
Abstract
Kidney transplantation is a lifesaving option for patients with end-stage kidney disease. In Taiwan, urothelial carcinoma (UC) is the most common de novo cancer after kidney transplantation (KT). UC has a greater degree of molecular heterogeneity than do other solid tumors. Few studies have explored genomic alterations in UC after KT. We performed whole-exome sequencing to compare the genetic alterations in UC developed after kidney transplantation (UCKT) and in UC in patients on hemodialysis (UCHD). After mapping and variant calling, 18,733 and 11,093 variants were identified in patients with UCKT and UCHD, respectively. We excluded known single-nucleotide polymorphisms (SNPs) and retained genes that were annotated in the Catalogue of Somatic Mutations in Cancer (COSMIC), in the Integrative Onco Genomic cancer mutations browser (IntOGen), and in the Cancer Genome Atlas (TCGA) database of genes associated with bladder cancer. A total of 14 UCKT-specific genes with SNPs identified in more than two patients were included in further analyses. The single-base substitution (SBS) profile and signatures showed a relative high T > A pattern compared to COMSIC UC mutations. Ingenuity pathway analysis was used to explore the connections among these genes. GNAQ, IKZF1, and NTRK3 were identified as potentially involved in the signaling network of UCKT. The genetic analysis of posttransplant malignancies may elucidate a fundamental aspect of the molecular pathogenesis of UCKT.
Similar content being viewed by others


Introduction
Kidney transplantation is a lifesaving option for patients with end-stage kidney disease (ESKD) because it significantly improves survival; it has a mortality rate that is 63–80% lower than that of continued dialysis [1, 2]. Modern immunosuppressive agents have tremendously reduced the incidence of acute rejection within the first year and achieved outstanding short-term patient and graft survival [3]. However, the long-term survival rate of patients who receive kidney transplants is low. Recipients of a kidney transplant experience various complications, including cardiovascular disease and posttransplant malignancies (PTMs), that lead to morbidity and mortality [3].
The incidence of cancer in recipients of a kidney transplant is at least 2 to 4 times higher than that of their age-matched and gender-matched counterparts in the general population [4,5,6]. Studies from Western countries have reported that nonmelanoma skin cancer is the most common PTM [7]. The incidence of skin cancer is lower in Asian countries. Urothelial carcinoma (UC) of the bladder and urinary tract is the most common PTM in Taiwan and Mainland China, comprising approximately 40% of PTMs [8, 9]. In a nationwide cohort population-based study, Tsai et al. reported that recipients of a heart, lung, kidney, or liver transplant exhibited a risk of de novo cancer that was three times higher than that of the general population [10]. The standardized incidence ratio of urinary tract malignancies (among which bladder cancer was the most common) among recipients of a kidney transplant was 10.93 (95% CI, 9.20–12.99) [10]. Furthermore, in our previous single-center retrospective study, the most common PTMs were UC and hepatocellular carcinoma [11].
UC has a high mutational burden and exhibits greater molecular heterogeneity than do other solid tumors [12]. Different genetic alterations and pathogenic pathways occur in UCs at various anatomical locations [13, 14]. Somatic genetic mutation is a highly influential factor in UC tumorigenesis and progression. An abundance of information regarding somatic alteration in UC has been published due to rapid progress in next-generation sequencing [15]. According to the Cancer Genome Atlas (TCGA) database, the genes with the most common mutations in bladder UC are TP53, KDM6A, and ARID1A [15, 16]. TP53 mutations were commonly observed in high-grade tumors, whereas FGFR3, CREBBP, and STAG2 mutations were more commonly observed in low-grade tumors [15].
Recipients of a kidney transplant are prone to developing cancer due to underlying diseases leading to renal failure, chronic infections by oncogenic viruses, immunocompromised status related to treatment with immunosuppressive drugs, nutritional deficiencies, or altered deoxyribonucleic acid (DNA) repair. Knowledge of the underlying somatic genomic alterations in UC developed after kidney transplantation (UCKT) is limited. To our knowledge, no unbiased systematic effort has been made to describe genomic alterations in UCKT.
Whole-exome sequencing (WES) has been extensively applied to identify drivers and somatic alterations in diseases, including cancer genome profiles, in both research and clinical settings [17, 18]. WES is an effective tool for medical genetic research because it targets almost all protein-coding regions in the human genome. In this study, we performed WES and analyzed UC tissues from recipients of a kidney transplant (the UCKT group) and patients on hemodialysis (the UCHD group) to further characterize the unique genomic landscape of UCKT.
Materials and methods
Sample collection and DNA extraction
The sample collection and study protocol were conducted under the Institutional Review Board of Kaohsiung Medical University Hospital (KMUHIRB-G(I)-20150030). Written informed consent was obtained from all the patients, and all clinical investigations were conducted in accordance with the Declaration of Helsinki.
A total of 12 tumor samples, comprising 7 formalin-fixed archival UC samples from 6 recipients of a kidney transplant and 5 UC samples from patients on hemodialysis, were collected. Recipients of a kidney transplant who had a diagnosis of UC before their respective transplants were excluded. Because chronic kidney disease (CKD) and ESKD are risk factors for UC, five patients on hemodialysis who received a diagnosis of UC after the commencement of dialysis were selected to form the control group. Each patient’s medical history was confirmed by chart review. None of the patients had previously received chemotherapy or radiotherapy.
After a pathologist reviewed all the samples, the tumor samples from the paraffin blocks were macrodissected. All the specimens were placed in Eppendorf containers to prevent tissue cross-contamination. Genomic DNA was extracted using a FavorPrep formalin-fixed paraffin-embedded (FFPE) Tissue DNA Extraction Micro Kit (Favorgen Biotech, Pingtung, Taiwan) from FFPE tumor tissue. The DNA was quantified and qualified using Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, USA), 4150 TapeStation System (Agilent, Santa Clara, USA), and Nanodrop spectrophotometer (ASP-2680, Celltagen, Seoul, Republic of Korea) according to the manufacturer’s protocol. One microgram DNA from each tumor sample was subjected to run the WES analysis.
Whole-exome sequencing
Library enrichment for WES was conducted using a SureSelectXT V6_r2 reagent kit (Agilent Technologies, Santa Clara, CA, USA). The enriched samples were sequenced using an Illumina HiSeq2500 system (San Diego, CA, USA) with a 2 × 76–base pair (bp) paired-end sequencing approach. The mean coverage of the exomes was 79.76 × , and more than 96% of the exomes had a coverage higher than10 × . The sequencing data were annotated according to the GRCh37/hg19 reference genome.
The data were examined using a data analysis pipeline developed at the Cologne Center for Genomics (CCG) [19]. Initially, raw sequencing reads were mapped to human genome reference (hg19) using Burrows-Wheeler Alignment tool (BWA) [20] followed by duplication marking using Picard [21].
Subsequently, base quality score recalibration and local indel realignment were performed according to Genomic Analysis Toolkit (GATK) practice guideline [22, 23]. After the post-alignment improvements, variant calling was performed using GATK’s unified-genotyper and SAMtools’ mpileup [24]. The variant lists from both callers were merged and annotated using various public databases: dbSNP [25], 1000 Genomes Project [26], Exome Variant Server [27], HGDM professional database [28], dbVAR and DGVa [29], GERP, and Ensembl [30]. The functional effect of variants were predicted using PolyPhen [31], SIFT [32], and RVIS [33]. All of these downstream analysis has been assembled together with a few scripts developed in-house. Splice site analysis based on Yeo [34] et al. was performed. Finally, the annotated variant list is uploaded to CCG’s web interface and VARBANK database [35]. Scripts developed in-house at the Cologne Center for Genomics were applied to detect protein changes, affected donor and acceptor splice sites, and overlaps with known variants. Our analysis focused on single-nucleotide variants/polymorphisms (SNVs or SNPs) and insertions or deletions (InDels) that may result in alterations in primary protein structure or in strong splice site effects [34].
Identifying specific genes in UCKT
The processed results of all 12 samples were then downloaded from VARBANK analysis platform. A series of steps were performed to identify cancer driver genes that contain novel mutations and are not reported previously in UCKT patients. First, mutations with dbSNPs annotations and with low allele frequencies (≤ 5%) were excluded from the data sets. Mutations at the same genomic location that were annotated as different transcripts were counted as one occurrence.
To ensure that these genetic alterations identified in our study were associated with tumor development, we identified genes with these mutations and using these gene names to search against the Catalogue of Somatic Mutations in Cancer (COSMIC) [36]. If in our list, a gene is also a reported cancer driver gene in COSMIC, the gene and its mutations was retained for further analysis. In other words, these newly identified mutations in our samples were considered potential driver mutations.
Subsequently, we utilized previous studies or databases that had analyzed bladder cancer including the COSMIC [36], the Integrative Onco Genomic (IntOGen) cancer mutations browser [37], the TCGA bladder tumor database [15], and the Chinese bladder cancer genome [38] to compare with our data. Cancer driver genes that were retained in the previous step but had not been reported in these bladder cancer studies were of great interest.
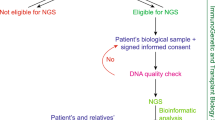
Then we selected the above driver genes which were identified in UCKT but not in UCHD groups. Finally, we reported genes that were found in two or more UCKT patients. Figure 1 illustrates the data processing steps and corresponding mutations and gene numbers for UCKT and UCHD groups. COSMIC UC single-base mutation data was retrieved from the Cancer Browser in COSMIC [39] by using the selection of Primary site (Urinary tract), Sub site (Bladder), Histology (Carcinoma) and Sub histology (Transitional cell carcinoma). The mutational signatures were conducted using the SigProfiler tools (MatrixGenerator, Extractor and Plotting) [40]. Data from UCKT, UCHD and COSMIC UC were applied separately.

Flow chart of data analysis. The WES data from each patient is subjected to genome mapping and variant calling (Variant calling). Then the data was divided into 2 groups. UCKT; N = 7 and UCHD; N = 5. The mutations were then pooled together in each group and known dbSNPs were excluded (Novel variations). The COSMIC-annotated cancer driver genes were selected (Retain cancer driver genes), but next, bladder cancer-related genes were removed from the list (Exclude bladder cancer genes). Genes that were unique in each group were identified (Overlap). Final list of genes contains those occurred in two or more UCKT patients (Multiple occurrences in UCKT group). Numbers represent gene counts, except those in parentheses, which are nucleotide mutations
Sanger sequencing analysis for confirmation of somatic mutations
Genomic DNA from patients’ peripheral blood mononuclear cells (PBMC) was used as reference for detecting somatic mutations. We used a FavorPrep Blood / Cultured Cell Genomic DNA Extraction Mini Kit (Favorgen Biotech, Pingtung, Taiwan) to extract the genomic DNA from PBMC. All the somatic mutations of the selected genes were independently verified through Sanger sequencing of the PBMC genomic DNA. Primer pairs were designed using the University of California, Santa Cruz Genome Browser [41] and Primer-BLAST Browser of the National Institutes of Health [42].
The target regions were investigated using a touchdown polymerase chain reaction (PCR) protocol. The PCR mixture contained primers at 5 μmol L−1 each, Taq DNA polymerase (Vazyme Biotech, China), and 50 ng/uL genomic DNA. The following touchdown PCR protocol was executed in a T100 thermal cycler (Bio-Rad Laboratories, CA, USA): initial denaturation at 94 °C for 5 min; annealing temperature decreased by 0.5 °C every other cycle: 94 °C for 30 s, annealing from 72 to 55 °C for 30 s, 72 °C for 30 s; followed by a cycle of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and extension at 72 °C for 10 min. The PCR products were resolved in 1.5% (w/v) agarose gel for 20 min at 100 V in 1 × TAE buffer and stained with Gel Red biosafety dye (Biotium, CA, USA). The gel was documented on a UV transilluminator (Major Science, Taiwan). The DNA sequencing was performed at the core facility of the National Yang Ming Chiao Tung University (NYCU) Genome Center.
Analysis of gene expression profile by ingenuity pathway analysis
The Ingenuity Pathway Analysis (IPA version 68752261; QIAGEN) tool was used to establish protein networks. The main molecules detected from WES analysis and Sanger sequencing verified from the PBMC genomic DNA PCR products were set as the focusing molecules and were analyzed using the build tool to determine how their relationships were affected by UC. The molecules from the QIAGEN knowledge base were then added to the network according to their specific ontological associations.
Copy number variation (CNV) detection
CNV analysis was performed using ExomeDepth (ver 1.1.15) R software package, which uses read depth data to call CNVs from exome sequencing experiments. We generated read count data from WES BAM files, and compare the count results with an aggregate reference made up of samples from the same sequencing run to determine copy number at exon level resolution. Data were analyzed according to the ExomeDepth standard procedure [43].
Results
Baseline characteristics
The baseline characteristics of all the patients and their clinical and pathological features are listed in Table 1. All patients were female and sporadic, with no family history of UC nor any history of smoking. The majority (10 out of 12) had high-grade tumors. The KT80 and KT721 samples were from the same patient, a woman who underwent surgical removal of different parts of her genitourinary tract system over 5 years (Table 1). In the KT recipients, all the tumors occurred in their native genitourinary systems. The patients underwent induction treatments with steroids and IL-2 receptor antagonists. Tacrolimus, mycophenolate mofetil, and steroids were selected as maintenance agents. The average age at UC diagnosis was younger in the UCKT group than in the UCHD group (55 vs. 61.6 years, respectively; Table 2).
Whole-exome sequencing result analysis of UC
After WES was performed, the results were aligned to the human reference genome (hg19) and further used in a bioinformatics analysis. At least 100 million reads were collected from each WES sample. In the UCKT and UCHD groups, the mean coverages were 81 and 77 reads for UCKT and UCHD, respectively, and 96.18% and 95.78% of the coding exons, respectively, were covered by at least 10 reads (Table 2). A total of 18,733 and 11,093 variants were identified from the UCKT and UCHD cohorts (Fig. 1).
The majority of the mutations were unique to individual patients. Mutations were slightly more common in the UCKT group than in the UCHD group. An analysis of the WES data revealed an average of 564 known SNPs per individual. To identify new UC-related alterations, the mutations with dbSNP annotations were excluded.
We used the COSMIC cancer driver genes database to identify new SNPs in our cohorts. During this step, 278 and 235 genes in the UCKT and UCHD groups, respectively, were retained (Fig. 1). To identify oncodriver genes specific to our cohorts (i.e., previously unknown to be related to UC), we first collected UC-related studies from the COSMIC [36], IntOGen cancer mutations browser [37], TCGA bladder tumor database [15], and Chinese bladder cancer genome [38] and we excluded genes that had already been reported in these studies. Ultimately, we identified 150 and 121 genes in the UCKT and UCHD groups, respectively, that had never previously been known to be associated with UC. Of these cancer driver genes, 79 were uniquely identified in the UCKT cohort, and 17 of them (BTK, CARD11, ELL, FNBP1, GNAQ, HOXD13, IKZF1, MAX, MLLT10, NTRK3, PAX5, SEPTIN6, SEPTIN9, SH3GL1, SLC34A2, TAL1, and TRAF7) exhibited mutations that occurred in two or more patients (Table 3).
Table 4 showed the related genes with non-synonymous mutations. After deleting the genes without amino acid change, only 14 genes (CARD11, FNBP1, GNAQ, HOXD13, IKZF1, MAX, MLLT10, NTRK3, SEPTIN6, SEPTIN9, SH3GL1, SLC34A2, TAL1, and TRAF7) were left.
To verify the tumorigenic nature of the 14 genes in the UCKT cohort (CARD11, FNBP1, GNAQ, HOXD13, IKZF1, MAX, MLLT10, NTRK3, SEPTIN6, SEPTIN9, SH3GL1, SLC34A2, TAL1, and TRAF7), we developed a reliable PCR system to validate the mutations in the patients’ PBMC genomic DNA and subjected to Sanger sequencing analysis. The results revealed that mutations in GNAQ, SEPTIN 6, NTRK3, and IKZF1 occur only in tumor samples. The possible interactions among these genes deserved further pathway analysis.
SBS and CNV analysis
The mutations were classified into two categories: single-base substitutions (SBSs) and InDels (insertions/deletions) (Table 2). The single-base substitution category was further divided into six subcategories: synonymous, missense, nonsense, frameshift, splice acceptor or donor, and nonstop (Table 2). The frameshift subcategory comprising one-base-pair differences between the alleles and reference sites, whereas the InDels category comprises mutations involving longer regions. Missense mutations were the most common type in both the UCKT and UCHD groups (74.75% and 79.40%, respectively; Table 2). These mutations are associated with amino acids changes in protein sequences, thereby affecting the structures and functions of the proteins that their respective genes encode. Interestingly, synonymous substitutions were much more common among the patients with UCKT than among those with UCHD (10.57% vs. 0.59%), which might indicate that the overall mutation rate increases after kidney transplantation, resulting in a higher risk of cancer. Among the nucleotide changes, T > A was the most common one in both the UCKT and UCHD groups (Table 2). In addition, in a TCGA study, 51% of all the mutations in bladder cancers were TpC > T or G mutations and 3.8% were TpC > A mutations [15]. Figure 2 illustrates the mutational features of the patients. Although missense mutations were the most common somatic mutations, followed by nonsense mutations, splice site mutations were also common. The effects of splice site mutations are difficult to determine; however, they may affect protein function by causing alternative splicing, intron retention, or exon skipping.

The mutational features of the individual UC patients. The mutational features of individual patients were plotted according to the percentages of nucleotide changes
To further analyze the mutation signatures and compare to other cohort, total of 95,657 unique mutations were obtained from the Cancer browser in COSMIC for UC. The distribution of six classes of SBS showed distinct patterns among UCKT, UCHD and COMIC UC data (Fig. 3). The UCKT and UCHD showed relative high percentage in T > A (40.03% and 44.63%) while COSMIC showed high C > T (42.12%) and C > G (35.17%) mutations.

The comparison of single-base substitution (SBS) profile of datasets. The SBS data from COSMIC UC was retrieved from COMSIC Cancer browser. The six classes of SBS pattern from UCKT, UCHD and COSMIC UC were calculated and showed as the frequency of total events, respectively
The results from SigProfiler tool [44] showed that both SBSs in UCKT and UCHD presented highly similar to the SBS22 signature (83.6% and 81.12%) with the proposed aetiology “Aristolochic acid exposure”, SBS5 (10.44% and 11.18%) with “unknown aetiology mutational burden is increased in bladder cancer samples with ERCC2 mutations and in many cancer types due to tobacco smoking” and SBS1 (5.96% and 7.7%) with “an endogenous mutational process initiated by spontaneous or enzymatic deamination” while COSMIC UC is similar to SBS5 (27.12%) SBS1 (4.48%), SBS13 (40.36%) and SBS2 (28.04%), respectively (Additional file 1: Fig. S1). These findings might indicate the possible unique aetiology for the UC development in Taiwan.
In addition, CNV data was retrieved from the WES results and divided into two groups. We observed 1495 and 1033 events in UCKT and UCHD groups. Using the start site as a reference, events occur in two or more patients in either group were selected. We removed records that were found in both groups and only reported those that were specific in UCKT. We identified 208 events with 92 unique CNVs in UCKT group (Additional file 2: Table S1). None of 14 genes of interest in Table 4 were found in this CNV analysis.
IPA network analysis
Through IPA, we explored the interactions and pathways among GNAQ, SEPTIN6, NTRK3, and IKZF1 (Fig. 4). SEPTIN6 exhibited no connections in the protein network. GNAQ, NTRK3, and IKZF1 were analyzed using the build tool to determine how their relationships are affected by UC. The protein network associated with the three main molecules is involved in the canonical signaling pathways of bladder cancer, PI3K/AKT, and mTOR. Each molecule was treated as a seed, and the network was constructed by connecting these seeds, thereby enabling us to explore the relationships among them. As indicated in Fig. 4, GNAQ, NTRK3, and IKZF1 may regulate UC through AKT1, MTOR, PI3KR1, HRAS, SRC, TP53, CCDN1, and RUNX3. The relationships among molecules involve activation, causation, correlation, expression, phosphorylation, protein–protein binding, were indicated in the network.

IPA based protein networks involved in target protein and disease. The focused molecules represented in grey color and bold were NTRK3, GNAQ and NTRK3 which related to Urothelial cancer (UC). The solid lines indicate direct interaction whereas dashed lines correspond to indirect relationship among the interacting proteins, pathways and disease. The node shapes of the protein, molecular classes and relationship labels as outlined in the legends in the inserted boxes
As indicated in Fig. 5, the network analysis revealed that GNAQ, NTRK3, and IKZF1 are related to UC because they are involved in PI3K/AKT and bladder cancer signaling. The 25 interacting proteins in the network (AKT1, CCND1, CDKN1A, CDKN1B, CDKN2A, CXCL8, EGFR, ERBB2, FOXO1, HDAC5, HRAS, HSP90AA1, HSP90AB1, MAP2K1, MAPK1, MMP2, MTOR, PIK3CA, PIK3R1, RB1, SIN3A, TP53, TSC2, VEGFA, and RUNX3) warranted further investigation. Thus, the result from IPA analysis explored the target proteins and networks that potentially contribute to UC and bladder cancer signaling pathways, which may inform future studies of the underlying mechanisms of UCKT.

Protein network of Urothelial cancer (UC) relevant target protein with canonical pathway (CP). The molecules represented in CP were bladder cancer signaling and PI3K/AKT signaling. The relationships represented in light blue lines were genes involved in CP related to NTRK3, IKZF1, GNAQ, respectively
Discussion
Mutational profile analysis is the key to understanding the tumorigenic mechanisms of UC. The mutational profiles of UC that develop after transplantation have not been thoroughly investigated. In this study, WES analysis allowed us to characterize the mutational signatures of UCKT in detail. The comparison of CNV data in UCKT and UCHD revealed a subset of unique events and was warranted further investigation. Specific nucleotide conversion patterns that develop in certain cancers can elucidate the establishment of these mutations [45, 46]. In a TCGA study, 51% of all the mutations in bladder cancers were TpC > T or G mutations and 3.8% were TpC > A mutations [15]. In addition, TP53, PIK3CA, and FGFR3 are the most common molecular alterations associated with UC [15, 47].
Tumors developed after kidney transplants are believed to be related to various factors, including immunosuppressive agents, viral infections, geographic area, and uremic environment, in patients with CKD/ESKD [48,49,50]. In the present study, missense mutations were the most common somatic mutations, followed by nonsense mutations. Furthermore, T > A replacements were the most common point mutations. Aristolochic acid (AA), which originates from plants in the genus Aristolochia that were used in herbal medicine, is a powerful nephrotoxin and human carcinogen associated with CKD and upper urinary tract UC (UUC) [51]. Aristolactam–DNA adducts, which are found in urothelial tissues [52], initiate mutation in the TP53 tumor suppressor gene, creating a biomarker specific to AA-induced UUC [52, 53]. Taiwan has an extraordinarily high incidence of UUC due to the widespread use of Aristolochia herbal therapies [54], which suggests that AA might play a central role in the etiology of UUC. The mutation pattern discovered in our patients suggests that a possible history of exposure to Aristolochia remedies led them to develop ESKD and UC.
Four somatic mutations (GNAQ, SEPTIN6, NTRK3, and IKZF1) identified in current study may be related to UCKT. All these mutations were single-base substitutions specifically identified in the UCKT group, and none of the genes had been reported in the genomes of patients with UC. In our study, these mutations were detected in more than one UCKT genome, which suggests they did not occur randomly and potentially played roles in the pathogenesis of UCKT. According to the protein network in IPA, GNAQ, NTRK3, and IKZF1 may regulate UC through AKT1, MTOR, PI3KR1, HRAS, SRC, TP53, CCDN1, and RUNX3. Therefore, PI3K/AKT and bladder cancer signaling and their associated pathways must be further investigated in the future.
G proteins play an essential role in cellular signal transduction. The Gq protein alpha subunit (encoded by GNAQ) couples with a seven-transmembrane receptor to activate phospholipase C-beta, which generates second messengers, and to activate kinase cascades in the cytoplasm of the cells [55]. These signals ultimately control gene transcription and cell survival, motility, and growth. However, signals transmitted by G-protein-coupled receptors/G proteins and downstream targets are involved in the initiation and progression of cancer [56]. GNAQ is located on chromosome 9q21. The role of GNAQ as an oncogene has been observed in uveal melanoma [57], blue nevi [58], and malignancies affecting the meninges [59].
NTRK3 is a member of the NTRK-encoding family, which comprises the genes NTRK1, NTRK2, and NTRK3 that encode TrkA, TrkB, and TrkC, respectively [60]. NTRK3 is located on chromosome 15q25. NTRK receptors signal through the JAK/STAT, PI3K/AKT, and MEK/ERK pathways to promote cell proliferation, differentiation, and survival [61]. Although numerous studies have investigated the key roles of these receptors in the development and function of the central and peripheral nervous system [62], alterations in NTRK genes have been observed in patients with colon [63], thyroid [64, 65], lung [66], glial [67], and breast [68] cancers. These alterations occur at relatively low frequencies (< 1%) among patients with each of these types of tumors, but NTRK-driven cancers affect numerous patients, making them key therapeutic targets [69,70,71]. NTRK gene fusions have been implicated in several diseases, and numerous therapeutic inhibitors have been developed [72]. Exploring the possible effect of NTRK3 fusion on the development of UCKT among our cohort was worthwhile.
IKZF1 encodes a transcription factor that belongs to the family of zinc finger DNA-binding proteins associated with chromatic remodeling. IKZF1 is located on chromosome 7p12. IKZF1 expression was highly conserved, which suggests its fundamental role in the ontogeny of the lymphopoietic system across species [73]. Ikaros genes are a major determinant of hematopoietic lineage, especially that of lymphocytes [74]. IKZF1 deletions and mutations affect B-cell precursor acute lymphoblastic leukemia and contribute to its poor prognosis [75]. Constitutional and acquired genetic changes in Ikaros genes have been associated with human diseases, including lung, ovarian, liver, and colorectal cancers [74, 76,77,78].
Cell signaling pathways play key roles in tumorigenesis. Genetic and protein alterations in these pathways can modify cell cycle control, DNA repair, and carcinogen metabolism [79]. The mammalian target of rapamycin (mTOR) pathway is central to the development of multiple cancers, including UC [80]. mTOR is involved in complex signaling cascades that regulate cell growth and angiogenesis under both normal and cancerous conditions. Other key molecules involved in this pathway include upstream activators, such as PI3K; AKT; negative regulators, such as the tuberous sclerosis complex (TSC) 1/2; and downstream effectors, such as p70 S6 kinase and 4EBP1 [81]. The functional roles of our candidate genes (GNAQ, NTRK3, and IKZF1) in these pathways identified through IPA require further validation.
This study has several limitations. First, this was a retrospective study with a limited sample size. Second, due to limitations in sample collection, all the patients in our study groups were female; nonetheless, this is consistent with the finding that most UCKT individuals in the Taiwanese population are female [11, 82]. Additional studies with larger sample sizes are required to achieve conclusive results. Third, the contribution of the dbSNPs were not thoroughly discussed. Lastly, functional validation is necessary to identify the characteristics of each of the candidate genes.
Conclusion
In conclusion, we identified 14 novel mutations of CARD11, FNBP1, GNAQ, HOXD13, IKZF1, MAX, MLLT10, NTRK3, SEPTIN6, SEPTIN9, SH3GL1, SLC34A2, TAL1, and TRAF7 in a group of patients with UCKT. Among the affected genes, GNAQ, IKZF1, and NTRK3 were potentially involved in the signaling network of UCKT. These findings could elucidate the development of UCKT and serve as a basis for the discovery of new potential biomarkers and the development of more effective treatments for UCKT.
Availability of data and materials
The data that support the findings presented in this manuscript are available from the corresponding author upon request.
Abbreviations
- ESKD:
-
End-stage kidney disease
- PTMs:
-
Post-transplant malignancies
- KT:
-
Kidney transplantation
- UC:
-
Urothelial carcinoma
- TCGA:
-
The Cancer Genome Atlas
- UCKT:
-
Urothelial carcinoma post kidney transplantation
- WES:
-
Whole-exome sequencing
- UCHD:
-
Urothelial carcinoma of hemodialysis patients
- CKD:
-
Chronic kidney disease
- SNVs or SNPs:
-
Single-nucleotide variants/polymorphisms
- InDels:
-
Insertions or deletions
- FFPE:
-
Formalin-fixed paraffin-embedded
- DNA:
-
Deoxyribonucleic acid
- CCG:
-
Cologne Center for Genomics
- BWA:
-
Burrows-wheeler Alignment
- SBS:
-
Single-base substitution
- GATK:
-
Genomic Analysis Toolkit
- COSMIC:
-
Catalogue of Somatic Mutations in Cancer
- IntOGen:
-
The Integrative Onco Genomic
- PCR:
-
Polymerase chain reaction
- PBMC:
-
Peripheral blood mononuclear cell
- IPA:
-
Ingenuity Pathway Analysis
- GNAQ:
-
Guanine nucleotide-binding protein G(q)
- CNV:
-
Copy number variations
- NTRK3:
-
Neurotrophic tyrosine receptor kinase 3
- IKZF1:
-
Ikaros family zinc finger protein 1
- AA:
-
Aristolochic acid
- UUC:
-
Upper urinary tract urothelial carcinoma
- mTOR:
-
Mammalian target of rapamycin
References
Butler AM, Olshan AF, Kshirsagar AV, Edwards JK, Nielsen ME, Wheeler SB, Brookhart MA. Cancer incidence among US Medicare ESRD patients receiving hemodialysis, 1996–2009. Am J Kidney Dis. 2015;65(5):763–72.
Gill JS, Lan J, Dong J, Rose C, Hendren E, Johnston O, Gill J. The survival benefit of kidney transplantation in obese patients. Am J Transplant. 2013;13(8):2083–90.
Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011;11(3):450–62.
Engels EA, Pfeiffer RM, Fraumeni JF Jr, Kasiske BL, Israni AK, Snyder JJ, Wolfe RA, Goodrich NP, Bayakly AR, Clarke CA, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA. 2011;306(17):1891–901.
Yanik EL, Clarke CA, Snyder JJ, Pfeiffer RM, Engels EA. Variation in cancer incidence among patients with ESRD during kidney function and nonfunction intervals. J Am Soc Nephrol. 2016;27(5):1495–504.
Au E, Wong G, Chapman JR. Cancer in kidney transplant recipients. Nat Rev Nephrol. 2018;14(8):508–20.
Agraharkar ML, Cinclair RD, Kuo YF, Daller JA, Shahinian VB. Risk of malignancy with long-term immunosuppression in renal transplant recipients. Kidney Int. 2004;66(1):383–9.
Xiao J, Zhu X, Hao GY, Zhu YC, Hou HJ, Zhang J, Ma LL, Tian Y, Zhang YH. Association between urothelial carcinoma after kidney transplantation and aristolochic acid exposure: the potential role of aristolochic acid in HRas and TP53 gene mutations. Transplant Proc. 2011;43(10):3751–4.
Li WH, Chen YJ, Tseng WC, Lin MW, Chen TJ, Chu SY, Hwang CY, Chen CC, Lee DD, Chang YT, et al. Malignancies after renal transplantation in Taiwan: a nationwide population-based study. Nephrol Dial Transplant. 2012;27(2):833–9.
Tsai HI, Lee CW, Kuo CF, See LC, Liu FC, Chiou MJ, Yu HP. De novo malignancy in organ transplant recipients in Taiwan: a nationwide cohort population study. Oncotarget. 2017;8(22):36685–95.
Lim LM, Kung LF, Kuo MC, Huang AM, Kuo HT. Timing of mTORI usage and outcomes in kidney transplant recipients. Int J Med Sci. 2021;18(5):1179–84.
Morrison CD, Liu P, Woloszynska-Read A, Zhang J, Luo W, Qin M, Bshara W, Conroy JM, Sabatini L, Vedell P, et al. Whole-genome sequencing identifies genomic heterogeneity at a nucleotide and chromosomal level in bladder cancer. Proc Natl Acad Sci USA. 2014;111(6):E672-681.
Frantzi M, Makridakis M, Vlahou A. Biomarkers for bladder cancer aggressiveness. Curr Opin Urol. 2012;22(5):390–6.
Pandith AA, Shah ZA, Siddiqi MA. Oncogenic role of fibroblast growth factor receptor 3 in tumorigenesis of urinary bladder cancer. Urol Oncol. 2013;31(4):398–406.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22.
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–8.
Nakagawa H, Wardell CP, Furuta M, Taniguchi H, Fujimoto A. Cancer whole-genome sequencing: present and future. Oncogene. 2015;34(49):5943–50.
Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11(10):685–96.
Kawalia A, Motameny S, Wonczak S, Thiele H, Nieroda L, Jabbari K, Borowski S, Sinha V, Gunia W, Lang U, et al. Leveraging the power of high performance computing for next generation sequencing data analysis: tricks and twists from a high throughput exome workflow. PLoS ONE. 2015;10(5): e0126321.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
http://broadinstitute.github.io/picard/. Accessed 2 Jun 2022.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. 2013;43:11 10 11-11 10 33.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
http://evs.gs.washington.edu/EVS/. Accessed 2 Jun 2022.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9.
Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41(Database issue):D936-941.
Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2014. Nucleic Acids Res. 2014;42(Database issue):D749-755.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013, Chapter 7:Unit7 20.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8): e1003709.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2–3):377–94.
http://varbank.ccg.uni-koeln.de. Accessed 28 Jul 2016.
Yang H, Wang Q, Zheng L, Lin M, Zheng XB, Lin F, Yang LY. Multiple genetic modifiers of bilirubin metabolism involvement in significant neonatal hyperbilirubinemia in patients of Chinese descent. PLoS ONE. 2015;10(7): e0132034.
Mutational Cancer Driver Genes. Accessed Jan 2018 from https://www.intogen.org/search. 2014.
Wang M, Li Z, Chu H, Lv Q, Ye D, Ding Q, Xu C, Guo J, Du M, Chen J, et al. Genome-wide association study of bladder cancer in a chinese cohort reveals a new susceptibility locus at 5q12.3. Cancer Res. 2016;76(11):3277–84.
https://cancer.sanger.ac.uk/cosmic/browse/tissue. Accessed 13 Jun 2022.
Bergstrom EN, Kundu M, Tbeileh N, Alexandrov LB. Examining clustered somatic mutations with SigProfilerClusters. Bioinformatics. 2022;38(13):3470–3.
https://genome.ucsc.edu/. Accessed 12 Dec 2017.
https://www.ncbi.nlm.nih.gov/tools/primer-blast/. Accessed 12 Dec 2017.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO, Thrasher AJ, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–54.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101.
Glaser AP, Fantini D, Shilatifard A, Schaeffer EM, Meeks JJ. The evolving genomic landscape of urothelial carcinoma. Nat Rev Urol. 2017;14(4):215–29.
Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14(12):786–800.
Sfakianos JP, Cha EK, Iyer G, Scott SN, Zabor EC, Shah RH, Ren Q, Bagrodia A, Kim PH, Hakimi AA, et al. Genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2015;68(6):970–7.
Maisonneuve P, Agodoa L, Gellert R, Stewart JH, Buccianti G, Lowenfels AB, Wolfe RA, Jones E, Disney AP, Briggs D, et al. Cancer in patients on dialysis for end-stage renal disease: an international collaborative study. Lancet. 1999;354(9173):93–9.
Vajdic CM, McDonald SP, McCredie MR, van Leeuwen MT, Stewart JH, Law M, Chapman JR, Webster AC, Kaldor JM, Grulich AE. Cancer incidence before and after kidney transplantation. JAMA. 2006;296(23):2823–31.
Karami S, Yanik EL, Moore LE, Pfeiffer RM, Copeland G, Gonsalves L, Hernandez BY, Lynch CF, Pawlish K, Engels EA. Risk of renal cell carcinoma among kidney transplant recipients in the United States. Am J Transplant. 2016;16(12):3479–89.
Grollman APSJ, Jelakovi CB. Aristolochic acid nephropathy: an environmental and iatrogenic disease. 3rd ed. Amsterdam: Elsevier; 2009.
Grollman AP, Shibutani S, Moriya M, Miller F, Wu L, Moll U, Suzuki N, Fernandes A, Rosenquist T, Medverec Z, et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc Natl Acad Sci U S A. 2007;104(29):12129–34.
Jelakovic B, Karanovic S, Vukovic-Lela I, Miller F, Edwards KL, Nikolic J, Tomic K, Slade N, Brdar B, Turesky RJ, et al. Aristolactam-DNA adducts are a biomarker of environmental exposure to aristolochic acid. Kidney Int. 2012;81(6):559–67.
Lai MN, Wang SM, Chen PC, Chen YY, Wang JD. Population-based case-control study of Chinese herbal products containing aristolochic acid and urinary tract cancer risk. J Natl Cancer Inst. 2010;102(3):179–86.
Choi E, Park SJ, Lee G, Yoon SK, Lee M, Lee SK. The GNAQ T96S mutation affects cell signaling and enhances the oncogenic properties of hepatocellular carcinoma. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22063284.
Larribere L, Utikal J. Update on gna alterations in cancer: implications for uveal melanoma treatment. Cancers (Basel). 2020. https://doi.org/10.3390/cancers12061524.
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–9.
Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602.
Kusters-Vandevelde HV, van Engen-van Grunsven IA, Kusters B, van Dijk MR, Groenen PJ, Wesseling P, Blokx WA. Improved discrimination of melanotic schwannoma from melanocytic lesions by combined morphological and GNAQ mutational analysis. Acta Neuropathol. 2010;120(6):755–64.
Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30(Suppl8):viii5–15.
Tognon C, Garnett M, Kenward E, Kay R, Morrison K, Sorensen PH. The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res. 2001;61(24):8909–16.
Calella AM, Nerlov C, Lopez RG, Sciarretta C, von Bohlen und Halbach O, Bereshchenko O, Minichiello L. Neurotrophin/Trk receptor signaling mediates C/EBPalpha, -beta and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Dev. 2007;2:4.
Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319(6056):743–8.
Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol. 2010;321(1):44–9.
Brzezianska E, Karbownik M, Migdalska-Sek M, Pastuszak-Lewandoska D, Wloch J, Lewinski A. Molecular analysis of the RET and NTRK1 gene rearrangements in papillary thyroid carcinoma in the Polish population. Mutat Res. 2006;599(1–2):26–35.
Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–72.
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–50.
Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, Mathers JA, Becker L, Carneiro F, MacPherson N, Horsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2(5):367–76.
Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–47.
Jin W. Roles of TrkC signaling in the regulation of tumorigenicity and metastasis of cancer. Cancers (Basel). 2020. https://doi.org/10.3390/cancers12010147.
Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol. 2018. https://doi.org/10.1200/PO.18.00183.
Hechtman JF. NTRK insights: best practices for pathologists. Mod Pathol. 2021. https://doi.org/10.1038/s41379-021-00913-8.
Molnar A, Wu P, Largespada DA, Vortkamp A, Scherer S, Copeland NG, Jenkins NA, Bruns G, Georgopoulos K. The Ikaros gene encodes a family of lymphocyte-restricted zinc finger DNA binding proteins, highly conserved in human and mouse. J Immunol. 1996;156(2):585–92.
Chen Q, Shi Y, Chen Y, Ji T, Li Y, Yu L. Multiple functions of Ikaros in hematological malignancies, solid tumor and autoimmune diseases. Gene. 2019;684:47–52.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80.
Javierre BM, Rodriguez-Ubreva J, Al-Shahrour F, Corominas M, Grana O, Ciudad L, Agirre X, Pisano DG, Valencia A, Roman-Gomez J, et al. Long-range epigenetic silencing associates with deregulation of Ikaros targets in colorectal cancer cells. Mol Cancer Res. 2011;9(8):1139–51.
Keita M, Wang ZQ, Pelletier JF, Bachvarova M, Plante M, Gregoire J, Renaud MC, Mes-Masson AM, Paquet ER, Bachvarov D. Global methylation profiling in serous ovarian cancer is indicative for distinct aberrant DNA methylation signatures associated with tumor aggressiveness and disease progression. Gynecol Oncol. 2013;128(2):356–63.
Zhang Z, Xu Z, Wang X, Wang H, Yao Z, Mu Y, Ma Z, Liu Z. Ectopic Ikaros expression positively correlates with lung cancer progression. Anat Rec (Hoboken). 2013;296(6):907–13.
Chen M, Cassidy A, Gu J, Delclos GL, Zhen F, Yang H, Hildebrandt MA, Lin J, Ye Y, Chamberlain RM, et al. Genetic variations in PI3K-AKT-mTOR pathway and bladder cancer risk. Carcinogenesis. 2009;30(12):2047–52.
Garcia JA, Danielpour D. Mammalian target of rapamycin inhibition as a therapeutic strategy in the management of urologic malignancies. Mol Cancer Ther. 2008;7(6):1347–54.
Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab Invest. 2010;90(10):1406–14.
Chou YH, Chang WC, Wu WJ, Li CC, Yeh HC, Hou MF, Chou PS, Lee MH, Chang WP, Li WM. The association between gender and outcome of patients with upper tract urothelial cancer. Kaohsiung J Med Sci. 2013;29(1):37–42.
Acknowledgements
The authors acknowledge the Cologne Center for Genomics, University of Cologne, Germany and the Clinical and Industrial Genomic Application Development Service Center of National Core Facility for Biopharmaceuticals, Taiwan (MOST 107-2319-B-010-002) for sequencing. The authors also thank the Center for Research Resources and Development in Kaohsiung Medical University, Kuan-Wen Chen and Tzu-Mao Hung in GGA Corporation, Molecular Science and digital innovation center (Taiwan) for the assistance in performing the QIAGEN IPA analysis. This study was supported by grants from the Kaohsiung Medical University Hospital (KMUH104-4M09, KMUH110-0R18), Taiwan Ministry of Science and Technology (MOST) 109-2635-B-037 -009 and MOST 110-2314-B-037 -071.
Funding
This study was supported by research grants from the Kaohsiung Medical University Hospital (KMUH104-4M09, KMUH110-0R18), Taiwan Ministry of Science and Technology (MOST) 109-2635-B-037-009 and MOST 110-2314-B-037-071.
Author information
Authors and Affiliations
Contributions
Conceptualization, H.T.K., D.Y.H and L.M.L.; methodology, H.T.K., D.Y.H. and A.M.H; software, W.Y.C.; formal analysis, W.Y.C, D.Y.H, C.C.Y, P.I.L. and S.M.C; resources, H.L.K.; writing—original draft preparation, L.M.L.; writing—review and editing, L.M.L, W.Y.C, A.M.H and T.W.L.; supervision: A.M.H and H.T.K.; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The sample collection and study protocol were conducted under the Institutional Review Board of Kaohsiung Medical University Hospital (KMU-HIRB-G(I)-20150030). Written informed consent was obtained from all the patients, and all clinical investigations were conducted in accordance with the Declaration of Helsinki.
Consent for publication
The publication of this manuscript has been approved by all authors.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The comparison of SBS signature. Single-base substitution (SBS) signatures were analyzed by SigProfiler tool. The SBS96 Decomposition results of (A) UCKT, (B) UCHD and (C) COMIC UC were shown. The signatures and percentages of the SBS signatures were indicated, respectively.
Additional file 2: Table S1.
Unique CNV in UCKT samples. CNV data was extracted from the WES results and divided into two groups. Total 1495 and 1033 events were observed in UCKT and UCHD groups. Using the start site as a reference, events occur in two or more patients in either group were selected. The records that were found in both groups were removed and only reported those that were specific in UCKT was retained.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lim, LM., Chung, WY., Hwang, DY. et al. Whole-exome sequencing identified mutational profiles of urothelial carcinoma post kidney transplantation. J Transl Med 20, 324 (2022). https://doi.org/10.1186/s12967-022-03522-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03522-4