano F, Taguchi Y, Sugawara T, Nishizono T, Nishikawa K, et al. JNK1/2-dependent phosphorylation of angulin-1/LSR is required for the exclusive localization of angulin-1/LSR and tricellulin at tricellular contacts in EpH4 epithelial sheet. Genes Cells. 2014;19(7):565–81.
Kim NK, Higashi T, Lee KY, Kim AR, Kitajiri S, Kim MY, et al. Downsloping high-frequency hearing loss due to inner ear tricellular tight junction disruption by a novel ILDR1 mutation in the Ig-like domain. PLoS ONE. 2015;10(2): e0116931.
Mesli S, Javorschi S, Berard AM, Landry M, Priddle H, Kivlichan D, et al. Distribution of the lipolysis stimulated receptor in adult and embryonic murine tissues and lethality of LSR-/- embryos at 12.5 to 14.5 days of gestation. Eur J Biochem. 2004;271(15):3103–14.
Steed E, Rodrigues NT, Balda MS, Matter K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009;10:95.
Steed E, Elbediwy A, Vacca B, Dupasquier S, Hemkemeyer SA, Suddason T, et al. MarvelD3 couples tight junctions to the MEKK1-JNK pathway to regulate cell behavior and survival. J Cell Biol. 2014;204(5):821–38.
Vacca B, Sanchez-Heras E, Steed E, Balda MS, Ohnuma SI, Sasai N, et al. MarvelD3 regulates the c-Jun N-terminal kinase pathway during eye development in Xenopus. Biol Open. 2016;5(11):1631–41.
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142(1):117–27.
Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem. 2000;275(27):20520–6.
Arrate MP, Rodriguez JM, Tran TM, Brock TA, Cunningham SA. Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J Biol Chem. 2001;276(49):45826–32.
Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19(5):1862–72.
Monteiro AC, Luissint AC, Sumagin R, Lai C, Vielmuth F, Wolf MF, et al. Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol Biol Cell. 2014;25(10):1574–85.
Aurrand-Lions M, Johnson-Leger C, Wong C, Du Pasquier L, Imhof BA. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood. 2001;98(13):3699–707.
Tomi M, Hosoya K. Application of magnetically isolated rat retinal vascular endothelial cells for the determination of transporter gene expression levels at the inner blood-retinal barrier. J Neurochem. 2004;91(5):1244–8.
Kummer D, Ebnet K. Junctional Adhesion Molecules (JAMs): the JAM-Integrin Connection. Cells. 2018;7(4):25.
Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, et al. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood. 2007;110(6):1848–56.
Daniele LL, Adams RH, Durante DE, Pugh EN Jr, Philp NJ. Novel distribution of junctional adhesion molecule-C in the neural retina and retinal pigment epithelium. J Comp Neurol. 2007;505(2):166–76.
Economopoulou M, Avramovic N, Klotzsche-von Ameln A, Korovina I, Sprott D, Samus M, et al. Endothelial-specific deficiency of Junctional Adhesion Molecule-C promotes vessel normalisation in proliferative retinopathy. Thromb Haemost. 2015;114(6):1241–9.
Van Itallie CM, Tietgens AJ, Anderson JM. Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol Biol Cell. 2017;28(4):524–34.
Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103(3):755–66.
Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci U S A. 1991;88(8):3460–4.
Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;141(1):199–208.
Inoko A, Itoh M, Tamura A, Matsuda M, Furuse M, Tsukita S. Expression and distribution of ZO-3, a tight junction MAGUK protein, in mouse tissues. Genes Cells. 2003;8(11):837–45.
Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275(36):27979–88.
Fanning AS, Anderson JM. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165:113–20.
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126(4):741–54.
Ooshio T, Kobayashi R, Ikeda W, Miyata M, Fukumoto Y, Matsuzawa N, et al. Involvement of the interaction of afadin with ZO-1 in the formation of tight junctions in Madin-Darby canine kidney cells. J Biol Chem. 2010;285(7):5003–12.
Katsuno T, Umeda K, Matsui T, Hata M, Tamura A, Itoh M, et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell. 2008;19(6):2465–75.
Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28(5):1669–78.
Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim Biophys Acta. 2009;1788(4):761–7.
Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, et al. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208(6):821–38.
Pulgar VM. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front Neurosci. 2018;12:1019.
Hosoya K, Tachikawa M. The inner blood-retinal barrier: molecular structure and transport biology. Adv Exp Med Biol. 2012;763:85–104.
Dermietzel R, Krause D, Kremer M, Wang C, Stevenson B. Pattern of glucose transporter (Glut 1) expression in embryonic brains is related to maturation of blood-brain barrier tightness. Dev Dyn. 1992;193(2):152–63.
Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, et al. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet. 2006;15(7):1169–79.
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. 2015;18(4):521–30.
Leung SWS, Shi Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol Sin. 2022;43(2):251–9.
Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14(2):98–112.
Gu X, Fliesler SJ, Zhao YY, Stallcup WB, Cohen AW, Elliott MH. Loss of caveolin-1 causes blood-retinal barrier breakdown, venous enlargement, and mural cell alteration. Am J Pathol. 2014;184(2):541–55.
Schlingemann RO, Hofman P, Anderson L, Troost D, van der Gaag R. Vascular expression of endothelial antigen PAL-E indicates absence of blood-ocular barriers in the normal eye. Ophthalmic Res. 1997;29(3):130–8.
Shue EH, Carson-Walter EB, Liu Y, Winans BN, Ali ZS, Chen J, et al. Plasmalemmal vesicle associated protein-1 (PV-1) is a marker of blood-brain barrier disruption in rodent models. BMC Neurosci. 2008;9:29.
Keuschnigg J, Henttinen T, Auvinen K, Karikoski M, Salmi M, Jalkanen S. The prototype endothelial marker PAL-E is a leukocyte trafficking molecule. Blood. 2009;114(2):478–84.
Wisniewska-Kruk J, van der Wijk AE, van Veen HA, Gorgels TG, Vogels IM, Versteeg D, et al. Plasmalemma Vesicle-associated protein has a key role in blood-retinal barrier loss. Am J Pathol. 2016;186(4):1044–54.
Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, et al. Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest. 2014;124(9):3825–46.
Benz F, Wichitnaowarat V, Lehmann M, Germano RF, Mihova D, Macas J, et al. Low wnt/beta-catenin signaling determines leaky vessels in the subfornical organ and affects water homeostasis in mice. Elife. 2019;8.
Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. 2014;509(7501):503–6.
Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509(7501):507–11.
Guemez-Gamboa A, Nguyen LN, Yang H, Zaki MS, Kara M, Ben-Omran T, et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat Genet. 2015;47(7):809–13.
Chow BW, Gu C. Gradual suppression of transcytosis governs functional blood-retinal barrier formation. Neuron. 2017;93(6):1325–33.
Lobanova ES, Schuhmann K, Finkelstein S, Lewis TR, Cady MA, Hao Y, et al. Disrupted blood-retina lysophosphatidylcholine transport impairs photoreceptor health but not visual signal transduction. J Neurosci. 2019;39(49):9689–701.
Wong BH, Chan JP, Cazenave-Gassiot A, Poh RW, Foo JC, Galam DL, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid (DHA) in eye and is important for photoreceptor cell development. J Biol Chem. 2016;291(20):10501–14.
Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron. 2017;94(3):581–94.
Coelho-Santos V, Shih AY. Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip Rev Dev Biol. 2020;9(2): e363.
Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325(6101):253–7.
Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail–chick transplantation chimeras. Dev Biol. 1981;84(1):183–92.
Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443(7112):700–4.
Lendahl U, Nilsson P, Betsholtz C. Emerging links between cerebrovascular and neurodegenerative diseases-a special role for pericytes. EMBO Rep. 2019;20(11): e48070.
Frank RN, Turczyn TJ, Das A. Pericyte coverage of retinal and cerebral capillaries. Invest Ophthalmol Vis Sci. 1990;31(6):999–1007.
Mae MA, He L, Nordling S, Vazquez-Liebanas E, Nahar K, Jung B, et al. Single-cell analysis of blood-brain barrier response to pericyte loss. Circ Res. 2021;128(4):e46–62.
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153(3):543–53.
Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277(5323):242–5.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–61.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–27.
Park DY, Lee J, Kim J, Kim K, Hong S, Han S, et al. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun. 2017;8:15296.
Ornelas S, Berthiaume AA, Bonney SK, Coelho-Santos V, Underly RG, Kremer A, et al. Three-dimensional ultrastructure of the brain pericyte-endothelial interface. J Cereb Blood Flow Metab. 2021;41(9):2185–200.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–405.
Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, et al. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20(3):291–302.
Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem. 2004;89(2):503–13.
Pfister F, Wang Y, Schreiter K, vom Hagen F, Altvater K, Hoffmann S, et al. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta Diabetol. 2010;47(1):59–64.
Rangasamy S, Srinivasan R, Maestas J, McGuire PG, Das A. A potential role for angiopoietin 2 in the regulation of the blood-retinal barrier in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(6):3784–91.
Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016;131(5):753–73.
Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61(12):1939–58.
Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330(6005):774–8.
Dorrell MI, Aguilar E, Friedlander M. Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest Ophthalmol Vis Sci. 2002;43(11):3500–10.
O’Sullivan ML, Punal VM, Kerstein PC, Brzezinski JAt, Glaser T, Wright KM, et al. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia. 2017;65(10):1697–716.
Tout S, Dreher Z, Chan-Ling T, Stone J. Contact-spacing among astrocytes is independent of neighbouring structures: in vivo and in vitro evidence. J Comp Neurol. 1993;332(4):433–43.
Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007;10(2):77–88.
Ashraf M, Sampani K, Clermont A, Abu-Qamar O, Rhee J, Silva PS, et al. Vascular density of deep, intermediate and superficial vascular plexuses are differentially affected by diabetic retinopathy severity. Invest Ophthalmol Vis Sci. 2020;61(10):53.
Nagase T, Nagase M, Machida M, Fujita T. Hedgehog signalling in vascular development. Angiogenesis. 2008;11(1):71–7.
Osterlund T, Kogerman P. Hedgehog signalling: how to get from Smo to Ci and Gli. Trends Cell Biol. 2006;16(4):176–80.
Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334(6063):1727–31.
Milsted A, Barna BP, Ransohoff RM, Brosnihan KB, Ferrario CM. Astrocyte cultures derived from human brain tissue express angiotensinogen mRNA. Proc Natl Acad Sci U S A. 1990;87(15):5720–3.
Wosik K, Cayrol R, Dodelet-Devillers A, Berthelet F, Bernard M, Moumdjian R, et al. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci. 2007;27(34):9032–42.
Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413.
Chen ZL, Yao Y, Norris EH, Kruyer A, Jno-Charles O, Akhmerov A, et al. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J Cell Biol. 2013;202(2):381–95.
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106(2):641–6.
Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183(3):409–17.
Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322(5905):1247–50.
Moon RT. Wnt/beta-catenin pathway. Sci STKE. 2005;2005(271):cm1.
Guerit S, Fidan E, Macas J, Czupalla CJ, Figueiredo R, Vijikumar A, et al. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog Neurobiol. 2021;199: 101937.
Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330(6006):985–9.
Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U S A. 2011;108(7):2807–12.
Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci U S A. 2011;108(14):5759–64.
Posokhova E, Shukla A, Seaman S, Volate S, Hilton MB, Wu B, et al. GPR124 functions as a WNT7-specific coactivator of canonical beta-catenin signaling. Cell Rep. 2015;10(2):123–30.
Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev Cell. 2014;31(2):248–56.
Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. Elife. 2015;4.
Cho C, Smallwood PM, Nathans J. Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron. 2017;95(5):1056–73.
Shi Q, Chen YG. Regulation of Dishevelled protein activity and stability by post-translational modifications and autophagy. Trends Biochem Sci. 2021;46(12):1003–16.
Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, Dumitru AC, et al. A molecular mechanism for Wnt ligand-specific signaling. Science. 2018;361(6403).
Smallwood PM, Williams J, Xu Q, Leahy DJ, Nathans J. Mutational analysis of Norrin-Frizzled4 recognition. J Biol Chem. 2007;282(6):4057–68.
Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, et al. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139(2):299–311.
Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, et al. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139(2):285–98.
Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116(6):883–95.
Nikopoulos K, Venselaar H, Collin RW, Riveiro-Alvarez R, Boonstra FN, Hooymans JM, et al. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum Mutat. 2010;31(6):656–66.
Wang Y, Rattner A, Zhou Y, Williams J, Smallwood PM, Nathans J. Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell. 2012;151(6):1332–44.
Wang Z, Liu CH, Huang S, Fu Z, Tomita Y, Britton WR, et al. Wnt signaling activates MFSD2A to suppress vascular endothelial transcytosis and maintain blood-retinal barrier. Sci Adv. 2020;6(35):eaba7457.
Paes KT, Wang E, Henze K, Vogel P, Read R, Suwanichkul A, et al. Frizzled 4 is required for retinal angiogenesis and maintenance of the blood-retina barrier. Invest Ophthalmol Vis Sci. 2011;52(9):6452–61.
Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Gothert JR, et al. Endothelial beta-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation. 2016;133(2):177–86.
Corada M, Orsenigo F, Bhat GP, Conze LL, Breviario F, Cunha SI, et al. Fine-tuning of Sox17 and canonical Wnt coordinates the permeability properties of the blood-brain barrier. Circ Res. 2019;124(4):511–25.
Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, et al. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell. 2010;18(6):938–49.
Zhu X, Yang M, Zhao P, Li S, Zhang L, Huang L, et al. Catenin alpha 1 mutations cause familial exudative vitreoretinopathy by overactivating Norrin/beta-catenin signaling. J Clin Invest. 2021;131(6).
Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20(12):1752–60.
Greene C, Hanley N, Reschke CR, Reddy A, Mae MA, Connolly R, et al. Microvascular stabilization via blood-brain barrier regulation prevents seizure activity. Nat Commun. 2022;13(1):2003.
Nehra G, Bauer B, Hartz AMS. Blood-brain barrier leakage in Alzheimer’s disease: from discovery to clinical relevance. Pharmacol Ther. 2022;234: 108119.
Greene C, Kealy J, Humphries MM, Gong Y, Hou J, Hudson N, et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol Psychiatry. 2018;23(11):2156–66.
Sundstrom JM, Tash BR, Murakami T, Flanagan JM, Bewley MC, Stanley BA, et al. Identification and analysis of occludin phosphosites: a combined mass spectrometry and bioinformatics approach. J Proteome Res. 2009;8(2):808–17.
Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284(31):21036–46.
Murakami T, Frey T, Lin C, Antonetti DA. Protein kinase cbeta phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes. 2012;61(6):1573–83.
Muthusamy A, Lin CM, Shanmugam S, Lindner HM, Abcouwer SF, Antonetti DA. Ischemia-reperfusion injury induces occludin phosphorylation/ubiquitination and retinal vascular permeability in a VEGFR-2-dependent manner. J Cereb Blood Flow Metab. 2014;34(3):522–31.
Goncalves A, Dreffs A, Lin CM, Sheskey S, Hudson N, Keil J, et al. Vascular expression of permeability-resistant occludin mutant preserves visual function in diabetes. Diabetes. 2021;70(7):1549–60.
Goncalves A, Su EJ, Muthusamy A, Zeitelhofer M, Torrente D, Nilsson I, et al. Thrombolytic tPA-induced hemorrhagic transformation of ischemic stroke is mediated by PKCbeta phosphorylation of Occludin. Blood. 2022.
Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. The NINDS t-PA Stroke Study Group. Stroke. 1997;28(11):2109–18.
Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. 2016;7:10523.
Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. 2017;114(7):E1243–52.
Ma F, Sun P, Zhang X, Hamblin MH, Yin KJ. Endothelium-targeted deletion of the miR-15a/16-1 cluster ameliorates blood-brain barrier dysfunction in ischemic stroke. Sci Signal. 2020;13(626).
Toyama K, Spin JM, Deng AC, Huang TT, Wei K, Wagenhauser MU, et al. MicroRNA-mediated therapy modulating blood-brain barrier disruption improves vascular cognitive impairment. Arterioscler Thromb Vasc Biol. 2018;38(6):1392–406.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–9.
Storck SE, Meister S, Nahrath J, Meissner JN, Schubert N, Di Spiezio A, et al. Endothelial LRP1 transports amyloid-beta(1–42) across the blood-brain barrier. J Clin Invest. 2016;126(1):123–36.
Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal-DeMotta MA, et al. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein, and impairs cognition. J Alzheimers Dis. 2009;17(3):553–70.
Hartz AM, Miller DS, Bauer B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol. 2010;77(5):715–23.
Keaney J, Walsh DM, O’Malley T, Hudson N, Crosbie DE, Loftus T, et al. Autoregulated paracellular clearance of amyloid-beta across the blood-brain barrier. Sci Adv. 2015;1(8): e1500472.
Campbell M, Hanrahan F, Gobbo OL, Kelly ME, Kiang AS, Humphries MM, et al. Targeted suppression of claudin-5 decreases cerebral oedema and improves cognitive outcome following traumatic brain injury. Nat Commun. 2012;3:849.
Campbell M, Cassidy PS, O’Callaghan J, Crosbie DE, Humphries P. Manipulating ocular endothelial tight junctions: applications in treatment of retinal disease pathology and ocular hypertension. Prog Retin Eye Res. 2018;62:120–33.
Acknowledgements
The authors would like to thank Tiago Figueiredo for the scientific illustrations.
Funding
This research was supported by Grants from the National Institutes of Health, HL055374 (DAA), EY012021 (DAA); and Research to Prevent Blindness Stein Innovation Award (DAA).
Author information
Authors and Affiliations
Contributions
All authors, AG and DAA, contributed to the writing, illustration, and reviewing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
DAA acts as a consultant for EyeBiotech
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Goncalves, A., Antonetti, D.A. Transgenic animal models to explore and modulate the blood brain and blood retinal barriers of the CNS. Fluids Barriers CNS 19, 86 (2022). https://doi.org/10.1186/s12987-022-00386-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-022-00386-0