
Abstract
Background
Soluble epoxide hydrolase (sEH) is a bifunctional enzyme with COOH-terminal hydrolase and NH2-terminal lipid phosphatase activities. It is expressed in various cell types in the brain and is involved in the pathogenesis of inflammatory and neurodegenerative diseases. Alzheimer’s disease (AD) is a progressive neuroinflammatory and neurodegenerative disease. However, the pathological significance of sEH and underlying molecular mechanism in AD remain unclear.
Methods
To examine the role of sEH in pathogenesis of AD, we used wild-type (WT) mice, soluble epoxide hydrolase deficient (sEH−/−) and two mouse models of AD, including amyloid precursor protein (APP)/presenilin 1 (PS1) transgenic (APP/PS1 Tg) and APP/PS1 Tg/sEH−/− mice. Western blotting analysis and immunohistochemistry assay were performed to evaluate the protein expression. Locomotion, nesting building ability, Y-maze, and Morris water maze tests were conducted to study mouse behavior. The levels of interleukin (IL)-1β, IL-4, IL-6, and IL-10 and the activities of NF-κB and nuclear factor of activated T cells (NFAT) were measured by commercial assay kits. The quantitative protein level profiling in the brain lysate was analyzed using LC-MS/MS approaches.
Results
We demonstrated that the level of sEH was increased in the brain and predominantly appeared in hippocampal astrocytes of APP/PS1 Tg mice. Genetic ablation of sEH in APP/PS1 Tg mice delayed the progression of AD as evidenced by the alleviation in behavior outcomes and Aβ plaque deposition. In addition, loss of the function of sEH in APP/PS1 Tg mice increased astrogliosis and the production of astrocyte-derived anti-inflammatory cytokines including IL-1β, IL-4, and IL-10, as well as the activity of NF-kB and NFAT. Moreover, analysis of gene ontology in the AD brain revealed that important signaling pathways and processes related to AD pathogenesis such as translational regulation, oxidative stress, cytoskeleton reorganization, and small GTPase signal transduction were altered in APP/PS1 Tg/sEH−/− mice compared with APP/PS1 Tg mice.
Conclusion
Our results suggest that sEH is a crucial regulator in the progression of AD and might be a potential therapeutic target for the treatment of AD.
Similar content being viewed by others


Introduction
Soluble epoxide hydrolase (sEH), an enzyme with COOH-terminal hydrolase (EH) and NH2-terminal lipid phosphatase (PT) activities, is expressed in several tissues, including the brain, heart, and kidney [1,2,3,4]. In the brain, sEH is predominantly expressed in cortex, hippocampus, white matter, substantia nigra, and striatum [4,5,6]. Apart from the endothelial cells (ECs) of vessels, sEH appears in neurons and glial cells [4, 7]. The precise role of sEH in the pathogenesis of inflammatory and brain diseases has been well defined [8, 9]. Recent studies demonstrated that inhibition of the EH activity of sEH confers protection from ischemia-induced brain injury in rodents, suggesting that sEH is a crucial mediator in cerebrovascular and neuronal function upon the pathological insults [6, 7, 10,11,12,13]. In addition, several studies indicated the potential function of sEH in age-related cognitive decline and neurodegenerative disease [14,15,16]. However, the role of sEH and its underlying mechanism in neurodegenerative diseases are not fully understood.
Alzheimer’s disease (AD) is a progressive neurodegeneration disease with typical symptoms, including impairment of learning, memory, and cognition abilities [17, 18]. Recently evidence has indicated that amyloid-β (Aβ) plaque deposition is one of the pathological hallmarks in AD [17, 19, 20]. The Aβ peptide is generated from amyloid precursor protein (APP) by β-site APP cleavage enzyme 1 (BACE1) and presenilin 1 (PS1) [19, 21]. Particularly, neurotoxic Aβ triggers inflammation in glial cells and induces neurodegeneration, ultimately leading to the progression of AD [19, 21,22,23,24]. Moreover, astrocyte-derived inflammatory responses play a crucial role in AD pathology [25, 26]. Upon Aβ stimulation, reactive astrocytes can release pro-inflammatory cytokines including interleukin (IL)-1β, IL-6, and anti-inflammatory cytokines, IL-4 and IL-10 [24, 27]. Two different transcription factors NF-kB and nuclear factor of activated T cells (NFAT) are involved in the astrocyte-mediated cytokines production; NF-κB regulates the production of IL-1β and IL-6, and NFAT regulates the expression of IL-4 and IL-10 production [28,29,30,31]. However, the critical role and the molecular mechanisms underlying astrocyte-mediated inflammatory response upon Aβ stimulation are not fully elucidated.
In this study, we aimed to investigate the potential role of sEH and possible molecular mechanisms during AD pathogenesis by using the mouse models. Here, we report that the protein level of sEH was increased in astrocytes of 6-month-old APP/PS1 Tg mice. We demonstrated that genetic deletion of sEH in the APP/PS1 Tg mice rescued the impairment of AD pathologies, including Aβ plaque deposition, cytokines production, and dysfunction of behavioral outcomes. The quantitative proteomic analysis of brain samples from APP/PS1 Tg/sEH−∕− and APP/PS1 Tg mice was performed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Mapping of the deregulated proteins with bioinformatics tools revealed that differentially abundant proteins were significantly enriched for pathways and processes known to be important in AD pathology, including translational regulation, oxidative stress, cytoskeleton reorganization, and small GTPase-mediated signal transduction. Collectively, our results suggest that sEH is a key regulator of astrocytes-derived inflammation in AD progression.
Methods
Reagents
Rabbit anti-sEH (sc-25797), mouse anti-von Willebrand factor (vWF) (sc-365712), goat anti-rabbit IgG FITC-conjugated (sc-2012), Texas red-conjugated (sc-2780), and goat anti-mouse IgG Texas red-conjugated (sc-2781) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-glial fibrillary acidic protein (GFAP) (AB5804), mouse anti-GFAP (MAB360), anti-NeuN (MAB377B), and anti-ionized calcium-binding adapter molecule 1 (IBA-1) (MABN92) antibodies were obtained from Millipore (Darmstadt, Germany). Rabbit anti-LDLR-related protein 1 (LRP-1) (L2170) antibody, mouse anti-α-tubulin (T9026) antibody, bovine serum albumin (BSA), and phosphatase inhibitor cocktails 1 and 2 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Mouse anti-Aβ (SIG-39320) antibody was from Covance (Dedham, MA, USA). Rabbit anti-β-APP C-terminal fragment (βCTF) (802801) antibody was obtained from BioLegend (San Diego, CA, USA). Mouse anti-ATP-binding cassette transporter A1 (ABCA1) (ab18180), rabbit anti-BACE1 (ab2077), anti-IL-1β (ab9722), anti-IL-4 (ab9622), anti-IL-6 (ab6672), and anti-IL-10 (ab9969) antibodies were obtained from Abcam (Cambridge, MA, UK). Rabbit anti-apolipoprotein E (apoE) (1930-1) antibody was obtained from Epitomics (Burlingame, CA, USA). Retrieval buffer was from Biocare Medical (Concord, CA, USA). The mounting medium with DAPI was from Vector Laboratories (Burlingame, CA, USA). The ELISA kit for NF-κB activity was from Cayman Chemical (Ann Arbor, MI, USA) and for NFAT activity from Active Motif (Carlsbad, CA, USA). ELISA kits for IL-1β, IL-4, IL-6, and IL-10 were obtained from R&D systems (Minneapolis, MN, USA).
Mice
The investigation conformed to the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, eighth edition, 2011), and all animal experiments were performed in accordance with the approved guidelines by the Animal Care and Utilization Committee of the National Yang-Ming University. C57BL/6 J WT, Ephx2tm1/Gon2/J (sEH−∕−) mice, and B6.Cg-Tg(APPswe, PSEN1dE9)85Dbo/J (APP/PS1 Tg) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). For the generation of APP/PS1 Tg/sEH−∕− mice, sEH−∕− mice were crossed with APP/PS1 Tg background, and the genotypes were confirmed by PCR of genomic DNA isolated from these animals. Mice were housed in barrier facilities, maintained in a 12-h/12-h light and dark cycle. Temperature (22 °C) and humidity (40–60%) of the vivarium were tightly controlled. Mice were group-housed 3–4 per cage and fed a regular chow diet, which contained 4.5% fat by weight (0.02% cholesterol) (Newco Distributors, Redwood, CA, USA). At the end of the experiments, mice were euthanized with CO2, and then brain tissues were collected for histological analysis or were stored at − 80 °C. The isolated brain tissues were homogenized, and lysates were subjected to western blot analysis.
Western blot analysis
Brain tissues were lysed in immunoprecipitation lysis buffer (50 mmol/L Tris pH 7.5, 5 mmol/L EDTA, 300 mmol/L NaCl, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin, and 10 μg/mL aprotinin). Aliquots of brain lysates were separated on SDS-PAGE, transferred to membranes, incubated with primary antibodies, and then horseradish peroxidase-conjugated secondary antibody. Bands were detected using an enzyme-linked chemiluminescence detection kit (Perkin Elmer, Waltham, MA) and the band signal was quantified by Imagequant 5.2 (Healthcare Bio-Sciences, Philadelphia, PA).
Immunohistochemistry staining
The brain tissues were fixed in 4% paraformaldehyde and 15-μm cross sections were prepared. Sections were incubated with retrieval buffer for 10 min, blocked with 2% BSA for 60 min, and incubated with primary antibody overnight at 4 °C, then FITC- or Texas Red-conjugated secondary antibody for 1 h at 37 °C. Antigenic sites were visualized under a Nikon TE2000-U microscope (Tokyo) with QCapture Pro 6.0 software (QImaging, BC, Canada).
Measurement of inflammatory cytokines
The concentrations of pro-inflammatory cytokines, including IL-1β, IL-4, IL-6, and IL-10 in brain lysates, were measured by using the ELISA kits according to the manufacturer’s instructions.
Measurement of DNA-binding activity on NF-κB and NFAT
The DNA-binding activities of NF-κB and NFAT in brain lysates were measured by using the ELISA kit according to the manufacturer’s instructions.
Nest building
The protocol of nest building has been described [32]. Each mouse was housed in a single cage containing two pieces of cotton (5 × 5 cm) from the first day. The presence and quality of nesting were recorded by the nesting score which separated 5-point scale ranging from 1 to 5 as follows: 1 = nest not noticeably touched, 2 = cotton partially torn up, 3 = mostly shredded cotton, but often no identifiable nest location, 4 = a markedly nesting site, but flat nest, and 5 = a (near) perfect nest. Nesting score was recorded manually at 24 h.
Y-maze
The protocol of Y-maze was followed as previously described with modifications [33]. The Y-maze apparatus consisted of three arms made of stainless steel joined in the middle to form a “Y” shape. The mice were placed into one of the arms of the maze (star arm) and allowed to explore the maze with only one of the arms closed for 10 min (training trial). After 3 h, mice were returned to the Y-maze by placing them in the start arm. Then, the mice were allowed to explore freely all three arms of the maze for 5 min (test trial). The number of entries into and the distance in each arm, and the first choice of entry were registered from video recordings.
Morris water maze
The protocol of Morris water maze (MWM) has been described [33] and modified for this study. A large circular tank (diameter 0.8 m and depth 0.4 m) was filled with water (25 ± 1 °C, depth 20 cm) and the escape platform (8 cm × 4 cm) was submerged 1 cm below the surface. The training section was monitored by a video recording system. The escape latency and trajectory of swimming were recorded for each mouse. The hidden platform was located at the center of one of the four quadrants to the pool. The location of the platform was fixed throughout testing. Mice had to navigate using extra-maze cues that were placed on the walls of the maze. From day 1 to day 4, mice went through three trials with an inter-trial interval of 5 min. The mice were placed into the pool facing the sidewall randomly at one of four start locations and allowed to swim until they found the platform, or for a maximum of 2 min. Any mouse that failed to find the platform within 2 min was then guided to the platform. The animal then remained on the platform for 20 s, and then they were removed from the pool. The day after the completion of the hidden platform training, a probe trial was conducted to determine whether mice used a spatial strategy to find the platform. In day 5, the platform was removed from the pool, and the mice were allowed to swim freely for 2 min. The percentage of time spent in each quadrant of the pool and the number of times the mice crossed the former position of the hidden platform were recorded.
LC-MS/MS analysis
The LC-MS/MS analysis of brain tissues from APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice was performed by researchers of Visual Protein Company (Taipei, Taiwan).
Gene ontological analysis
The gene ontological analysis of brain tissues from APP/PS1 Tg and APP/PS1 Tg/sEH−∕− mice was performed by researchers of Visual Protein Company (Taipei, Taiwan).
Statistical analysis
Results are presented as mean ± SEM. Mann-Whitney U test was used to compare two independent groups. Kruskal-Wallis followed by Bonferroni post hoc analyses were used to account for multiple testing. SPSS v20.0 (SPSS Inc., Chicago, IL) was used for analysis. Differences were considered statistically significant at P < 0.05.
Results
The level of sEH is upregulated in hippocampal astrocytes during AD progression
To explore the role of sEH in the pathogenesis of AD, we first investigated the levels of sEH in mouse brain from WT and APP/PS1 Tg mice. The results of western blot analysis showed that the protein level of sEH was increased in 6-month-old APP/PS1 Tg mice (Fig. 1). Immunohistochemistry analysis revealed that sEH was primarily localized in ECs of vessels, hippocampal neurons, and astrocytes in WT or APP/PS1 Tg mice. However, such an increase in sEH expression was remarkably restricted to astrocytes in the brain of APP/PS1 Tg mice (APP/PS1 Tg mice vs. WT mice = 4.23 ± 1.35 vs. 1.14 ± 0.28-fold) (Fig. 2a and b). These data suggest that sEH might play an important role in the development of AD.

The protein expression of sEH is increased in the brain of APP/PS1 Tg mice. Brains were harvested from 6-month-old WT mice and APP/PS1 Tg mice. Western blot analysis of sEH and α-tubulin. Data are mean ± SEM from 6 mice in each group. *P < 0.05 vs. WT mice

The level of sEH protein is increased in hippocampal astrocytes of APP/PS1 Tg mice. a, b Brain tissue sections from 6-month-old WT mice and APP/PS1 Tg mice were immunostained with anti-sEH, anti-vWF (for endothelial cells), anti-NeuN (for neurons), anti-GFAP (for astrocytes), anti-IBA-1 (for microglia), and their corresponding secondary antibodies. Scale bar = 50 μm. Arrowheads indicate sEH-positive astrocytes
Genetic deletion of sEH improves the nesting building ability, learning, and memory in the APP/PS1 Tg mice
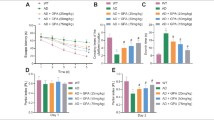
To assess the role of sEH on the impairment of AD-related behaviors in APP/PS1 Tg mice, we generated the APP/PS1 Tg/sEH−∕− mice by an inbred of sEH−∕− mice with APP/PS Tg mice (Fig. 3a). The analysis of nesting building activity showed that APP/PS1 Tg/sEH−∕− mice constructed higher quality of nest shape along with increased quantification of score compared with age-matched 6-month-old APP/PS1 Tg mice (Fig. 3b and c). These data indicated that ablation of sEH improved the nesting building ability in APP/PS1 Tg mice. In addition, spatial learning and memory were examined with Y-maze and MWM. In Y-maze test, loss of sEH expression increased the preference of first entry to new arm, the entry numbers ration, and distance in new arm in 6-month-old APP/PS1 Tg mice (Fig. 3d–g). Next, the MWM test was performed to evaluate the ability of spatial learning and memory of mice. With the hidden platform test, APP/PS1 Tg mice learned the location of the hidden platform since day 3, but APP/PS1 Tg/sEH−∕− mice learned the hidden platform location since day 2 (Fig. 3h). However, there were no statistically significant differences between APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice in escape latency during the 4 days of training. Moreover, APP/PS1 Tg/sEH−∕− mice showed an increased number of times crossing the hidden platform and retention times in the target quadrant at day 5 after training (Fig. 3i and j). The genotypes did not differ in swimming to the visible platform test (Fig. 3k). These findings suggest that the sEH might be a critical regulator in AD-related cognitive performance and spatial learning and memory ability.

Deletion of sEH recovers nesting building ability, spatial learning, and memory in APP/PS1 Tg mice. a PCR analysis for genotyping of representative animals depicting WT, sEH−∕−, APP/PS1 Tg, and APP/PS1 Tg/sEH−∕−. PCR bands of 608 bp for PS1, 377 bp for APP, 338 bp for sEH+/+, and 295 bp for sEH−∕− were amplified for genotypes. b, c Representative examples and the score for nest building for 6-month-old WT, sEH−∕−, APP/PS1 Tg, and APP/PS1 Tg/sEH−∕− mice. d–g Schematic diagram of Y-maze experimental design and ratio of first entry, number of entries, and distance moved in the novel arm. h Morris water maze (MWM) test of learning patterns verified on days 1 to 4. i–j At day 5 after training, the number of times of crossing the hind platform and retention times in the target quadrants. k The latency of arrival to the visible platform. Data are mean ± SEM from 8 mice in each group. c, e–g, i, and j *P < 0.05 vs. WT mice. #P < 0.05 vs. APP/PS1 Tg mice (i). *P < 0.05 vs. day 1. #P < 0.05 vs. WT mice
Loss of sEH not only diminishes Aβ deposition, but also decreases apoE expression in brain lesions
Next, we further examined the level of Aβ plaque formation with or without sEH under APP/PS1 Tg background. Immunohistochemistry analysis indicated that the Aβ accumulation was dramatically decreased on hippocampus and cortex of APP/PS1 Tg/ sEH−∕− mice (Fig. 4a). Similar results of decreased Aβ levels were obtained by western blotting analysis in the brain lysates of APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice. Western blot results of Aβ in both oligomers and monomers, BACE1, and βCTF, within brain lysates from APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice supported the decreased Aβ plaque formation in APP/PS1 Tg/sEH−∕− mice (Fig. 4b–f). Further, the levels of apoE, but not ABCA1 and LRP-1, were decreased in APP/PS1 Tg/sEH−∕− mice (Fig. 4g–j).

a–j Loss of function of sEH decreases Aβ deposition in APP/PS1 Tg mice. a Brain tissue sections from 6-month-old APP/PS1 Tg and APP/PS1 Tg/sEH−∕− mice were immunostained with anti-Aβ and FITC-conjugated secondary antibody, and then the FITC intensity was assessed. Scale bar = 100 μm. b–j Western blot analysis of protein levels of oligomer (oligo) or monomer (mono) Aβ, BACE1, βCTF, ABCA1, LRP-1, apoE, and α-tubulin. Data are mean ± SEM from 8 mice in each group. *P < 0.05 vs. APP/PS1 Tg mice
Genetic ablation of sEH in an APP/PS1 Tg background aggravates astrogliosis and increases the production of astrocyte-derived cytokines
Subsequently, we observed the unexpected phenomena that the level of astrogliosis was more obvious in APP/PS1 Tg/sEH−∕− mice than that in APP/PS1 Tg mice (Fig. 5a and b). However, deficiency of sEH markedly increased the production of pro-inflammatory cytokine IL-1β, and anti-inflammatory cytokines IL-4 and IL-10 in APP/PS1 Tg mice (Fig. 5c–f). We next focused on the activities of transcription factor NF-κB and NFAT, which predominantly regulate the expression of the IL-1β, IL-6, IL-4, and IL-10 genes [28,29,30,31]. We demonstrated that deletion of sEH increased the DNA-binding activity of NF-κB and NFAT in APP/PS1 Tg mice (Fig. 5g and h). Next, immunohistochemistry analysis revealed that the increased levels of IL-1β, IL-4, and IL-10 were restricted in hippocampal astrocytes in APP/PS1 Tg/sEH−∕− mice (Fig. 6). Thus, loss of sEH may facilitate astrogliosis, but remarkably increase the production of anti-inflammatory cytokines IL-4 and IL-10 in APP/PS1 Tg mice.

Loss of function of sEH increases astrogliosis and the production of cytokines in APP/PS1 Tg mice. a Brain tissue sections from 6-month-old APP/PS1 Tg and APP/PS1 Tg/sEH−∕− mice were immunostained with anti-GFAP (for astrocyte) and FITC-conjugated secondary antibody. Scale bar = 50 μm. b Western blot analysis of protein levels of GFAP and α-tubulin in the brain. c–f ELISA of the production of IL-1β, IL-6, IL-4, and IL-10 in brain specimens. g, h The DNA-binding activity of NF-κB and NFAT in brain specimens. Data are mean ± SEM from 8 mice in each group. *P < 0.05 vs. APP/PS1 Tg mice

Astrocyte-derived cytokine production is increased in APP/PS1 Tg/sEH−∕− mice. a–d Immunostaining of hippocampus specimens from 6-month-old APP/PS1 Tg and APP/PS1 Tg/sEH−∕− mice with anti-IL-1β, IL-6, IL-4, or IL-10 and anti-GFAP antibodies, then FITC- or Texas Red-conjugated secondary antibody. Scale bar = 50 μm. IL-1β-, IL-6-, IL-4-, and IL-10-positive astrocytes (green color) and GFAP-positive cells (red color) were co-localized, respectively
Analysis of the gene ontology in APP/PS1 Tg mice and APP/PS1 Tg/sEH −∕− mice
In order to investigate the precise role of sEH in AD pathogenesis, we used LC-MS/MS and analyze gene ontology to address the possible pathway and mechanisms during AD progression. The quantitative proteomics analysis between the brain tissue samples of APP/PS1 Tg/sEH−∕− mice and APP/PS1 Tg mice was performed following the LC-MS/MS method. Bioinformatics analyses of the deregulated proteins revealed significant pathways and processes known to be associated with AD progression. Upregulated proteins in APP/PS1 Tg/sEH−∕− mice are known factors that are involved in translation, oxidation, and cytoskeleton organization (Table 1). Moreover, downregulated proteins in APP/PS1 Tg/sEH−∕− mice indicated their key role in small GTPase-mediated signal transduction (Table 2). Thus, these findings provide the novel evidence that sEH plays a crucial role in regulating the biological process in AD pathogenesis.
Discussion
In this study, we identified the detrimental effect of sEH in neuroinflammation and dysfunction of behavior outcomes in AD progression. The protein level of sEH was increased in 6-month-old APP/PS1 Tg mice. Based on the immunohistochemistry results, we found that the increased levels of sEH in brain glial cells, especially in astrocytes. Besides, our findings that the loss of sEH retarded the progression of AD pathology by regulating Aβ plaque deposition, astrogliosis, and cytokines production, leading to the alleviation in the AD-impaired nesting building ability, spatial learning, and memory formation in APP/PS1 Tg mice. Interestingly, we demonstrated that loss of sEH in APP/PS1 Tg mice decreased Aβ plaque formation, but increased astrogliosis in the brain. Importantly, the production of anti-inflammatory cytokines and the activity of NF-κB and NFAT were also increased in APP/PS1 Tg/sEH−∕− mice, suggesting the crucial role of sEH in the regulation of neuroinflammation in AD pathology.
The quantitative proteomics analysis of brain tissue lysates from APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice revealed that several significant pathways and cellular processes, including translational regulation, oxidative stress, and cytoskeleton reorganization, are possibly associated with the function of sEH as well as the pathogenesis of AD. Taken together, our results strongly suggest that sEH has an impact in regulating biological processes of the brain during the progression of AD pathology.
sEH is a bifunctional enzyme with EH and PT activities. One of the functions of sEH is to degrade cytochrome P450 products, such as arachidonic acid and epoxyeicosatrienoic acids (EETs) [1,2,3,4]. Recently, several lines of evidence suggest that the distribution of sEH plays an important role in the development of inflammatory diseases and brain diseases [1,2,3,4, 9,10,11,12,13]. Inhibition of the EH activity of sEH confers the protection from ischemia-induced brain injury [6, 7, 10,11,12,13]. Hung et al. reported that sEH in astrocytes is a critical regulator in inflammatory response brain injury in temporal lobe epilepsy [34], which implies that sEH is a crucial regulator in cerebrovascular and neuronal function upon the pathological insults. Additionally, Sarkar et al. addressed the detrimental effect of Aβ on degradation of EETs in the cerebellum, suggesting the possible involvement of sEH in AD pathology [16]. Moreover, Yao et al. showed that pharmacological inhibition of EH activity of sEH protects from tauopathy by regulating PI3K/AKT/GSK-3β pathway [35]. sEH has also been shown to be involved in age-related vascular cognitive decline [15]. Very recently, Ren et al. and Ma et al. reported that sEH plays a crucial role in the pathogenesis of Parkinson’s disease and neurodevelopmental disorders [36, 37]. These previous studies highlight the important role of sEH in the neurodegenerative diseases; therefore, the impact and detailed regulatory mechanism of sEH are worth elucidating. In this study, our findings further support this notion that deletion of sEH in APP/PS1 Tg mice improved AD-related behavior outcomes and the progression of pathology.
APP can be rapidly metabolized by posttranslational proteolysis via an amyloidogenic or non-amyloidogenic pathway in neurons [38]. In the amyloidogenic pathway, APP is cleaved by BACE1 and results in the release of the N-terminal APP fragments and βCTF [38]. βCTF is then cleaved by γ-secretase to generate an amyloid precursor protein intracellular domain and a toxic Aβ peptide (37–49 amino acids), which triggers inflammatory responses of astrocytes and causes and neuron dysfunction [19, 23, 38]. On the other hand, the astrocyte-derived high-density lipoprotein (HDL)-like particles comprising cholesterol and apoE are known to play a central role in regulating neuron function and Aβ metabolism [39,40,41]. These secreted HDL-like particles can bind with insoluble β-amyloid, following uptake by glial cells through ABCA1 and LRP-1 to avoid the Aβ deposition [41]. Our data showed that the levels of Aβ peptides, BACE1, βCTF, and apoE were decreased in APP/PS1 Tg/sEH−∕− mice. However, the protein levels of ABCA1 and LRP-1 were not changed. These results suggest that the decreased levels of neurotoxic Aβ in the brain of APP/PS1 Tg/sEH−∕− mice could be attributed to a decrease in Aβ biosynthesis but not an increase in Aβ clearance. Nevertheless, the detailed mechanism underlying sEH-mediated regulation in regulating Aβ generation and its clearance needs further investigation.
Astrocyte-mediated inflammation has been shown to play a key role in the progression of AD pathology [25, 26, 42, 43]. Previous studies suggest that Aβ-elicited production of pro-inflammatory cytokines IL-1β and IL-6 is involved in the initiation and progression of AD [27, 44]. In contrast, anti-inflammatory cytokines IL-4 and IL-10 can limit inflammation by decreasing the production of pro-inflammatory cytokines during the development of AD [27]. In line with these findings, our findings demonstrated that the levels of anti-inflammatory cytokines, including IL-4 and IL-10, were increased in APP/PS1 Tg/sEH−∕− mice. We also found that deletion of sEH in APP/PS1 Tg mice increased DNA-binding activity of NF-κB and NFAT, two key transcriptional factors for in the expression of IL-4 gene [28,29,30,31]. Thus, we suggest that improved AD-related behavior outcomes in APP/PS1 Tg/sEH−∕− mice were mediated by the increased anti-inflammatory responses. Nevertheless, further investigations are required for evaluating the exact mechanism underlying sEH-mediated inflammation and AD pathology.
Astrogliosis (referred to as reactive astrocytes) occurs prominently in the central nervous system in response to pathological insults [26, 45]. Although the biological significance of astrogliosis is not fully understood, reactive astrocytes are intimately associated with Aβ plaque and involved in the regulation of neural protection and repair, glial scarring, and neuroinflammation in the pathogenesis of AD [45]. In addition, the reactive astrocytes promote the degradation and clearance of Aβ in AD brain and consequently limit the formation of Aβ plaque [46]. Interestingly, we found greater astrogliosis in the brain of APP/PS1 Tg/sEH−∕− mice than that in APP/PS1 Tg mice. In parallel, the levels of the anti-inflammatory cytokines were increased in the brain of APP/PS1 Tg/sEH−∕− mice. We thus suggest that sEH plays a critical role in regulating astrogliosis, which protects the neuronal cells from Aβ stimulation and delays the progression of AD.
To examine the possible molecular mechanism of sEH in AD pathogenesis, we used LC-MS/MS and gene ontology analysis to evaluate the involvement of sEH in cellular pathways during AD progression. Our results showed that the pathways of oxidative stress, cytoskeleton reorganization, small GTPase-mediated signal transduction, and translational regulation are regulated in the brain of APP/PS1 Tg/sEH−∕− mice as compared with that of APP/PS1 Tg mice. Oxidative stress is an event that precedes the appearance of the pathologic hallmark of AD and forming a vicious cycle that promotes the initiation and progression of AD [47]. Besides tau protein, several histopathological cytoskeleton structures, including actin and the actin-binding protein, have recently been recognized in AD pathology [48]. Numerous studies showed that pathways governing actin cytoskeleton stability have deleterious effects on AD pathologies in human and mouse [48, 49]. The stability of actin cytoskeleton is important for regulating the normal structures and functions of dendritic spines [48,49,50]. For example, Rho GTPase, one of the small GTPase family proteins, acts as the key regulator in actin cytoskeleton remodeling and dendritic spine maintenance [50]. Overall, perturbed synaptic efficacy in dendritic spines finally causes the impairment of neuron functions in AD progression [47]. In addition, our data showed that genetic deletion of sEH deregulates the ribosome-related functions in APP/PS1 Tg mice, which are agreement with Ding et al. who found that a posttranslational regulation-dependent impairment in ribosome function was detected in cortical areas of AD subjects [50].
Furthermore, several lines of evidence support the close relationship between oxidative stress and AD pathology [47, 51]. According to our LC-MS/MS results, the pathways of oxidative stress, cytoskeleton reorganization, small GTPase-mediated signal transduction, and translational regulation were modulated in APP/PS1 Tg/sEH−∕− mice compared with APP/PS1 Tg mice. Our data also showed that APP/PS1 Tg/sEH−∕− mice exhibited improved AD-related behavior outcomes, Aβ production, and aggregation compared with APP/PS1 Tg mice. Collectively, our findings suggested that sEH is critical in regulating the biological response of oxidative stress, cytoskeleton-dependent synaptic efficacy, and translational regulation during AD progression. However, extensive investigations are required for clarifying the participation and the regulatory mechanisms of sEH in these signaling pathways.
Nevertheless, our study contains several limitations because we only examined the effect of genetic deletion of sEH on the progression of AD pathology. We did not examine the levels of epoxide fatty acids in the brain of APP/PS1 Tg mice and APP/PS1 Tg/sEH−∕− mice due to the technical limitation. In addition, from the point in search of drug targets, the roles of EH activity or PT activity of sEH in the progression of AD should be investigated by using pharmacological inhibitors targeting EH activity and PT activity of sEH in AD mice. To this end, further investigations describing the implications of sEH in AD are warranted.
Conclusions
We provide several lines of novel evidence for the role of sEH in the pathogenesis of AD, which advances our knowledge for better understanding of the biological significance of sEH in AD. Our findings suggest a broader biological role of sEH and the underlying molecular mechanisms in the initiation and progression of neuroinflammatory and neurodegenerative diseases. Overall, sEH might be a potential pharmacological target for treating AD and related neurological diseases.
Availability of data and materials
The datasets used in the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABCA1:
-
ATP-binding cassette transporter A1
- AD:
-
Alzheimer’s disease
- apoE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid-β
- BACE1:
-
β-site APP cleavage enzyme 1
- EH:
-
hydrolase
- GFAP:
-
Glial fibrillary acidic protein
- IBA-1:
-
Ionized calcium-binding adapter molecule 1
- IL:
-
Interleukin
- LC-MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- LRP-1:
-
LDLR-related protein 1
- NFAT:
-
Nuclear factor of activated T cells
- NF-κB:
-
Nuclear factor-κB
- PS1:
-
Presenilin 1
- PT:
-
Phosphatase
- sEH:
-
Soluble epoxide hydrolase
- vWF:
-
von Willebrand factor
- βCTF:
-
β-APP C-terminal fragment
References
Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2 J2 in human tissues. J Histochem Cytochem. 2004;52(4):447–54.
Gill SS, Hammock BD. Distribution and properties of a mammalian soluble epoxide hydrase. Biochem Pharmacol. 1980;29(3):389–95.
Johansson C, Stark A, Sandberg M, Ek B, Rask L, Meijer J. Tissue specific basal expression of soluble murine epoxide hydrolase and effects of clofibrate on the mRNA levels in extrahepatic tissues and liver. Arch Toxicol. 1995;70(1):61–3.
Sura P, Sura R, Enayetallah AE, Grant DF. Distribution and expression of soluble epoxide hydrolase in human brain. J Histochem Cytochem. 2008;56(6):551–9.
Wang SB, Pang XB, Gao M, Fang LH, Du GH. Pinocembrin protects rats against cerebral ischemic damage through soluble epoxide hydrolase and epoxyeicosatrienoic acids. Chin J Nat Med. 2013;11(3):207–13.
Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27(12):1931–40.
Wang J, Fujiyoshi T, Kosaka Y, Raybuck JD, Lattal KM, Ikeda M, et al. Inhibition of soluble epoxide hydrolase after cardiac arrest/ cardiopulmonary resuscitation induces a neuroprotective phenotype in activated microglia and improves neuronal survival. J Cereb Blood Flow Metab. 2013;33(10):1574–81.
Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, et al. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105(48):18901–6.
Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43(1):55–90.
Iliff JJ, Alkayed NJ. Soluble epoxide hydrolase inhibition: targeting multiple mechanisms of ischemic brain injury with a single agent. Future Neurol. 2009;4(2):179–99.
Poli G, Corda E, Martino PA, Dall’ara P, Bareggi SR, Bondiolotti G, et al. Therapeutic activity of inhibition of the soluble epoxide hydrolase in a mouse model of scrapie. Life Sci. 2013;92(23):1145–50.
Wang SB, Pang XB, Zhao Y, Wang YH, Zhang L, Yang XY, et al. Protection of salvianolic acid on rat brain from ischemic damage via soluble epoxide hydrolase inhibition. J Asian Nat Prod Res. 2012;14(11):1084–92.
Zhang W, Davis CM, Edin ML, Lee CR, Zeldin DC, Alkayed NJ. Role of endothelial soluble epoxide hydrolase in cerebrovascular function and ischemic injury. PLoS One. 2013;8(4):e61244.
Badin AS, Eraifej J, Greenfield S. High-resolution spatio-temporal bioactivity of a novel peptide revealed by optical imaging in rat orbitofrontal cortex in vitro: possible implications for neurodegenerative diseases. Neuropharmacology. 2013;73:10–8.
Nelson JW, Young JM, Borkar RN, Woltjer RL, Quinn JF, Silbert LC, et al. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014;113-115:30–7.
Sarkar P, Narayanan J, Harder DR. Differential effect of amyloid β on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience. 2011;194:241–9.
Kawas CH. Clinical practice. Early Alzheimer’s disease. N Engl J Med. 2003;349(11):1056–63.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44.
Lee KI, Lee HT, Lin HC, Tsay HJ, Tsai FC, Shyue SK, et al. Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J Neuroinflammation. 2016;13(1):92.
Jack CR Jr. Alzheimer disease: new concepts on its neurobiology and the clinical role imaging will play. Radiology. 2012;263(2):344–61.
Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–36.
Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120(4):545–55.
Zhang F, Jiang L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2015;11:243–56.
Ebrahimi T, Rust M, Kaiser SN, Slowik A, Beyer C, Koczulla AR, et al. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1-42-stimulated murine astrocytes. J Neuroinflammation. 2018;15(1):282.
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer’s disease. Neurotherapeutics. 2010;7(4):399–12.
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 2012;2012:756357.
Cogswell JP, Godlevski MM, Wisely GB, Clay WC, Leesnitzer LM, Ways JP, et al. NF-kappa B regulates IL-1 beta transcription through a consensus NF-kappa B binding site and a nonconsensus CRE-like site. J Immunol. 1994;153(2):712–23.
Fric J, Zelante T, Wong AY, Mertes A, Yu HB, Ricciardi-Castagnoli P. NFAT control of innate immunity. Blood. 2012;120(7):1380–9.
Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–34.
Rincón M, Flavell RA. Transcription mediated by NFAT is highly inducible in effector CD4+ T helper 2 (Th2) cells but not in Th1 cells. Mol Cell Biol. 1997;17(3):1522–34.
Filali M, Lalonde R. Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer’s disease. Brain Res. 2009;1292:93–9.
Jung WR, Kim HG, Kim KL. Ganglioside GQ1b improves spatial learning and memory of rats as measured by the Y-maze and the Morris water maze tests. Neurosci Lett. 2008;439(2):220–5.
Hung YW, Hung SW, Wu YC, Wong LK, Lai MT, Shih YH, et al. Soluble epoxide hydrolase activity regulates inflammatory responses and seizure generation in two mouse models of temporal lobe epilepsy. Brain Behav Immun. 2015;43:118–29.
Yao ES, Tang Y, Liu XH, Wang MH. TPPU protects tau from H2O2-induced hyperphosphorylation in HEK293/tau cells by regulating PI3K/AKT/GSK-3β pathway. J Huazhong Univ Sci Technolog Med Sci. 2016;36(6):785–90.
Ren Q, Ma M, Yang J, Nonaka R, Yamaguchi A, Ishikawa KI, et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc Natl Acad Sci U S A. 2018;115(25):E5815–23.
Ma M, Ren Q, Yang J, Zhang K, Xiong Z, Ishima T, et al. Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation. Proc Natl Acad Sci U S A. 2019;116(14):7083–8.
Lazarov O, Demars MP. All in the family: how the APPs regulate neurogenesis. Front Neurosci. 2012;6:81.
Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–96.
Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12(2):105–12.
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18.
Bouvier DS, Murai KK. Synergistic actions of microglia and astrocytes in the progression of Alzheimer’s disease. J Alzheimers Dis. 2015;45(4):1001–14.
Fakhoury M. Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis. 2015;15(2):63–9.
Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging. 2001;22(6):903–8.
Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94(4):1077–98.
Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27(1):187–98.
Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxidative Med Cell Longev. 2013;2013:316523.
Bamburg JR, Bloom GS. Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton. 2009;66(8):635–49.
Penzes P, Vanleeuwen JE. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res Rev. 2011;67(1-2):184–92.
Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25(40):9171–5.
Huang WJ, Zhang X, Chen WW. Role of oxidative stress in Alzheimer’s disease. Biomed Rep. 2016;4(5):519–22.
Acknowledgements
Authors thank Professor Huey-Jen Tsay for her help in the acquisition of research materials.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (104-2320-B-010-041-MY3, 105-2320-B-010-036, 105-2811-B-010-022, 106-2320-B-002-057-MY3, 106-2320-B-002-056, 106-2811-B-002-146, and 108-2320-B-002-032-MY3), Yen Tjing Ling Medical Foundation (CI-105-15), and Cheng Hsin General Hospital (CY10606 and CY10708).
Author information
Authors and Affiliations
Contributions
H-TL, K-IL, and C-HC performed the experiments and analyzed the data; T-SL designed the experiments and wrote the paper. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed in accordance with the approved guidelines by the Animal Care and Utilization Committee of the National Yang-Ming University.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, HT., Lee, KI., Chen, CH. et al. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J Neuroinflammation 16, 267 (2019). https://doi.org/10.1186/s12974-019-1635-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1635-9