
Abstract
Recent research demonstrates a reciprocal relationship between gut microbiota-derived metabolites and the host in controlling the energy homeostasis in mammals. On the one hand, to thrive, gut bacteria exploit nutrients digested by the host. On the other hand, the host utilizes numerous products of gut bacteria metabolism as a substrate for ATP production in the colon. Finally, bacterial metabolites seep from the gut into the bloodstream and interfere with the host’s cellular bioenergetics machinery. Notably, there is an association between alterations in microbiota composition and the development of metabolic diseases and their cardiovascular complications. Some metabolites, like short-chain fatty acids and trimethylamine, are considered markers of cardiometabolic health. Others, like hydrogen sulfide and nitrite, demonstrate antihypertensive properties. Scientific databases were searched for pre-clinical and clinical studies to summarize current knowledge on the role of gut microbiota metabolites in the regulation of mammalian bioenergetics and discuss their potential involvement in the development of cardiometabolic disorders. Overall, the available data demonstrates that gut bacteria products affect physiological and pathological processes controlling energy and vascular homeostasis. Thus, the modulation of microbiota-derived metabolites may represent a new approach for treating obesity, hypertension and type 2 diabetes.
Similar content being viewed by others


Introduction
Metabolic diseases and their cardiovascular complications are the primary cause of morbidity and mortality in affluent societies. The last few decades have seen a fundamental transformation of dietary patterns. There is a notable increase in the consumption of high saturated fat, simple carbohydrates and salt. A decrease in physical activity accompanies these dietary changes. The two together disrupt the energy homeostasis in humans, resulting in a dramatic increase in the prevalence of metabolic and cardiovascular disorders such as obesity, diabetes and hypertension [1]. Interestingly, recent studies suggest that alterations in gut microbiota composition and increased gut permeability accompany these lifestyle-associated disorders [2,3,4]. These changes lead to excessive leakage of gut metabolites into the circulation [5, 6].
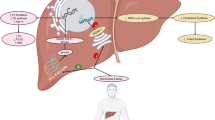
The mammalian gut is colonized by a complex microbial community composed of bacteria, archaea, fungi, helminths and protozoa. Most abundant are bacteria (~ 1012 cells/g of stool), followed by archaea (~ 109 cells/g of stool) and fungi (~ 104 cells/g of stool) [7, 8]. The composition of gut microbes is shaped more by environmental factors, such as mode of delivery, diet and geography than genetics [9]. The relationship between microbiota and host is commensal under physiological conditions. On the one hand, the host provides habitat and nutrition for the growth of gut microbes. On the other, commensal microbes regulate the gut's homeostasis, metabolize nutrients, bile acids, xenobiotics, and toxins [10]. Importantly, microbiota represents the largest endocrine organ in the human body by producing various biologically active compounds (Fig. 1). Gut-derived metabolites may act locally in the gut or signal distant organs via systemic circulation.

Metabolism of gut microbiota products from dietary substrates. Dietary subtrates enter the gastrointestinal tract and are further processed by gut microbes to various metabolites, including SCFAs (acetate, propionate, butyrate), H2S and TMA. The majority of butyrate is oxidized to acetyl-CoA in the mitochondria of colonic epithelial cells. Propionate and acetate are partially metabolized in the gut. Though, the majority of propionate is oxidized to succinyl-CoA in the liver. From SCFAs, acetate reaches the highest concentrations in peripheral blood, thus contributing mostly to the systemic production of energy in the peripheral tissues. Additionally, colonic mucosa oxidizes H2S by the action of mitochondrial SQR to thiosulfate. Thiosulfate may be either recycled to H2S by the action of microbial TR in the lumen of the gut or reach liver for further oxidation to sulfite and sulfate by mitochondrial TST and SO respectively. Likewise, the fraction of TMA that reaches the liver is oxidized to TMAO by cytosolic FMO3. CoA; coenzyme A, SQR; sulfide quinone reductase, TR; thiosulfate reductase, TST; thiosulfate sulfurtransferase, SO; sulfite oxidase, TMA; trimethylamine, TMAO; trimethylamine N-oxide, FMO3; flavin-containing monooxygenase 3
Accumulating evidence points to the prominent role of gut microbiota products in the regulation of energy metabolism [11, 12]. Gut metabolites use both passive and active transport to reach the intracellular space. Once inside the cell, they can interfere with mitochondrial respiration, thereby controlling ATP production [11, 13,14,15]. In addition, gut products may alter the activity of membrane channels and receptors involved in regulating blood pressure, glucose and lipid metabolism, thus playing a key role in developing cardiometabolic disorders [16,17,18]. This review summarizes recently published studies investigating the influence of gut microbiota-derived metabolites on the regulation of cellular energy homeostasis and the development of cardiometabolic disorders.
Microbiota metabolites as energy substrates and biological mediators
The colonization of the human gut by microbes results in creating a complex ecosystem unique to every individual. This living coating of the intestines is referred to as biofilm. Polymicrobial biofilms are formed at the surface of the gut epithelium or in the gut lumen attached to the mucus or food particles [19]. In comparison to tissue-forming cells, members of polymicrobial community have distinctive structures and metabolism, requiring a higher level of organization. The metabolic rate of gut bacteria depends on several factors, but mainly on substrate availability, intestinal transit time and redox balance [20, 21]. Therefore, diet plays an essential role in the formation and modulation of gut biofilms. Firstly, the composition of the microbial communities depends on the interactions occurring between members of the intestinal flora. For instance, some bacterial strains compete for the same substrate, making co-colonization difficult. Thus, co-colonization occurs between those species which produce substrates other strains can utilize. Secondly, polymicrobial biofilms are in direct contact with mucosal tissues of the colon. Hence, gut microbes and their products affect the metabolism of the colonic mucosa and vice versa. The mutual interactions between polymicrobial phenotypes and whole-body homeostasis have been intensively studied [19]. In the following paragraphs, we present several metabolites involved in the regulation of host energy metabolism. We provide physiological ranges of these metabolites in the intestines, their absorption rates and major commensal microbial producers (Table 1). We also discuss how they affect mitochondrial bioenergetics. For a detailed description of the effects of these metabolites on membrane channels and receptors involved in the regulation of energy and vascular homeostasis, please refer to previous reviews [17, 22,23,24,25,26].
Short-chain fatty acids
Dietary fermentation in the mammalian gut produces SCFAs. Since the pKa of the SCFAs is ~ 4.8, at the colonic pH ~ 6, more than 90% is in its un-protonated form [27]. These small molecules may contain up to seven carbon atoms, straight or branched chain and include formate, acetate, propionate, butyrate, isobutyrate, valerate, isovalerate, 2-methylbutyrate, hexanoate and heptanoate. The substrates for the production of straight-chain fatty acids originate primarily from plants and include cellulose, pectins and oligosaccharides. The branched-chain fatty acids are formed by the degradation of amino acids valine, leucine and isoleucine. Fermentation of pectins and xylans produces acetate, while that of starch produces butyrate [28, 29]. The metabolism of arabinoglycans results in the production of acetate and propionate. The concentrations of SCFAs are highest in the colon and determined by substrate availability and microbiota composition in different segments, reaching 20–70 mmol/l in the distal colon and 70–140 mmol/l in the proximal colon [30]. With the increase in SCFAs concentration, pH value is decreasing due to the accumulation of H+. Importantly, pH value regulates microbiota composition and reduce the colonization by pathogenic species [31]. For instance, at pH 5.5, butyrate producers of Firmicutes phylum are dominant. In contrast, acetate and propionate producers of Bacteroidetes dominate at higher pH values over butyrate producers [32].
Furthermore, the availability of substrates regulates the fermentation process. For instance, Bacteroides ovatus accumulates succinate under high carbohydrate levels. However, it utilizes succinate for energy production and accumulates propionate under low carbohydrate levels. Similarly, Clostridium perfringens and Bifidobacterium breve generate lactate as an electron sink under excess carbohydrates and acetate or formate as products of energy metabolism under carbohydrate-limiting conditions [33]. The levels of SCFAs decrease from millimolar ranges in the colon to nanomolar ranges in the portal and peripheral blood. In healthy humans, the acetate, propionate and butyrate ratio is ~ 60/20/20 in the colon, ~ 69/23/8 in the portal blood and ~ 89/6/5 in the peripheral blood [27]. Diet composition affects the ratios of different SCFAs. For instance, a diet rich in carbohydrates and sugars increases the propionate/acetate ratio, whereas a high-fiber diet increases acetate levels [34, 35].
Butyrate
Various gut bacteria, particularly the gram-positive anaerobic bacteria, Faecalibacterium, Eubacterium and Roseburia species from Clostridial clusters of Firmicutes generate butyrate [36,37,38]. These bacteria condense two molecules of acetyl-CoA to acetoacetyl-CoA, which subsequently converts to β-hydroxybutyryl-CoA and crotonyl-CoA. The final product is butyryl-CoA (Fig. 2A). This process consumes NADH. Butyryl-CoA may be further converted to butyrate by a butyryl-CoA:acetate Co-A transferase or phosphorylated to butyryl-phosphate and converted to butyrate by a butyrate kinase. Colon-derived SCFAs represent ~ 10% of the daily human caloric requirement [27]. SCFAs provide ~ 60–75% of energy for colonic epithelial cells, with butyrate as the primary contributor, followed by propionate and acetate [39]. Butyrate is converted in the mitochondria to butyryl-CoA by a butyryl-CoA synthetase. This process consumes ATP. Butyryl-CoA is subsequently transformed to crotonyl-CoA by a butyryl-CoA dehydrogenase. Crotonase catalyzes the formation of hydroxybutyryl-CoA, which then forms acetoacetyl-CoA, producing NADH, and then acetyl-CoA by an acetoacetyl-CoA thiolase. Colonic epithelial cells oxidize most of the colon-derived butyrate. The remaining butyrate is absorbed through the gut-blood barrier into the portal vein and metabolized by the liver. The absorption occurs from all colon segments at a rate of ~ 6–12 µmol/cm2/h, primarily by monocarboxylate transporters (MCT) [40]. These transporters exchange SCFAs for bicarbonates. This process neutralizes protons formed during SCFAs production and plays an essential role in regulating luminal pH. A minor portion of SCFAs is co-transported with H+ and Na+ cations. Only a fraction of the colon-derived butyrate reaches the peripheral circulation.

Schematic diagram of SCFAs and gaseous molecules production by gut microbes. A The fermentation of saccharides to PEP by gut microbes is coupled with the reduction of NAD+ to NADH. The metabolism of PEP to SCFAs (acetate, propionate and butyrate) is coupled with the regeneration of NAD+ and H2 production by Fd B Hydrogenotrophs utilize H2 to produce gaseous signaling molecules (H2S, CH4 and NO). Pathways have been simplified to highlight key end-products. PEP; phosphoenolpyruvate, Fd; ferredoxin oxidoreductase
Propionate
Propionate may be produced in the gut in the process of carbohydrate fermentation either by succinate or acrylate pathway (Fig. 2A). The conversion of succinate to methylmalonate and further release of CO2 results in propionate production and is present in e.g., Bacteroides, Prevotella, Alistipes, Ruminococcus, Phascolarctobacterium, Dialister, Akkermansia species [38]. The conversion of pyruvate to lactate and acrylate is associated with the consumption of NADH. Acrylate may be further metabolized to propionate by the utilization of another NADH. The acrylate pathway is active in Megasphaera or Coprococcus species [38]. Propionate may also be derived from deoxy-sugars and other monosaccharides by the propanediol pathway mediated by various microbes, e.g., Roseburia, Eubacterium and Blautia species [38]. The gut partially metabolizes colon-derived propionate, and the liver uses the majority of it. Propionate is converted to propionyl-CoA by propionyl-CoA synthetase and then further metabolized to succinyl-CoA entering the TCA. Propionate is transported into the hepatocytes by organic anion transporters [41].
Acetate
Acetate may be formed from pyruvate after the release of CO2 (Fig. 2A). In addition, acetogenic bacteria utilize H2 to reduce CO2 to acetate. A wide range of gut microbes, e.g., Bacteroides, Bifidobacterium, Clostridium, Ruminococcus species, produce acetate [42, 43]. Acetate directly converts to acetyl-CoA that subsequently enters the mitochondrial TCA cycle to produce substrates for oxidative phosphorylation. The liver utilizes acetate to produce energy, cholesterol, long-chain fatty acids, glutamine and glutamate. From SCFA, acetate reaches the highest concentrations in the peripheral blood and thus contributes mostly to the systemic production of energy for the muscles, heart, kidney and adipose tissues [23, 44].
Hydrogenotrophic products
Hydrogen sulfide
Gut bacteria can produce H2S from several substrates, particularly sulfate, sulfite, cysteine or taurine. The availability of colonic sulfate is determined mainly by the amount of substrates in the diet, e.g., inorganic sulfate, sulfur amino acids, and the absorption of substrates by the small intestine [45]. Sulfate reducing bacteria (SRB), predominantly Desulfovibrio species, generate H2S by the reduction of sulfate in the presence of electron donor to acetate or CO2 by incomplete and complete oxidation, respectively [46]. H2 is predominantly used as an electron donor, although other simple organic compounds (lactate, propionate, butyrate) may be utilized as well (Fig. 2B) [20]. The metabolism of sulfate is mediated by dissimilatory reduction coupled with the production of energy. In detail, sulfate is converted to adenosine phosphosulfate and further reduced to sulfite. Next, H2S is generated by the reduction of sulfite. The activation of sulfate is coupled with the consumption of ATP, while the reduction is coupled with the pumping of protons, thereby generating a proton gradient fueling the production of ATP. Dissimilatory sulfite reductase is a protein of very ancient origin. It is believed that sulfite metabolism occurred earlier than sulfate reduction and probably served to conserve energy in the first prokaryotes in the early earth [47].
Several bacterial species, e.g., Escherichia, Salmonella, Enterobacter, possess specific enzymes to produce H2S [48]. A phosphate-sulfite reductase maintains the redox balance by reducing sulfite to H2S and NADP+ or oxidizing H2S to sulfite and recovering NADPH [49]. Bacteria expressing cysteine desulfhydrase convert cysteine to H2S, pyruvate and ammonia [50]. Bilophila wadsworthia utilizes taurine by taurine:pyruvate aminotransferase to sulfoacetaldhyde and alanine, which is further metabolized by sulfoacetaldhyde sulfolyase to sulfite and acetate. Subsequent sulfite reduction by sulfite reductase produces H2S and CO2 [51]. Tissue enzymes also metabolize methionine to homocysteine, which is coupled with serine to form cystathionine by cystathionine β-synthase (CBS). Cystathionine γ-lyase (CSE) subsequently converts cystathionine to cysteine, which CBS or CSE may further transform to H2S. Half of the luminal H2S is produced by gut microbiota and half by colonic tissues [52]. Interestingly, the inhibition of the colonic tissue enzymes promotes the production of H2S by gut microbiota [52].
The proportion of dissociated HS− to total sulfide is ~ 60% in the colon (pH ~ 7), the pKa1 = 6.8 and pKa2 > 12 [53]. The range of total sulfide in human feces is 0.2–3.4 mmol/l, averaging at 0.74 mmol/l [20]. H2S may exist in a free form or be bound to cysteine thiols as sulfane sulfur [54]. In addition, feces have large sulfide-binding activity and therefore, less than 8% of total sulfide (~ 60 µmol/l) is in the free form [20]. H2S is lipophilic and rapidly diffuses through lipid bilayers of cell membranes. Colonic mucosa effectively oxidizes H2S to thiosulfate [55]. A significant fraction of thiosulfate is recycled in the gut to H2S by the action of thiosulfate reductase expressed in several microbes, e.g., Citrobacter, Proteus and Edwardsiella [56, 57]. In addition, the oxidation products may be absorbed into the portal blood. We have shown that intracolonic injection of H2S increases thiosulfate in rats' portal blood. The fraction of thiosulfate that reached peripheral blood 30 min after the administration was ~ 80% lower than the fraction in the portal blood, suggesting a major role of the liver in the metabolism of gut-derived thiosulfate. In addition, the administration of antibiotics lowered the concentration of thiosulfate metabolites in the portal blood [58]. Shen et al. studied the concentrations of free sulfide and bound sulfane sulfur in the peripheral blood and tissues of germ-free mice. They found significantly lower levels of free H2S in the cecum, colon and peripheral blood plasma. The levels of sulfane sulfur were decreased in the blood plasma and adipose tissue of germ-free mice. Furthermore, lower activity of CSE and higher cysteine levels were found in gastrointestinal and extraintestinal tissues of germ-free mice [59].
The toxicological effects of H2S on mammals were studied over the centuries, with the first reports occurring in the eighteenth century. Toxic effects of H2S were described near swamps, volcanoes or industrial accidents [60]. H2S at ≥ 10 ppm causes eye and skin irritation and other neurological and cardiovascular disorders. The ability to sense the H2S odor disappears at ≥ 100 ppm due to the olfactory nerve's paralysis. H2S at ≥ 500 ppm causes respiratory arrest and death [61]. The toxic-effect mechanism is based on the reversible binding of H2S to the heme center of cytochrome c oxidase (complex IV). Thus, H2S competes with oxygen for the active site, resulting in the inhibition of the mitochondrial electron transport, loss of the transmembrane electrochemical gradient, and the reduction of ATP production (Fig. 3A) [62].

Effects of hydrogen sulfide (H2S) on mitochondrial bioenergetics. A At higher concentrations, H2S binds to complex IV (cytochrome c oxidase), thereby inhibiting mitochondrial electron transport and blocking ATP production. B A persulfide (-SSH) is formed on the cysteine residue (-SH) of coenzyme A in the presence of H2S, which blocks the oxidation and utilization of butyrate. C At low concentrations, mitochondria oxidize H2S by the action of sulfide SQR and transfer electrons through coenzyme Q and complexes III and IV to oxygen, thereby promoting ATP production at complex V (ATP synthase). Pathways have been simplified to highlight key end-products. CI-V; complexes I-V, e−; electrons, Q; coenzyme Q, cyt C; cytochrome c, CoA; coenzyme A, SQR; sulfide quinone reductase
Additionally, H2S may block the β-oxidation of butyrate in the gut. Babidge et al. showed that H2S (1.5 mmol/l) strongly reduced the formation of CO2 from butyrate in colonocytes. In the presence of CoA and ATP, H2S decreased the formation of crotonyl-CoA and acetyl-CoA. It also increased the formation of butyryl-CoA, thus inhibiting the activity of butyryl-CoA dehydrogenase [63]. It should be stressed that the concentration used in this study was far over the physiological levels of free H2S reported in the rat colon (~ 120 µmol/l) [64]. Landry et al. showed that the oxidation of H2S by mitochondrial enzyme sulfide quinone reductase (SQR) results in the production of CoA persulfide (CoA-SSH), which inhibits the activity of butyryl-CoA dehydrogenase (Fig. 3B) [65].
Recent studies describe a bell-shaped effect of H2S on cellular functions. In detail, lower concentrations of H2S support cellular bioenergetics, whereas higher H2S concentrations block cellular respiration. First reports describing the utilization of H2S for ATP production in eukaryotic cells were performed in non-mammals. Recently, the promotion of bioenergetics was confirmed in various mammalian cells and isolated mitochondria [66,67,68,69]. The positive effect of H2S on functions of mitochondria isolated from rat liver was observed at 0.1–1 µmol/l H2S, whereas concentrations of H2S ≥ 3 µmol/l inhibited respiration and generation of ATP [66]. The concentrations of H2S promoting mitochondrial function in colonocytes were much higher (20–40 µmol/l) in comparison to other cell types [14]. The inhibition of the colonocytes' oxygen consumption occurred at a concentration ≥ 65 µmol/l [14]. In addition, exogenous H2S promoted the production of colonic mucus and the formation of microbial biofilms [70]. These reports suggest that the colon is adapted to the H2S-rich environment and utilizes H2S to protect the integrity of the gut-blood barrier. Since the average concentration of free H2S in human colon lumen was estimated to be 60 µmol/l, most individuals may employ H2S to produce energy [20]. However, in individuals with a concentration of free H2S over 65 µmol/l, inhibition of colonocytes respiration may occur. It should be noted that several common detection methods used in biological samples overestimate physiological levels of H2S due to the additional liberation of H2S from the bound stores [71]. Interestingly, mitochondrial enzymes can also oxidize H2S, a function that can be coupled with ATP generation [72]. One of the evolution theories posits that eukaryotes evolved from an endosymbiotic ancestral mitochondrion living in a prokaryotic host. The symbiotic relationship was based on the oxidation of H2S by the ancestral mitochondrion coupled with the reduction of oxidized sulfur forms to H2S by the host prokaryote [73]. This hypothesis is also supported by the fact that mitochondria share common features with a purple sulfur bacterium Rhodobacter [74]. In modern eukaryotic cells, mitochondria utilize electrons from H2S through SQR to coenzyme Q [66]. Electrons are subsequently driven through complex III and IV to oxygen, resulting in ATP production at complex V (Fig. 3C). The oxidation of H2S by SQR results in the formation of sulfane sulfur at the SQR cysteine group (SQR-SSH). The sulfane sulfur may be further transferred to glutathione to form glutathione persulfide or to sulfite to form thiosulfate. Persulfide dioxygenase (ETHE1) and thiosulfate sulfurtranferase subsequently oxidize the persulfides to reduce coenzyme Q and regenerate sulfite, thereby consuming oxygen. Finally, sulfite oxidase converts sulfite to sulfate [75]. Bound sulfane sulfur forms have recently attracted significant attention as an H2S pool and signaling molecules. Akaike’s group identified an endogenous source of cysteine persulfide (Cys-SSH) in the mitochondria, the mammalian cysteinyl-tRNA synthetase 2 (CARS2). They showed that mitochondrial respiration is lower in CARS2 KO cells. They provided further evidence of the reduction of CARS2 derived Cys-SSH by accepting an electron from the mitochondrial electron transport chain to produce H2S [76].
Nitric oxide-species
Nitric oxide species (NOx) are produced and utilized by commensal bacteria via various pathways. Firstly, oral bacteria can convert nitrate (NO3−) to nitrite (NO2−) by nitrate reductases [77]. Subsequently, under the stomach's acidic environment, the chemical reduction of nitrite generates NO. Secondly, gut bacteria can reduce nitrate either by respiratory denitrification or by dissimilatory and assimilatory nitrate reduction (Fig. 2B). In the denitrification process, the reduction of nitrate by membrane-bound nitrate reductases to nitrogen oxides (NO and N2O) and N2 gas leads to the translocation of protons and generation of ATP [77]. The dissimilatory nitrate reduction to ammonium (DNRA) by nitrate reductases located in the periplasm is not directly associated with energy conservation [78]. The coupling of DNRA with the oxidation of substrates, such as formate or lactate, generates ATP [79]. Assimilatory nitrate reduction by cytosolic nitrate reductases to ammonium is coupled with incorporating the derived ammonium to glutamine or glutamate. The reduction of nitrate may be either non-enzymatic or catalyzed by nitrite/nitrate reductases [80, 81]. Several microbial species express bacterial nitric oxide synthase (bNOS) [82,83,84]. Compared to eukaryotic isoforms, bNOS is smaller and lacks a reductase domain, which supplies electrons for NO synthesis. Therefore, other bacterial reductases must couple with bNOS to provide electrons for NO synthesis [84].
Analysis of human fecal samples determined the concentration of 5–19 µmol/kg and 0–14 µmol/kg for nitrite and nitrate, respectively [85]. The colon tissue of rats contained 1–1.5 µmol/l of nitrite and 4–32 µmol/l of nitrate [86]. Whitter et al. studied the distribution of labeled nitrate and nitrite in rats. The labeled 13N was found in the liver, kidney, cecum, large intestine, and blood, however not in rats' feces after gavage of labeled nitrate and nitrite. The absorption from the stomach was faster for nitrate (70%) than for nitrite (39%) [87]. The fraction of absorbed nitrite from the colonic lumen either entered the circulation or was oxidized in the colonocytes and partly reentered the colonic lumen as nitrate [88]. The plasmatic levels of nitrate in germ-free mice were strongly reduced [89]. Surprisingly, nitrite levels in plasma were increased in germ-free mice after nitrate supplementation, probably due to the activation of nitrate reduction in the liver as a compensatory response to the absence of microbiota [90]. Also, the distribution of labeled nitrate administered by gavage was altered in germ-free rats, with lower levels of 13N found in the gut and higher levels in the stomach and bladder [87]. The absorption of labeled nitrate administered into the lower intestines of germ-free rats was faster than the absorption in conventional rats. In reverse, the absorption of nitrate from circulation to the intestinal lumen was lower in germ-free rats than in control animals [87].
Similar to H2S, the inhibitory effect of NO on mitochondrial respiration has been known for centuries. NO interacts at physiological levels (1–200 nmol/l) with the oxygen-binding site of the cytochrome c oxidase, thereby rapidly but reversibly inhibiting the enzyme's activity. The binding of NO may occur at the reduced heme center of the cytochrome c oxidase, thus competing with oxygen binding or at the oxidized copper center by nitrite. The inhibition of mitochondrial functions at higher concentrations of NO (≥ 1 µmol/l) and by reactive nitrogen species, particularly peroxynitrite, is slow and non-selective but irreversible due to the modification of proteins. On the other hand, NO/cGMP signaling stimulates mitochondrial biogenesis through the activation of PGC-1α [91]. Nitrite (5 mmol/l) promotes the oxidation of butyrate in rat colonic cells [92]. It should be stressed that the concentration used in this study was far over the physiological levels of nitrite reported in the rat colon (~ 1 µmol/l). Larsen et al. showed that supplementation with nitrate decreased whole-body oxygen consumption during exercise. This finding strongly correlated with the increased efficiency of oxidative phosphorylation in mitochondria isolated from human skeletal muscle. The basal respiration, proton leak and expression of ATP/ADP translocase were decreased in human skeletal muscle after nitrate supplementation, suggesting that the increase in mitochondrial efficiency could be mediated by reduced leakage of protons across the inner mitochondrial membrane.
Interestingly, nitrate supplementation increased the plasmatic levels of nitrite. However, the possible involvement of commensal microbes in the reduction of nitrate to nitrite was not examined. In contrast to nitrate, nitrite did not affect basal respiration or oxidative phosphorylation and decreased oxygen affinity to skeletal muscle mitochondria at pH 6.7, but not at pH 7.2. Since hypoxia and low pH facilitate the conversion of nitrite to NO, it is assumed that NO-dependent mitochondrial inhibition mediates the effect [93].
Methane and methylamines
Methane derivates are produced in the human gut by methanogenic, anaerobic archaea. Methanogens express methyl-coenzyme M reductase, which catalyzes the formation of methane. The majority of methanogens are hydrogenotrophic and utilize H2 to reduce CO2 to methane (Fig. 2B). Alternatively, methylotrophs convert methylated compounds, e.g., methanol, methylamines and methyl-sulfides to methane by substrate-specific methyltransferases [94]. The human gut's dominant methanogen is Methaninobrevibacter smithii, which preferentially colonizes the distal portion of the colon [95]. Methane is excreted in the breath and flatus or metabolized in the liver after absorption through the gut-blood barrier. Interestingly, despite changes in diet over a period of 35 years, human methanogens' activity remained stable [96]. The determination of methane in human breath divides the subject into the group of methane producers (~ 35%), with the mean level of methane ~ 15 ppm, and methane nonproducers, with concentrations of < 1 ppm [96]. The methane production status is similar between family members. In addition, methane excretion was not detected in germ-free rats nor infants up to 6 months of age, nor was it significantly altered by administering antibiotics [97].
In addition, dietary carnitine and choline degradation results in the production of trimethylamine (TMA) in the gut. Various members of human gut microbiota mediate the conversion of substrates to TMA, e.g., Anaerococcus, Clostridium, Desulfitobacterium, Escherichia, Proteus, Providencia, Edwardsiella, Yokenella and Citrobacter species [98, 99]. TMA may be further utilized by methylotrophs from Methanomassiliicoccales genera in the gut or pass through the gut-blood barrier to the portal vein and reach the liver [100, 101]. Flavin monooxygenase 3 (FMO-3) oxidizes TMA to trimethylamine N-oxide (TMAO) in the liver, which the circulation transports for excretion in the urine, sweat and breath. The concentration of TMA in the colon and TMAO in the plasma of healthy humans ranges between 1–200 μmol/l and 3–4 μmol/l, respectively [102, 103]. The administration of antibiotics significantly reduced plasma TMAO. Moreover, TMA and TMAO were below the detection level in germ-free mice [104].
Alcohol intake reduced mitochondrial respiration and increased whole-body methane emission in humans and rats [105]. However, incubation of liver mitochondria with methane did not alter mitochondrial respiration [13]. A protective effect of methane was observed in an experimental model of ischemia–reperfusion injury of the liver. Methane inhalation reduced cytochrome c oxidase release and improved basal respiration and maximal respiratory capacity in liver mitochondria during ischemia–reperfusion [13]. Makrecka-Kuka et al. studied the effect of TMAO on the respiration of cardiac mitochondria. They showed that acute exposure to TMAO decreased oxidative phosphorylation in cardiac fibers. The activity of pyruvate dehydrogenase was also reduced. Additionally, treatment of mice with TMAO for 8 weeks impaired cardiac mitochondrial energy metabolism [106]
Co-colonization and competition between gut microbes
SRB co-colonize with other species, which produce substrates for sulfate reduction. For instance, SRB are abundant in the presence of Bacteroides that generate sulfate from sulfomucin and mucopolysaccharides by sulfatases (B. thetaiotaomicron, B. fragilis) [107]. The levels of D. piger positively correlate with the levels of an H2 producer Collinsella aerofaciens. This co-colonization is beneficial for both strains because the removal of H2 by D. piger increases the efficiency of dietary fermentation by C. aerofaciens [108].
A negative correlation was observed between SRB and hydrogenotrophic methanogens, which compete for the common substrate H2 [109]. Co-incubation of human SRB with methanogens reduced the production of H2S in the feces; however, it did not alter the production of methane [110]. Supplementing the diet with sulfate increased both sulfate reduction in the subjects' feces and the number of SRB, which were undetectable during control diets. The number of methanogenic bacteria and methane expiration decreased after supplementing a sulfate diet [111]. Another mechanism is the conversion of choline to TMA by Desulfovibrio desulfuricans, which promotes the metabolism of methylotrophic methanogens over hydrogenotrophic methanogens [112].
Several studies suggest that SRB may utilize nitrate and nitrite as electron acceptors. The data implies that SRBs generate ATP and, at the same time, compete with denitrifying and nitrate fermenting bacteria for these substrates [113]. On the other hand, the presence of nitrate and nitrite suppresses the reduction of sulfate by SRB [113]. The dissimilatory reduction of nitrate (DNRA) and nitrite to ammonium are not generally attributed to SRB. Only a few strains of Desulfovibrio species possess this ability. It was reported that Desulfovibrio desulfuricans express nitrite reductase constitutively, and this expression of nitrate reductase is inducible by nitrate or nitrite and suppressed by sulfate [113]. The reduction of nitrate to ammonia by D. desulfuricans and Sulfurospirillum deleyianum was coupled with the oxidation of H2S to sulfate or elemental sulfur, respectively [114, 115]. Interestingly, M. smithii may utilize ammonium as a primary nitrogen source, competing with B. thetaiotaomicron for this substrate [116].
Gut dysbiosis, energy accumulation and related diseases
Excessive energy accumulation in the body is associated with the development of cardiometabolic disorders, such as obesity, diabetes and metabolic syndrome [117]. Gut microbes play an essential role in the regulation of the host's energy homeostasis. Alterations in the composition of gut microbes are associated with disturbed energy balance resulting in the development of various diseases [118]. These pathologies or phenotypes may be related to an overall reduction in microbial diversity and an imbalance of microbial composition. The result of which may be either an overproduction of some metabolites and/or suppression of others. Below, we review evidence on the involvement of gut microbiota metabolites in the pathogenesis of metabolic diseases and their cardiovascular complications.
Obesity and diabetes
Mitochondrial dysfunction is a major factor leading to the accumulation of lipids and decreased sensitivity to insulin [117]. Based on metabolomic studies, Samczuk et al. suggested a relationship between mitochondria, gut microbiota metabolites and clinical outcome in obese patients with diabetes undergoing bariatric surgery. In this group, rapid improvement in blood glucose control was associated with changes in mitochondrial citrate cycle pathway, ketone bodies synthesis and degradation, butyrate and propionate metabolism whereas patients with slower improvement of glucose control showed alternations in nitrogen metabolism, branched-chain fatty acids (BCFAs) synthesis and degradation [119]. In animal studies, administration of butyrate and nitrate improved mitochondrial functions in liver, skeletal muscle and adipose tissue of insulin-resistant obese mice [120,121,122,123,124]. In addition, accumulating evidence suggests that gut metabolites affect lipid, glucose and cholesterol metabolism in peripheral tissues [18, 22, 125,126,127,128]. Germ-free mice colonized with microbiota from obese donors show higher weight gain and total body fat than germ-free mice colonized with microbiota from lean mice [129]. The oxidation of SCFAs is associated with the inhibition of lipolysis and de novo synthesis of fatty acids resulting in reduced plasmatic concentrations of free fatty acids in the plasma [130]. Besides the direct effect on mitochondrial respiration, SCFAs regulate energy homeostasis via G-protein coupled receptors 41 (GPR41) and 43 (GPR43), also known as free fatty acid receptor 3 (FFAR3) and 2 (FFAR2). In addition, SCFAs stimulate the secretion of gut hormones, peptide YY (PYY) and glucagon-like peptide-1 (GLP-1), which modulate the response of brain centers responsible for food intake (Fig. 4). Lower levels of acetate were found in the liver and brain of mice on a high-fat diet [131]. The administration of acetate reduced the accumulation of lipids in the adipose tissue and liver and improved glucose tolerance in obese animals [132, 133]. Acetate is taken up by the brain and directly modulates appetite in hypothalamic centers [134].

Transport and signaling of gut microbiota products. A SCFAs are transported by MCT, while, gaseous molecules diffuse to the colonic cells and are oxidized in the mitochondria or in the unmetabolized form enter portal vein. B SCFAs stimulate GPR41 and GPR43 and C the secretion of gut hormones, GLP-1 and PYY, which regulate energy homeostasis in the body. MCT; monocarboxylate transporters, GPR; G-protein coupled receptor, GLP-1; glucagon-like peptide-1, PYY; peptide YY
On the other hand, propionate stimulates the accumulation of PYY and GLP-1 in the portal vein and the expression of GPR43; thus, promoting intestinal gluconeogenesis and reducing food intake [135]. Obesity and diabetes are also associated with lower bioavailability of NO [22]. Moreover, the administration of nitrate or nitrite reduces the accumulation of fat in adipose tissues and improves insulin signaling in mouse models of diabetes and metabolic syndrome [136, 137].
Various studies suggest that the levels of circulating metabolites positively correlate with the development of metabolic disorders [22, 138,139,140]. Higher levels of BCFAs were detected in obese individuals and subjects on a high-fat diet. Similarly, a more significant release of H2S from adipose tissue was detected in animals on a high-fat diet [141]. Administering H2S stimulated lipolysis, promoted energy storage in adipocytes via the peroxisome proliferator-activated receptor γ (PPARγ) pathway, and marginally increased fat accumulation in visceral tissues of animals on a high-fat diet [142]. Several reports also describe the inhibitory effect of H2S on glucose-stimulated insulin secretion as a factor contributing to the development of diabetes. This effect is mediated via the opening of ATP-sensitive potassium channels (KATP) [22]. TMAO attracted attention in recent years as a marker of cardiovascular health. A systemic review showed that the risk of major cardiovascular events and all-cause mortality is higher in patients with high TMAO plasma levels than patients with low TMAO levels [143]. One of the mechanisms responsible for higher cardiovascular risk involves the suppression of cholesterol metabolism by TMAO. In detail, TMAO inhibits the reverse transport of cholesterol and the synthesis of bile acids in the liver [144]. It should be pointed out that the changes in the plasmatic levels of metabolites were not associated with changes in the gut microbiota composition and could have resulted from altered tissue metabolism.
Hypertension
High blood pressure is one of the hallmarks of metabolic syndrome. It increases the risk of developing diabetes and obesity. Recent studies suggest that gut microbes are involved in the regulation of blood pressure. Hypotension and reduced vascular contractility were observed in germ-free rats [145]. In addition, angiotensin II-induced increase in blood pressure was lower in germ-free mice than conventionally raised mice [146], suggesting that gut microbiota promotes the development of hypertension. Several other gut-derived products were shown to exert antihypertensive actions. Acute or chronic administration of SCFAs, acetate, propionate, butyrate and valerate, lowered blood pressure in mice and rats [147,148,149,150]. The activation of GPR41 was proposed as the underlying mechanism of the SCFAs-mediated hypotensive effects [148, 151].
NO and H2S mediated vasodilation and blood pressure-lowering effects are known over decades. However, the role of microbiota-derived H2S and NO-species in the regulation of blood pressure remains unclear. We have shown that the administration of H2S into the colon lowered arterial blood pressure several times longer than parenteral administration. Furthermore, the hypotensive effect of colonic H2S was more pronounced in hypertensive animals than normotensive controls [58]. Daliri et al. reported that consumption of soy protein decreased blood pressure, possibly by increasing the colonization of the gut by H2S-producing bacteria in hypertensive rats [152]. Similarly, dietary nitrate reduced blood pressure in healthy volunteers and hypertensive patients [18]. In contrast, hypertensive rats showed a greater permeability of the colon to TMA, and TMA administration increased blood pressure in rats [153, 154]. Although plasma TMAO levels correlate with a higher risk of hypertension and preeclampsia, growing evidence suggests that gut bacteria-produced TMA exerts toxic effects; that is, TMAO precursor is the culprit rather than TMAO itself [103, 155, 156].
Conclusion
During dietary fermentation, gut microbes metabolize dietary substrates to gain energy most efficiently, thereby maintaining redox balance. This process generates a wide variety of products. The most abundant are SCFAs, which represent the primary fuel for colonic epithelial cells. SCFAs also play a crucial role in the regulation of energy homeostasis in peripheral tissues.
H2S, together with other gut-derived gases and their precursors, emerged as signaling molecules capable of influencing cellular bioenergetics. These compounds may diffuse into the cell and directly affect energy production. They may also interact with the production and metabolism of other gut metabolites, particularly SCFAs. Despite the accumulating evidence demonstrating the involvement of gut-derived metabolites in cellular energy production, more research is needed to elucidate the role of these compounds in regulating host energy metabolism. More specifically, changes in the composition of gut microbes and their products should be studied in relation to different lifestyle disorders to establish a link between gut dysbiosis and the development of metabolic diseases. In order to create therapeutics based on gut microbial interventions, detailed mechanisms by which these organisms contribute to the regulation of energy metabolism need to be described.
Availability of data and materials
Not applicable.
Abbreviations
- BCFAs:
-
Branched-chain fatty acids
- bNOS:
-
Bacterial nitric oxide synthase
- CARS2:
-
Cysteinyl-tRNA synthetase 2
- CBS:
-
Cystathionine β-synthase
- CoA:
-
Coenzyme A
- CSE:
-
Cystathionine γ-lyase
- cyt C:
-
Cytochrome c
- DNRA:
-
Dissimilatory nitrate reduction to ammonium
- ETHE1:
-
Persulfide dioxygenase
- FFAR:
-
Free fatty acid receptor
- FMO-3:
-
Flavin monooxygenase 3
- GLP-1:
-
Glucagon-like peptide-1
- GPR:
-
G-protein coupled receptor
- H2S:
-
Hydrogen sulfide
- KATP :
-
ATP-sensitive potassium channels
- MCT:
-
Monocarboxylate transporters
- NO:
-
Nitric oxide
- NOx:
-
Nitric oxide species
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PYY:
-
Peptide YY
- SQR:
-
Sulfide quinone reductase
- SRB:
-
Sulfate-reducing bacteria
- TMA:
-
Trimethylamine
- TMAO:
-
Trimethylamine N-oxide
References
Sattar N, Gill JMR, Alazawi W. Improving prevention strategies for cardiometabolic disease. Nat Med. 2020;26(3):320–5.
Amoroso C, Perillo F, Strati F, Fantini M, Caprioli F, Facciotti F. The role of gut microbiota biomodulators on mucosal immunity and intestinal inflammation. Cells. 2020;9(5):1234.
Giuffrè M, Campigotto M, Campisciano G, Comar M, Crocè LS. A story of liver and gut microbes: how does the intestinal flora affect liver disease? A review of the literature. Am J Physiol Gastrointest Liver Physiol. 2020;318(5):G889-g906.
Schiattarella GG, Sannino A, Esposito G, Perrino C. Diagnostics and therapeutic implications of gut microbiota alterations in cardiometabolic diseases. Trends Cardiovasc Med. 2019;29(3):141–7.
Drapala A, Szudzik M, Chabowski D, Mogilnicka I, Jaworska K, Kraszewska K, et al. Heart failure disturbs gut-blood barrier and increases plasma trimethylamine, a toxic bacterial metabolite. Int J Mol Sci. 2020;21(17):6161.
Zhao L, Lou H, Peng Y, Chen S, Fan L, Li X. Elevated levels of circulating short-chain fatty acids and bile acids in type 2 diabetes are linked to gut barrier disruption and disordered gut microbiota. Diabetes Res Clin Pract. 2020;169:108418.
Wampach L, Heintz-Buschart A, Hogan A, Muller EEL, Narayanasamy S, Laczny CC, et al. Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Front Microbiol. 2017;8:738.
Raimondi S, Amaretti A, Gozzoli C, Simone M, Righini L, Candeliere F, et al. Longitudinal survey of fungi in the human gut: ITS profiling, phenotyping, and colonization. Front Microbiol. 2019;10:1575.
Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555(7695):210–5.
Hajiagha MN, Taghizadeh S, Asgharzadeh M, Dao S, Ganbarov K, Köse Ş, et al. Gut microbiota and human body interactions; its impact on health: a review. Curr Pharm Biotechnol. 2021. https://doi.org/10.2174/1389201022666210104115836.
Schönfeld P, Wojtczak L. Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res. 2016;57(6):943–54.
Heiss CN, Olofsson LE. Gut microbiota-dependent modulation of energy metabolism. J Innate Immun. 2018;10(3):163–71.
Strifler G, Tuboly E, Szel E, Kaszonyi E, Cao C, Kaszaki J, et al. Inhaled methane limits the mitochondrial electron transport chain dysfunction during experimental liver ischemia-reperfusion injury. PLoS ONE. 2016;11(1):e0146363.
Beaumont M, Andriamihaja M, Lan A, Khodorova N, Audebert M, Blouin JM, et al. Detrimental effects for colonocytes of an increased exposure to luminal hydrogen sulfide: the adaptive response. Free Radical Biol Med. 2016;93:155–64.
Roediger WE, Babidge WJ. Nitric oxide effect on colonocyte metabolism: co-action of sulfides and peroxide. Mol Cell Biochem. 2000;206(1–2):159–67.
Lee Y, Lee HY. Revisiting the bacterial phylum composition in metabolic diseases focused on host energy metabolism. Diabetes Metab J. 2020;44(5):658–67.
Onyszkiewicz M, Jaworska K, Ufnal M. Short chain fatty acids and methylamines produced by gut microbiota as mediators and markers in the circulatory system. Exp Biol Med (Maywood). 2020;245(2):166–75.
Lundberg JO, Carlström M, Weitzberg E. Metabolic effects of dietary nitrate in health and disease. Cell Metab. 2018;28(1):9–22.
Motta JP, Wallace JL, Buret AG, Deraison C, Vergnolle N. Gastrointestinal biofilms in health and disease. Nat Rev Gastroenterol Hepatol. 2021;18(5):314–34.
Blachier F, Davila AM, Mimoun S, Benetti PH, Atanasiu C, Andriamihaja M, et al. Luminal sulfide and large intestine mucosa: friend or foe? Amino Acids. 2010;39(2):335–47.
Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40(3):235–43.
Gheibi S, Samsonov AP, Gheibi S, Vazquez AB, Kashfi K. Regulation of carbohydrate metabolism by nitric oxide and hydrogen sulfide: implications in diabetes. Biochem Pharmacol. 2020;176:113819.
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325–40.
Tomasova L, Konopelski P, Ufnal M. Gut bacteria and hydrogen sulfide: the new old players in circulatory system homeostasis. Molecules. 2016;21(11):1558.
Boros M, Keppler F. Methane production and bioactivity—a link to oxido-reductive stress. Front Physiol. 2019;10:1244.
Gui DD, Luo W, Yan BJ, Ren Z, Tang ZH, Liu LS, et al. Effects of gut microbiota on atherosclerosis through hydrogen sulfide. Eur J Pharmacol. 2021;896:173916.
Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70(2):567–90.
Kaur A, Chen T, Green SJ, Mutlu E, Martin BR, Rumpagaporn P, et al. Physical inaccessibility of a resistant starch shifts mouse gut microbiota to butyrogenic firmicutes. Mol Nutr Food Res. 2019;63(7):e1801012.
Zhang S, Hu H, He W, Muhammad Z, Wang L, Liu F, et al. Regulatory roles of pectin oligosaccharides on immunoglobulin production in healthy mice mediated by gut microbiota. Mol Nutr Food Res. 2019;63(14):e1801363.
Bugaut M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp Biochem Physiol B: Comp Biochem. 1987;86(3):439–72.
Cherrington CA, Hinton M, Pearson GR, Chopra I. Short-chain organic acids at pH 5.0 kill Escherichia coli and Salmonella spp. without causing membrane perturbation. J Appl Bacteriol. 1991;70(2):161–5.
Walker AW, Duncan SH, McWilliam Leitch EC, Child MW, Flint HJ. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol. 2005;71(7):3692–700.
Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc. 2003;62(1):67–72.
Fleming SE, Rodriguez MA. Influence of dietary fiber on fecal excretion of volatile fatty acids by human adults. J Nutr. 1983;113(8):1613–25.
Spiller GA, Chernoff MC, Hill RA, Gates JE, Nassar JJ, Shipley EA. Effect of purified cellulose, pectin, and a low-residue diet on fecal volatile fatty acids, transit time, and fecal weight in humans. Am J Clin Nutr. 1980;33(4):754–9.
Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. 2000;66(4):1654–61.
De Vuyst L, Leroy F. Cross-feeding between bifidobacteria and butyrate-producing colon bacteria explains bifdobacterial competitiveness, butyrate production, and gas production. Int J Food Microbiol. 2011;149(1):73–80.
Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29–41.
Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83(2):424–9.
Ritzhaupt A, Ellis A, Hosie KB, Shirazi-Beechey SP. The characterization of butyrate transport across pig and human colonic luminal membrane. J Physiol. 1998;507(Pt 3):819–30.
Islam R, Anzai N, Ahmed N, Ellapan B, Jin CJ, Srivastava S, et al. Mouse organic anion transporter 2 (mOat2) mediates the transport of short chain fatty acid propionate. J Pharmacol Sci. 2008;106(3):525–8.
Macfarlane GT, Gibson GR. Metabolic activities of the normal colonic flora. In: Gibson SAW, editor. Human health: the contribution of microorganisms. London: Springer; 1994. p. 17–52.
Crost EH, Le Gall G, Laverde-Gomez JA, Mukhopadhya I, Flint HJ, Juge N. Mechanistic insights into the cross-feeding of Ruminococcus gnavus and Ruminococcus bromii on host and dietary carbohydrates. Front Microbiol. 2018;9:2558.
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28(10):1221–7.
Lewis S, Cochrane S. Alteration of sulfate and hydrogen metabolism in the human colon by changing intestinal transit rate. Am J Gastroenterol. 2007;102(3):624–33.
Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IA. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol. 2015;66:55–321.
Dhillon A, Goswami S, Riley M, Teske A, Sogin M. Domain Evolution and functional diversification of sulfite reductases. Astrobiology. 2005;5(1):18–29.
Kushkevych I, Cejnar J, Treml J, Dordević D, Kollar P, Vítězová M. Recent advances in metabolic pathways of sulfate reduction in intestinal bacteria. Cells. 2020;9(3):698.
Siegel LM, Murphy MJ, Kamin H. Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. I. The Escherichia coli hemoflavoprotein: molecular parameters and prosthetic groups. J Biol Chem. 1973;248(1):251–64.
Guarneros G, Ortega MV. Cysteine desulfhydrase activities of Salmonella typhimurium and Escherichia coli. Biochem Biophys Acta. 1970;198(1):132–42.
Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc Natl Acad Sci USA. 2019;116(8):3171–6.
Flannigan KL, McCoy KD, Wallace JL. Eukaryotic and prokaryotic contributions to colonic hydrogen sulfide synthesis. Am J Physiol Gastrointest Liver Physiol. 2011;301(1):G188–93.
Hughes MN, Centelles MN, Moore KP. Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radical Biol Med. 2009;47(10):1346–53.
Sawa T, Motohashi H, Ihara H, Akaike T. Enzymatic regulation and biological functions of reactive cysteine persulfides and polysulfides. Biomolecules. 2020;10(9):1245.
Furne J, Springfield J, Koenig T, DeMaster E, Levitt MD. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: a specialized function of the colonic mucosa. Biochem Pharmacol. 2001;62(2):255–9.
Barrett EL, Clark MA. Tetrathionate reduction and production of hydrogen sulfide from thiosulfate. Microbiol Rev. 1987;51(2):192–205.
Barton LL, Ritz NL, Fauque GD, Lin HC. Sulfur cycling and the intestinal microbiome. Dig Dis Sci. 2017;62(9):2241–57.
Tomasova L, Dobrowolski L, Jurkowska H, Wróbel M, Huc T, Ondrias K, et al. Intracolonic hydrogen sulfide lowers blood pressure in rats. Nitric Oxide Biol Chem. 2016;60:50–8.
Shen X, Carlström M, Borniquel S, Jädert C, Kevil CG, Lundberg JO. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radical Biol Med. 2013;60:195–200.
Szabo C, Ransy C, Módis K, Andriamihaja M, Murghes B, Coletta C, et al. Regulation of mitochondrial bioenergetic function by hydrogen sulphide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2014;171(8):2099–122.
Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92(2):791–896.
Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem J. 1984;224(2):591–600.
Babidge W, Millard S, Roediger W. Sulfides impair short chain fatty acid beta-oxidation at acyl-CoA dehydrogenase level in colonocytes: implications for ulcerative colitis. Mol Cell Biochem. 1998;181(1–2):117–24.
Levitt MD, Springfield J, Furne J, Koenig T, Suarez FL. Physiology of sulfide in the rat colon: use of bismuth to assess colonic sulfide production. J Appl Physiol (Bethesda, Md: 1985). 2002;92(4):1655–60.
Landry AP, Moon S, Kim H, Yadav PK, Guha A, Cho US, et al. A catalytic trisulfide in human sulfide quinone oxidoreductase catalyzes coenzyme a persulfide synthesis and inhibits butyrate oxidation. Cell Chem Biol. 2019;26(11):1515-25.e4.
Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J: Off Publ Fed Am Soc Exp Biol. 2013;27(2):601–11.
Módis K, Ju Y, Ahmad A, Untereiner AA, Altaany Z, Wu L, et al. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol Res. 2016;113(Pt A):116–24.
Untereiner AA, Oláh G, Módis K, Hellmich MR, Szabo C. H(2)S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem Pharmacol. 2017;136:86–98.
Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J: Off Publ Fed Am Soc Exp Biol. 2007;21(8):1699–706.
Motta JP, Flannigan KL, Agbor TA, Beatty JK, Blackler RW, Workentine ML, et al. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm Bowel Dis. 2015;21(5):1006–17.
Nagy P, Dóka É, Ida T, Akaike T. Measuring reactive sulfur species and thiol oxidation states: challenges and cautions in relation to alkylation-based protocols. Antioxid Redox Signal. 2020;33(16):1174–89.
Augustyn KD, Jackson MR, Jorns MS. Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry. 2017;56(7):986–96.
Searcy DG. Metabolic integration during the evolutionary origin of mitochondria. Cell Res. 2003;13(4):229–38.
Dickerson RE. Evolution and gene transfer in purple photosynthetic bacteria. Nature. 1980;283(5743):210–2.
Zaorska E, Tomasova L, Koszelewski D, Ostaszewski R, Ufnal M. Hydrogen sulfide in pharmacotherapy, beyond the hydrogen sulfide-donors. Biomolecules. 2020;10(2):323.
Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun. 2017;8(1):1177.
Koch CD, Gladwin MT, Freeman BA, Lundberg JO, Weitzberg E, Morris A. Enterosalivary nitrate metabolism and the microbiome: Intersection of microbial metabolism, nitric oxide and diet in cardiac and pulmonary vascular health. Free Radical Biol Med. 2017;105:48–67.
Brittain T, Blackmore R, Greenwood C, Thomson AJ. Bacterial nitrite-reducing enzymes. Eur J Biochem. 1992;209(3):793–802.
Potter L, Angove H, Richardson D, Cole J. Nitrate reduction in the periplasm of gram-negative bacteria. Adv Microb Physiol. 2001;45:51–112.
Gilberthorpe NJ, Poole RK. Nitric oxide homeostasis in Salmonella typhimurium: roles of respiratory nitrate reductase and flavohemoglobin. J Biol Chem. 2008;283(17):11146–54.
Sobko T, Reinders CI, Jansson E, Norin E, Midtvedt T, Lundberg JO. Gastrointestinal bacteria generate nitric oxide from nitrate and nitrite. Nitric Oxide Biol Chem. 2005;13(4):272–8.
Adak S, Aulak KS, Stuehr DJ. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J Biol Chem. 2002;277(18):16167–71.
Adak S, Bilwes AM, Panda K, Hosfield D, Aulak KS, McDonald JF, et al. Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc Natl Acad Sci USA. 2002;99(1):107–12.
Gusarov I, Starodubtseva M, Wang ZQ, McQuade L, Lippard SJ, Stuehr DJ, et al. Bacterial nitric-oxide synthases operate without a dedicated redox partner. J Biol Chem. 2008;283(19):13140–7.
Saul RL, Kabir SH, Cohen Z, Bruce WR, Archer MC. Reevaluation of nitrate and nitrite levels in the human intestine. Can Res. 1981;41(6):2280–3.
Saijo F, Milsom AB, Bryan NS, Bauer SM, Vowinkel T, Ivanovic M, et al. On the dynamics of nitrite, nitrate and other biomarkers of nitric oxide production in inflammatory bowel disease. Nitric Oxide Biol Chem. 2010;22(2):155–67.
Witter JP, Balish E, Gatley SJ. Distribution of nitrogen-13 from labeled nitrate and nitrite in germfree and conventional-flora rats. Appl Environ Microbiol. 1979;38(5):870–8.
Roediger WE, Radcliffe BC. Role of nitrite and nitrate as a redox couple in the rat colon. Implications for diarrheal conditions. Gastroenterology. 1988;94(4):915–22.
Moretti C, Zhuge Z, Zhang G, Haworth SM, Paulo LL, Guimaraes DD, et al. The obligatory role of host microbiota in bioactivation of dietary nitrate. Free Radical Biol Med. 2019;145:342–8.
Huang L, Borniquel S, Lundberg JO. Enhanced xanthine oxidoreductase expression and tissue nitrate reduction in germ free mice. Nitric Oxide Biol Chem. 2010;22(2):191–5.
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science (New York, NY). 2003;299(5608):896–9.
Roediger WE, Deakin EJ, Radcliffe BC, Nance S. Anion control of sodium absorption in the colon. Q J Exp Physiol (Camb, Engl). 1986;71(2):195–204.
Larsen FJ, Schiffer TA, Borniquel S, Sahlin K, Ekblom B, Lundberg JO, et al. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011;13(2):149–59.
Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–89.
Pochart P, Lemann F, Flourie B, Pellier P, Goderel I, Rambaud JC. Pyxigraphic sampling to enumerate methanogens and anaerobes in the right colon of healthy humans. Gastroenterology. 1993;105(5):1281–5.
Levitt MD, Furne JK, Kuskowski M, Ruddy J. Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements. Clin Gastroenterol Hepatol: Off Clin Pract J Am Gastroenterol Assoc. 2006;4(2):123–9.
Bond JH Jr, Engel RR, Levitt MD. Factors influencing pulmonary methane excretion in man. An indirect method of studying the in situ metabolism of the methane-producing colonic bacteria. J Exp Med. 1971;133(3):572–88.
Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio. 2015;6(2):e0248114.
Fennema D, Phillips IR, Shephard EA. Trimethylamine and trimethylamine N-oxide, a flavin-containing monooxygenase 3 (FMO3)-mediated host-microbiome metabolic axis implicated in health and disease. Drug Metab Dispos: Biol Fate Chem. 2016;44(11):1839–50.
de la Cuesta-Zuluaga J, Spector TD, Youngblut ND, Ley RE. Genomic insights into adaptations of trimethylamine-utilizing methanogens to diverse habitats, including the human gut. mSystems. 2021;6(1):e0093920.
Jaworska K, Huc T, Gawrys M, Onyszkiewicz M, Samborowska E, Ufnal M. An in vivo method for evaluating the gut-blood barrier and liver metabolism of microbiota products. J Vis Exp: JoVE. 2018;140:58456.
Borrel G, McCann A, Deane J, Neto MC, Lynch DB, Brugère JF, et al. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. ISME J. 2017;11(9):2059–74.
Jaworska K, Hering D, Mosieniak G, Bielak-Zmijewska A, Pilz M, Konwerski M, et al. TMA, a forgotten uremic toxin, but not TMAO, is involved in cardiovascular pathology. Toxins. 2019;11(9):490.
Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575–84.
Tuboly E, Molnár R, Tőkés T, Turányi RN, Hartmann P, Mészáros AT, et al. Excessive alcohol consumption induces methane production in humans and rats. Sci Rep. 2017;7(1):7329.
Makrecka-Kuka M, Volska K, Antone U, Vilskersts R, Grinberga S, Bandere D, et al. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol Lett. 2017;267:32–8.
Willis CL, Cummings JH, Neale G, Gibson GR. In vitro effects of mucin fermentation on the growth of human colonic sulphatereducing bacteria. Anaerobe. 1996;2:117–22.
Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci USA. 2013;110(33):13582–7.
Gibson GR, Cummings JH, Macfarlane GT. Competition for hydrogen between sulphate-reducing bacteria and methanogenic bacteria from the human large intestine. J Appl Bacteriol. 1988;65(3):241–7.
Strocchi A, Furne J, Ellis C, Levitt MD. Methanogens outcompete sulphate reducing bacteria for H2 in the human colon. Gut. 1994;35(8):1098–101.
Christl SU, Gibson GR, Cummings JH. Role of dietary sulphate in the regulation of methanogenesis in the human large intestine. Gut. 1992;33(9):1234–8.
Gaci N, Borrel G, Tottey W, O’Toole PW, Brugere JF. Archaea and the human gut: new beginning of an old story. World J Gastroenterol. 2014;20(43):16062–78.
Seitz H-J, Cypionka H. Chemolithotrophic growth of Desulfovibrio desulfuricans with hydrogen coupled to ammonification of nitrate or nitrite. Arch Microbiol. 1986;146(1):63–7.
Dannenberg S, Kroder M, Dilling W, Cypionka H. Oxidation of H2, organic compounds and inorganic sulfur compounds coupled to reduction of O2 or nitrate by sulfate-reducing bacteria. Arch Microbiol. 1992;158(2):93–9.
Eisenmann E, Beuerle J, Sulger K, Kroneck PMH, Schumacher W. Lithotrophic growth of Sulfurospirillum deleyianum with sulfide as electron donor coupled to respiratory reduction of nitrate to ammonia. Arch Microbiol. 1995;164(3):180–5.
Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci USA. 2007;104(25):10643–8.
Bullon P, Marin-Aguilar F, Roman-Malo L. AMPK/Mitochondria in metabolic diseases. Exp Suppl. 2012;2016(107):129–52.
Marzullo P, Di Renzo L, Pugliese G, De Siena M, Barrea L, Muscogiuri G, et al. From obesity through gut microbiota to cardiovascular diseases: a dangerous journey. Int J Obes Suppl. 2020;10(1):35–49.
Samczuk P, Hady HR, Adamska-Patruno E, Citko A, Dadan J, Barbas C, et al. In-and-out molecular changes linked to the type 2 diabetes remission after bariatric surgery: an influence of gut microbes on mitochondria metabolism. Int J Mol Sci. 2018;19(12):3744.
Mollica MP, Mattace Raso G, Cavaliere G, Trinchese G, De Filippo C, Aceto S, et al. Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin-resistant obese mice. Diabetes. 2017;66(5):1405–18.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17.
Jia Y, Hong J, Li H, Hu Y, Jia L, Cai D, et al. Butyrate stimulates adipose lipolysis and mitochondrial oxidative phosphorylation through histone hyperacetylation-associated β(3)-adrenergic receptor activation in high-fat diet-induced obese mice. Exp Physiol. 2017;102(2):273–81.
Henagan TM, Stefanska B, Fang Z, Navard AM, Ye J, Lenard NR, et al. Sodium butyrate epigenetically modulates high-fat diet-induced skeletal muscle mitochondrial adaptation, obesity and insulin resistance through nucleosome positioning. Br J Pharmacol. 2015;172(11):2782–98.
Peleli M, Ferreira DMS, Tarnawski L, Haworth SM, Xuechen L, Zhuge Z, et al. Dietary nitrate attenuates high-fat diet-induced obesity via mechanisms involving higher adipocyte respiration and alterations in inflammatory status. Redox Biol. 2020;28:101387.
Weitkunat K, Schumann S, Nickel D, Kappo KA, Petzke KJ, Kipp AP, et al. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol Nutr Food Res. 2016;60(12):2611–21.
Granér M, Gustavsson S, Nyman K, Siren R, Pentikäinen MO, Lundbom J, et al. Biomarkers and prediction of myocardial triglyceride content in non-diabetic men. Nutr Metab Cardiovasc Dis. 2016;26(2):134–40.
Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JM, et al. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metab Cardiovasc Dis. 2014;24(6):606–13.
Ufnal M, Zadlo A, Ostaszewski R. TMAO: a small molecule of great expectations. Nutrition. 2015;31:1317–23.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31.
Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen JL, et al. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology. 2008;149(9):4519–26.
Yau YF, El-Nezami H, Galano JM, Kundi ZM, Durand T, Lee JC. Lactobacillus rhamnosus GG and oat beta-glucan regulated fatty acid profiles along the gut-liver-brain axis of mice fed with high fat diet and demonstrated antioxidant and anti-inflammatory potentials. Mol Nutr Food Res. 2020;64(18):e2000566.
Yamashita H, Fujisawa K, Ito E, Idei S, Kawaguchi N, Kimoto M, et al. Improvement of obesity and glucose tolerance by acetate in Type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Biosci Biotechnol Biochem. 2007;71(5):1236–43.
Kondo T, Kishi M, Fushimi T, Kaga T. Acetic acid upregulates the expression of genes for fatty acid oxidation enzymes in liver to suppress body fat accumulation. J Agric Food Chem. 2009;57(13):5982–6.
Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. 2014;5:3611.
Chambers ES, Preston T, Frost G, Morrison DJ. Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr Nutr Rep. 2018;7(4):198–206.
Jiang H, Torregrossa AC, Potts A, Pierini D, Aranke M, Garg HK, et al. Dietary nitrite improves insulin signaling through GLUT4 translocation. Free Radical Biol Med. 2014;67:51–7.
Khalifi S, Rahimipour A, Jeddi S, Ghanbari M, Kazerouni F, Ghasemi A. Dietary nitrate improves glucose tolerance and lipid profile in an animal model of hyperglycemia. Nitric Oxide Biol Chem. 2015;44:24–30.
Nowiński A, Ufnal M. Trimethylamine N-oxide: a harmful, protective or diagnostic marker in lifestyle diseases? Nutrition. 2018;46:7–12.
Park JE, Miller M, Rhyne J, Wang Z, Hazen SL. Differential effect of short-term popular diets on TMAO and other cardio-metabolic risk markers. Nutr Metab Cardiovasc Dis. 2019;29(5):513–7.
Haufe S, Engeli S, Kaminski J, Witt H, Rein D, Kamlage B, et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr Metab Cardiovasc Dis. 2017;27(10):858–64.
Bełtowski J, Jamroz-Wiśniewska A. Hydrogen sulfide in the adipose tissue-physiology, pathology and a target for pharmacotherapy. Molecules. 2016;22(1):63.
Cai J, Shi X, Wang H, Fan J, Feng Y, Lin X, et al. Cystathionine γ lyase-hydrogen sulfide increases peroxisome proliferator-activated receptor γ activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochem Biophys Acta. 2016;1861(5):419–29.
Guasti L, Galliazzo S, Molaro M, Visconti E, Pennella B, Gaudio GV, et al. TMAO as a biomarker of cardiovascular events: a systematic review and meta-analysis. Intern Emerg Med. 2020;16:201–7.
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–85.
Joe B, McCarthy CG, Edwards JM, Cheng X, Chakraborty S, Yang T, et al. Microbiota introduced to germ-free rats restores vascular contractility and blood pressure. Hypertension (Dallas, TX : 1979). 2020;76(6):1847–55.
Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, et al. Gut microbiota promote angiotensin ii-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. 2016;5(9):e003698.
Onyszkiewicz M, Gawrys-Kopczynska M, Sałagaj M, Aleksandrowicz M, Sawicka A, Koźniewska E, et al. Valeric acid lowers arterial blood pressure in rats. Eur J Pharmacol. 2020;877:173086.
Onyszkiewicz M, Gawrys-Kopczynska M, Konopelski P, Aleksandrowicz M, Sawicka A, Koźniewska E, et al. Butyric acid, a gut bacteria metabolite, lowers arterial blood pressure via colon-vagus nerve signaling and GPR41/43 receptors. Pflugers Arch. 2019;471(11–12):1441–53.
Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135(10):964–77.
Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110(11):4410–5.
Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. 2016;48(11):826–34.
Daliri EB, Ofosu FK, Chelliah R, Lee BH, An H, Elahi F, et al. Influence of fermented soy protein consumption on hypertension and gut microbial modulation in spontaneous hypertensive rats. Biosci Microbiota Food Health. 2020;39(4):199–208.
Jaworska K, Bielinska K, Gawrys-Kopczynska M, Ufnal M. TMA (trimethylamine), but not its oxide TMAO (trimethylamine-oxide), exerts haemodynamic effects: implications for interpretation of cardiovascular actions of gut microbiome. Cardiovasc Res. 2019;115(14):1948–9.
Jaworska K, Huc T, Samborowska E, Dobrowolski L, Bielinska K, Gawlak M, et al. Hypertension in rats is associated with an increased permeability of the colon to TMA, a gut bacteria metabolite. PLoS ONE. 2017;12(12):e0189310.
Wen Y, Peng L, Xu R, Zang N, Huang Q, Zhong M. Maternal serum trimethylamine-N-oxide is significantly increased in cases with established preeclampsia. Pregnancy Hypertens. 2019;15:114–7.
Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut microbiota metabolites and risk of major adverse cardiovascular disease events and death: a systematic review and meta-analysis of prospective studies. J Am Heart Assoc. 2017;6(7):e004947.
Acknowledgements
Not applicable
Funding
This work was supported by the Medical University of Warsaw, the Slovak Research and Development Agency [Grant No. APVV-19-0154] and the VEGA Grant Agency of the Slovak Republic [Grant No. VEGA-2/0079/19].
Author information
Authors and Affiliations
Contributions
L.T. and M.U. wrote the paper. L.T. and M.G. prepared the figures. M.G. and K.O. have read and edited the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tomasova, L., Grman, M., Ondrias, K. et al. The impact of gut microbiota metabolites on cellular bioenergetics and cardiometabolic health. Nutr Metab (Lond) 18, 72 (2021). https://doi.org/10.1186/s12986-021-00598-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-021-00598-5