
Abstract
β-Site amyloid precursor protein (APP) cleaving enzyme-1 (BACE1) is the major described β-secretase to generate Aβ peptides in Alzheimer’s disease (AD). However, all therapeutic attempts to block BACE1 activity and to improve AD symptoms have so far failed. A potential candidate for alternative Aβ peptides generation is the metalloproteinase meprin β, which cleaves APP predominantly at alanine in p2 and in this study we can detect an increased meprin β expression in AD brain. Here, we report the generation of the transgenic APP/lon mouse model of AD lacking the functional Mep1b gene (APP/lon × Mep1b−/−). We examined levels of canonical and truncated Aβ species using urea-SDS-PAGE, ELISA and immunohistochemistry in brains of APP/lon mouse × Mep1b−/−. Additionally, we investigated the cognitive abilities of these mice during the Morris water maze task. Aβ1-40 and 1–42 levels are reduced in APP/lon mice when meprin β is absent. Immunohistochemical staining of mouse brain sections revealed that N-terminally truncated Aβ2–x peptide deposition is decreased in APP/lon × Mep1b−/− mice. Importantly, loss of meprin β improved cognitive abilities and rescued learning behavior impairments in APP/lon mice. These observations indicate an important role of meprin β within the amyloidogenic pathway and Aβ production in vivo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The β-site APP cleaving enzyme 1 (BACE1) has been identified as a membrane bound aspartyl protease and it is considered the main β-secretase to generate amyloid β peptides (Aβ) in neurons. This became particularly evident when BACE1 knockout (BACE1−/−) mice showed a dramatic decrease in Aβ production once crossed to APP-overexpressing transgenic mouse lines such as Tg2576 [1] or to 5× FAD mice [2]. This decrease was accompanied by improvements in Alzheimer’s disease (AD)-related cognitive deficits, such as memory and learning impairment [3]. Based on this observation, several BACE1 inhibitors were developed, reaching clinical trials as potential new treatments for AD [4]. Strikingly, many of those therapeutic strategies have been suspended due to their failure to improve AD symptoms [5]. Furthermore, recent evidence indicated that complete blockage of BACE1 can lead to considerable side-effects, e.g., interfering with neural progenitor cell proliferation, resulting in a decreased number of neurons [6] and synaptic plasticity impairment [7]. These effects can be explained by observations from analysis of BACE1−/− mice, such as elevated pain sensitivity, reduced grip strength [8] and epileptic seizures [9]. BACE1 depletion leads to accumulation of unprocessed type 3 neuregulin 1 (NRG1) and to hypomyelination in the hippocampus and peripheral nerves [10]. Moreover, BACE1 inhibition increases neuronal cell surface levels of seizure protein 6 (Sez6), which controls synaptic connectivity and motor coordination in mice [11]. Due to its importance in the brain, these data suggest that BACE1 might not be the best therapeutic target since its chronic or acute pharmacological manipulation would give rise to many deleterious consequences.
Aβ peptides are generated through the proteolytic processing of the Amyloid Precursor Protein (APP) by the sequential action of β- and γ-secretase. Apart from the canonical ‘full-length’ Aβ (1–40, 1–42), multiple Aβ species bearing different N and C-termini have been described [12,13,14] The exact physiological role of the N-terminally truncated forms of Aβ (Aβx–40/ x–42) is still debated, but they are detected as major components in extracellular amyloid deposits in human brain. In fact, N-terminal truncations accounted for more than 70% of total Aβ species detected in human brain tissues from patients diagnosed with severe AD [15, 16], thus emphasizing their important role in the pathology of the disease (reviewed in [17]). Since BACE1 can only generate Aβ peptides starting at position 1 or 11 (Aβ1–x/11–x), Aβ species starting at other N-terminal positions, e.g., 4 (Aβ4–x) or 2 (Aβ2–x), cannot be assigned to BACE1 activity [16, 18]. BACE1 knockout neurons do not secrete Aβ1–40/42 and Aβ11–40/42 [1] while N-terminally truncated Aβ peptides can still be detected under these conditions [19]. The existence of different Aβ species in the absence of BACE1 indicates an independent pathway of APP processing by alternative β-secretases. Over the last few years other enzymes, such as ADAMTS4 [20], Aminopeptidase A [21] and cathepsin B [22] have been described to be involved in BACE1–independent N-terminal truncated Aβ generation.
Another potential candidate for alternative Aβ production is the metalloproteinase meprin β. Meprin β was recently identified as a potential risk gene associated with AD through exome-wide association analysis in a cohort of patients clinically diagnosed with AD [23]. Fittingly, meprin β mRNA levels were shown to be increased in brains of AD patients compared to age-matched controls [24, 25]. Meprin β is predominantly membrane-bound and co-localizes with APP in the late secretory pathway or at the cell membrane [26]. Of note, it was demonstrated that meprin β cleaves APP and it is capable of liberating not only Aβ1–x , but also N-truncated Aβ peptides starting mainly at position 2 and 3 [24, 27], indicating a possible role of meprin β within the amyloidogenic pathway in addition to BACE1. Moreover, we have shown that meprin β even has a higher affinity to wild-type APP compared to BACE1 [24] and that the various familiar APP mutations differently affect meprin β cleavage specificity. The protective A673T APP mutant, which reduces BACE1 cleavage, also induced a 70% decrease in the Aβ2-40 generated by meprin β in vitro [26]. Importantly, the change in amino acid composition around the β-site in the ‘Swedish’ APP mutation (K670N/M671L) almost fully abolished generation of N-terminally truncated Aβ2-40/42 variants [26]. However, this Swedish mutation (APPswe) is commonly introduced into mouse models of AD because it strongly enhances overall Aβ production by BACE1, thus facilitating studies focusing on the role of Aβ in AD. As a consequence, though these transgenic mouse models have substantially broadened our knowledge on the alterations underlying AD pathology, they might have also supported to oversight certain other potentially relevant pathophysiological mechanisms. In this respect, the strong affinity of BACE1 for APPswe might have concealed effects from other important enzymes such as meprin β that could be mechanistically involved in AD pathogenesis. Based on increasing evidence for alternative β-secretases, we aimed at analyzing meprin β-dependent Aβ generation in vivo using an animal model for AD that does not harbor the Swedish mutation on the APP transgene. We now demonstrate that soluble Aβ levels are decreased in the brain of the AD-mouse model APP/lon mice when meprin β is absent. More specifically, we found a decrease in the deposition of the N-terminally truncated Aβ2–x in the cortex of these animals. Furthermore, we show that loss of meprin β improved cognitive impairments such as memory and learning.
Materials and methods
Human brain tissue
All post mortem brain samples of frontal isocortex from neuropathologically confirmed AD patients (n = 21, 10 females, 11 males; mean age ± SD: 77.9±10.1 years) and non-demented control patients (n = 17, 8 females, 9 males; 75.8 ± 11.6 years) were obtained from University Medical Center HH-Eppendorf (UKE). AD cases were diagnosed as CERAD B-C, Braak stages III–VI. None of the controls suffered from dementia or any other neurodegenerative disease.
Histological and immunohistochemical analysis of formalin-fixed paraffin embedded (FFPE) human brains
Sections of FFPE frontal isocortex from healthy control and AD were stained with a Bielschowsky staining kit according to standard laboratory procedures. Immunostainings of Aβ (using monoclonal antibody 6E10; 1:100, #39320, Biolegend, CA, USA), meprin β (using rabbit polyclonal anti-meprin β antibody; 1:500) and phosphorylated Tau protein (using monoclonal antibody AT8 (MN1020); 1:5000; Thermo Scientific) were performed with a Ventana Benchmark XT system according to common protocols. Meprin β-positive neurons were counted in three representative microscopic fields of approximately 1.87 mm2 (taken along cortical layers III to VI) per patient. The mean value of meprin β-positive cells derived from these three pictures per patient (corresponding to n = 1) was divided by the field area analysed in each experimental group and used for statistical analysis.
Human brain lysates
Brain tissue of human frontal isocortex (3 controls and 3 AD) were lysed in RIPA buffer [50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% Octylphenoxy polyethoxyethanol (IGEPAL), 0.1% Sodium dodecyl sulfate (SDS), 0.5% deoxycholate, 10 mM NaF, protease and phosphatase inhibitor cocktails] by homogenization in a glass homogenizer. Homogenates were incubated on ice for 30 min and centrifuged at 120,000×g at 4 °C for 30 min (TLA120.2 rotor, Beckman Coulter ultracentrifuge).
Animals
hAPP[V717I] (APP/lon) [28], BACE1−/− [29], meprin β knockout (Mep1b−/−) [30] and wild-type (WT) mouse strains were maintained on a 12 h light/dark cycle with food and water ad libitum. APP/lon mice lacking Mep1b were generated by crossing Mep1b−/− mice into hAPP[V717I] mice, which we will refer to as APP/lon × Mep1b−/−.
Mouse brain lysates
Animals were sacrificed by cervical dislocation and brains were removed. For the meprin β characterization, brains were dissected into cortex and hippocampus. For each animal group, we pooled the specific brain regions from five mice and homogenized in homogenization buffer (320 mM Sucrose, 10 mM HEPES, 1 mM EDTA) on a 1:6 ratio (weight:volume). Homogenates were centrifuged at 18,000×g for 30 min, the soluble extract was removed and the pellet was re-supended in 1% NP-40 lysis buffer (150 mM NaCl, 50 mM Tris–OH pH 8.0). Lysates were centrifuged at 18,000×g for 30 min and supernatants were collected for further analysis. For Aβ analysis, snap frozen hemispheres were mechanically homogenized in 0.01M PBS on the same above mentioned ratio and subsequently ultracentrifuged at 120,000×g for 30 min (TLA120.2 rotor, Beckman Coulter). The PBS-extracted supernatant (soluble fraction) was collected and the pellet was re-suspended in 800 µL of 0.01M PBS containing 2% SDS. Homogenates were ultracentrifuged at 120,000×g for 30 min and the SDS-extracted supernatant (insoluble fraction) was kept for further analysis. PBS and SDS protein extracts were stored at – 80 °C until use. All buffers were supplemented with protease (cOmplete, Roche) and phosphatase inhibitor cocktails (PhosSTOP, Roche).
Immunoprecipitation of amyloid β
Soluble fractions from mouse protein extracts were used for immunoprecipitation (IP). Magnetic Dynabeads (M-280 Sheep Anti-Mouse IgG, 11201D, Invitrogen) containing sheep anti-mouse IgG attached to their surface were activated with the monoclonal antibody (mAb) IC16 according to the protocol of the manufacturer (direct IP method) and added to the samples. IC16 recognizes residues 1–16 of the human Aβ sequence and it was used to a final 1:100 concentration [31]. Briefly, total Aβ was immunoprecipitated from soluble brain lysates by mixing fivefold concentrated detergent buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 0.5% [v/v] Nonidet P-40, 0.05% [w/v] SDS and protease inhibitor cocktail [Roche Applied Science]) with each sample. After overnight incubation at 4 °C, samples were washed three times in 1× PBS, 0.1% (w/v) BSA, and once in 10 mM Tris–HCl, pH 7.5. After heating the samples to 95 °C in 15 μL sample buffer (0.36 M Bis–Tris, 0.16 M bicine, 1% [w/v] SDS, 15% [w/v] sucrose, and 0.0075% [w/v] bromophenol blue) the supernatants were loaded in “Urea-SDS-PAGE” section.
Urea-SDS-PAGE
Immunoprecipitated Aβ peptides from soluble fractions were separated in a 0.75 mm 10% polyacrylamide 8 M urea SDS-gels as described [26, 32]. For separation of Aβx–40 from Aβx–42, a final concentration of 0.3 M H2SO4 was used in resolving gels. Peptides were transferred to an Immobilon‑P PVDF membrane via semi-dry western blotting (Bio-Rad) at 47 mA for 30 min. Membranes were boiled for 3 min in PBS and then blocked in 5% powder milk in TBST (20 mM Tris, 137 mM NaCl, 0.1% [v/v] Tween-20) for 30 min afterwards. IC16 antibody was used for overnight immunostaining. After washings in TBS-T, membranes were incubated with appropriate secondary anti-mouse antibody (1:5000, Sigma). Immunoreactive bands were visualized using an ECL enhanced chemiluminescence system (Millipore, MA, USA).
SDS-PAGE
Pooled samples, soluble and insoluble fractions were prepared in SDS loading buffer (4X Roti-Load, Carl Roth, Germany) and boiled at 95 °C. Protein extracts were separated by SDS-PAGE, transferred onto nitrocellulose membranes (Amersham Hybond ECL), and then blocked in 5% (w/v) milk powder in TBST. Soluble fractions were used to detect total soluble N-APP (sAPP), using an anti-N-terminal APP antibody (22C11, MAB348, Millipore). Moreover, the following antibodies were used for detection in insoluble fractions: anti-CT15 1:10,000 (APP and CTFs) [33], anti-PSEN1 (7H8, C-terminal), anti-Nicastrin (N1660, Sigma), anti-ADAM10 (Ab19026, Millipore, Massachusetts, USA), anti-Notch1 (ab27526, Abcam), anti-Notch intracellular domain (NICD, New England Bioscence #2421S), and anti-mep1b (AF2895, R&D Systems, Minneapolis, USA).
Sandwich enzyme-linked immunosorbent assay (ELISA)
Human Aβ1–40 and Aβ1–42 concentrations were measured using commercially available ELISA kits (IBL International, Hamburg, Germany) according to the manufacturer’s protocol. Briefly, soluble or insoluble fractions were diluted 1:50 or 1:200, respectively, then added to wells coated either with anti-human Aβx–42 (44A3) or Aβx–40 (1A10) and incubated overnight at 4 °C. Wells were washed and incubated with HRP-conjugated anti-human Aβ1–x (82E1) for 1 h. After final washing steps, TMB solution was added as a substrate. The reaction was stopped with 1N H2SO4. Optical density (OD) values were measured at 450 nm on a microplate reader (Anthos 2010). For total Aβx–40/42 levels including N-terminally truncated peptides, samples were diluted 1:10 and the ELISA was performed according to the manufacturer’s instructions of LEGEND MAX™ β‐Amyloid x‐40 ELISA Kit (Biolegend, Cat. no.: 842301) and LEGEND MAX™ β‐Amyloid x‐42 ELISA Kit (Biolegend, Cat. no.: 842401).
Immunohistochemistry on FFPE mouse brain sections
15-month-old mice were sacrificed by cervical dislocation; right hemispheres were washed in 0.01M PBS and immerged in 4% PFA 0.01M PBS for fixation for 48 h. Tissue was dehydrated and embedded in paraffin. Immunohistochemistry was performed on 4 µm sagittal paraffin sections as previously described [34]. In brief, sections were deparaffinized in xylene and rehydrated in a descending series of ethanol. To block endogenous peroxidases, we treated the sections with 0.3% H2O2 in PBS and antigen retrieval was achieved by boiling sections in 0.01 M citrate buffer pH 6.0, followed by 3-min incubation in 88% formic acid. Non-specific binding sites were blocked by treatment with 4% skim milk and 10% fetal calf serum in PBS for 1 h prior to the addition of the primary antibodies. We used the following antibodies: rabbit anti-polyclonal antibody 77 (pAb77, 1:500) [35], raised against residues 2–14 of the Aβ peptide, detecting N-truncated Aβ peptides (Aβ2–x); mouse anti-mAb80C2 (1:1000, 218 231 Synaptic Systems) detecting Aβ1–x peptides [36], and mouse anti-GFAP (1:500, 173 004 Synaptic Systems). Primary antibodies were incubated overnight in a humid chamber at room temperature followed by incubation with corresponding biotinylated secondary antibodies (1:200, Dianova). Staining was visualized using the ABC method using a Vectastain kit (Vector Laboratories, Burlingame, USA) and 3,3ʹ-diaminobenzidine (DAB) as chromogen providing a reddish-brown color. Counterstaining was carried out with hematoxylin. In case of Aβ load analysis, the counterstaining was omitted. Serial images were captured with an Olympus Bx–51 microscope equipped with a Moticam pro 282 camera (Motic, Wetzlar, Germany) on three sections per animal, which were at least 50 µm afar from each other.
Morris water maze
For behavior analysis, 7-month-old female mice of were tested (n = 5–10 animals/group). Spatial learning and memory were assessed by the Morris water maze hidden platform task performed as previously described [37] with minor modifications. Briefly, water maze (120 cm) was filled with clear water (23 ºC). Prominent symbols around the maze provided abundant extra-maze cues. The platform stayed in the same quadrant from day 1 to 4 and the animals were released from four different positions at the pool perimeter. Mice performed four trials per day on 4 consecutive days with a maximum length of 90 s and an inter-trial interval of 90 s. If mice did not find the platform within the given time, they were guided to the platform. Mice were allowed to stay on the platform for 10 s. On the 5th water maze day, a probe trial (60 s) without platform was performed. Basal motor activity was evaluated by swim speed. Learning was assessed by measuring the escape latency to find the platform. Memory capabilities were characterized by the number each mouse crossed the former platform location at probe trial and the latency to reach the location. For vision abilities, a visible platform task was done after the learning assessment at day 6. It consisted of three trials in a row starting the mice opposite to the platform which was indicated by a table tennis ball 15 cm above the platform.
Monitoring of behavior
A computerized video recording system registered moving-path and duration in water maze tests automatically. The hardware consisted of an IBM-type AT computer combined with a video digitizer and a CCD video camera. The software used for data acquisition and analysis was EthoVision XT® release 8.0 (Noldus Information Technology, Utrecht, the Netherlands).
Quantification and statistical analysis
All graphs and statistical analyses were prepared using GraphPad Prism 6 software (La Jolla, CA). Western blots were quantified by densitometry using ImageJ v.1.52 (NIH, USA) and data were analyzed by t test. For human and mouse brain immunohistochemical analysis, all statistical comparisons were made using two-tailed unpaired Student’s t test. Using ImageJ, Aβ load and GFAP staining pictures were binarized to 8-bit images and a fixed intensity threshold was applied defining the DAB staining. Measurements were performed for a percentage area covered by DAB staining. Behavioral data were analyzed by two-way analysis of variance (ANOVA) with genotype factor one and time factor two by repeated measures. Other parameters without repeated measures were analyzed by one-way ANOVA followed by Tukey’s post hoc test. For the analysis, p values less than or equal to 0.05 were considered as statistically significant.
Results
Meprin β protein levels are increased in sporadic AD brain
We previously identified the metalloprotease meprin β as an APP-cleaving enzyme with β-secretase activity. Therefore, we wanted to investigate, whether meprin β brain protein levels differ between healthy control and AD patients. Herein, we performed immunohistochemical staining of human brain sections using anti-6E10, Bielschowsky and anti-tau to confirm the diagnosis of AD patients showing amyloid plaques and neurofibrillary tangles (Fig. 1A). Upon immunohistochemical assessment, a subpopulation of neurons in cortical layers III to VI showed prominent DAB signal of soma and neurites in all samples and was thus defined as meprin β positive. Generation and specificity of meprin β antibodies are described in the supplementary information (Fig. S1). Immunohistological analyses of sections of frontal cortex show an increase in the number of meprin β-positive cortical neurons in AD (n = 21) compared to control brains of non-demented patients (n = 17) (Fig. 1A, B) of nearly 50% (see Supplementary Fig. S2, for all cases). Additionally, we performed western blot analysis with brain tissue homogenates of sporadic AD patients (n = 3) versus age matched non-demented controls (n = 3) (Fig. 1C). We found significantly increased signals of meprin β in brains of AD patients compared to controls (Fig. 1C, D). Moreover, we were able to detect meprin β in the brains of wild-type and in the AD-mouse model APP/lon mice using western blot analysis. Interestingly, expression of meprin β seems to be higher in the hippocampus compared to cortex (Fig. 1E). In summary, the morphometric analysis revealed a significant up-regulation of meprin β specifically in AD compared to controls, thus confirming previous observations by Schlenzig and colleagues [27].

Meprin β expression in brains of AD patients. A Representative images of human brain sections from an AD patient and an age-matched, non-demented individual (control). Immunostainings were performed with anti-meprin β antibody, anti-Aβ1–16 (6E10), for the detection of Aβ aggregates, Bielschowsky silver stain, which impregnates both amyloid senile plaques and neurofibrillary tangles, and anti-phosphorylated Tau protein (AT8) antibody. Scale bars = 100 µm. Note Aβ accumulation, neuritic plaques and pTau stainings confirming AD diagnosis. B Densitometric analysis of meprin β-positive neurons in cortical sections revealed a significantly higher number of meprin β positive neurons in AD compared to control brains from non-AD individuals. Graph shows mean ± SEM (n = 21 AD, n = 17 Control; **p < 0.05, t-test). Representative comparison of all cases analyzed can be found in supplementary Fig. S2. C Human brain lysates were tested with a polyclonal anti-meprin β antibody (supplementary Fig. S1) and actin as loading control. Control and AD samples were on the same gel but are rearranged for better presentation. D Quantification of meprin β protein levels normalized to actin levels of AD versus control brains. Graph shows mean ± SEM (n = 3 AD, n = 3 Control; p < 0.05 t-test). E Mouse kidney and different brain region samples were tested using anti-meprin β antibodies in Western blot analysis. Signals of meprin β were detected in cortex and hippocampus of wild-type and APP/lon mice. Analysis of meprin β knockout kidney and brain samples confirmed the absence of meprin β in the APP/lon × Mep1b−/− mice (KO). Kidney and brain samples were on the same gel but they had highly different intensities and were arranged for better presentation (dotted line)
Aβ levels are reduced in APP/lon mice with a knockout for meprin β
Given that meprin β can generate N-terminally truncated Aβ peptides in vitro [24], we hypothesized that Aβ levels would also be altered by meprin β knockout in vivo. To test this assumption, Mep1b−/− mice were crossed to an APP-overexpressing mouse model. We have shown previously that meprin β generates predominantly N-terminal truncated Aβ2–x peptides, which are fully blocked when APP carries the Swedish familial mutation close to the β-secretase cleavage site [26]. Since most commonly used AD mouse models harbor the Swedish mutation in the APP sequence, these mice are not a useful model to investigate the generation of N-terminally truncated Aβ2–x peptides by meprin β. Hence, Mep1b−/− animals were crossed into mice expressing APP only with a ‘London’ mutation (V717I) close to the γ-secretase cleavage site. This mutation also increases the amount of Aβx–42 but leaves the N-terminal region of the Aβ sequence unaltered. This model reflects more appropriately the situation for sporadic AD in terms of the accessibility and involvement of enzymes close to the N-terminus of Aβ. Brains from 15-month-old APP/lon and APP/lon × Mep1b−/− mice were dissected and processed to isolate soluble (PBS) Aβ fractions. To analyze the meprin β-dependent generation of Aβ peptides we applied two different detection methods. First, we performed an immunoprecipitation assay followed by Urea-SDS-PAGE (Fig. 2A, B). For the purpose of detecting different Aβ species we used here the IC16 antibody. A significant decrease in levels of Aβ was detected in soluble fractions of APP/lon mice lacking Mep1b (n = 10), compared to APP/lon mice (n = 10) (Fig. 2A, B).

Aβ levels are reduced in APP/lon mice knockout for meprin β. A Soluble-extracted brain fractions from 15-month-old APP/lon and APP/lon × Mep1b−/− mice were analyzed by immunoblot for Aβ peptides species. Samples were on the same gel but are rearranged for better presentation. B Densitometric analysis of Aβ1–42 levels show a decrease in soluble fractions (****p < 0.0001, t-test, n = 10). C ELISA analysis showed decreased levels of 1–40 (*p < 0.05, t-test, n = 8) and 1–42 (*p < 0.05, t-test, n = 8) in soluble fractions of APP/lon × Mep1b−/− compared to APP/lon mice. D No differences were detected in insoluble brain fractions (n = 9–10). E A following ELISA approach revealed reduced levels of Aβ x–40 (p < 0.005, t-test, n = 7) and x–42 (p < 0.05, t-test, n = 4) in soluble brain fractions of APP/lon × Mep1b−/− (F) No obvious differences were identified in total Aβ species x–40 and x–42 in insoluble brain fractions (n = 6)
In a second approach, we used sandwich enzyme-linked immunosorbent assays (sELISA) for the specific detection of Aβ1–40 and Aβ1–42 (Fig. 2C, D), followed by another ELISA to detect total Aβx–40 and Aβx–42 levels (Fig. 2E). We detected a remarkable reduction in levels of soluble Aβ1–40 and Aβ1–42 in the brains of APP/lon × Mep1b−/− compared to APP/lon mice alone (n = 8 animals/group). However, the Aβ1–40 and Aβ1–42 concentrations observed in the insoluble fractions were not significantly altered amongst the different genotypes (Fig. 2D). We have previously shown that meprin β generated Aβ2–x peptides exhibit a higher aggregation rate than Aβ1–x [26]. Hence, we would expect a reduction of Aβ2–x peptides in insoluble fractions of APP/lon × Mep1b−/− mice. However, the antibody 82E1 used in this setup specifically detects the very N-terminus of Aβ peptides (1–x) generated by BACE1 cleavage [38]. Recognition of Aβ1–40 and Aβ1–42, but not Aβ2-40, was further confirmed by immunoblot of 82E1 against synthetic peptides (Fig. S3). Reprobing of the same membrane with the IC16 antibody revealed the Aβ2-40 detection.
The amounts of Aβx–40 and Aβx–42 were even more strongly decreased when meprin β was lacking compared to APP/lon mice alone (Fig. 2E). In general, we observed that the absolute levels of x–40 and x–42 detected were almost twofold higher than the corresponding Aβ1–40 and Aβ1–42 (Fig. 2C and E for comparison). Therefore, we conclude that the remaining portion of x–40 and x–42 might correspond to all species of N-terminally truncated Aβ peptides, which have been documented previously [16, 24] No significant differences could be detected in insoluble fractions for Aβx–40 or Aβx–42 (2F)
General APP processing and secretase activities are not altered in APP/lon mice lacking meprin β
Previously, we have shown that meprin β participates in the processing of APP in vitro, generating soluble N-APP fragments and Aβ peptides [24, 39]. To evaluate the impact of meprin β on general APP processing in vivo, we analyzed both soluble and insoluble brain fractions of 15-month-old APP/lon and APP/lon × Mep1b−/− mice. In soluble fractions, analysis by SDS-PAGE and subsequent western blotting revealed no obvious differences in total sAPP (Fig. 3A). Similar, in insoluble fractions, full length unprocessed APP or CTFs levels were unaltered between the two groups (Fig 3A). This result is in part consistent with BACE1 knockout data, where also no measurable effect on full-length APP expression was detected [40, 41].

The meprin β knockout has no evident effect on the overall APP processing and its secretases. A Soluble brain fractions of 15-month-old APP/lon and APP/lon × Mep1b−/− mice (n = 10/group) were analyzed by immunoblot for sAPP forms using the monoclonal antibody 22C11. Further immunoblot analysis of insoluble brain fractions using an antibody against the C-terminus of full-length APP and CTFs in 15-month-old APP/lon and APP/lon × Mep1b−/− mice (n = 10/group). B Immunoblot analysis shows protein levels for Notch1 and (n = 8/group) its fragments (n = 4/group). C The protein levels for ADAM10 in insoluble brain fractions show no differences between the groups (n = 8/group). D γ-Secretase subunits PSEN1 and Nicastrin in insoluble fractions of APP/lon and APP/lon × Mep1b−/− mice (n = 5/group). E Total brain lysates from double knockout mice for BACE1 and meprin β, as well as Mep1b−/− and WT mice were analyzed by immunoblot for Sez6. A–D Densitometric analyses normalized to β-tubulin revealed no significant differences. E Densitometric analysis normalized to GAPDH detected an increase in levels of endogenous Sez6 in the double knockout mice lacking BACE1, but no significant differences were found in meprin β knockout compared to WT brain homogenates
As γ-secretase is considered a key target for Aβ generation and previous work showed meprin β influence on α-secretase [42], we wanted to investigate whether the knockout of meprin β may lead to decreased secretases activities. Notch1 is a type I transmembrane protein involved in numerous pathways such as cell fate and morphogenesis in both embryonic and adult brain [43]. Upon ligand binding, Notch1 is processed initially by ADAM10, by cleavage at its extracellular juxtamembrane domain. This is followed by intramembrane cleavage through γ-secretase, which generates the cytoplasmic domain of Notch (NICD). We analysed the production of these two fragments in the insoluble fraction by western blot. Loss of meprin β did not change the processing or levels of Notch1 (Fig. 3B), indicating that the metalloproteinase has no significant effect on α- or γ-secretase activity in this model. ADAM10 protein levels were evaluated in insoluble fractions and results confirmed there were no significant difference between the two groups (Fig. 3C).
γ-Secretase is a multiprotein complex consisting of Presenilin (PSEN), Nicastrin, Aph-1, and Pen-2 and all four proteins are necessary for full intramembrane proteolytic activity [44]. Exemplary we evaluated γ-secretase expression through PSEN1 and Nicastrin levels, which showed no significant difference between APP/lon (n = 5) and APP/lon × Mep1b−/− (n = 5) mice (Fig 3D). In sum, the meprin β knockout has no significant effect on the expression of APP cleaving secretases.
Having ruled out alterations in α- and γ-secretase activities with regard to APP processing, we wanted to investigate whether the knockout of meprin β may lead to decreased BACE1 activity. As the majority of Aβ generation has been attributed to BACE1, lower Aβ levels observed in APP/lon × Mep1b−/− mice could be due to decreased BACE1 activity. To investigate this, we analyzed the well-described BACE1 substrate Sez6, which is involved in maintenance of dendritic arborisation and enhancing synaptic connectivity [45]. The full-length Sez6 is cleaved by BACE1 and it generates a secreted soluble ectodomain and a C-terminal transmembrane fragment, which is further cleaved by γ-secretase [11]. To evaluate whether meprin β affects BACE1 activity, we analyzed the meprin β knockout effect on processing of Sez6. Therefore, we used total brain lysates from Mep1b−/− mice, double knockout mice for BACE1 and meprin β compared to wild-type (WT) mice. As expected, increased levels of Sez6 were detected in the double knockout mice lacking BACE1. In contrast, levels of endogenous Sez6 from meprin β knockout brains remained the same as in WT animals (Fig. 3E), suggesting that BACE1 activity is not altered in the absence of meprin β.
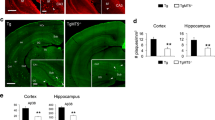
N-terminally truncated Aβ2–x peptide deposition is decreased in APP/lon mice lacking meprin β
Meprin β is capable of generating Aβ2–x peptides in vitro [24, 26]. Subsequently we wanted to determine whether the Mep1b knockout had any effect on Aβ2–x deposition in vivo. Thus, we evaluated plaque pathology in brains of APP/lon mice using antibodies specifically detecting Aβ peptides starting at position p1 or p2. Amyloid plaques arise in APP/lon mice at the age of 10–12 months [28]. To analyze plaque deposition, sagittal brain sections of 15-month-old APP/lon and APP/lon × Mep1b mice were stained using pAb77, specifically detecting Aβ2–x and mAb 80C2, which binds to Aβ1–x. The selectivity of the Aβ2–x (Aβ2–x pAb77) and Aβ1–x (80C2) antibodies have been confirmed pr−/−eviously [35, 36]. In general, both antibodies detected mainly diffuse plaques of different sizes in cortex and hippocampus. While we observed amyloid plaques spread throughout all cortical layers, plaques in the hippocampal formation seemed to be restricted to CA1 and subiculum. Similarly to AD patients, the subiculum is the first brain region to present amyloid deposits in APP/lon mice [46]. A significant decrease in Aβ2–x plaque load of about 40% was detected in the cortex of APP/lon mice lacking Mep1b compared to APP/lon mice alone (Fig. 4A, 4C). However, no significant difference between the two groups was evident for Aβ1–x staining (Fig. 4A, B). This result correlates well with the observed effect on insoluble Aβ1–40 and Aβ1–42 levels documented by ELISA (Fig. 2). Taken together, these results indicate that meprin β is involved in the generation of soluble Aβ peptides in vivo and that it has an impact on plaque deposition through the generation of Aβ2–x peptides.

N-terminally truncated Aβ2–x peptide deposition is decreased in APP/lon mice lacking meprin β. A Representative image of cortices of sagittal brain sections from 15-month-old APP/lon and APP/lon × Mep1b−/− which were embedded in paraffin and stained for Aβ1–x (mAb 80C2) and Aβ2–x (pAb77) plaque load. Detection was made possible through hydrogen peroxidase and 3,3ʹ-diaminobenzidine (DAB) reaction. B Quantitative analysis showing the percentage of stained area for Aβ1–x peptides (n = 5). C Quantitative analysis showing percentage of stained area for N-terminally truncated Aβ2–x peptides (*p < 0.05, t-test, n = 5–6). Scale bar = 100 µm
Reactive astrocytes are decreased in the cortex of APP/lon × Mep1b −/− mice
Severity of AD pathology strongly correlates with the density of activated astrocytes [47]. Numerous AD mouse models have demonstrated a correlation of astrocytic gliosis with plaque pathology [48, 49]. Such gliosis can be identified by staining of glial fibrillary acidic protein (GFAP) in reactive astrocytes surrounding amyloid plaques [50]. Since we have demonstrated decreased levels of soluble Aβ peptides and reduced Aβ2–x-derived extracellular plaques, we sought to determine whether gliosis in the brains of APP/lon × Mep1b−/− was also reduced. We analyzed cortical sections immunohistochemically stained for GFAP in 15-month-old APP/lon and APP/lon × Mep1b−/− mice. Images revealed extensive gliosis throughout the cortex of APP/lon mice (Fig. 5A) thus confirming previous work [48]. However, APP/lon × Mep1b−/− mice showed a significant decrease in the area of GFAP-positive immunostaining (Fig. 5B). This result further strengthens our observation that decreased meprin β expression reduces Aβ-peptide generation and subsequently improves brain pathology.

GFAP immunoreactivity is increased in the cortex of APPlon × Mep1b−/− mice. A Representative image of sagittal brain sections from 15-month-old APP/lon and APP/lon × Mep1b−/− embedded in paraffin and stained for GFAP and hematoxylin. B Quantitative analysis showing the percentage area covered by DAB staining (*p < 0.05, t-test, n = 5/group). Scale bar = 100 µm
Meprin β knockout rescues learning behavior impairments in APP/lon mice
As we have demonstrated that absence of meprin β has no detectable influence on other APP processing enzymes, but significantly alters amounts of soluble Aβ peptides, which are strongly associated with a decline in cognitive ability, we tested whether meprin β knockout had any influence on learning behavior of APP/lon mice. As control groups, we included age-matched WT and Mep1b−/− mice. In APP/lon mice, behavior alterations start at the age of 6 months [28]. To assess hippocampus-dependent learning we chose the Morris water maze hidden platform task [37]. All genotypes showed the same basal motor activity indicated by unaltered swimming speed (F(3;29) = 2.489, p = 0.0801) (Fig. 6A) and had no specific deficits of finding a visible platform (F(3;29) = 0.3073 p = 0.8199) in the pool (Fig. 6B), allowing for evaluation of cognitive performance.

Meprin β knockout rescues cognitive impairments in APP/lon mice. Spatial learning and memory tests were carried out on 7-month-old APP/lon, APP/lon × Mep1b−/−, Mep1b−/−, and wild-type mice using the Morris water maze setting. A Swimming speed analysis showed no motor impairment in any of the groups. B Latency to find visible platform was evaluated after probe trial and no specific deficits were detected. C The average escape latency in each trial was measured for 4 days and 24 h later all experimental groups were subjected to a probe trial (PT) in which the platform was removed. On day 4, APP/lon showed significant learning deficit compared to WT mice (*p < 0.05) and to Mep1b−/− (*p < 0.05), but not to APP/lon × Mep1b−/− mice. Data shown is the mean ± SEM of four different trials performed on day 4. Statistical analysis performed with one-way ANOVA followed by Tukey’s post hoc test. D Memory was evaluated by measuring the numbers of crossings over the former platform location. APP/lon mice showed significant differences when compared to Mep1b−/− or APP/lon × Mep1b−/− (*p < 0.05, **p = 0.0016). E Latency to reach platform location in all four groups. On PT day, cognitive deficits are prominent in APP/lon when compared to WT (*p < 0.05), Mep1b-/- (****p < 0.0001) and APP/lon × Mep1b−/− mice (***p < 0.0001). Data are presented as mean ± SEM (n = 5–10). Graph shows latency to find platform location in one probe trial
As expected, APP/lon mice showed a significant learning deficit compared to wild-type (WT) mice (Fig. 6C) (p < 0.05 on day 4). Escape latency to find the visible platform from day 4 shows also a significant difference when comparing APP/lon to Mep1b−/− mice (day 4 p < 0.05) WT mice showed no different escape latency when compared to either APP/lon × Mep1b−/− or Mep1b−/−. Memory abilities were characterized on the probe trial (PT) by the number each mouse crossed the former platform location and the latency to reach the location. In line with the observations on learning, APP/lon mice showed the fewest crossings of the former platform location (Fig. 6D) [F(3;29) = 6.587, p = 0.0016], showing significant differences to all other genotypes (p < 0.05). Most interestingly, on PT day, APP/lon mice showed the highest average latency [F(3;29) = 15.32, p < 0.0001] to find the platform location and with significant difference to all other three genotypes (Fig. 6E). This effect is also seen when comparing APP/lon to APP/lon × Mep1b−/−(p < 0.0001). Taken together, these findings indicate that the absence of meprin β in APP/lon mice ameliorates the cognitive impairments since there were no differences observed in learning behavior when compared to WT animals. APP/lon mice deficient for meprin β were able to learn the location of the hidden platform, and to find the platform location when it was removed. Hence, the present data suggest that meprin β contributes to the development of cognitive impairments in APP/lon mice.
Discussion
Generation of Aβ peptides is a hallmark of AD pathology. BACE1 was identified as the major β-secretase, however, so far, all therapeutic attempts to block BACE1 activity to hold AD progression or cure the symptoms in AD patients have not been successful. There are many possible explanations, one of which is the existence of alternative proteases acting complementary to BACE1. Here we demonstrate that loss of the alternative β-secretase meprin β is capable of rescuing the learning deficits in the APP/lon AD mouse model. Importantly, most AD mouse models carry the APPswe mutation, which is a variant that is highly prone to BACE1 activity but strongly impairs meprin β cleavage [26]. We hypothesize that the wide use of APPswe models in the Alzheimer field opened just a very small window to investigate alternative amyloidogenic APP processing. Of note, the potential relevance for meprin β in AD was recently supported by a genetic study where a Mep1b variant was identified as one of the AD-associated genes in a British dementia cohort [23].
Previously, we and others have identified APP as a substrate for meprin β in vitro [27, 39]. Moreover, mass spectrometry analysis of peptides incubated with meprin β revealed cleavage sites not only at the aspartate p1, but also in alanine p2 and the peptide bond in p3, resulting in an N-terminal glutamate residue [24]. Together, these observations identify meprin β as a protease candidate for the generation of not only the canonical full-length Aβ peptides, but also N-terminally truncated Aβ2–x and Aβ3–x species commonly found in AD brain. Of particular interest, Aβ3–x generated by meprin β serve as a substrate for glutaminyl cyclase (QC) which converts the N-terminus into pyroglutamic acid to produce pyroglutamate Aβ peptides (pGlu-3Aβ) [27]. This particular peptide is found in amyloid plaques in abundance, comprising 15–45% of total Aβ in AD brains [51]. Preclinical immunotherapy aiming at the reduction of pGlu-3Aβ have shown remarkable results in cognitive improvement of AD-like mouse models [52, 53]. Recently Eli Lilly & Company announced that the Phase 2 TRAILBLAZER-ALZ study using “Donanemab”, a monoclonal antibody that recognizes pyroglutamated Aβ, slowed cognitive decline in people with early AD. To provide further evidence for the relevance of meprin β in vivo, we generated mouse lines to analyze whether meprin β plays an important role in the pathogenesis of AD. The present work describes the in vivo effect of meprin β gene knockout on APP processing, Aβ generation and behavioral phenotype in APP/lon mice.
The APP/lon animal model is characterized by an age-dependent, progressive increase of soluble Aβ levels [28]. Interestingly, homozygous knockout of meprin β led to a decrease in soluble Aβ in this mouse model. In APP/lon mice, the absolute levels of Aβx–40 and x–42 detected by ELISA were around twofold higher than the corresponding specific levels of Aβ1–40 and Aβ1–42. Hence, the remaining amount of x–40 and x–42 likely corresponds to all species of N-terminally truncated Aβ peptides. [38]. N-terminal truncations make up the majority of Aβ species in AD [12, 54] but, of note, are much less abundant in the transgenic mouse models that overexpress APPswe, which might explain the differences in amyloid deposition mechanisms between human and rodents [55]. There are several known mutations within the APP sequence, but the majority of AD research is based on mouse models harboring the APPswe (K670N/M671L) mutation. However, cells overexpressing meprin β and APPswe showed a lack of Aβ2–x variants [26]. This result indicates that meprin β is responsible for generating N-terminally truncated Aβ2–x almost exclusively from the APPwt sequences. Essentially, wild-type APP is considered as a relatively poor substrate for BACE1, which cleaves preferentially APPswe [56]. In terms of the relative abundance of specific variants of Aβ peptides, such as Aβ1–42, and N-terminally truncated Aβ2/3–42, clear differences between AD brain and APP23 mice were observed [54, 57], with Aβ1–40 being the most abundant species detected in brain lysates of the latter model.
Previously, we and others detected increased levels of meprin β mRNA in brains of demented versus non-demented control patients [24, 27]. In this study, we could further strengthen this observation by providing evidence for increased meprin β protein expression levels in AD brains. We found significantly elevated amounts of strongly meprin β-positive cells in brains of sporadic AD patients, compared to brains of age-matched non-demented control patients. While cause and relevance of this clearly deserves further studies, increased meprin β-positive cell count (in combination with previously published elevated mRNA levels found in AD) supports our hypothesis of a potential role of meprin β in AD pathology. We already showed a different APP cleavage pattern in brains of meprin β knockout compared to WT mice, suggesting that meprin β is involved in N-terminal processing of endogenous APP in vivo [26]. Moreover, we could clearly show that meprin β is expressed in the mouse brain, particularly in the hippocampus. Although meprin β expression is lower in the brain compared to kidneys, we were able to show that the neural processing of APP is directly affected by its knockout.
It has been shown that there is a difference in the Aβ pattern composition between brain samples from non-demented and demented individuals. Regarding the species composition, soluble Aβ aggregates that accumulate in AD differ from those generated during normal ageing. The higher neurotoxicity (more prone to aggregate) correlates with the predominance of N-terminally truncated species over the full-length forms [58]. Overall, brain tissue of AD patients had more N-terminally truncated and pyroglutamate-modified Aβ accumulation than healthy elderly patients, suggesting that they might play a critical role in plaque formation [59]. In AD temporal lobe samples, Aβ1–42 was the predominant form and the second most abundant species comigrated with synthetic Aβ2-42 [54]. The concentration of soluble Aβ peptides (probably in the form of neurotoxic oligomeric assemblies) is strongly associated with cognitive decline and pathological synaptic changes in neurons [60]. Because the loss of charged amino acids in the Aβ truncated forms contribute to their enhanced insolubility and resistance to enzymatic degradation, they are considered highly neurotoxic and induce faster aggregation. Thus, N-terminally truncated species might be associated with more severe neurodegeneration [58]. For example, N-terminally truncated Aβ4–x peptides seem to be restricted mainly to amyloid plaque cores and cerebral amyloid angiopathy in AD patients and in AD mouse models [61]. Transgenic mice expressing Aβ4-42 (Tg4-42 transgenic line) developed a massive CA1 pyramidal neuron loss in the hippocampus [62]. Aβ4-42 was also identified as the major N-truncated species in postmortem brain samples from aged controls, patients with vascular dementia, and AD patients [63, 64].
AD is characterized by accumulation of different forms of Aβ peptides and their deposition into extracellular amyloid plaques. Plaque deposition in APP/lon mice arise at the age of 10–12 months and it shows patterns that are reminiscent of AD brain. Using an antibody specific to Aβ2–x peptides we were able to detect plaque load by immunohistochemistry in aged APP/lon mice in the presence or absence of meprin β. Peptides starting at p2 in the form of small round and diffuse plaques were detected in cortex and hippocampus. More interestingly, a 40% decrease in Aβ2–x plaque load was detected in the cortex of APP/lon mice lacking meprin β. However, meprin β knockout decreased but not fully abolished Aβ2–x production as we expected. These results further support the idea that in addition to BACE1, meprin β influences the generation of Aβ peptides to some extent. As a diagnostic tool, assessment of Aβ peptides in CSF has been used to support the diagnosis of AD and to identify patients who might be at risk of developing AD. Along with other truncated species, Aβ2–42, for example, is decreased in CSF [65] and enriched in AD brains [16]. Moreover, deficits in learning and memory, as assessed by Morris water maze tasks, were detected at 7 months of age in APP/lon in accordance with a previous report [66]. We now demonstrate that APP/lon mice lacking meprin β showed cognitive abilities similar to wild-type mice. Further accumulation of brain plaques is usually associated with increased brain inflammation that eventually lead to cognitive impairment [67]. Amyloid β increased generation and accumulation lead to an extensive proliferation of reactive astrocytes that can secrete an excess of proinflammatory cytokines [68]. Previous work analyzing astroglial activation demonstrated that GFAP-positive cells were mostly localized surrounding amyloid plaques in aged APP/lon [48]. We have shown here decreased levels of GFAP signal in the absence of meprin β in vivo. This result supports the in vivo effect of meprin β gene knockout on amyloid pathology. Since APP/lon × Mep1b−/− mice present decreased levels of soluble Aβx–40 and Aβx–42 and particularly reduced Aβ2–x plaque load, the astroglial response is lower compared to APP/lon alone.
Additionally, supporting the relevance in murine AD models, the group of Dennis Selkoe [69] detected meprin β in microsomal fractions from mouse brain lysates that are responsible for the majority of Aβ production. Although a portion of BACE1 co-fractionated with ADAM10 in this higher molecular weight fractions, the majority of BACE1 protein was found in low molecular weight pools that did not contribute to Aβ generation. Instead, meprin β showed a much stronger co-fractionation in the high molecular weight fractions, which may indicate its importance for Aβ generation.
Of note, truncated Aβ fragments favor seeding and aggregation of other Aβ species [70]. Truncated fragments such as Aβ2–x peptides generated by meprin β promote and seed aggregation of less prone Aβ species. Although we could not detect obvious differences in Aβ1–x plaque deposition, we observed significant reduced Aβ2–x plaque load and decreased levels of soluble Aβ1–x species. While the presence of extracellular plaque deposition is a hallmark of AD pathology, soluble amyloid β species present much more evidence as a source of primary synaptoxicity and memory loss in transgenic mice [71]. Moreover, plaque deposition is not clearly associated with disease severity. In our conditions, the fact that we could not detect alterations in Aβ1–x deposition might be related to the age of animals. While most AD mice models bearing APPswe mutations begin plaque deposition around 2-month-old, APP/lon mice show first signs of amyloid deposition at age of 10 months old. By the age of 15 month-old plaques are more abundant but there are no signs of neurodegeneration yet. It is possible that the full impact of lack of functional meprin β and reduced 2–x production will show more pronounced effects at a later age.
We cannot exclude, though, that some N-truncated amyloid β peptides might arise secondarily by exopeptidase activity towards Aβ1–40/42 originally produced by BACE1. For example, it was recently shown that aminopeptidase A has the ability to truncate Aβ1–40 at its N-terminus, thereby yielding Aβ2-40 [72]. Moreover, dipeptidyl peptidase 4 releases the N-terminus of Aβ, converting Aβ1–40 into Aβ3–40 [73]. In both studies, genetic reduction and pharmacological blockade of these enzymes reduced full-length Aβ production. However, many findings point to an important role of meprin β within initial Aβ production. Here we report the generation of the mouse line APP/lon × Mep1b−/− which presented improved cognitive abilities and a decrease of total Aβ levels. We believe that our findings offer several new pathways that may improve our understanding of AD pathogenesis and, in particular, roles of previously underestimated molecules and mechanisms therein.
Availability of data and materials
All primary data and materials in the manuscript are available upon reasonable request.
Code availability
Not applicable.
References
Luo Y, Bolon B, Kahn S et al (2001) Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci 4:231–232. https://doi.org/10.1038/85059
Ohno M, Cole SL, Yasvoina M et al (2007) BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis 26:134–145. https://doi.org/10.1016/j.nbd.2006.12.008
Ohno M, Sametsky EA, Younkin LH et al (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41:27–33. https://doi.org/10.1016/S0896-6273(03)00810-9
Hsiao CC, Rombouts F, Gijsen HJM (2019) New evolutions in the BACE1 inhibitor field from 2014 to 2018. Bioorg Med Chem Lett 29:761–777
Moussa-Pacha NM, Abdin SM, Omar HA et al (2020) BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med Res Rev 40:339–384
Chatila ZK, Kim E, Berlé C et al (2018) BACE1 regulates proliferation and neuronal differentiation of newborn cells in the adult hippocampus in mice. eNeuro. https://doi.org/10.1523/ENEURO.0067-18.2018
Lombardo S, Chiacchiaretta M, Tarr A et al (2019) BACE1 partial deletion induces synaptic plasticity deficit in adult mice. Sci Rep 9:1–14. https://doi.org/10.1038/s41598-019-56329-7
Hu X, Hicks CW, He W et al (2006) Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 9:1520–1525. https://doi.org/10.1038/nn1797
Barão S, Moechars D, Lichtenthaler SF, De Strooper B (2016) BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci 39:158–169
Willem M, Garratt AN, Novak B et al (2006) Control of peripheral nerve myelination by the β-secretase BACE1. Science 314:664–666. https://doi.org/10.1126/science.1132341
Pigoni M, Wanngren J, Kuhn PH et al (2016) Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol Neurodegener 11:1–18. https://doi.org/10.1186/s13024-016-0134-z
Sergeant N, Bombois S, Ghestem A et al (2003) Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J Neurochem 85:1581–1591. https://doi.org/10.1046/j.1471-4159.2003.01818.x
Dunys J, Valverde A, Checler F (2018) Are N- and C-terminally truncated Aβ species key pathological triggers in Alzheimer’s disease? J Biol Chem 293:15419–15428. https://doi.org/10.1074/JBC.R118.003999
Bayer TA (2021) N-truncated Aβ starting at position four-biochemical features, preclinical models, and potential as drug target in Alzheimer’s disease. Front Aging Neurosci. https://doi.org/10.3389/FNAGI.2021.710579
Wildburger NC, Esparza TJ, Leduc RD et al (2017) Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Sci Rep. https://doi.org/10.1038/s41598-017-10422-x
Wiltfang J, Esselmann H, Cupers P et al (2001) Elevation of β-amyloid peptide 2–42 in sporadic and Familial Alzheimer’s disease and its generation in PS1 knockout cells. J Biol Chem 276:42645–42657. https://doi.org/10.1074/jbc.M102790200
Wirths O, Zampar S (2019) Emerging roles of N- and C-terminally truncated Aβ species in Alzheimer’s disease. Expert Opin Ther Targets 23:991–1004
Vassar R, Kovacs DM, Yan R, Wong PC (2009) The β-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 29(41):12787–12794
Nishitomi K, Sakaguchi G, Horikoshi Y et al (2006) BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice2. J Neurochem 99:1555–1563. https://doi.org/10.1111/j.1471-4159.2006.04178.x
Walter S, Jumpertz T, Hüttenrauch M et al (2019) The metalloprotease ADAMTS4 generates N-truncated Aβ4–x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol 137:239–257. https://doi.org/10.1007/s00401-018-1929-5
Sevalle J, Amoyel A, Robert P et al (2009) Aminopeptidase a contributes to the N-terminal truncation of amyloid β-peptide. J Neurochem 109:248–256. https://doi.org/10.1111/j.1471-4159.2009.05950.x
Hook V, Toneff T, Bogyo M et al (2005) Inhibition of cathepsin B reduces β-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: evidence for cathepsin B as a candidate β-secretase of Alzheimer’s disease. Biol Chem 386:931–940. https://doi.org/10.1515/BC.2005.108
Patel T, Brookes KJ, Turton J et al (2018) Whole-exome sequencing of the BDR cohort: evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol Appl Neurobiol 44:506–521. https://doi.org/10.1111/nan.12452
Bien J, Jefferson T, Čaušević M et al (2012) The metalloprotease meprin β generates amino terminal-truncated amyloid β peptide species. J Biol Chem 287:33304–33313. https://doi.org/10.1074/jbc.M112.395608
Medoro A, Bartollino S, Mignogna D et al (2019) Proteases upregulation in sporadic Alzheimer’s disease brain. J Alzheimer’s Dis 68:931–938. https://doi.org/10.3233/JAD-181284
Schönherr C, Bien J, Isbert S et al (2016) Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site. Mol Neurodegener 11(1):1–6
Schlenzig D, Cynis H, Hartlage-Rübsamen M et al (2018) Dipeptidyl-peptidase activity of meprin β links N-truncation of Aβ with glutaminyl cyclase-catalyzed pGlu-Aβ formation. J Alzheimer’s Dis 66:359–375. https://doi.org/10.3233/JAD-171183
Tanghe A, Termont A, Merchiers P et al (2010) Pathological hallmarks, clinical parallels, and value for drug testing in Alzheimer’s disease of the APP [V717I] London transgenic mouse model. Int J Alzheimer’s Dis. https://doi.org/10.4061/2010/417314
Cai H, Wang Y, McCarthy D et al (2001) BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci 4:233–234. https://doi.org/10.1038/85064
Norman LP, Jiang W, Han X et al (2003) Targeted disruption of the meprin β gene in mice leads to underrepresentation of knockout mice and changes in renal gene expression profiles. Mol Cell Biol 23:1221–1230. https://doi.org/10.1128/mcb.23.4.1221-1230.2003
Jäger S, Leuchtenberger S, Martin A et al (2009) α-Secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Aβ generation. J Neurochem 111:1369–1382. https://doi.org/10.1111/j.1471-4159.2009.06420.x
Klafki HW, Wiltfang J, Staufenbiel M (1996) Electrophoretic separation of betaA4 peptides (1–40) and (1–42). Anal Biochem 237:24–29. https://doi.org/10.1006/abio.1996.0195
Soriano S, Lu DC, Chandra S et al (2001) The amyloidogenic pathway of amyloid precursor protein (APP) is independent of its cleavage by caspases. J Biol Chem 276:29045–29050. https://doi.org/10.1074/jbc.M102456200
Wirths O, Multhaup G, Czech C et al (2002) Intraneuronal APP/Aβ trafficking and plaque formation in β-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol 12:275–286. https://doi.org/10.1111/j.1750-3639.2002.tb00442.x
Savastano A, Klafki H, Haußmann U et al (2015) N-truncated Aβ2-X starting with position two in sporadic Alzheimer’s disease cases and two Alzheimer mouse models. J Alzheimer’s Dis 49:101–110. https://doi.org/10.3233/JAD-150394
Zampar S, Klafki HW, Sritharen K et al (2020) N-terminal heterogeneity of parenchymal and vascular amyloid-β deposits in Alzheimer’s disease. Neuropathol Appl Neurobiol 46:673–685. https://doi.org/10.1111/nan.12637
Schmitt U, Hiemke C, Fahrenholz F, Schroeder A (2006) Over-expression of two different forms of the α-secretase ADAM10 affects learning and memory in mice. Behav Brain Res 175:278–284. https://doi.org/10.1016/j.bbr.2006.08.030
Horikoshi Y, Sakaguchi G, Becker AG et al (2004) Development of Aβ terminal end-specific antibodies and sensitive ELISA for Aβ variant. Biochem Biophys Res Commun 319:733–737. https://doi.org/10.1016/j.bbrc.2004.05.051
Jefferson T, Čaušević M, Auf Dem Keller U et al (2011) Metalloprotease meprin β generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J Biol Chem 286:27741–27750. https://doi.org/10.1074/jbc.M111.252718
Dominguez D, Tournoy J, Hartmann D et al (2005) Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem 280:30797–30806. https://doi.org/10.1074/jbc.M505249200
Devi L, Ohno M (2015) Effects of BACE1 haploinsufficiency on APP processing and Aβ concentrations in male and female 5XFAD Alzheimer mice at different disease stages. Neuroscience 307:128–137. https://doi.org/10.1016/j.neuroscience.2015.08.037
Wichert R, Scharfenberg F, Colmorgen C et al (2019) Meprin β induces activities of a disintegrin and metalloproteinases 9, 10, and 17 by specific prodomain cleavage. FASEB J 33:11925–11940. https://doi.org/10.1096/fj.201801371R
Weber S, Niessen MT, Prox J et al (2011) The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 138:495–505. https://doi.org/10.1242/dev.055210
De Strooper B (2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38:9–12
Gunnersen JM, Kim MH, Fuller SJ et al (2007) Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron 56:621–639. https://doi.org/10.1016/j.neuron.2007.09.018
Dorpe JV, Smeijers L, Dewachter I et al (2000) Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol 157:1283–1298. https://doi.org/10.1016/s0002-9440(10)64644-5
Acosta C, Anderson HD, Anderson CM (2017) Astrocyte dysfunction in Alzheimer disease. J Neurosci Res 95:2430–2447. https://doi.org/10.1002/jnr.24075
Heneka MT, Sastre M, Dumitrescu-Ozimek L et al (2005) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2:22. https://doi.org/10.1186/1742-2094-2-22
Perez-Nievas BG, Serrano-Pozo A (2018) Deciphering the astrocyte reaction in Alzheimer’s disease. Front Aging Neurosci 10:114. https://doi.org/10.3389/fnagi.2018.00114
Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. https://doi.org/10.1016/j.neuron.2013.12.034
Piechotta A, Parthier C, Kleinschmidt M et al (2017) Structural and functional analyses of pyroglutamate-amyloid-β-specific antibodies as a basis for Alzheimer immunotherapy. J Biol Chem 292:12713–12724. https://doi.org/10.1074/jbc.M117.777839
Antonios G, Borgers H, Richard BC et al (2015) Alzheimer therapy with an antibody against N-terminal Abeta 4-X and pyroglutamate Abeta 3-X. Sci Rep 5:17338. https://doi.org/10.1038/srep17338
Crehan H, Liu B, Kleinschmidt M et al (2020) Effector function of anti-pyroglutamate-3 Aβ antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimer’s Res Ther 12:12. https://doi.org/10.1186/s13195-019-0579-8
Schieb H, Kratzin H, Jahn O et al (2011) β-amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: comparison with human Alzheimer amyloid. J Biol Chem 286:33747–33758. https://doi.org/10.1074/jbc.M111.246561
Kalback W, Watson MD, Kokjohn TA et al (2002) APP transgenic mice Tg2576 accumulate Aβ peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry 41:922–928. https://doi.org/10.1021/bi015685+
Dislich B, Lichtenthaler SF (2012) The membrane-bound aspartyl protease BACE1: molecular and functional properties in Alzheimer’s disease and beyond. Front Physiol 3:8
Sturchler-Pierrat C, Abramowski D, Duke M et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94:13287–13292. https://doi.org/10.1073/pnas.94.24.13287
Piccini A, Russo C, Gliozzi A et al (2005) β-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem 280:34186–34192. https://doi.org/10.1074/jbc.M501694200
Portelius E, Lashley T, Westerlund A et al (2015) Brain amyloid-beta fragment signatures in pathological ageing and alzheimer’s disease by hybrid immunoprecipitation mass spectrometry. Neurodegener Dis 15:50–57. https://doi.org/10.1159/000369465
Lue LF, Kuo YM, Roher AE et al (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155:853–862. https://doi.org/10.1016/S0002-9440(10)65184-X
Wirths O, Walter S, Kraus I et al (2017) N-truncated Aβ4-x peptides in sporadic Alzheimer’s disease cases and transgenic Alzheimer mouse models. Alzheimer’s Res Ther 9:80. https://doi.org/10.1186/s13195-017-0309-z
Bouter Y, Dietrich K, Wittnam JL et al (2013) N-truncated amyloid β (Aβ) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol 126:189–205. https://doi.org/10.1007/s00401-013-1129-2
Lewis H, Beher D, Cookson N et al (2006) Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-β(42) peptide in vascular dementia. Neuropathol Appl Neurobiol 32:103–118. https://doi.org/10.1111/j.1365-2990.2006.00696.x
Shinohara M, Koga S, Konno T et al (2017) Distinct spatiotemporal accumulation of N-truncated and full-length amyloid-β42 in Alzheimer’s disease. Brain 140:3301–3316. https://doi.org/10.1093/brain/awx284
Bibl M, Gallus M, Welge V et al (2012) Cerebrospinal fluid amyloid-β 2–42 is decreased in Alzheimer’s, but not in frontotemporal dementia. J Neural Transm 119:805–813. https://doi.org/10.1007/s00702-012-0801-3
Moechars D, Dewachter I, Lorent K et al (1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274:6483–6492. https://doi.org/10.1074/jbc.274.10.6483
Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18:1539–1545. https://doi.org/10.1038/nn.4142
Jo S, Yarishkin O, Hwang YJ et al (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896. https://doi.org/10.1038/nm.3639
Liu L, Ding L, Rovere M et al (2019) A cellular complex of BACE1 and γ-secretase sequentially generates Aβ from its full-length precursor. J Cell Biol 218:644–663. https://doi.org/10.1083/jcb.201806205
Kummer MP, Heneka MT (2014) Truncated and modified amyloid-beta species. Alzheimer’s Res Ther 6:28
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. https://doi.org/10.1101/CSHPERSPECT.A006338
Valverde A, Dunys J, Lorivel T et al (2021) Aminopeptidase a contributes to biochemical, anatomical and cognitive defects in Alzheimer’s disease (AD) mouse model and is increased at early stage in sporadic AD brain. Acta Neuropathol 141:823–839. https://doi.org/10.1007/S00401-021-02308-0/FIGURES/9
Valverde A, Dunys J, Lorivel T et al (2021) Dipeptidyl peptidase 4 contributes to Alzheimer’s disease–like defects in a mouse model and is increased in sporadic Alzheimer’s disease brains. J Biol Chem 297:100963. https://doi.org/10.1016/J.JBC.2021.100963
Acknowledgements
The authors thank Dr. Stefan Lichtenthaler for providing us the SEZ6 antibody, Dr. Jens Wiltfang for providing the Aβ2–x antibody and Johanna Wesselowski for excellent technical assistance. We also thank the UKE Mouse Pathology Core Facility (Kristin Hartmann) for IHC of human samples. We thank KUL and reMYND for sharing the APP (V717I) mouse model as well as Paul Saftig for providing the BACE1-/- mice model.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grant SFB877, “Proteolysis as a Regulatory Event in Pathophysiology” (A9 [CBP], A12 [MG], A15 [CUP]) and the Alzheimer Forschung Initiative e.V. to CBP, HA and CUP.
Author information
Authors and Affiliations
Contributions
LM performed experiments, analyzed data and wrote the manuscript with SW, CUP and CBP. CUP and CBP conceived and designed the study. FA performed ELISA x–40/x–42 and analyzed data. SZ and OW provided Aβ2–x antibody and performed Aβ2–x immunohistochemistry. HA and MG provided access to human brain tissue. CS and HA performed immunohistochemistry in human brains and analyzed data. SES and US performed behavioral experiments. CK provided Notch antibodies, Notch1 stably transfected cell line and contributed to interpretation of Notch results.
Corresponding author
Ethics declarations
Ethics approval
Usage of anonymized human post mortem brain tissues is in accordance with regulations at the University Medical Center Hamburg–Eppendorf and approved by the Ethical Committee at the University Medical Center Hamburg–Eppendorf. The collection and its use are in agreement with ethical regulations of the 1964 Helsinki declaration, its later amendments or comparable ethical standards. All animal studies were conducted in compliance with European and German guidelines for the care and use of laboratory animals and were approved by the Central Animal Facility of the University of Mainz and the ethical committee on animal care and use of Rhineland–Palatinate, Germany.
Consent for publication
All authors read and approved the final manuscript.
Conflicts of interest
The authors declare they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marengo, L., Armbrust, F., Schoenherr, C. et al. Meprin β knockout reduces brain Aβ levels and rescues learning and memory impairments in the APP/lon mouse model for Alzheimer’s disease. Cell. Mol. Life Sci. 79, 168 (2022). https://doi.org/10.1007/s00018-022-04205-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04205-5