
Abstract
The choroid plexus is situated at an anatomically and functionally important interface within the ventricles of the brain, forming the blood-cerebrospinal fluid barrier that separates the periphery from the central nervous system. In contrast to the blood–brain barrier, the choroid plexus and its epithelial barrier have received considerably less attention. As the main producer of cerebrospinal fluid, the secretory functions of the epithelial cells aid in the maintenance of CNS homeostasis and are capable of relaying inflammatory signals to the brain. The choroid plexus acts as an immunological niche where several types of peripheral immune cells can be found within the stroma including dendritic cells, macrophages, and T cells. Including the epithelia cells, these cells perform immunosurveillance, detecting pathogens and changes in the cytokine milieu. As such, their activation leads to the release of homing molecules to induce chemotaxis of circulating immune cells, driving an immune response at the choroid plexus. Research into the barrier properties have shown how inflammation can alter the structural junctions and promote increased bidirectional transmigration of cells and pathogens. The goal of this review is to highlight our foundational knowledge of the choroid plexus and discuss how recent research has shifted our understanding towards viewing the choroid plexus as a highly dynamic and important contributor to the pathogenesis of neurological infections. With the emergence of several high-profile diseases, including ZIKA and SARS-CoV-2, this review provides a pertinent update on the cellular response of the choroid plexus to these diseases. Historically, pharmacological interventions of CNS disorders have proven difficult to develop, however, a greater focus on the role of the choroid plexus in driving these disorders would provide for novel targets and routes for therapeutics.
Similar content being viewed by others


Introduction
The choroid plexus (CP) is a highly vascularized complex found within each of the four ventricles of the brain. It is comprised of a monolayer of polarized secretory epithelial cells whose primary role is the production and secretion of 60–75% of total cerebrospinal fluid (CSF) [1]. The anatomy of the CP reflects this role. The epithelial surface, which is a continuation of the ependyma that lines the ventricles, becomes highly folded and forms villi throughout its structure. On their apical side, the cells present a brush-border of microvilli that greatly increases surface area to enable high rates of exchange of water and solutes. At its core lies a stroma of connective tissue and highly fenestrated capillaries [2, 3] that permit the diffusion of fluid and small molecules into the stroma. Together, this allows for the rapid production of CSF in which, for humans, the total volume (150 ml) of CSF is circulated and replaced approximately three to four times per day [4]. As there are no tight junctions between the endothelia of the CP, the epithelium functions similarly to the blood–brain barrier in regulating the passage of peripheral substances into the central nervous system (CNS).
The blood-cerebrospinal fluid barrier (BCSFB) is formed by the epithelial layer of the CP through the expression of junctional complexes forming a tight barrier segregating the highly vascularized stroma and the CSF of the ventricles. Like the blood–brain barrier (BBB), the BCSFB aids in the separation of the peripheral and central systems, maintaining a highly regulated environment. Although the ependymal cells that line the ventricles express some junctional components, they are loosely connected by desmosomes and any tight junctions tend to be discontinuous, representing a leaky CSF-brain interface [5]. Thus, any pathogens that cross this barrier into the CSF can greatly influence the homeostatic state and have direct access to the brain parenchyma. Moreover, disruption of the BCSFB can lead to indirect damage to the nervous system through inflammatory mechanisms and autoimmunity within the CSF [6]. As such, the choroid plexus houses many types of immune cells primarily within the stroma, including macrophages, dendritic cells, and T cells, all of which contribute to local immunosurveillance. Additionally, the choroid plexus has been shown to provide a point of entry into the CSF for peripheral immune cells. Historically, considerable attention has focused on the BBB for its role in the pathogenesis of infection into the CNS, in part due to its large interface with the brain [7, 8]. However, there has also been substantial evidence showing that the BCSFB provides a site of entry for many pathogens and may play a major role in modulating the immune response of the CNS. Suggesting that the BCSFB may not just be a potential site of entry but a preferential site for some infections.
In recent years, our understanding of the CP and its role in CNS immunity has been greatly improved. The wide-spread use of barrier model systems has provided insight into the bidirectional crosstalk between the peripheral and central systems at the CP interface and, with new understandings in CSF production and reabsorption, novel mechanisms that underlie disorders such as post-infectious hydrocephalus (PIH) are being uncovered. Novel model systems such as the choroid plexus organoid system allows for the manipulation and study of CSF production in conjunction to barrier functions [9]. This review covers experiments that utilize diverse model systems that have unique benefits and limitations [10,11,12,13]. Furthermore, with the use of single-cell sequencing, detailed mapping of the CP across brain ventricles and developmental ages has been performed, providing insight on the cellular makeup and transcriptional shifts throughout maturation [14]. The goal of this review is to highlight anatomical and functional aspects of the choroid plexus that are relevant to its role in the pathogenesis of neurological infections, and to bring attention to future research and potential therapeutics.
Anatomy and function of the choroid plexus
CSF production
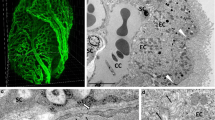
The choroid plexus epithelial cells are responsible for the primary production of CSF which occurs through the cotransportation of water and ions [15]. The production of CSF is a complex process that requires the active and passive transport of solutes and water which rely on changes in an osmotic pressure gradient. As such, there is still debate on the mechanisms that underlie CSF production by the CP, and in fact opposing hypotheses suggest that the CP may not be the main producer of CSF [16]. However, the current generally accepted process of CSF production and secretion is thought to occur in two discrete stages. First, the fenestrated capillaries within the CP allow for an ultrafiltrate of plasma to passively permeate into the basal lamina. The fenestrae permit the free flow of water, ions, and small molecules dependent upon the pressure gradient between the blood and interstitial fluid. Secondly, the fluid then must undergo active transport across the epithelium barrier (1C) [17].
The secretion of fluid by the choroid plexus epithelial cells is dependent on the unidirectional transport of specific ions because of polarized expression of transporters. This leads to an osmotic gradient that causes water to move from the stroma, across the epithelial barrier in either a paracellular or transcellular fashion and enter the ventricular lumen. The primary determinants of this exchange are Na+, K+, Cl−, and HCO3− [18]. Following the diffusion of water and CO2 into the epithelial cells from the interstitial fluid, cytoplasmic carbonic anhydrases catalyze the production of HCO3− and H+ [19]. The accumulation of intracellular HCO3− and H+ is then exchanged for Na+ and Cl− through the SLC4 (anion exchange proteins [AEs] and sodium bicarbonate cotransporters [NBCs]) and SLC9 (sodium-hydrogen exchanger [NHEs]) family of transporters located on the basolateral membrane [20,21,22]. Na+-K+ ATPase (NKA) pumps play a vital role in the secretion of Na+ into the CSF, and conversely, the uptake of K+ into the epithelial cells. These ATPases are localized at the apical brush border which exchange three Na+ ions for two K+ ions at the expense of one ATP [23]. The importance of these have been observed through inhibitor studies that reduce CSF production by up to 80% [24]. Furthermore, a family of electroneutral cotransporters (SLC12 family—NKCCs) are responsible for the movement of Cl− with Na+ and/or K+ across the cell membrane. The movement of Cl− via the SLC12 family members is driven by changes in the chemical gradients of Na+ and K+ [18]. Ultimately, this series of passive diffusion and active ion exchange across the polarized epithelial membrane creates an osmotic gradient that drives the secretion of water into the CSF that is facilitated by aquaporins [15]. Figure 1C highlights this process.

Overview of CSF production and junctional properties of the blood-CSF barrier. A A diagram indicating the bulk flow of CSF following its production from the CP. The CSF flows in a unilateral direction through the ventricular system, and multidirectionally throughout the subarachnoid space. B A coronal section showing the drainage of CSF. Three distinct sites of CSF drainage are highlighted—through dural lymphatics, arachnoid granulations, and through perivascular space (inset) where it enters the brain parenchyma and exits via perivenous routes. C A molecular view of CSF production and several important transporters. Passive diffusion of water and solutes occur through the fenestrae of the blood vessels. Differential transport of ions across the membrane creates an electrochemical gradient that drives water transport via AQP1. Direct coupling of water with transporters occurs, NKCC1. D An overview of the polarized junctional proteins found in the CP epithelium – tight junctions, adherens junctions, desmosomes, and gap junctions are present, along with their scaffolding proteins. Created with BioRender.com
Recent studies have shown that knockout of AQP1 results in decline of CSF production by only 20% and that the observed osmolarity across the membrane (5–10 mOsm) does not reach the calculated amount (250 mOsm) required for the observed rate of CSF production [4, 25,26,27]. In fact, the choroid plexus has been shown to be able to transport water against the direction of the osmolarity gradient through cotransporter channels [26, 28, 29]. Therefore, it is suggested that one of the main contributors for water secretion and thus CSF production occurs through the direct coupling of water to the Na+/K+/2Cl− and K+/Cl− cotransporters (NKCC1 and KCC respectively) [27, 29, 30].
Following secretion, CSF generally flows directionally through the ventricular system in a pulsatile fashion that corresponds to the CP systolic pulse wave of the arteries and aided by the movement of ependymal cilia [1, 31]. CSF produced within the lateral ventricles will pass through the interventricular foramina and into the third ventricle. From here it then passes through the cerebral aqueduct and enters the fourth ventricle. The CSF then leaves the ventricular cavities and flows multidirectionally throughout the subarachnoid space and spinal canal through the central canal of the spinal cord, the foramen of Magendie, and the foramina of Luschka [1, 32]. Classically, CSF drainage into the vascular system was thought to occur through the absorption of CSF by arachnoid granulations that protrude into the dural venous sinuses. However, the predominance of this pathway has come into question with new routes being discovered [33]. For example, meningeal lymphatics play an important role in the transport and drainage of CSF passing through the cribriform plate into the nasal lymphatics and cervical lymph nodes [34,35,36,37,38]. Recent research has emphasized the significance of this lymphatic drainage system due to its potential role in modulating the pathophysiology of many neurological diseases and disorders, as well as regulating neuroinflammation through the transport of CSF immune cells and antigens into lymph nodes [39, 40]. This lymphatic drainage allows for constant immune surveillance through the flow of CSF antigens and immune cells. In the healthy CNS, this allows for self-tolerance of brain-derived antigens; however, during neuroinflammation, this allows for antigen presenting cells and circulating T cells to relay inflammatory information to the periphery during tissue damage or innate immunity activation [40, 41]. This migration of immune cells to the draining lymphatics occurs in a CCR7-dependent manner, that is enhanced during immune activation [40,41,42]. In addition, the expression of CCL19/21 chemotaxis is regulated by proximal lymphatic vessels. Importantly, during experimental autoimmune encephalomyelitis, the ablation of meningeal lymphatics reduces pathology and inflammation [40,41,42]. The role of the meningeal lymphatics system in modulating and communicating CNS inflammatory and immunity to the periphery has received considerable attention in regards to autoimmunity. A common theme amongst CNS infections is the invasion of peripheral immune cells, specifically T cells into the CSF, that may be activated through initial drainage of antigens or antigen presenting cells through the meningeal lymphatics. The importance of this lymphatic system during CNS infection should not be understated and requires greater research to determine potential avenues in modulating CNS pathology.
In addition to the lymphatics system, CSF and solutes have been observed to also flow through the perivascular space (Virchow–Robin space) of penetrating blood vessels and entering the brain parenchyma through aquaporin-4 on astrocytic endfeet [43]. Along with the interstitial fluid, the CSF is then drained via perivenous pathways [43]. Figure 1A and B illustrate the bulk flow and drainage of CSF.
Barrier properties and junctional proteins
The choroid plexus epithelia also form the blood–cerebrospinal fluid barrier. Like the structural organization of transporters, the junctional proteins that comprise the BCSFB are highly polarized. As indicated in Fig. 1D, these proteins are spatially oriented from the apical to basolateral side. [44,45,46,47]. Together, these protein complexes allow for a highly regulated selectively permeable barrier to paracellular diffusion and cellular movement. Importantly, the barrier can maintain the semi-immune privileged state of the CNS by limiting the movement of peripheral immune cells and pathogens across the membrane. Thus, an uncompromised barrier is integral to maintaining a healthy homeostatic environment and modulating CNS immunity.
The primary functions of tight junctions are to act as a gate-like barrier between adjacent cells that regulates the paracellular movement of water, ions, and macromolecules, and to establish and maintain cellular polarity by preventing the redistribution of lipids and membrane bound proteins between the apical and basal surfaces [48]. The junctions are formed by transmembrane proteins that include occludins, claudins, and junctional adhesion molecules (JAMs) and can be found throughout the choroid plexus epithelium [45, 49,50,51]. The protein’s extracellular domains from adjacent cells bind directly to each other and seal the paracellular pathway, allowing for the regulation of ions and solutes. However, the permeability of these tight junctions, including JAMs, can change depending on their conformation and can be disassociated based on their phosphorylated state [52, 53].
Gap junctions provide minimal adhesive properties and do not form a tight seal between adjacent cells, but instead they form intercellular channels that directly connect the cytoplasm of cells [54]. These channels, formed by proteins called connexins, provide a route of communication to coordinate and maintain a homeostatic environment that are often required in many blood-tissue barriers, including the blood–brain barrier [55]. This form of communication allows for dynamic changes to occur in response to cellular stress, inflammation, or infection [55]. Their roles within the BCSFB have been largely ignored, which represents a major gap in our understanding of the barrier properties of the choroid plexus and how pathogens may compromise these complexes, especially during fetal development.
Receptors and adhesion molecules
The choroid plexus is a highly vascularized structure, and in combination with the fenestrae of the blood vessels, provides an interface between the peripheral circulation and the CNS. Thus, like many epithelial barriers, the cells of the BCSFB contain many types of immune receptors and adhesive molecules to surveil and sample the microenvironment of the stroma (Fig. 2). A significant feature of the innate immune response is pattern recognition receptors (PRRs) to identify pathogen-associated molecular patterns (PAMPS). These receptors induce an innate immune response and give rise to inflammatory pathways through the release of cytokines which can lead to greater BCSFB permeability. Inappropriate stimulation of these receptors by infection and subsequent inflammation at the choroid plexus or in adjacent tissue can lead to devastating outcomes such as meningitis, hydrocephalus, hemorrhage, and death [56,57,58,59]. Thus, many studies have focused on the impact of peripheral infection and systemic inflammation on BCSFB integrity and the transmigration of both pathogens and immune cells.

An illustration of the choroid plexus, receptors, and resident immune cells. Three distinct immune cells can be found within the CP–dendritic cells, macrophages, and CD4 + T cells. Kolmer cells can be found adhering to the apical surface of the epithelium. CD4 + T cells are seen to constantly surveil both the CP and CSF, and regular traverse the barrier bidirectionally through the aid of adhesion molecules. The CP epithelium express many receptors and can be activated by resident immune cells, pathogens, or circulating cytokines from the periphery. Dendritic cells and macrophages phagocytize pathogens and present antigens to naïve T-cells. This allows for differentiation of CD4 + T cells into Th1 or Th2 dependent upon the current cytokine environment. The epithelial cells can release chemokines in the stroma and CSF. This can alter the CSF composition and promotes homing of peripheral immune cells to and from the CSF. The epithelial cells can also release extracellular vesicles into the CSF that contain inflammatory miRNA and potentially pathogens. Created with BioRender.com
The most studied PAMP receptor within the choroid plexus are the toll-like receptors (TLRs). Several studies have sought to determine the expression pattern of TLRs in the CP in mice, rats, ewes, and in vitro human systems in different physiological states [60,61,62,63,64,65,66,67]. There has been a total of 13 TLRs identified so far and it is important to note that the presence of TLRs vary between species, with TLR1-10 being found in humans, and all except TLR10 being found in mice. Furthermore, tissue localization of TLRs varies substantially and as such the expression of TLRs within specific model systems of the choroid plexus, such as human choroid plexus papilloma cells, should be carefully considered. Nevertheless, considerable research has shown that TLRs of the CP epithelial cells play a fundamental role in CNS immunity and inflammation. Lipopolysaccharide (LPS) and Pam3CSK4 (PAM), ligands for TLR-4 and TLR-1/2 respectively, are commonly used to study the effects of systemic inflammation on barrier function. Challenging with TLR agonists in animals and in in vitro models commonly show an increase in the expression of inflammatory genes and a downregulation of junctional proteins, including claudins and cadherins [64, 68, 69]. Many of the inflammatory genes promote the activation of resident immune cells and act as chemoattractants for circulating immune cells, suggesting that stimulation of TLRs leads to cytoskeleton restructuring and loss of junctional complexes, and promotes peripheral immune cell migration to the choroid plexus [64, 67,68,69]. In fact, the activation of the TLR1/2 complex by PAM has been shown to induce a significant increase in both cytokine levels and leukocyte count within the CSF [68, 70]. These studies have provided mechanistic insight into disease pathogenesis of the CNS as their effects tend to reflect those observed in many infectious models. Other types of PAMP receptors that have been shown to be important in mediating immunity, including NOD-like receptors, C-type lectin receptors, and RIG-I-like receptors have received considerably less attention within the CP, but may play important roles in the exacerbation of CNS inflammation as many have been shown to be associated with TLR-signaling [71,72,73,74]. In fact, a study by our laboratory showed an increase in the RIG-I-like receptor and components within its signaling pathway when human CP epithelial cells were stimulated by the Lyme disease bacterium, suggesting a potential role for this receptor in the CP immune response [73].
In addition to PRRs, CP epithelial cells contain a myriad of cytokine receptors that can respond to ligands in circulation or from the CP itself if challenged. TNFα, a pro-inflammatory cytokine that is produced in periphery tissue as well as by the CP epithelia, can cause deleterious effects on tight junctions, an increase in cell-adhesion molecules utilized by immune cells (ICAM and VCAM), and an increase in matrix metalloproteases (MMPs) [75,76,77]. Collectively, MMPs are enzymes that are capable of cleaving and remodeling many components of the extracellular matrix. The dysregulation of MMPs is associated with many neuroinflammatory disorders and in fact, some MMPs, such as MMP-2 and MMP-9, are indicative of specific infectious diseases as they show a consistent elevation in CSF concentrations [78,79,80,81]. Furthermore, with the use of an MMP inhibitor, Zeni et al. showed that alterations of the BCSFB induced by TNFα were in part dependent on MMPs [82]. More recent studies corroborate and expand upon the role of MMPs showing that barrier integrity is compromised via the NF-κB pathway and subsequently led to the degradation of claudins [75, 83]. Several cytokines that are found in peripheral circulation following infection, including IL-1β, IL-6, and IFNs have been shown to stimulate similar inflammatory profiles that, in turn, may lead to compromised barrier integrity [84,85,86]. As the choroid plexus itself can produce these inflammatory signals, it can be suggested that a positive feed-back loop, if not properly regulated, would lead to a more deleterious outcome in disease pathology.
While it is well-known that CP epithelial cells release cytokines into the CSF following PRR activation or other stressors and can influence brain inflammation, a novel mechanism of blood–brain communication at the CP was discovered [87]. CP epithelial cells can release extracellular vesicles into the CSF, and their release can be increased through systemic peripheral inflammation (LPS injection). These vesicles contained miRNAs that are then taken up by astrocytes and microglia, inducing an inflammatory program [87]. This pathway has the potential to shuttle pathogens between the periphery and brain parenchyma, a subject that has seen very limited research—recently the John Cunningham virus was shown to have the ability to utilize this pathway in infecting glial cells, which typically lack the necessary viral receptors [88].
Resident immune cells
Under physiological conditions, there are typically 3 types of immune cells that can be commonly found within the CP stroma or adhered to the apical side of the CP epithelia. These are macrophages (Epiplexus/Kolmer cells), dendritic cells, and T-cells (Fig. 2) [89,90,91,92,93,94]. Two subtypes of macrophages can be found to be in contact with the CP–stromal CP macrophages and Kolmer cells that adhere to the apical side of the epithelial barrier and ventricular wall. Both cells share similar functional characteristics in that they are phagocytically active, and are antigen presenting cells, aiding in the activation of T-cells [90, 95]. Kolmer first reported macrophage-like cells on the surface of the CP epithelium of amphibians in 1921, and subsequent studies further reinforced these findings in other vertebrates, including humans [90]. It wasn’t until the 1970’s in which electron microscopy aided in the functional and morphological characterization of these cells, with later studies identifying phagocytic activity [96, 97]. Immediately after their discovery, the ontology of these cells came into question. The prevailing theory was that of myeloid origin in which circulating monocytes infiltrated the CP stroma, differentiated into tissue macrophages, and subsequently migrated across the epithelial barrier to become Kolmer cells [90]. As such, and due to the lack of differentiating markers and characteristics, stromal macrophages and Kolmer cells have commonly been evaluated together in many CP studies. However, a recent study utilizing single-cell sequencing determined distinct profiles between Kolmer and stromal cells. It was shown that while both populations are originally yolk sac-derived, stromal macrophages are gradually replaced by circulating monocytes while Kolmer cells were capable of repopulation, independent of bone marrow progenitors [98, 99]. Interestingly, while Kolmer cells showed distinct clustering and shared many characteristics with stromal macrophages, these cells also expressed several signature genes typically found in microglia, including and Sparc and may suggest that they are instead a subset of microglia [98, 100, 101]. However, further characterization is needed in order to fully delineate the lineage of these macrophages throughout development. A recent review by Cui and Xu et al. detail the immunological role and heterogeneity of macrophages in the CP [102].
Dendritic cells (DCs) are functionally similar to macrophages in that they may act as antigen presenting cells and are capable of phagocytosis. As DCs are some of the first cells to encounter a pathogen when invading a host, and are present at the interface of the BCSFB, they play an integral role in bridging the innate and adaptive immune response. Following activation, dendritic cells are able to secrete a range of inflammatory cytokines including TNF-α, IL-1, IL-6 which stimulate and promote the release of chemotactic chemokines by the CP epithelium leading to T-cell activation and differentiation [103]. Through these mechanisms, DCs initiate an innate immune response, leading to CP inflammation and the further migration and activation of peripheral immune cells. This creates an environment that is conducive to the transmigration of both immune cells and pathogens across the BCSFB, and gives rise to an increase of cytokines and immune cells within the CSF, a hallmark of many neurological infections [104, 105].
Under normal physiological state, the choroid plexus has been found to be populated with T-cells, with the majority being effector memory CD4+T Cells, and a smaller population of CD8+T cells [106]. During infection or inflammation, MHCII + resident immune cells have been shown to closely associate with T cells, suggesting antigen presentation and activation prior to T-cell invasion of the CSF [93, 107]. Additionally, the CP epithelium plays an important role in the activation and transmigration of T-cells across its border. Aside from secreted chemokines that activate and attract immune cells, the epithelial cells constitutively express cellular adhesion molecules, including VCAM-1 and ICAM-1, both of which are important for adhesion of T-cells and required for transmigration into the CSF [108, 109]. Furthermore, polar expression of these adhesion molecules can be observed on the apical side of the epithelial cells and expression is found to increase during inflammation and infection [110, 111].
The presence of B cells within the CP during infection is poorly understood. However, in many forms of autoimmunity (multiple sclerosis and lupus), deposits of immunoglobulins can be observed, as well as the accumulation of B cell subsets within the CP and CSF [112,113,114]. There is evidence showing that AIDS patients and a patient with subacute bacterial endocarditis form immune complex deposition at the CP [115, 116]. In 75% of patients with AIDS, immunoglobulin deposits were observed, however, as circulating immune complexes are common in AIDS patients and there was a lack of CP pathology, it is suggested that their origin stems from the bloodstream, as opposed to B cell infiltration [115]. In a rodent model of malaria, circulating immune complexes and depositions were also found within the CP [117]. These observations make it clear that the antibody immune response impacts the CP in many circumstances in both autoimmunity and infection and may in fact be a common occurrence that is poorly studied. A study of MS patients showed preferential localization of antibodies at CP epithelium [118]. While antibodies can be observed circulating throughout the CSF during disease, it is not clear how the choroid plexus plays a role in their transport. Some studies suggest that the localization of antibodies at the CP epithelium is due to the efflux (CSF to blood) of these immunoglobulins via the FcRn-dependent IgG transcytosis pathway [118,119,120,121]. Our current understanding suggests that the CP may act as a trap, pooling immunoglobulins within the stromal matrix, and protecting the brain from immune-mediated damage [118].
The cellular response of the choroid plexus to specific pathogens—an update
The goal of this review is to bring attention to recent major developments in our understanding of the CP immunity during infection. In the past several years, thorough reviews have been published describing the impact of specific pathogens and their molecular pathways for migration (reviewed here: [6, 92, 122,123,124]). Since then, there has been an exponential focus on the CP, in part due to several high impact diseases that have affected the world, including Zika and SARS-CoV-2. Notable advancements in our knowledge of CP function during infection has prompted the need for an update. Figure 3 highlights our current understanding of these diseases that are being discussed.

Summary of pathogens and their interactions with the choroid plexus. (Hydrocephalus) Inflammation can cause obstruction of CSF flow from the lateral ventricles leading to increased intraventricular pressure. Ventricular hemorrhage and infection can induce inflammation dependent hypersecretion of CSF, promoting hydrocephalus. The hypersecretion occurs via TLR4-NF-κB and the NKCC1 transporter. (ZIKA) The ZIKA virus is shown to preferentially infect pericytes within the CP. This infection precedes CSF and brain invasion—the mechanism of transmigration unknown. Depending on the developmental stage, neural progenitor cells may be exposed to the CSF and thus susceptible to infection. (JC Virus) During the lytic phase, JC virus may disseminate hematogenously to the CP or possibly by B cells. The CP epithelial cells express viral receptors and are susceptible to infection. Extracellular vesicles secreted by the CP into the CSF can contain JC virion and transport these to glia in the brain. (HIV) HIV can be found within the CP, which may act as a reservoir for viral replication. Phylogenetic analysis indicates distinct clustering of HIV in the brain and spleen, whereas the CP contains virus from both clusters. This suggests there is unique selective pressure within the CP towards CNS tropism of HIV. FIV is capable of transmigrating across the BCSFB. (Borrelia burgdorferi) CSF findings indicate increased inflammatory cytokines, lymphocytic pleocytosis, and Bb. Infection of CP epithelial cells with Bb induce an increase in secretion of inflammatory and chemotactic cytokines, as well as the downregulation of junctional proteins. (SARS-CoV-2) Early findings suggest that viral presence in CNS is rare but neurological complications more common, characterized by increased cytokines and lymphocytes in the CSF. The CP expresses binding receptors for viral fusion. The CP may provide a route of entry for SARS-CoV-2 in rare circumstances, or more likely, relay inflammatory signaling to the CNS. Created with BioRender.com
Hydrocephalus
Hydrocephalus is a condition in which abnormal amounts of CSF accumulates within the CNS, typically resulting in increased intracranial pressure, macrocephaly, cognitive dysfunction, and death, if not properly diagnosed and treated. Although hydrocephalus has a wide range of etiologies, including congenital malformations, trauma, or tumor formation, research has shown that the most common cause world-wide is induced by infection, termed post-infectious hydrocephalus (PIH) [125, 126]. Furthermore, recent research has shown that post-hemorrhagic hydrocephalus (PHH), another common cause, shares many similar pathophysiological pathways with PIH, namely through shared inflammatory mechanisms [125, 127, 128]. The build-up of CSF has been commonly attributed to intraventricular obstruction of both CSF flow and reabsorption (choroid plexitis, tumor formation, narrowing of passages such as the foramen of Monro, or ablation of arachnoid granulations) [128,129,130]. As such, the standard practice for hydrocephalus amelioration is through surgical methods such as placement of intraventricular shunts or endoscopic third ventriculostomy (ETV) to restore CSF flow. ETV may also be combined with choroid plexus cauterization to reduce CSF production. However, due to the invasive nature of treatment, complications often occur due to shunt failure or infections, and continued dependence on surgical intervention is required [131, 132]. Until recently, the role of CSF hypersecretion by the CP in the pathogenesis of hydrocephalus was minimally researched. Other secretory epithelial tissues have been shown to drastically increase the rate of fluid secretion when encountered by a pathogen or inflammatory environment, including intestinal and respiratory epithelial surfaces, suggesting the CP epithelium may respond in a similar manner [133, 134]. In 2017, Karimy et al. reported an inflammatory mechanism for the hypersecretion of CSF by the CP epithelium in PHH. The CSF hypersecretion observed in their rat model of PHH was mediated through the upregulation of the SPAK-NKCC1 co-transporter complex, which interestingly was dependent on the upstream signaling of the TLR4-NF-κB pathway [127]. This suggests that this mechanism of action may underlie CSF hypersecretion in PIH as TLR4 has been shown to activate under pathogenic conditions, specifically through the binding of LPS [62, 64, 122]. However, in contrast, the role of NKCC1 to mediate CSF clearance was shown in a mouse model of obstructive hydrocephalus and that overexpression of NKCC1 resulted in the reduction of ventriculomegaly [135]. The hypothesis of inflammatory induced CSF hypersecretion following infection and thus inducing PIH warrants further investigation as it may provide alternative, non-surgical methods for amelioration of hydrocephalus by pharmaceutical interventions such as TLR4 or NKCC1 inhibition.
Zika virus
In adults, Zika virus (ZIKV) infection typically leads to mild symptoms including fever and joint pain; however, the ability of ZIKV to cross the placental barrier and infect the fetus has garnered considerable attention due to the severity of neurological injuries that can arise in the fetus. Early in development, the fetal brain is highly susceptible to infection once a pathogen has reached the CSF. This is because at early gestational periods, the ependymal lining of the ventricles have yet to fully form—in humans, ependymal differentiation occurs until 22 weeks of gestation, with complete maturation occurring postnatally [136, 137]. Prior to ependymal formation, neural progenitor cells (NPCs), which are found within the ventricular and subventricular zones, are in direct contact with the CSF. Several studies have shown that the ZIKV infects NPCs, inhibiting cellular differentiation and neurogenesis, and inducing cell death, leading to microcephaly [138,139,140,141]. While the impacts of ZIKV on neurodevelopment have been well studied, the route of entry into the CNS is less understood. Several lines of evidence have implicated the CP as a site of ZIKV trafficking from the blood to CSF. In in vivo non-human primate models, periventricular injury and damage to the ependymal lining is commonly found in congenital ZIKV infection [142,143,144,145]. Such injury patterns are also seen during human fetal neuroimaging and include ventriculomegaly and hypertrophy/cyst of the choroid plexus [146,147,148]. Although ZIKV infection is commonly associated with microcephaly, severe postnatal hydrocephalus can occur, suggesting that damage to the CSF and ventricular system may be involved [149]. However, there is no current understanding on how ZIKV impacts CSF production at a mechanistic level that provides insight into the pathogenesis of hydrocephalus. In a human cerebral organoid model system, the choroid plexus was found to be infected by the ZIKV [150]. A recent article by Kim et al. provided further evidence in CP involvement for ZIKV dissemination. In a mouse model for ZIKV brain infection, they observed that the ZIKV establishes a presence within the CP through infecting resident pericytes that adhere to the vasculature of the CP. This infection is soon followed by the emergence of the ZIKV within the CSF, and importantly precedes parenchymal infection [151]. In the same study, using an in vitro blood-CSF barrier model, they demonstrated that infected pericytes greatly enhanced the transmigration of ZIKV across the epithelial barrier and that secreted factors from the infected cells induced barrier disruption through a reduction in ZO-1 at cellular junctions [151]. The exact mechanism of entry for ZIKV to enter the CNS is still a work in progress (BBB vs BCSFB; direct transmigration vs “Trojan horse”), and as disease prognosis depends on key developmental milestones, additional research is needed to determine preferential dissemination pathways [152].
John Cunningham virus
The John Cunningham virus (JCV, also known as Human polyomavirus 2), is widespread among the general population, with infection rates varying between 50 and 90% [153, 154]. The majority of infected individuals will show no clinical manifestations of infection as the virus tends to remain latent in secondary tissue sites such as the gastrointestinal tract or kidneys [154, 155]. In some cases, occasional shedding of JCV in the urine occurs [156,157,158]. However, JCV is the pathological agent responsible for progressive multifocal leukoencephalopathy (PML), an often fatal disease of the CNS that is characterized by progressive inflammation of the white matter, and through infection of oligodendrocytes and astrocytes, leads to demyelination [159, 160]. Although PML is rare, it almost exclusively occurs in the context of immunocompromised individuals, and as such up to 80% of PML patients occur in those with HIV [160, 161]. The progression of latent JCV to lytic invasion of the CNS and thus PML is poorly understood. It is suspected that at some point, reactivation of the latent virus within the secondary infected tissue leads to dissemination into the CNS, infecting glial cells responsible for inflammation and demyelination [154]. However, in some cases, latent JCV virus has been found to reside within the brain tissue of patients without PML [162]. Nevertheless, the route of dissemination into the CNS either during the lytic stage or prior to an established latent infection is not well understood. Although the blood–brain barrier has been suggested as a route of transmigration, and potentially through the infection of B-cells, the CP and the BCSFB have only recently been implicated in PML pathogenesis [163]. The JCV entry into cells has been shown to be dependent on two receptors, lactoseries tetrasaccharide c (LSTc) for initial cellular attachment, and a serotonin (5-HT)-2 receptor for cellular entry [164, 165]. Interestingly, distribution mapping of these receptors in both healthy and PML human samples indicated a lack of LSTc and thus no viral binding on oligodendrocytes and astrocytes; this is in contrast to both receptors and subsequently binding of the JCV occurring on kidney and CP epithelium tissue [166]. Furthermore, a case of fatal JCV meningitis with symptoms atypical of PML were characterized by communicating hydrocephalus and choroid plexus epithelial cells harboring productive JCV which is thought to be the cause of the high levels of viral load in the CSF [167]. These findings suggest viral infection of astrocytes and oligodendrocytes occurs independent of LSTc and that dissemination into the parenchyma may involve infection of CP epithelial cells. Indeed, the novel identification of extracellular vesicles derived by the CP epithelium has been shown to bridge CP involvement and LSTc-independent infection of parenchymal glia [87, 88]. Recent studies by O’Hara et al. highlight the susceptibility of CP epithelial cells to JCV infection and viral transmission via extracellular vesicles [88, 168]. In their investigation, they observed infected CP epithelial cells produced extracellular vesicles that contained JCV virions. Furthermore, it was shown that JCV is readily transmitted to glial cells through the uptake of these viral loaded vesicles in a receptor independent manner [88]. However, it is not well understood how JCV impacts CP epithelial cells following infection—studies aimed at barrier function and CP inflammatory relay to the brain are needed to attain a full picture of the effects of CP infection and CNS health.
Human immunodeficiency virus
In 2021, it was estimated that 38 million people globally were living with human immunodeficiency virus (HIV; assume HIV-1 unless otherwise stated), with 1.5 million newly infected people reported that year [169]. HIV commonly infects host immune cells such as CD4 + T cells, macrophages, and dendritic cells through the binding of viral glycoproteins (gp160 and gp120) to host CD4 and chemokine receptors such as CCR5 or CXCR4 [170]. Due to the viral tropism towards host immune cells, HIV infection leads to critically low levels of CD4 + T cells, thus causing acquired immunodeficiency syndrome (AIDS) and increasing susceptibility to opportunistic infections. [171] Through the use of combination antiretroviral therapy (CART), suppression of HIV replication is possible, and many patients are capable of living with HIV without the progression towards AIDS. In fact, the development and administration of CART led to a substantial decrease in deaths while also halting the progression of HIV-associated neurocognitive disorder (HAND) in up to 77% of patients [172,173,174,175,176]. However, while some studies indicate beneficial outcomes of HAND following CART, the impact of the therapy on neurocognitive impairment is still poorly understood. Despite the most severe aspects of HAND [HIV-associated dementia (HAD)] being greatly reduced following the introduction of CART, the overall prevalence of HAND has not changed [177,178,179,180,181,182]. This is mainly caused by an increase in milder forms of HAND [asymptomatic neurocognitive impairment (ANI) and mild neurocognitive disorder (MND)] [177,178,179,180,181,182]. The prevalence of HAND can range from 20 to 50% and can occur in patients receiving CART, even when HIV RNA levels in the plasma are undetectable [177, 183, 184]. Although plasma levels of HIV RNA can be successfully controlled, other regions have been shown to act as reservoirs for HIV, including the genitourinary system, lymphoid tissue, and the CNS [185,186,187,188].
The phenomenon of HIV to escape into regions such as the CSF, termed CSF viral escape, has been observed to occur in 5–15% of CART patients [189,190,191]. While this may suggest a reservoir role for the brain, in recent years, the role of the choroid plexus has been largely ignored and needs to be revisited. Several studies in the 90’s and early 2000’s using post-mortem tissue found HIV infected immune cells situated within the stroma and supra-epithelial areas of the choroid plexus in approximately half of HIV cases [192,193,194]. These cells comprised of T lymphocytes, dendritic cells, and macrophages, and due to their apposition to capillaries, initial establishment of infection is thought to be of hematogenous origin [192,193,194]. Further evidence of the choroid plexus acting as a reservoir and key player in CNS pathogenesis comes from phylogenetic analysis of HIV in the brain, CP, and spleen of post-mortem tissue. In these studies, genotyping indicated that HIV from the brain and spleen formed distinct clusters based on the mutations of the HIV env and pol sequences, while HIV from the CP were found within each of the clusters, but with greater similarity towards brain sequences [195, 196]. Similarly, while spleen isolates displayed CCR5 and CXCR4 utilization, brain and CP isolates showed preferential utilization of CCR5, a major coreceptor for the infection of microglia [195,196,197]. In a more recent study, 44% of HIV-positive individuals showed BCSFB dysfunction, and similarly, individuals with CSF pleocytosis showed significant elevation in CSF inflammatory markers [198]. This suggests that the unique environment of the CP may provide selective pressure on mutations that confer drug resistance (mutations in pol sequence) and viral tropism towards the CNS through preferential utilization of host coreceptors (mutations in env sequence). Evolution of HIV within reservoir sites has seen on-going research, however, the CP has received little attention—such an important interface between the periphery and CNS requires greater focus in order to understand HIV evolution towards CNS tropism and drug resistance. While there has been research aimed at CNS penetrance of antiretroviral drugs and identification of transport systems at the CP, a greater focus on the CP would allow us to differentiate between the blood–CSF barrier and the BBB. This would provide insight into antiretroviral drug penetrance specifically across the blood-CSF barrier, allowing for more effective combinations of drugs that target the CP and CSF.
Research into feline immunodeficiency virus (FIV) and simian immunodeficiency virus (SIV) provides further contextual evidence that the CP may play a significant role in HIV neuropathology due to their similar mechanisms of infection [199, 200]. In an in vitro model system and in vivo, macrophages of the feline choroid plexus are infected by FIV, and effectively transfer the infection to T cells [201]. Furthermore, in a barrier model system of the feline CP, enhanced transmigration of macrophages and T cells are observed following FIV infection [202]. Similarly, in studies utilizing SIV in rhesus macaques, SIV is found within the CP, with an increased presence of macrophages and T cells within the stroma [203]. When rhesus macaques are infected with SIV of differing tropisms, lymphocyte- and macrophage-tropic viruses showed preferential infection of microglia [204].
Borrelia burgdorferi
Borrelia burgdorferi (Bb), the etiological agent of Lyme disease, is estimated to infect up to 300,000 individuals in the US each year, with 30,000 cases being annually reported to the CDC [205,206,207,208]. Bb is transmitted to humans through the bite of a tick, and within a few days elicits symptoms similar to the flu, as well as the hallmark “bulls-eye” rash [209]. The bacteria invade secondary tissue through hematogenous dissemination, commonly residing in the extracellular matrix of joints – unilateral knee pain is typical in the manifestation of Lyme arthritis [209, 210]. Antibiotic treatment is highly effective in removing active infection, however, the efficacy can be time dependent. Following late or no treatment, persistent symptoms can occur even in the absence of infection [209, 211, 212]. This is noteworthy as it suggests that inflammatory mechanisms that were induced during infection persist–either through dysregulation in inflammatory pathways or continued induction through bacterial debris [213]. As Bb does not produce any known toxins, the inflammatory response is assumed to be the cause of tissue damage. Neurological manifestations, termed neuroborreliosis, are considered a late-stage symptom that leads to Bell’s palsy, lymphocytic meningitis, behavioral disorders (depression, fatigue, sleep disturbances), and overall cognitive decline [214,215,216]. Bb is not known to penetrate into the brain parenchyma; however, the bacteria can be found within the CSF of patients and colonize within the dura mater of mice [217,218,219]. It can be inferred that a likely mechanism of neurological manifestations occurs through the indirect induction of inflammation in the brain parenchyma. This may occur through invasion of peripheral immune cells or inflammatory cytokines released from the meninges or choroid plexus into the CSF that prompts inflammation within the brain. While the CNS pathology of neuroborreliosis is well-studied, it is unknown how Bb is able to enter the CNS. Traversal across the BBB has been studied as a potential route; however, there is limited evidence of parenchymal invasion. Recently, our lab sought to determine the effects of Bb on human CP epithelial cells in vitro [73]. Similar to other infections, we found that infection with Bb induced the production and secretion of inflammatory and chemotactic cytokines. Transcriptome analysis revealed reduced expression of barrier and scaffolding proteins, which may lead to a loss in barrier integrity. This suggests that infection with Bb and subsequent alterations to the BCSFB would promote an environment that allows for the migration of Bb and peripheral immune cells into the CSF. It is important to note such findings still need to be explored in vivo and the mechanism of CNS entry is yet to be determined. Furthermore, it is known that while Bb may not enter the brain parenchyma, the brain still undergoes inflammation during infection [220]. The mechanisms that underlie this indirect transmission of inflammatory signals is still unknown.
SARS-CoV-2
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the virus that causes COVID-19, a respiratory illness that has led to the deaths of millions world-wide and is responsible for the on-going pandemic since late 2019. SARS-CoV-2 gains cellular entry through the binding of its spike protein S1 subunit to the ACE2 receptor which facilitates viral attachment to the cellular surface; cellular entry occurs through cleavage of the S1 subunit by the host cell protease TMPRSS2, exposing the S2 subunit that is needed for fusion [221, 222]. The virus predominantly infects epithelial tissue of the respiratory tract, but can be found to reside in other tissue including kidneys, intestines, and the brain [223, 224]. Recent studies have begun to examine and identify neurological complications in COVID-19 patients [225, 226]. Research into the prevalence of SARS-CoV-2 in the CNS of patients is ongoing, however, early studies suggest that viral presence in the CSF and brain parenchyma is a rare occurrence despite neurological symptoms being common [223,224,225,226,227,228,229,230,231,232,233,234,235]. Several possible routes of dissemination into the CNS have been suggested and studied, including transmigration across the olfactory mucosa, the enteric nervous system, the BBB, as well as the CP [236,237,238,239,240]. CP involvement in the pathogenesis of neurological manifestations of COVID-19 patients, with or without direct viral invasion of the CNS, is supported by several lines of evidence. Distribution mapping of the cellular components ACE2 and TMPRSS2 have shown a wide variety of expression in different organs, with expression levels within the brain being lower compared to other organs such as the lungs or small intestines [237, 241, 242]. Within the brain though, ACE2 was found to be expressed mainly on neurons, astrocytes, oligodendrocytes, and endothelial cells in distinct regions of the brain [243]. However, expression of ACE2 was notably higher within the choroid plexus of humans and mice [243]. Histological observations further substantiates the protein expression of ACE2 and TMPRSS2 on choroid plexus epithelial cells [237]. In human brain organoid models, SARS-CoV-2 shows neurotropic affinity to the choroid plexus epithelium with minimal or no infection of glia or neurons, and leads to disruption of the BCSFB [244, 245]. Furthermore, in a study of MS patients with COVID-19, researchers found SARS-CoV-2 (as well as ACE2) within CP epithelial cells and ependymal cells of both MS and non-MS patients with no evidence of neuronal and glial infection [246]. In contrast, single-cell transcriptome analysis of brain and CP samples from patients who had severe COVID-19 showed no molecular signs of SARS-CoV-2 [247]. However, results indicated robust expression in genes required for viral infection and substantial CP inflammation that is potentially relayed to the brain, resulting in inflammation [247]. These findings suggest that neurological complications from COVID-19 does not require the direct invasion of SARS-CoV-2 into the CNS and, in fact, it appears that neurological complications may more commonly arise as a result of aberrant inflammation throughout regions of the CNS, perhaps relayed by the CP. Findings in CSF samples from patients corroborate this hypothesis of a cytokine release syndrome, with rare observations of the virus but abnormal CSF findings that include increased CSF protein levels, elevated inflammatory factors (IL-8, IL-6, TNF-α), and CSF pleocytosis (neutrophilic and/or lymphocytic most commonly)—a systemic review of these findings was performed by Lewis et al. and Tandon et al. [227,228,229, 233, 248, 249]. As the pandemic continues, continued research into the neurological consequences of COVID-19 is needed in order to provide the necessary and potentially long-term care of patients.
Conclusion—a target for therapeutics
Historically, therapeutics targeting the CNS have failed at much greater rates compared to non-CNS drugs and are further plagued by greater approval times and developmental times [250]. Nevertheless, the CP has received renewed interest for its therapeutic potential in part due to its unique anatomical position and its role as an immunological niche. As the CP is situated at the blood-CSF interface and is the main producer of CSF, designing vectors that target the CP epithelium would allow for the delivery of drugs to the CSF as well as providing a method to modulate its molecular composition [251]. This would allow for the bypass of the BBB and impact therapeutic targets that are located in regions such as the subarachnoid space and perivascular space, as well as the regions within the ventricular system. While this would lead to shallower penetration of drugs to the brain parenchyma, it would allow for broader penetration through acting upon the glia limitans. This would have the potential to control and ameliorate CNS inflammation [252, 253]. Although it is desirable to have a highly selective BCSFB, this presents a challenge in drug design as it may be difficult for the drug to cross into the CSF. The BBB and BCSFB contain transporters that actively efflux a range of drugs, one of which is P-glycoprotein I (P-gp)—inhibition of this transporter allows for the penetration of nelfinavir, an HIV antiviral, into the brain parenchyma [254, 255]. The importance of P-gp and similar transporters are not limited to drug penetration. In T cells, P-gp has been shown to be involved in the transmembrane transport of inflammatory and activating cytokines, IL-2, IL-4, and IFN-γ [256]. Following exposure to HIV pseudotype virus, T cells enhanced their expression of P-gp, leading to enhanced expression of TNFα, IFN-γ, and IL-6 [257]. Understanding the role of P-gp and other multidrug resistant proteins within immune cells and the CP would provide understanding to differential drug penetration and the relay of inflammatory mediators across the BCSFB. While studies have shown that the CP expresses several types of influx/efflux transporters necessary for the transport of metabolites, there have been minimal studies targeting these transporters to enhance the efficacy of drug delivery or regulation of inflammatory mediators [6, 258]. Additionally, as treatment options for hydrocephalus are limited to surgical interventions that focus on CSF flow obstruction, pharmaceutical inhibition of CSF production may prove efficacious in scenarios were surgical options fail or are not available.
During infection, the accumulation and activation of resident immune cells can promote deleterious inflammation leading to a compromise in barrier integrity. Modulation of the cytokine milieu, such as IFN-γ, has the potential to alter the immune cell population and inflammatory state during healthy and disease states [106, 127, 259]. Additionally, therapeutics targeting junctional proteins may protect against the breakdown of the BCSFB and thus prevent pathogen and immune cell invasion. Dexamethasone has been shown to prevent tight junction alterations during S. suis infection in vitro; however, its use in meningitis patients has seen mixed results [260, 261]. Nevertheless, a class of therapeutics that target the BCSFB would prove highly beneficial in maintaining CSF homeostasis.
The choroid plexus is a highly complex system whose cellular and molecular composition is still being unraveled. Understanding these complexities opens an entirely new route for therapeutic interventions. As the CSF surrounds the entirety of the CNS, it represents a critical environment that must be closely maintained. The CP and ventricular system provide a major avenue for CNS modulation in which future studies are needed to explore the many options of pharmaceutical targets and their downstream applications.
Availability of data and materials
Not applicable.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- BBB:
-
Blood–brain barrier
- BCSFB:
-
Blood–cerebrospinal fluid barrier
- Bb :
-
Borrelia burgdorferi
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CP:
-
Choroid plexus
- CART:
-
Combination antiretroviral therapy
- DCs:
-
Dendritic cells
- ETV:
-
Endoscopic third ventriculostomy
- FIV:
-
Feline immunodeficiency virus
- HAND:
-
HIV-associated neurocognitive disorder
- HIV:
-
Human immunodeficiency virus
- JCV:
-
John Cunningham virus
- JAMs:
-
Junctional adhesion molecules
- LSTc:
-
Lactoseries tetrasaccharide c
- LPS:
-
Lipopolysaccharide
- MMPs:
-
Matrix metalloproteases
- NPCs:
-
Neural progenitor cells
- PAM:
-
Pam3CSK4
- PAMPS:
-
Pathogen-associated molecular patterns
- PRRs:
-
Pattern recognition receptors
- PHH:
-
Post-hemorrhagic hydrocephalus
- PIH:
-
Post-infectious hydrocephalus
- PML:
-
Progressive multifocal leukoencephalopathy
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SIV:
-
Simian immunodeficiency virus
- TLRs:
-
Toll-like receptors
References
Sakka L, Coll G, Chazal J. Anatomy and physiology of cerebrospinal fluid. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128(6):309–16.
Schmidley JW, Wissig SL. Anionic sites on the luminal surface of fenestrated and continuous capillaries of the CNS. Brain Res. 1986;363(2):265–71.
Redzic ZB, Segal MB. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Deliv Rev. 2004;56(12):1695–716.
Cserr HF. Physiology of the choroid plexus. Physiol Rev. 1971;51(2):273–311.
Del Bigio MR. Ependymal cells: biology and pathology. Acta Neuropathol. 2010;119(1):55–73.
Solár P, Zamani A, Kubíčková L, Dubový P, Joukal M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17(1):35.
Varatharaj A, Galea I. The blood–brain barrier in systemic inflammation. Brain Behav Immun. 2017;1(60):1–12.
Pulzova L, Bhide MR, Andrej K. Pathogen translocation across the blood–brain barrier. FEMS Immunol Med Microbiol. 2009;57(3):203–13.
Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, Lancaster MA. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science. 2020;369(6500):eaaz5626.
Pellegrini L, Lancaster MA. Breaking the barrier: in vitro models to study choroid plexus development. Curr Opin Cell Biol. 2021;1(73):41–9.
Redzic ZB. Studies on the human choroid plexus in vitro. Fluids Barriers CNS. 2013;10(1):10.
Angelow S, Zeni P, Galla HJ. Usefulness and limitation of primary cultured porcine choroid plexus epithelial cells as an in vitro model to study drug transport at the blood–CSF barrier. Adv Drug Deliv Rev. 2004;56(12):1859–73.
Jang A, Lehtinen MK. Experimental approaches for manipulating choroid plexus epithelial cells. Fluids Barriers CNS. 2022;19(1):36.
Dani N, Herbst RH, McCabe C, Green GS, Kaiser K, Head JP, et al. A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell. 2021;184(11):3056-3074.e21.
Damkier HH, Brown PD, Praetorius J. Cerebrospinal Fluid secretion by the choroid plexus. Physiol Rev. 2013;93(4):1847–92.
Oresković D, Klarica M. The formation of cerebrospinal fluid: nearly a hundred years of interpretations and misinterpretations. Brain Res Rev. 2010;64(2):241–62.
Khasawneh AH, Garling RJ, Harris CA. Cerebrospinal fluid circulation: what do we know and how do we know it? Brain Circ. 2018;4(1):14–8.
Brown PD, Davies SL, Speake T, Millar ID. Molecular mechanisms of cerebrospinal fluid production. Neuroscience. 2004;129(4):957–70.
Vogh BP, Godman DR, Maren TH. Effect of AlCl3 and other acids on cerebrospinal fluid production: a correction. J Pharmacol Exp Ther. 1987;243(1):35–9.
Praetorius J, Nejsum LN, Nielsen S. A SCL4A10 gene product maps selectively to the basolateral plasma membrane of choroid plexus epithelial cells. Am J Physiol Cell Physiol. 2004;286(3):C601-610.
Murphy VA, Johanson CE. Alteration of sodium transport by the choroid plexus with amiloride. Biochim Biophys Acta. 1989;979(2):187–92.
Lindsey AE, Schneider K, Simmons DM, Baron R, Lee BS, Kopito RR. Functional expression and subcellular localization of an anion exchanger cloned from choroid plexus. Proc Natl Acad Sci USA. 1990;87(14):5278–82.
Masuzawa T, Ohta T, Kawamura M, Nakahara N, Sato F. Immunohistochemical localization of Na+, K+-ATPase in the choroid plexus. Brain Res. 1984;302(2):357–62.
Pollay M, Hisey B, Reynolds E, Tomkins P, Stevens FA, Smith R. Choroid plexus Na+/K+-activated adenosine triphosphatase and cerebrospinal fluid formation. Neurosurgery. 1985;17(5):768–72.
Oshio K, Watanabe H, Song Y, Verkman AS, Manley GT. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005;19(1):76–8.
MacAulay N, Zeuthen T. Water transport between CNS compartments: contributions of aquaporins and cotransporters. Neuroscience. 2010;168(4):941–56.
Steffensen AB, Oernbo EK, Stoica A, Gerkau NJ, Barbuskaite D, Tritsaris K, et al. Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nat Commun. 2018;9(1):2167.
Hamann S, Herrera-Perez JJ, Bundgaard M, Alvarez-Leefmans FJ, Zeuthen T. Water permeability of Na+–K+–2Cl− cotransporters in mammalian epithelial cells. J Physiol. 2005;568(1):123–35.
Zeuthen T. Cotransport of K+, Cl− and H2O by membrane proteins from choroid plexus epithelium of Necturus maculosus. J Physiol. 1994;478(2):203–19.
Hamann S, Herrera-Perez JJ, Zeuthen T, Alvarez-Leefmans FJ. Cotransport of water by the Na+–K+–2Cl− cotransporter NKCC1 in mammalian epithelial cells. J Physiol. 2010;588(21):4089–101.
Kishimoto N, Sawamoto K. Planar polarity of ependymal cilia. Differentiation. 2012;83(2):S86-90.
Wright BLC, Lai JTF, Sinclair AJ. Cerebrospinal fluid and lumbar puncture: a practical review. J Neurol. 2012;259(8):1530–45.
Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS. 2014;11(1):10.
Cserr HF, Harling-Berg CJ, Knopf PM. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992;2(4):269–76.
Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol. 1993;19(6):480–8.
Johnston M. The importance of lymphatics in cerebrospinal fluid transport. Lymphat Res Biol. 2003;1(1):41–5.
Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8(1):1434.
Ahn JH, Cho H, Kim JH, Kim SH, Ham JS, Park I, et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. 2019;572(7767):62–6.
Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117(1):1–14.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21(10):1380–91.
Tavares GA, Louveau A. Meningeal lymphatics: an immune gateway for the central nervous system. Cells. 2021;10(12):3385.
Clarkson BD, Walker A, Harris MG, Rayasam A, Hsu M, Sandor M, et al. CCR7 deficient inflammatory dendritic cells are retained in the central nervous system. Sci Rep. 2017;7(1):42856.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra111.
Sarnat HB. Histochemistry and immunocytochemistry of the developing ependyma and choroid plexus. Microsc Res Tech. 1998;41(1):14–28.
Liddelow SA, Dziegielewska KM, Ek CJ, Habgood MD, Bauer H, Bauer HC, et al. Mechanisms That determine the internal environment of the developing brain: a transcriptomic, functional and ultrastructural approach. PLoS ONE. 2013;8(7):e65629.
Liddelow SA. Development of the choroid plexus and blood–CSF barrier. Front Neurosci. 2015. https://doi.org/10.3389/fnins.2015.00032/full.
Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol. 2015;209(4):493–506.
Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22(1):207–35.
Steinemann A, Galm I, Chip S, Nitsch C, Maly IP. Claudin-1, -2 and -3 are selectively expressed in the epithelia of the choroid plexus of the mouse from early development and into adulthood while claudin-5 is restricted to endothelial cells. Front Neuroanat. 2016. https://doi.org/10.3389/fnana.2016.00016/full.
Wolburg H, Wolburg-Buchholz K, Liebner S, Engelhardt B. Claudin-1, claudin-2 and claudin-11 are present in tight junctions of choroid plexus epithelium of the mouse. Neurosci Lett. 2001;307(2):77–80.
Kratzer I, Vasiljevic A, Rey C, Fevre-Montange M, Saunders N, Strazielle N, et al. Complexity and developmental changes in the expression pattern of claudins at the blood–CSF barrier. Histochem Cell Biol. 2012;138(6):861–79.
Van Itallie CM, Anderson JM. Phosphorylation of tight junction transmembrane proteins: many sites, much to do. Tissue Barriers. 2017;6(1):e1382671.
Redzic Z. Molecular biology of the blood–brain and the blood–cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS. 2011;8(1):3.
De Bock M, Vandenbroucke RE, Decrock E, Culot M, Cecchelli R, Leybaert L. A new angle on blood–CNS interfaces: a role for connexins? FEBS Lett. 2014;588(8):1259–70.
Li MWM, Mruk DD, Cheng CY. Gap junctions and blood–tissue barriers. In: Cheng CY, editor. Biology and regulation of blood–tissue barriers. New York: Springer; 2013. p. 260–80 (10.1007/978-1-4614-4711-5_13).
Mathews VP, Smith RR. Choroid plexus infections: neuroimaging appearances of four cases. Am J Neuroradiol. 1992;13(1):374–8.
Kovoor JME, Mahadevan A, Narayan JP, Govindappa SS, Satishchandra P, Taly AV, et al. Cryptococcal choroid plexitis as a mass lesion: MR imaging and histopathologic correlation. Am J Neuroradiol. 2002;23(2):273–6.
Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163(4):1379–93.
Hagiwara E, Nath J. Choroid plexitis in a case of systemic nocardiosis. Emerg Radiol. 2007;14(5):337–43.
Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15(1):155–63.
Laflamme N, Echchannaoui H, Landmann R, Rivest S. Cooperation between toll-like receptor 2 and 4 in the brain of mice challenged with cell wall components derived from gram-negative and gram-positive bacteria. Eur J Immunol. 2003;33(4):1127–38.
Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia independent of systemic cytokines. J Neurosci. 2005;25(7):1788–96.
Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–39.
Stridh L, Ek CJ, Wang X, Nilsson H, Mallard C. Regulation of toll-like receptors in the choroid plexus in the immature brain after systemic inflammatory stimuli. Transl Stroke Res. 2013;4(2):220–7.
Skipor J, Szczepkowska A, Kowalewska M, Herman A, Lisiewski P. Profile of toll-like receptor mRNA expression in the choroid plexus in adult ewes. Acta Vet Hung. 2014;63(1):69–78.
Borkowski J, Li L, Steinmann U, Quednau N, Stump-Guthier C, Weiss C, et al. Neisseria meningitidiselicits a pro-inflammatory response involving IκBζ in a human blood–cerebrospinal fluid barrier model. J Neuroinflammation. 2014;11(1):163.
Mottahedin A, Joakim Ek C, Truvé K, Hagberg H, Mallard C. Choroid plexus transcriptome and ultrastructure analysis reveals a TLR2-specific chemotaxis signature and cytoskeleton remodeling in leukocyte trafficking. Brain Behav Immun. 2019;1(79):216–27.
Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, et al. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab. 2009;29(5):921–32.
Shimada A, Hasegawa-Ishii S. Increased cytokine expression in the choroid plexus stroma and epithelium in response to endotoxin-induced systemic inflammation in mice. Toxicol Rep. 2021;1(8):520–8.
Mottahedin A, Smith PLP, Hagberg H, Ek CJ, Mallard C. TLR2-mediated leukocyte trafficking to the developing brain. J Leukoc Biol. 2017;101(1):297–305.
Quintana E, Fernández A, Velasco P, de Andrés B, Liste I, Sancho D, et al. DNGR-1+ dendritic cells are located in meningeal membrane and choroid plexus of the noninjured brain. Glia. 2015;63(12):2231–48.
Zhu L, Stein LR, Kim D, Ho K, Yu GQ, Zhan L, et al. Klotho controls the brain–immune system interface in the choroid plexus. Proc Natl Acad Sci USA. 2018;115(48):E11388–96.
Thompson D, Sorenson J, Greenmyer J, Brissette CA, Watt JA. The Lyme disease bacterium, Borrelia burgdorferi, stimulates an inflammatory response in human choroid plexus epithelial cells. PLoS ONE. 2020;15(7):e0234993.
Geijtenbeek TBH, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465–79.
Schwerk C, Rybarczyk K, Essmann F, Seibt A, Mölleken ML, Zeni P, et al. TNFα induces choroid plexus epithelial cell barrier alterations by apoptotic and nonapoptotic mechanisms. J Biomed Biotechnol. 2010;2010:307231.
Strazielle N, Khuth ST, Murat A, Chalon A, Giraudon P, Belin MF, et al. Pro-inflammatory cytokines modulate matrix metalloproteinase secretion and organic anion transport at the blood–cerebrospinal fluid barrier. J Neuropathol Exp Neurol. 2003;62(12):1254–64.
Szmydynger-Chodobska J, Gandy JR, Varone A, Shan R, Chodobski A. Synergistic interactions between cytokines and AVP at the blood–CSF barrier result in increased chemokine production and augmented influx of leukocytes after brain injury. PLoS ONE. 2013;8(11):e79328.
Giraudon P, Szymocha R, Buart S, Bernard A, Cartier L, Belin MF, et al. T lymphocytes activated by persistent viral infection differentially modify the expression of metalloproteinases and their endogenous inhibitors, TIMPs, in human astrocytes: relevance to HTLV-I-induced neurological disease. J Immunol. 2000;164(5):2718–27.
Pagenstecher A, Stalder AK, Kincaid CL, Shapiro SD, Campbell IL. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am J Pathol. 1998;152(3):729–41.
Khuth ST, Akaoka H, Pagenstecher A, Verlaeten O, Belin MF, Giraudon P, et al. Morbillivirus infection of the mouse central nervous system induces region-specific upregulation of MMPs and TIMPs correlated to inflammatory cytokine expression. J Virol. 2001;75(17):8268–82.
Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39(3):279–91.
Zeni P, Doepker E, Topphoff US, Huewel S, Tenenbaum T, Galla HJ. MMPs contribute to TNF-α-induced alteration of the blood–cerebrospinal fluid barrier in vitro. Am J Physiol Cell Physiol. 2007;293(3):C855–64.
Chiu PS, Lai SC. Matrix metalloproteinase-9 leads to claudin-5 degradation via the NF-κB pathway in BALB/c mice with eosinophilic meningoencephalitis caused by angiostrongylus cantonensis. PLoS ONE. 2013;8(3):e53370.
Marques F, Falcao AM, Sousa JC, Coppola G, Geschwind D, Sousa N, et al. Altered iron metabolism is part of the choroid plexus response to peripheral inflammation. Endocrinology. 2009;150(6):2822–8.
Liu CB, Wang R, Dong MW, Gao XR, Yu F. Expression of hepcidin at the choroid plexus in normal aging rats is associated with IL-6/Stat3 signaling pathway. Sheng Li Xue Bao. 2014;66(6):639–46.
Deczkowska A, Baruch K, Schwartz M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol. 2016;37(3):181–92.
Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, et al. Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med. 2016;8(10):1162–83.
O’Hara BA, Morris-Love J, Gee GV, Haley SA, Atwood WJ. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020;16(3):e1008371.
Matyszak MK, Lawson LJ, Perry VH, Gordon S. Stromal macrophages of the choroid plexus situated at an interface between the brain and peripheral immune system constitutively express major histocompatibility class II antigens. J Neuroimmunol. 1992;40(2):173–81.
Ling EA, Kaur C, Lu J. Origin, nature, and some functional considerations of intraventricular macrophages, with special reference to the epiplexus cells. Microsc Res Tech. 1998;41(1):43–56.
Strominger I, Elyahu Y, Berner O, Reckhow J, Mittal K, Nemirovsky A, et al. The choroid plexus functions as a niche for T-Cell stimulation within the central nervous system. Front Immunol. 2018;16(9):1066.
Meeker RB, Williams K, Killebrew DA, Hudson LC. Cell trafficking through the choroid plexus. Cell Adh Migr. 2012;6(5):390–6.
Hanly A, Petito CK. HLA-DR-positive dendritic cells of the normal human choroid plexus: a potential reservoir of HIV in the central nervous system. Hum Pathol. 1998;29(1):88–93.
Chinnery HR, Ruitenberg MJ, McMenamin PG. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J Neuropathol Exp Neurol. 2010;69(9):896–909.
Lu J, Kaur C, Ling EA. Up-regulation of surface antigens on epiplexus cells in postnatal rats following intraperitoneal injections of lipopolysaccharide. Neuroscience. 1994;63(4):1169–78.
Carpenter SJ, McCarthy LE, Borison HL. Electron microscopic study on the epiplexus (Kolmer) cells of the cat choroid plexus. Z Zellforsch. 1970;110(4):471–86.
Lu J, Kaur C, Ling EA. Intraventricular macrophages in the lateral ventricles with special reference to epiplexus cells: a quantitative analysis and their uptake of fluorescent tracer injected intraperitoneally in rats of different ages. J Anat. 1993;183(Pt 2):405–14.
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Antunes ARP, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22(6):1021–35.
Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. 2019;20(9):547–62.
Goldmann T, Jordão MJC, Wieghofer P, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at CNS interfaces. Nat Immunol. 2016;17(7):797–805.
Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol. 2016;17(12):1397–406.
Cui J, Xu H, Lehtinen MK. Macrophages on the margin: choroid plexus immune responses. Trends Neurosci. 2021;44(11):864–75.
Greter M, Lelios I, Croxford AL. Microglia versus myeloid cell nomenclature during brain inflammation. Front Immunol. 2015. https://doi.org/10.3389/fimmu.2015.00249/full.
Strazielle N, Creidy R, Malcus C, Boucraut J, Ghersi-Egea JF. T-Lymphocytes traffic into the brain across the blood–CSF barrier: evidence using a reconstituted choroid plexus epithelium. PLoS ONE. 2016;11(3):e0150945.
Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27(1):48–55.
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. PNAS. 2013;110(6):2264–9.
Petito CK, Adkins B. Choroid plexus selectively accumulates T-lymphocytes in normal controls and after peripheral immune activation. J Neuroimmunol. 2005;162(1):19–27.
Steffen BJ, Breier G, Butcher EC, Schulz M, Engelhardt B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am J Pathol. 1996;148(6):1819–38.
Carrithers MD, Visintin I, Kang SJ, Janeway CA. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain. 2000;123(Pt 6):1092–101.
Wolburg K, Gerhardt H, Schulz M, Wolburg H, Engelhardt B. Ultrastructural localization of adhesion molecules in the healthy and inflamed choroid plexus of the mouse. Cell Tissue Res. 1999;296(2):259–69.
Deckert-SchlÜter M, SchlÜter D, Hof H, Wiestler OD, Lassmann H. Differential expression of ICAM-1, VCAM-1 and their ligands LFA-1, Mac-1, CD43, VLA-4, and MHC class II antigens in murine toxoplasma encephalitis: a light microscopic and ultrastructural immunohistochemical study. J Neuropathol Exp Neurol. 1994;53(5):457–68.
Haas J, Rudolph H, Costa L, Faller S, Libicher S, Würthwein C, et al. The choroid plexus is permissive for a preactivated antigen-experienced memory b-cell subset in multiple sclerosis. Front Immunol. 2020;11:618544.
Boyer RS, Sun NC, Verity A, Nies KM, Louie JS. Immunoperoxidase staining of the choroid plexus in systemic lupus erythematosus. J Rheumatol. 1980;7(5):645–50.
McIntosh RM, Copack P, Chernack WB, Griswold WR, Weil R 3rd, Koss MN. The human choroid plexus and autoimmune nephritis. Arch Pathol. 1975;99(1):48–50.
Falangola MF, Castro-Filho BG, Petito CK. Immune complex deposition in the choroid plexus of patients with acquired immunodeficiency syndrome. Ann Neurol. 1994;36(3):437–40.
Davis JA, Weisman MH, Dail DH. Vascular disease in infective endocarditis: report of immune-mediated events in skin and brain. Arch Intern Med. 1978;138(3):480–3.
June CH, Contreras CE, Perrin LH, Lambert PH, Miescher PA. Circulating and tissue-bound immune complex formation in murine malaria. J Immunol. 1979;122(6):2154–61.
Moore GRW, Laule C, Leung E, Pavlova V, Morgan BP, Esiri MM. Complement and humoral adaptive immunity in the human choroid plexus: roles for stromal concretions, basement membranes, and epithelium. J Neuropathol Exp Neurol. 2016;75(5):415–28.
Strazielle N, Ghersi-Egea JF. Physiology of blood–brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharmaceutics. 2013;10(5):1473–91.
Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood–brain barrier. J Neurochem. 2002;81(1):203–6.
Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood–brain barrier. J Neuroimmunol. 2001;114(1):168–72.
Schwerk C, Tenenbaum T, Kim KS, Schroten H. The choroid plexus—a multi-role player during infectious diseases of the CNS. Front Cell Neurosci. 2015. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4357259/. Accessed 26 Mar 2019
Lauer AN, Tenenbaum T, Schroten H, Schwerk C. The diverse cellular responses of the choroid plexus during infection of the central nervous system. Am J Physiol Cell Physiol. 2018;314(2):C152–65.
Kaur C, Rathnasamy G, Ling EA. The choroid plexus in healthy and diseased brain. J Neuropathol Exp Neurol. 2016;75(3):198–213.
Robert SM, Reeves BC, Marlier A, Duy PQ, DeSpenza T, Kundishora A, et al. Inflammatory hydrocephalus. Childs Nerv Syst. 2021. https://doi.org/10.1007/s00381-021-05255-z.
Dewan MC, Rattani A, Mekary R, Glancz LJ, Yunusa I, Baticulon RE, et al. Global hydrocephalus epidemiology and incidence: systematic review and meta-analysis. J Neurosurg. 2018;130(4):1065–79.
Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med. 2017;23(8):997–1003.
Karimy JK, Reeves BC, Damisah E, Duy PQ, Antwi P, David W, et al. Inflammation in acquired hydrocephalus: pathogenic mechanisms and therapeutic targets. Nat Rev Neurol. 2020;16(5):285–96.
Strahle J, Garton HJL, Maher CO, Muraszko KM, Keep RF, Xi G. Mechanisms of hydrocephalus after neonatal and adult intraventricular hemorrhage. Transl Stroke Res. 2012;3(Suppl 1):25–38.
Tully HM, Dobyns WB. Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur J Med Genet. 2014;57(8):359–68.
Kahle KT, Kulkarni AV, Limbrick DD, Warf BC. Hydrocephalus in children. Lancet. 2016;387(10020):788–99.
Kulkarni AV, Drake JM, Kestle JRW, Mallucci CL, Sgouros S, Constantini S, et al. Endoscopic third ventriculostomy vs cerebrospinal fluid shunt in the treatment of hydrocephalus in children: a propensity score-adjusted analysis. Neurosurgery. 2010;67(3):588–93.
Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52(3):439–51.
Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35.
Xu H, Fame RM, Sadegh C, Sutin J, Naranjo C, Syau D, et al. Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development. Nat Commun. 2021;12(1):1–16.
Sarnat HB. Role of human fetal ependyma. Pediatr Neurol. 1992;8(3):163–78.
Bruni JE. Ependymal development, proliferation, and functions: a review. Microsc Res Tech. 1998;41(1):2–13.
Devhare P, Meyer K, Steele R, Ray RB, Ray R. Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis. 2017;8(10):e3106–e3106.
Liang Q, Luo Z, Zeng J, Chen W, Foo SS, Lee SA, et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell. 2016;19(5):663–71.
Wen Z, Song H, Ming G-L. How does Zika virus cause microcephaly? Genes Dev. 2017;31(9):849–61.
Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016;19(1):120–6.
Adams Waldorf KM, Nelson BR, Stencel-Baerenwald JE, Studholme C, Kapur RP, Armistead B, et al. Congenital Zika virus infection as a silent pathology with loss of neurogenic output in the fetal brain. Nat Med. 2018;24(3):368–74.
Coffey LL, Keesler RI, Pesavento PA, Woolard K, Singapuri A, Watanabe J, et al. Intraamniotic Zika virus inoculation of pregnant rhesus macaques produces fetal neurologic disease. Nat Commun. 2018;20(9):2414.
Gurung S, Reuter N, Preno A, Dubaut J, Nadeau H, Hyatt K, et al. Zika virus infection at mid-gestation results in fetal cerebral cortical injury and fetal death in the olive baboon. PLoS Pathog. 2019;15(1):e1007507.
Martinot AJ, Abbink P, Afacan O, Prohl AK, Bronson R, Hecht JL, et al. Fetal neuropathology in Zika virus infected pregnant female rhesus monkeys. Cell. 2018;173(5):1111-1122.e10.
Mulkey SB, Bulas DI, Vezina G, Fourzali Y, Morales A, Arroyave-Wessel M, et al. Sequential neuroimaging of the fetus and newborn with in utero Zika virus exposure. JAMA Pediatr. 2019;173(1):52–9.
Vouga M, Baud D. Imaging of congenital Zika virus infection: the route to identification of prognostic factors. Prenat Diagn. 2016;36(9):799–811.
Ximenes ASFC, Pires P, Werner H, Jungmann PM, Filho ELR, Andrade EP, et al. Neuroimaging findings using transfontanellar ultrasound in newborns with microcephaly: a possible association with congenital Zika virus infection. J Matern Fetal Neonatal Med. 2019;32(3):493–501.
van der Linden V, de Lima Petribu NC, Pessoa A, Faquini I, Paciorkowski AR, van der Linden H Jr, et al. Association of severe hydrocephalus with congenital Zika syndrome. JAMA Neurol. 2019;76(2):203–10.
Watanabe M, Buth JE, Vishlaghi N, de la Torre-Ubieta L, Taxidis J, Khakh BS, et al. Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep. 2017;21(2):517–32.
Kim J, Alejandro B, Hetman M, Hattab EM, Joiner J, Schroten H, et al. Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood–cerebrospinal fluid barrier. PLoS Pathog. 2020;16(5):e1008204.
Nelson BR, Roby JA, Dobyns WB, Rajagopal L, Gale M, Adams Waldorf KM. Immune evasion strategies used by Zika virus to infect the fetal eye and brain. Viral Immunol. 2020;33(1):22–37.
Wollebo HS, White MK, Gordon J, Berger JR, Khalili K. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann Neurol. 2015;77(4):560–70.
Pietropaolo V, Prezioso C, Bagnato F, Antonelli G. John Cunningham virus: an overview on biology and disease of the etiological agent of the progressive multifocal leukoencephalopathy. New Microbiol. 2018;41(3):179–86.
Ricciardiello L, Laghi L, Ramamirtham P, Chang CL, Chang DK, Randolph AE, et al. JC virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology. 2000;119(5):1228–35.
Kitamura T, Sugimoto C, Kato A, Ebihara H, Suzuki M, Taguchi F, et al. Persistent JC virus (JCV) infection is demonstrated by continuous shedding of the same JCV strains. J Clin Microbiol. 1997;35(5):1255–7.
Yogo Y, Kitamura T, Sugimoto C, Ueki T, Aso Y, Hara K, et al. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol. 1990;64(6):3139–43.
Berger JR, Miller CS, Mootoor Y, Avdiushko SA, Kryscio RJ, Zhu H. JC virus detection in bodily fluids: clues to transmission. Clin Infect Dis. 2006;43(1):e9-12.
Berger JR, Aksamit AJ, Clifford DB, Davis L, Koralnik IJ, Sejvar JJ, et al. PML diagnostic criteria: consensus statement from the AAN neuroinfectious disease section. Neurology. 2013;80(15):1430–8.
Tavazzi E, White MK, Khalili K. Progressive multifocal leukoencephalopathy: clinical and molecular aspects. Rev Med Virol. 2012;22(1):18–32.
Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 2009;60(12):3761–5.
White FA, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66(10):5726–34.
Chapagain ML, Nerurkar VR. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood–brain barrier. J Infect Dis. 2010;202(2):184–91.
Neu U, Maginnis MS, Palma AS, Ströh LJ, Nelson CDS, Feizi T, et al. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe. 2010;8(4):309–19.
Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–3.
Haley SA, O’Hara BA, Nelson CDS, Brittingham FLP, Henriksen KJ, Stopa EG, et al. Human polyomavirus receptor distribution in brain parenchyma contrasts with receptor distribution in kidney and choroid plexus. Am J Pathol. 2015;185(8):2246–58.
Agnihotri SP, Wuthrich C, Dang X, Nauen D, Karimi R, Viscidi R, et al. A fatal case of JC virus meningitis presenting with hydrocephalus in an HIV-seronegative patient. Ann Neurol. 2014;76(1):140–7.
O’Hara BA, Gee GV, Atwood WJ, Haley SA. Susceptibility of primary human choroid plexus epithelial cells and meningeal cells to infection by JC virus. J Virol. 2018;92(8):e00105-e118.
UNAIDS, IN DANGER: UNAIDS Global AIDS Update 2022. Geneva: Joint United Nations Programme on HIV/, AIDS; 2022. Licence: CC BY-NC-SA 3.0 IGO. 2022-global-aids-update-summary_en.pdf. 2022. https://www.unaids.org/sites/default/files/media_asset/2022-global-aids-update-summary_en.pdf. Accessed 15 Aug 2022
Moir S, Chun TW, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol. 2011;6(1):223–48.
Deeks SG, Overbaugh J, Phillips A, Buchbinder S. HIV infection. Nat Rev Dis Primers. 2015;1(1):1–22.
Sacktor N, Skolasky RL, Seaberg E, Munro C, Becker JT, Martin E, et al. Prevalence of HIV-associated neurocognitive disorders in the multicenter AIDS cohort study. Neurology. 2016;86(4):334–40.
The HIV-CAUSAL Collaboration. The effect of combined antiretroviral therapy on the overall mortality of HIV-infected individuals. AIDS. 2010;24(1):123–37.
Detels R, Muñoz A, McFarlane G, Kingsley LA, Margolick JB, Giorgi J, et al. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. Multicenter AIDS cohort study investigators. JAMA. 1998;280(17):1497–503.
Cole SR, Hernán MA, Robins JM, Anastos K, Chmiel J, Detels R, et al. Effect of highly active antiretroviral therapy on time to acquired immunodeficiency syndrome or death using marginal structural models. Am J Epidemiol. 2003;158(7):687–94.
Egger M, May M, Chêne G, Phillips AN, Ledergerber B, Dabis F, et al. Prognosis of HIV-1-infected patients starting highly active antiretroviral therapy: a collaborative analysis of prospective studies. Lancet. 2002;360(9327):119–29.
Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy. Neurology. 2010;75(23):2087–96.
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, et al. Dementia in AIDS patients: incidence and risk factors. Neurology. 1993;43(11):2245–2245.
Maschke M, Kastrup O, Esser S, Ross B, Hengge U, Hufnagel A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J Neurol Neurosurg Psychiatry. 2000;69(3):376–80.
Cysique LA, Maruff P, Brew BJ. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J Neurovirol. 2004;10(6):350–7.
Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(4):234–48.
Schouten J, Cinque P, Gisslen M, Reiss P, Portegies P. HIV-1 infection and cognitive impairment in the cART era: a review. AIDS. 2011;25(5):561–75.
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8(2):136–42.
Simioni S, Cavassini M, Annoni JM, Abraham AR, Bourquin I, Schiffer V, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243–50.
Blasi M, Carpenter JH, Balakumaran B, Cara A, Gao F, Klotman ME. Identification of HIV-1 genitourinary tract compartmentalization by analyzing the env gene sequences in urine. AIDS. 2015;29(13):1651–7.
Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. 2017;23(11):1271–6.
Fois AF, Brew BJ. The potential of the CNS as a reservoir for HIV-1 infection: implications for HIV eradication. Curr HIV/AIDS Rep. 2015;12(2):299–303.
Clements JE, Gama L, Graham DR, Mankowski JL, Zink MC. A simian immunodeficiency virus macaque model of highly active antiretroviral treatment: viral latency in the periphery and the central nervous system. Curr Opin HIV AIDS. 2011;6(1):37–42.
Hammond ER, Crum RM, Treisman GJ, Mehta SH, Clifford DB, Ellis RJ, et al. Persistent CSF but not plasma HIV RNA, is associated with increased risk of new-onset moderate-to-severe depressive symptoms; a prospective cohort study. J Neurovirol. 2016;22(4):479–87.
Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, Price RW. Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. AIDS. 2014;28(15):2251–8.
Pérez-Valero I, Ellis R, Heaton R, Deutsch R, Franklin D, Clifford DB, et al. Cerebrospinal fluid viral escape in aviremic HIV-infected patients receiving antiretroviral therapy: prevalence, risk factors and neurocognitive effects. AIDS. 2019;33(3):475–81.
Falangola MF, Hanly A, Galvao-Castro B, Petito CK. HIV infection of human choroid plexus: a possible mechanism of viral entry into the CNS. J Neuropathol Exp Neurol. 1995;54(4):497–503.
Hanly A, Petito CK. HLA-DR-positive dendritic cells of the normal human choroid plexus: a potential reservoir of HIV in the central nervous system. Hum Pathol. 1998;29(1):88–93.
Petito CK, Chen H, Mastri AR, Torres-Munoz J, Roberts B, Wood C. HIV infection of choroid plexus in AIDS and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection. J Neurovirol. 1999;5(6):670–7.
Chen H, Wood C, Petito CK. Comparisons of HIV-1 viral sequences in brain, choroid plexus and spleen: potential role of choroid plexus in the pathogenesis of HIV encephalitis. J Neurovirol. 2000;6(6):498–506.
Burkala EJ, He J, West JT, Wood C, Petito CK. Compartmentalization of HIV-1 in the central nervous system: role of the choroid plexus. AIDS. 2005;19(7):675–84.
Albright AV, Shieh JTC, Itoh T, Lee B, Pleasure D, O’Connor MJ, et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73(1):205–13.
de Almeida SM, Rotta I, Ribeiro CE, Smith D, Wang R, Judicello J, et al. Blood–CSF barrier and compartmentalization of CNS cellular immune response in HIV infection. J Neuroimmunol. 2016;15(301):41–8.
Sparger EE. FIV as a model for HIV: an overview. In: Friedman H, Specter S, Bendinelli M, editors. In vivo models of HIV disease and control. New York: Springer; 2006. p. 149–237.
Gardner MB. Simian and feline immunodeficiency viruses: animal lentivirus models for evaluation of AIDS vaccines and antiviral agents. Antiviral Res. 1991;15(4):267–86.
Bragg DC, Childers TA, Tompkins MB, Tompkins WA, Meeker RB. Infection of the choroid plexus by feline immunodeficiency virus. J Neurovirol. 2002;8(3):211–24.
Meeker RB, Bragg DC, Poulton W, Hudson L. Transmigration of macrophages across the choroid plexus epithelium in response to the feline immunodeficiency virus. Cell Tissue Res. 2012;347(2):443–55.
Lackner AA, Smith MO, Munn RJ, Martfeld DJ, Gardner MB, Marx PA, et al. Localization of simian immunodeficiency virus in the central nervous system of rhesus monkeys. Am J Pathol. 1991;139(3):609–21.
Czub S, Müller JG, Czub M, Müller-Hermelink HK. Impact of various simian immunodeficiency virus variants on induction and nature of neuropathology in macaques. Res Virol. 1996;147(2–3):165–70.
Centers for Disease Control and Prevention (CDC), Bacon RM, Kugeler KJ, Mead PS. Surveillance for Lyme disease—United States, 1992–2006. MMWR Surveill Summ. 2008;57(10):1–9.
Rosenberg R. Vital signs: trends in reported vectorborne disease cases—United States and Territories, 2004–2016. MMWR Morb Mortal Wkly Rep. 2018. https://www.cdc.gov/mmwr/volumes/67/wr/mm6717e1.htm. Accessed 21 Nov 2019
Nelson CA, Saha S, Kugeler KJ, Delorey MJ, Shankar MB, Hinckley AF, et al. Incidence of clinician-diagnosed Lyme disease, United States, 2005–2010. Emerging Infect Dis. 2015;21(9):1625–31.
Hinckley AF, Connally NP, Meek JI, Johnson BJ, Kemperman MM, Feldman KA, et al. Lyme disease testing by large commercial laboratories in the United States. Clin Infect Dis. 2014;59(5):676–81.
Arvikar SL, Steere AC. Diagnosis and treatment of Lyme arthritis. Infect Dis Clin North Am. 2015;29(2):269–80.
Wormser GP. Hematogenous dissemination in early Lyme disease. Wien Klin Wochenschr. 2006;118(21–22):634–7.
Aucott JN. Posttreatment Lyme disease syndrome. Infect Dis Clin. 2015;29(2):309–23.
Aucott JN, Rebman AW, Crowder LA, Kortte KB. Post-treatment Lyme disease syndrome symptomatology and the impact on life functioning: is there something here? Qual Life Res. 2013;22(1):75–84.
Wormser GP, Nadelman RB, Schwartz I. The amber theory of Lyme arthritis: initial description and clinical implications. Clin Rheumatol. 2012;31(6):989–94.
Hildenbrand P, Craven DE, Jones R, Nemeskal P. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. Am J Neuroradiol. 2009;30(6):1079–87.
Koedel U, Fingerle V, Pfister HW. Lyme neuroborreliosis-epidemiology, diagnosis and management. Nat Rev Neurol. 2015;11(8):446–56.
Knudtzen FC, Andersen NS, Jensen TG, Skarphédinsson S. Characteristics and clinical outcome of Lyme neuroborreliosis in a high endemic area, 1995–2014: a retrospective cohort study in Denmark. Clin Infect Dis. 2017;65(9):1489–95.
Casselli T, Divan A, Vomhof-DeKrey EE, Tourand Y, Pecoraro HL, Brissette CA. A murine model of Lyme disease demonstrates that Borrelia burgdorferi colonizes the dura mater and induces inflammation in the central nervous system. PLoS Pathog. 2021;17(2):e1009256.
Djukic M, Schmidt-Samoa C, Lange P, Spreer A, Neubieser K, Eiffert H, et al. Cerebrospinal fluid findings in adults with acute Lyme neuroborreliosis. J Neurol. 2012;259(4):630–6.
Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, et al. The spirochetal etiology of Lyme disease. N Engl J Med. 1983;308(13):733–40.
Coughlin JM, Yang T, Rebman AW, Bechtold KT, Du Y, Mathews WB, et al. Imaging glial activation in patients with post-treatment Lyme disease symptoms: a pilot study using [11C] DPA-713 PET. J Neuroinflammation. 2018;15(1):346.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78(4):779-784.e5.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271-280.e8.
Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. 2020;1(7):e290–9.
Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. 2020;383(6):590–2.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic Features in Severe SARS-CoV-2 Infection. N Engl J Med. 2020;382(23):2268–70.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683–90.
Destras G, Bal A, Escuret V, Morfin F, Lina B, Josset L. Systematic SARS-CoV-2 screening in cerebrospinal fluid during the COVID-19 pandemic. Lancet Microbe. 2020;1(4):e149.
Bellon M, Schweblin C, Lambeng N, Cherpillod P, Vazquez J, Lalive PH, et al. Cerebrospinal fluid features in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa1165.
Tandon M, Kataria S, Patel J, Mehta TR, Daimee M, Patel V, et al. A comprehensive systematic review of CSF analysis that defines neurological manifestations of COVID-19. Int J Infect Dis. 2021;1(104):390–7.
Huang YH, Jiang D, Huang JT. SARS-CoV-2 detected in cerebrospinal fluid by PCR in a case of COVID-19 encephalitis. Brain Behav Immun. 2020;87:149.
Kamal YM, Abdelmajid Y, Madani AARA. Cerebrospinal fluid confirmed COVID-19-associated encephalitis treated successfully. BMJ Case Rep CP. 2020;13(9):e237378.
Khatoon F, Prasad K, Kumar V. Neurological manifestations of COVID-19: available evidences and a new paradigm. J Neurovirol. 2020;26(5):619–30.
Lewis A, Frontera J, Placantonakis DG, Lighter J, Galetta S, Balcer L, et al. Cerebrospinal fluid in COVID-19: a systematic review of the literature. J Neurol Sci. 2021;15(421):117316.
Moriguchi T, Harii N, Goto J, Harada D, Sugawara H, Takamino J, et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int J Infect Dis. 2020;1(94):55–8.
Liu JM, Tan BH, Wu S, Gui Y, Suo JL, Li YC. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J Med Virol. 2021;93(3):1304–13.
Briguglio M, Bona A, Porta M, Dell’Osso B, Pregliasco FE, Banfi G. Disentangling the hypothesis of host dysosmia and SARS-CoV-2: the bait symptom that hides neglected neurophysiological routes. Front Physiol. 2020;11:671.
Deffner F, Scharr M, Klingenstein S, Klingenstein M, Milazzo A, Scherer S, et al. Histological evidence for the enteric nervous system and the choroid plexus as alternative routes of neuroinvasion by SARS-CoV2. Front Neuroanat. 2020;6(14):596439.
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2021;24(2):168–75.
Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. 2021;12:1494.
Erickson MA, Rhea EM, Knopp RC, Banks WA. Interactions of SARS-CoV-2 with the blood–brain barrier. Int J Mol Sci. 2021;22(5):2681.
Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol Syst Biol. 2020;16(7):e9610.
Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty. 2020;9(1):45.
Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol. 2021;11:1860.
Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood–CSF barrier in human brain organoids. Cell Stem Cell. 2020;27(6):951-961.e5.
Jacob F, Pather SR, Huang WK, Zhang F, Wong SZH, Zhou H, et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell. 2020;27(6):937-950.e9.
Fuchs V, Kutza M, Wischnewski S, Deigendesch N, Lutz L, Kulsvehagen L, et al. Presence of SARS-CoV-2 transcripts in the choroid plexus of MS and non-MS patients with COVID-19. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e957.
Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature. 2021;595(7868):565–71.
Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020;143(10):3104–20.
Pilotto A, Masciocchi S, Volonghi I, De Giuli V, Caprioli F, Mariotto S, et al. SARS-CoV-2 encephalitis is a cytokine release syndrome: evidences from cerebrospinal fluid analyses. Clin Infect Dis. 2021. https://doi.org/10.1093/cid/ciaa1933.
Gribkoff VK, Kaczmarek LK. The need for new approaches in CNS drug discovery: why drugs have failed, and what can be done to improve outcomes. Neuropharmacology. 2017;1(120):11–9.
Gonzalez AM, Leadbeater WE, Burg M, Sims K, Terasaki T, Johanson CE, et al. Targeting choroid plexus epithelia and ventricular ependyma for drug delivery to the central nervous system. BMC Neurosci. 2011;7(12):4.
Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–63.
Quintana FJ. Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation. J Clin Invest. 2017;127(8):2897–9.
Pardridge WM. Blood–brain barrier and delivery of protein and gene therapeutics to brain. Front Aging Neurosci. 2020;10(11):373.
Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, et al. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood–cerebrospinal-fluid drug-permeability barrier. Proc Natl Acad Sci USA. 1999;96(7):3900–5.
Drach J, Gsur A, Hamilton G, Zhao S, Angerler J, Fiegl M, et al. Involvement of P-glycoprotein in the transmembrane transport of interleukin-2 (IL-2), IL-4, and interferon-gamma in normal human T lymphocytes. Blood. 1996;88(5):1747–54.
Whyte-Allman SK, Kaul R, Bendayan R. Regulation of ABC drug efflux transporters in human T-cells exposed to an HIV pseudotype. Front Pharmacol. 2021. https://doi.org/10.3389/fphar.2021.711999.
Matsumoto K, Chiba Y, Fujihara R, Kubo H, Sakamoto H, Ueno M. Immunohistochemical analysis of transporters related to clearance of amyloid-β peptides through blood–cerebrospinal fluid barrier in human brain. Histochem Cell Biol. 2015;144(6):597–611.
Simard PF, Tosun C, Melnichenko L, Ivanova S, Gerzanich V, Simard JM. Inflammation of the choroid plexus and ependymal layer of the ventricle following intraventricular hemorrhage. Transl Stroke Res. 2011;2(2):227–31.
Tenenbaum T, Matalon D, Adam R, Seibt A, Wewer C, Schwerk C, et al. Dexamethasone prevents alteration of tight junction-associated proteins and barrier function in porcine choroid plexus epithelial cells after infection with Streptococcus suis in vitro. Brain Res. 2008;10(1229):1–17.
Brouwer MC, McIntyre P, Prasad K, van de Beek D. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2013;4(6):CD004405.
Acknowledgements
Not applicable.
Funding
National Institutes of Health Cobre Grant number P20GM104360. National Institutes of Health and National Institute of Allergy and Infectious Diseases 1R01AI158304-01A1.
Author information
Authors and Affiliations
Contributions
DT was responsible for the conceptualization of the article and was the principle author. JAW was responsible for the supervision of the article and was a major contributor to revision and editing. CAB was a major contributor to revision and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thompson, D., Brissette, C.A. & Watt, J.A. The choroid plexus and its role in the pathogenesis of neurological infections. Fluids Barriers CNS 19, 75 (2022). https://doi.org/10.1186/s12987-022-00372-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-022-00372-6