
Abstract
Background
Renal collecting duct carcinoma (CDC) is a rare and lethal subtype of renal cell carcinoma (RCC). The genomic profile of the Chinese population with CDC remains unclear. In addition, clinical treatments are contradictory. In this study, we aimed to identify the genomic mutation spectrum of CDC in the Chinese population.
Methods
Whole-exome sequencing was performed using the Illumina Novaseq™ 6000 platform. MuTect2 detects single-nucleotide variants (SNVs) and small scale insertions/deletions (INDELs). The identified mutations were annotated with ANNOVAR and validated by Sanger sequencing. Control-FREEC was used to detect copy number variation (CNV), and GISTIC was applied to detect frequently mutated altered regions. These data were compared with associated The Cancer Genome Atlas cohorts.
Results
Ten normal-matched CDC patients were included. The mean tumour mutation burden was 1.37 Mut/Mb. Six new recurrent somatic mutated genes were identified, including RBM14, MTUS1, GAK, DST, RNF213 and XIRP2 (20% and 2 of 10, respectively), and validated by Sanger sequencing. In terms of common mutated genes, SETD2 was altered in both CDC and other RCC subtypes but not in bladder urothelial carcinoma (BLCA); CDKN2A was a driver gene in both CDC (SNV: 10%, 1 of 10) and BLCA but not in other RCC subtypes. Next, 29 amplifications and 6 deletions of recurrent focal somatic CNVs were identified by GISTIC2.0, which displayed differences from kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP) and BLCA cohorts. Of note, CDKN2A (CNV alteration: 30%, 3 of 10) and CDKN2A-AS1 were the only overlapping genes of these four cohorts. Importantly, the CDKN2A mutation in our cohort differed from previous studies in urinary carcinomas. Moreover, CDKN2A-altered cases had significantly worse overall survival than wild-type cases in both KIRC and KIRP cohorts. In addition, the most frequently altered genomic pathway of our CDC cohort was the CDKN2A-mediated p53/RB1 pathway.
Conclusions
Our study offers the first genomic spectrum of the Chinese population with CDC, which differs from that of the Western population. The altered CDKN2A-mediated p53/RB1 pathway might provide new insight into potential therapeutic targets for CDC patients.
Similar content being viewed by others

Background
Collecting duct carcinoma (CDC) is a rare and lethal subtype of renal cell carcinoma [1] that is still mainly diagnosed based on pathological examination [2, 3]. Approximately half of CDC patients are initially diagnosed at an advanced stage with metastatic symptoms of the lymph node, bone, lung, or liver, and most die within 1–3 years [4, 5]. Moreover, the clinical treatments are contradictory. The kidney cancer part of the National Comprehensive Cancer Network (NCCN) guidelines recommend platinum-based chemotherapies due to some shared biological features with urothelial carcinoma [6]. However, 23 metastatic CDC patients treated with gemcitabine plus cisplatin or carboplatin showed only an objective response rate (ORR) of 26% and an overall survival (OS) of 10.5 months [7]. In addition, a systemic therapy for CDC, renal cell carcinoma (RCC), has been proposed according to a transcriptome sequencing study [8], yet the outcomes are unsatisfactory. Combined chemotherapies showed an ORR of 30.8% and an OS of 12.5 months [9], which was similar to single chemotherapies [10]. Targeted therapies for metastatic CDC have little clinical benefit [11], and no response to immunotherapy has been observed [4, 12].
Therefore, additional comparative studies are urgently needed to better distinguish the dominant molecular signature between CDC and other RCC subtypes or urothelial carcinomas, providing new insight into potential prognostic and therapeutic targets.
There are a few genomic studies to uncover genetic alterations in CDC patients. Seventeen advanced or metastatic American patients with CDC identified 36 genomic alterations by targeted next-generation sequencing of established rearrangement- and cancer-related genes, e.g., NF2 (29%), SETD2 (24%), SMARCB1 (18%), CDKN2A (12%), PIK3CA (6%), PIK3R2 (6%), FBXW7 (6%), BAP1 (6%), DNMT3A (6%), VHL (6%) and HRAS (6%). In the study, alterations in FH and SMARCB1 occurred in a mutually exclusive manner to NF2 alterations [13]. Moreover, a multicentre copy number variant calling study of 29 German CDC patients revealed recurrent DNA losses at 1p, 8p, 9p and 16p and gains at 13q [14]. Whole-exome sequencing (WES) of four normal-matched American CDC patients identified only one recurrent somatic single-nucleotide variant [15]. In addition, an integrative transcriptomic study of 17 CDC patients identified that CDC may originate from the distal convoluted tubules of the nephron. Moreover, CDC is considered not only a metabolic disease but also an oxidoreductase activity and pyruvate metabolism [16]. These studies have mainly focused on Western populations, whereas the genomic profile of CDC patients in the Chinese population remains unclear.
Therefore, in this study, we performed deep whole-exome sequencing of 10 paired CDC patient tumour tissues that matched normal kidney tissues to improve our understanding of the genomic profile of Chinese patients with CDC and compared the results with other RCC subtypes and bladder urothelial carcinoma. We found that CDC is not only characterised as a unique type of solid tumour but also shares some specific molecules with other RCC subtypes and urinary tract carcinoma. The CDKN2A alteration-mediated p53/RB1 pathway might provide new insight into potential prognostic and therapeutic targets for these patients.
Materials and methods
Study design and samples information
Patients were collected retrospectively from the First Affiliated Hospital (Changhai Hospital), Naval Military Medical University. Paired tumour and normal formaldehyde-fixed paraffin-embedded (FFPE) samples were obtained prior to any treatment. Pathological diagnoses were reconfirmed by two experienced uropathologists. Patients with urothelial carcinoma involving the upper tract, papillary RCC, clear cell carcinoma RCC, chromophobe RCC, unclassified RCCs and other malignant tumours were excluded.
Ethics approval was obtained from the institutional review board of Changhai Hospital (CHEC2021-064). The study was conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983, and the Good Clinical Practice guidelines. All research participants or their legal representatives signed informed consent forms for participation in the research.
DNA extraction and quantification
Genomic DNA was extracted using QIAamp DNA FFPE Tissue Kit (QIAGEN). DNA quality and yield were measured and assessed using a Qubit fluorometer and Qubit dsDNA HS Assay Kit (Thermo Fisher) following the manufacturer’s protocol.
WES library generation and sequencing
Before library generation, genomic DNA was fragmented by sonication to a median size of 350 bp. Then, the KAPA hyperprep kit (Roche) was used for library preparation, and xGen® Hybridization and Wash Kit (IDT) was used for exome capture before sequencing. Next, genomic DNA fragments were end-repaired, ligated with Illumina sequencing adapters, and amplified. Finally, DNA libraries were subjected to WES using the Illumina Novaseq™ 6000 platform (2 × 150-bp paired-end reads).
Sequencing data analysis
Paired-end reads were quality checked by FastQC (v0.11.9) and processed to high quality using Trimmomatic [17] (v0.36, parameters: SLIDINGWINDOW: 4: 15, LEADING: 3, TRAILING: 3, ILLUMINACLIP: adaptor.fa: 2: 30: 10: 8: ture, MINLEN: 36) to remove adapters and perform trimming. The reads were aligned to Human Genome Reference Consortium build 38 (GRCh38) using Burrows-Wheeler Aligner (BWA-MEM) v0.7.8.
Somatic mutations calling
Somatic mutations, including single-nucleotide variants (SNVs) and small-scale insertions/deletions (INDELs), were detected using the Mutect2 pipeline in Genome Analysis Toolkit (GATK, v4.1.9.0). ANNOVAR [18] was applied to annotate filtered variant call format files using multiple annotation databases. Briefly, mutations in segmental duplications (genomicSuperDups) or repetitive elements (RepeatMasker) were removed. Non-synonymous exonic mutations with minor allele frequency > 5% in the 1000 Genome Project, Exome Aggregation Consortium database with allele frequencies in East Asia (EAS), dbSNP 138, or exome sequencing project (ESP) were removed; all COSMIC variants were retained. Mutations with a variant allele frequency (VAF) greater than 0.03 after filtering were reviewed manually using integrated genomics viewer (IGV). Finally, mutations within the blacklist [19] were also filtered and removed. Next and importantly, recurrent mutated genes were experimentally validated by Sanger sequencing.
Further sequencing analyses, including the significantly mutated genes (SMGs), mutation signature pattern and tumour mutation burden, were also performed. Briefly, the prepared mutation annotation format file was analysed to determine SMGs using MutSigCV (v2.0), with a cut-off value of p < 0.05. The deconstructSigs R package [20] was adopted to calculate the ratios of 30 types of defined COSMIC mutation signatures [21] in each sample. Tumour mutation burden was defined as in our previous study [22], with 34.2 Mb of exonic region.
Somatic copy number alterations calling
Control-FREEC [23] (v.11.5) was used to detect genomic segments with somatic copy number variants (CNVs) under default parameters. The GISTIC2.0 [24] algorithm was applied to detect recurrently amplified and deleted genomic regions with the following modified parameters: -ta 0.1, -td 0.1, -js 100; -broad 1; -brlen 0.7; -conf 0.95; -genegistic 1; and -savegene 1.
Comparison of mutation landscapes and pathways across CDC and associated TCGA cohorts
The integrated SNV and CNV sequencing data of associated The Cancer Genome Atlas (TCGA) cohorts, including kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), and bladder urothelial carcinoma (BLCA), were downloaded from cBioPortal (https://www.cbioportal.org). Next, RCircos [25] and Venn diagrams were applied to visualise and compare the different distributions of the above CNV-based genes in our CDC, KIRC, KIRP and BLCA cohorts.
In addition, the mutation frequency of all key genes in 10 common and 3 specific cancer-related pathways related to SNV and CNV were used to compare the mutation landscape and pathway enrichment across these four study cohorts and to determine the putative critical pathway in our CDC cohort. The components of all the altered genes in each pathway were also calculated.
CDKN2A mutation spectrum in CDC and associated TCGA cohorts
Available SNV, CNV and overall survival data of CDKN2A for KIRC, KIRP, BLCA and breast cancer (BRCA) patients were downloaded from cBioPortal. The percentages of patients with SNVs, CNV alterations and wild-type CKDN2A were summarised, and the mutation spectrum of CDKN2A in these cohorts was analysed.
Results
Patient clinical information
Ten CDC patients with matched tumour and normal renal tissues were included, and their clinical information is summarised in Fig. 1 and Additional file 1: Table S1. Of these patients, eight were male and two female. The median age was 57.3 years old (range: 33–67). The ratio of left to right tumour side was 1: 1.5. Two patients were diagnosed with a high clinical T stage (T3 and T4), though most of the others were at T2. More than half of the patients had local lymph node infiltration, and two patients had distant metastases.

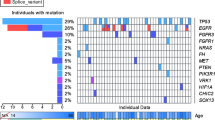
The landscape of somatic mutations. Left panel: The presence of 15 somatically altered genes in Cancer Gene Census and two previous collecting duct carcinoma (CDC) studies (Pal et al. and Wang et al.) as the heatmap on the left. Right panel: Each column represents an individual tumour patient (n = 10). Mutation profiles of the top 10 genes detected by maftools and five frequently mutated CDC-related genes using genome analysis toolkit (GATK) are displayed. Mutation frequencies and numbers are shown in the middle, and seven related clinicopathological characteristics for all 10 patients are shown at the bottom, including overall survival status, sex, age, tumour location, clinical T stage, clinical N stage, and clinical M stage. The p values of the mutated genes detected by MutSigCV2.0 are plotted on the right
The landscape of somatic mutations
The average sequencing depth was 534.78× (range: 302.30–794.62×) in tumours and 128× (range: 89.27–195.13×) in normal samples (Additional file 2: Table S2). We identified 471 filtered SNVs, including 359 nonsynonymous mutations, 31 stop-gain mutations, 2 stop-loss mutations, 50 frameshift INDELs and 29 nonframeshift INDELs (Additional file 3: Table S3). The median mutation burden of each sample was 1.37 Mut/Mb (range: 0.41–2.54).
As shown in Fig. 1, SETD2 was the most frequently mutated gene (30%). Some common mutated genes previously reported in CDC were also identified, including CDKN2A, TP53, FBXW7, ATM and KDM5C (10%). Moreover, eight novel recurrent somatic mutated genes were detected, including RBM14, MTUS1, GAK, DST, ASPM, CDC27, RNF213 and XIRP2 (20%). To validate these findings, Sanger sequencing was performed. Seven of nine recurrently mutated genes in our study were successfully validated; ASPM and CDC27 were not (Additional files 4 and 5: Figures S1 and S2). RBM14, GAK, DST and XIRP2 were selected to validate different variants of the same gene. However, some variant validations failed; variant allele frequencies (VAFs) were 11.0% (RBM14), 11.2% (DST) and 11.2% (XIRP2) (Additional file 4: Figure S1).
Next, compared with previous studies with CDC and associated TCGA cohorts, including KIRC, KIRP and BLCA, TP53 mutation was detected in all six studies. SETD2 was altered in Pal et al.’s study and our study and in KIRC and KIRP but not in BLCA. CDKN2A was a driver gene in both CDC and BLCA but not in KIRC and KIRP.
Further analysis identified SETD2, RBM14, MTUS1, GAK, DST, ASPM and CDKN2A as SMGs. However, SETD2 is the only SMG in Cancer Gene Census. The mutation signature patterns of each patient were examined, and those of 1, 3, 5, 8 and 9 were the most common (Additional file 6: Figure S3).
The landscape of copy number variants
We analysed somatic CNVs using Control-FREEC mutation-calling software. GISTIC2.0 analysis identified 29 amplified and 6 deleted recurrent focal CNVs (Fig. 2A). In terms of driver genes in Cancer Gene Census, focal amplified regions implicated the oncogenes PRDM16 and SKI at 1p36, MUC4 at 3q29, TERT at 5P15, TLX3, NPM1, FGFR4 and FLT4 at 1q35, CUX1 at 7p22, BRD3 and NOTCH1 at 9q34, HRAS at 11p15, CCND1 at 11q13, AKT1 at 14q32, KAT7 at 17q21, H3F3B at 17q25, GNA11, TCF3, MAP2K2, FSTL3 and SH3GL1 at 19p13, and U2AF1 at 21q22. However, only HRAS has been previously reported in CDC [13]; it belongs to the Ras oncogene family and induces GTPase activity [26]. Focally deleted regions identified the tumour-suppressor genes RHOA at 3p21, CDKN2A and CDKN2B at 9p21, RAD51B, MAX, DICER1, BCL11B, NKX2-1, CCNB1IP1 and BAZ1A at 9q33.

The landscape of copy number variants (CNVs). A Left: Significant (q < 0.25) recurrent focal amplified CNVs detected by Control-FREEC along all autosomes using GISTIC 2.0 are shown. Recurrent putative cancer driver genes of each SCNA are also highlighted. Right: Significant (q < 0.25) recurrent focal deleted CNVs detected by Control-FREEC along all autosomes using GISTIC 2.0 are shown. Recurrent putative cancer driver genes of each SCNA are also highlighted. B Venn diagram of the four included cohorts using the CNV-based deleted genes. C Rcircos results of the four included cohorts using the CNV-based deleted genes. KIRC, clear cell renal cell carcinoma; KIRP, papillary renal cell carcinoma; BLCA, bladder carcinoma; CDC, collecting duct carcinoma
At the CNV-based gene level, only CDKN2A and CDKN2A-AS1 were found using the deleted genes (Fig. 2B; Additional file 7: Table S4). The overlapping deleted genes were enriched on chromosomes 3 and 14 (Fig. 2C). However, no common genes were found among amplified genes (Additional file 8: Fig. S4A; Additional file 9: Table S5), and overlapping amplified genes were enriched on chromosomes 5 and 17 (Additional file 8: Fig. S4B).
CDKN2A alteration is important in CDC and associated TCGA cohorts
Since CDKN2A is a key mutated gene according to the somatic mutation and copy number alteration landscapes, we further explored CDKN2A alteration in CDC and associated TCGA cohorts, including BLCA, KIRC, KIRP and BRCA.
There was a higher percentage of CNV alterations than SNV alterations of CDKN2A in each cohort. The percentages of somatic mutated SNVs in CDC, BLCA, KIRC, KIRP and BRCA were 10%, 5.5%, 1.1%, 0.7% and 0.2%, respectively (Fig. 3A). Surprisingly, the CDKN2A mutation in our study, which encodes a protein change of p. R24Gfs*16 on exon 1, did not overlap with the previously reported CDKN2A mutation spectrum in urinary carcinomas and BRCA (Fig. 3B).

CDKN2A alteration is important in CDC and associated TCGA cohorts. A Components of single-nucleotide variants (SNV, green), copy number variants (CNV, orange), combinations of SNV and CNV (red), and wild-type (WT, blue) CDKN2A in different kinds of carcinomas (from left to right: CDC, BLCA, KIRC, KIRP and BRCA, respectively). B Lollipop plots of CDKN2A somatic variant mutation in different kinds of carcinomas. C Overall survival of patients with BLCA, BRCA, KIRC and KIRP between CDKN2A-altered and wild-type (WT) groups. KIRC, clear cell renal cell carcinoma; KIRP, papillary renal cell carcinoma; BLCA, bladder carcinoma; CDC, collecting duct carcinoma; BRCA, breast cancer
Moreover, patients with CDKN2A alteration displayed a significantly worse overall survival than patients with the wild-type gene in both the KIRC and KIRP cohorts. Despite no significant differences between CDKN2A-altered and wild-type cases in the BLCA and BRCA cohorts, these patients exhibited the same tendency in the early follow-up period (Fig. 3C). Overall, clinical data on CDKN2A in CDC are lacking due to its rarity.
The CDKN2A-mediated p53/RB1 pathway is mostly altered in the CDC population
Genomic alterations are known to target common cancer pathways, even though not all component genes are altered at an equal frequency [27]. Next, we compared the mutational landscape among the CDC, KIRC, KIRP and BLCA cohorts in a pathway-centric manner. We focused on 10 important common pathways and three cancer-specific pathways based on both the SNV and CNV data, which suggested that the overall pathway-level mutation burden was different among the four cohorts (Fig. 4A, B; Additional file 10 and 11: Table S6 and S7).

The CDKN2A-mediated p53/RB1 pathway is mostly altered in the CDC population. A Comparison of pathway-level alterations across the CDC, BLCA, KIRC and KIRP cohorts considering single-nucleotide variants (SNVs). B Comparison of pathway-level alterations across the CDC, BLCA, KIRC and KIRP cohorts considering copy number variants (CNVs). C SNVs and CNVs in components of the p53/RB1 pathway among the CDC, KIRC, KIRP and BLCA cohorts (from left to right in the box). Red, oncogene alterations; blue, tumour suppressor alterations. Percentages denote altered fractions. KIRC, clear cell renal cell carcinoma; KIRP, papillary renal cell carcinoma; BLCA, bladder carcinoma; CDC, collecting duct carcinoma
Notably, the most frequently altered cancer-specific pathway in both the CDC and BLCA cohorts was the p53/RB1 pathway (Fig. 4C). However, individual altered genes varied significantly. In our CDC cohort, CDKN2A was most frequently altered, whereas TP53 was the major mutated gene altered in the BLCA cohort. In addition, genes in the p53/RB1 pathway in the BLCA cohort were altered to different degrees. The KIRC cohort of TCGA typically showed specific VHL pathway alteration (Additional file 12: Fig. S5), which offers clinical drug targets, such as sunitinib and pazopanib. None of the key genes in the VHL pathway were altered in our CDC cohort, which might help to explain the limited efficacy of target therapies in CDC patients. In addition, MET was most frequently mutated in the specific MAPK pathway in the KIRP cohort but not in our CDC cohort (Additional file 13: Fig. S6).
Taken together, the CDKN2A alteration-mediated p53/RB1 pathway is most common in the CDC population, which might offer new insight into the clinical treatment of CDC patients.
Discussion
In this study, we identified eight recurrently somatically mutated genes: RBM14, MTUS1, GAK, DST, ASPM, CDC27, RNF213 and XIRP2. Except for ASPM and CDC27, six of these genes were validated by Sanger sequencing. In terms of the biological functions of these six genes [28,29,30,31,32,33,34,35], surprisingly, only RBM14 and MTUS1 have been reported to be associated with urothelial carcinomas. RBM14 encodes a ribonucleoprotein that functions as a general nuclear coactivator and an RNA splicing modulator in a PARP-dependent DSB repair process [29]. MTUS1, a tumour-suppressor gene encoding angiotensin-II type 2 receptor-interacting proteins, is downregulated in clear cell renal cell carcinoma [31]. However, whether these mutations alter the clinical treatment of CDC patients remains unclear. Hence, these six genes might be new targets for CDC molecular therapy.
For the remaining mutated genes shown in Fig. 1, TP53 is a known tumour-suppressor gene. SETD2 was altered in both CDC and RCC subtypes but not in BLCA. Additionally, CDKN2A was a driver gene in both the CDC and BLCA but not in other RCC subtypes. In one patient in our study, arotinib target therapy achieved good efficacy after gemcitabine plus cisplatin chemotherapy. Taken together, these results suggest that CDC might present some shared therapeutic target molecules with other RCC subtypes and BLCA, which offers new insight into the systemic treatment of CDC patients.
Next, when we compared copy number variants in our CDC cohort with other subtypes of renal cell carcinomas and bladder urothelial carcinoma and found a unique mutation spectrum. In total, 29 amplified and 6 deleted recurrent focal CNVs were identified. In terms of driver genes in Cancer Gene Census, oncogenes SKI, MUC4, TLX3, NPM1, KAT7, H3F3B, GNA11, MAP2K2, FSTL3, SH3GL1 and U2AF1 and tumour-suppressor genes RAD51B, DICER1, BCL11B, CCNB1IP1 and BAZ1A were detected. However, none of these genes are reported in association with renal cell carcinoma or urothelial carcinoma.
Notably, CDKN2A, a typical tumour-suppressor gene, was the shared gene in these four cohorts. Afterwards, we investigated the mutation spectrum of CDKN2A. CDKN2A encodes p16 and p14 to inactivate p53 [36]. Recently, a study reported 12.5% heterozygous losses and half homozygous losses in 16 patients with CDC [15]. Similar results were identified in 12% of 17 CDC patients, including one homozygous deletion and one truncation [13]. We also explored the role of CDKN2A in different subtypes of RCC, BLCA and BRCA. We detected a frameshift-deleted CDKN2A mutation in our CDC cohort, which suggested a novel mutated site different from any of the previously reported CDKN2A mutated sites in other urinary carcinomas and BRCA [37,38,39,40]. Furthermore, the most frequently cancer-specific altered pathway of this Chinese CDC cohort was the p53/RB1 pathway, which was consistent with our finding of CDKN2A. In addition, CDKN2A-altered patients displayed a significantly worse OS than patients without alterations in both the KIRC and KIRP cohorts, as validated by Girgis et al. [41], who found that KIRC with biallelic inactivation of CDKN2A indicates a poor prognosis. Importantly, CDKN2A upregulates expression of cyclin-dependent kinase 4 (CDK4)/CDK6 [15], the corresponding selective inhibitors of which, such as palbociclib, ribociclib, and abemaciclib, interfere with cell cycle progression, induce cell senescence, and promote cancer cell disruption through a cytotoxic CD8+ T cell-mediated effect [22]. No CDKN2A alteration was found to correlate with clinical outcome in patients with platinum-refractory metastatic urothelial carcinoma, and palbociclib did not show meaningful clinical efficacy [42]. Moreover, CDKN2A loss is not associated with further benefit from palbociclib in combination with letrozole in the Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 trial for patients with advanced ER+/HER2− breast cancer [43]. Nevertheless, a preclinical study of RCC cell lines reported that decreased CDKN2A is associated with sensitivity to CDK4/6 inhibitors [44], with an effective response in KIRC with wild-type VHL and CDKN2A mutations due to palbociclib [45]. Pal et al. [46] also observed a meaningful response to palbociclib in a patient with metastatic CDC harbouring a CDKN2A homozygous deletion, which suggests a direction for further study of CDC patients. Taken together, our findings indicate that this CDKN2A alteration-mediated pathway represents a rational and novel therapeutic strategy target for CDC, even for all urothelial carcinomas, which needs further validation in patients.
There are some limitations to our study. First, this was a single-centre, retrospective study with a small sample size due to the rare incidence of the cancer. Second, it was a study of single omics, which might also lack sufficient validation information. Third, there was a lack of collecting duct carcinoma cell lines and animal models to complete validations in vivo and in vitro. Hence, in the future, we will conduct a prospective, randomised and multicentre clinical trial to obtain multi-omics data to validate our findings.
Conclusion
In conclusion, our study offers a genomic spectrum of a Chinese population with CDC. CDC is not only characterised as a unique type of solid tumour, but it shares some specific molecules with other RCC subtypes and urinary tract carcinoma. The CDKN2A alteration-mediated p53/RB1 pathway might provide new insight into potential prognostic and therapeutic targets for CDC patients.
Availability of data and materials
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number HRA001344 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA001344).
Abbreviations
- BLCA:
-
Bladder urothelial carcinoma
- BRCA:
-
Breast cancer
- CDC:
-
Collecting duct carcinoma
- CGC:
-
Cancer gene census
- CNV:
-
Copy number variant
- COSMIC:
-
Catalogue of somatic mutations in cancer
- EAS:
-
East Asian
- ESP:
-
Exome sequencing project
- FFPE:
-
Formaldehyde-fixed paraffin-embedded
- GETUG:
-
Groupe d'Etudes des Tumeurs Uro-Génitales
- IGV:
-
Integrative genomics viewer
- NCCN:
-
National comprehensive cancer network
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- RCC:
-
Renal cell carcinoma
- SNV:
-
Single-nucleotide variant
- TCGA:
-
The cancer genome atlas
- UTUC:
-
Upper tract urothelial carcinoma
- WES:
-
Whole-exome sequencing
- WT:
-
Wild-type
References
Baniak N, Tsai H, Hirsch MS. The differential diagnosis of medullary-based renal masses. Arch Pathol Lab Med. 2021;145(9):1148–70.
Kobayashi N, Matsuzaki O, Shirai S, Aoki I, Yao M, Nagashima Y. Collecting duct carcinoma of the kidney: an immunohistochemical evaluation of the use of antibodies for differential diagnosis. Hum Pathol. 2008;39(9):1350–9.
Albadine R, Schultz L, Illei P, Ertoy D, Hicks J, Sharma R, Epstein JI, Netto GJ. PAX8 (+)/p63 (−) immunostaining pattern in renal collecting duct carcinoma (CDC): a useful immunoprofile in the differential diagnosis of CDC versus urothelial carcinoma of upper urinary tract. Am J Surg Pathol. 2010;34(7):965–9.
Tokuda N, Naito S, Matsuzaki O, Nagashima Y, Ozono S, Igarashi T. Collecting duct (Bellini duct) renal cell carcinoma: a nationwide survey in Japan. J Urol. 2006;176(1):40–3.
Dason S, Allard C, Sheridan-Jonah A, Gill J, Jamshaid H, Aziz T, Kajal B, Kapoor A. Management of renal collecting duct carcinoma: a systematic review and the McMaster experience. Curr Oncol. 2013;20(3):e223–32.
Motzer RJ, Jonasch E, Boyle S, Carlo MI, Manley B, Agarwal N, Alva A, Beckermann K, Choueiri TK, Costello BA, et al. NCCN guidelines insights: kidney cancer, version 1.2021. J Natl Compr Cancer Netw. 2020;18(9):1160–70.
Oudard S, Banu E, Vieillefond A, Fournier L, Priou F, Medioni J, Banu A, Duclos B, Rolland F, Escudier B, et al. Prospective multicenter phase II study of gemcitabine plus platinum salt for metastatic collecting duct carcinoma: results of a GETUG (Groupe d’Etudes des Tumeurs Uro-Génitales) study. J Urol. 2007;177(5):1698–702.
Zeng Y, Zhang W, Li Z, Zheng Y, Wang Y, Chen G, Qiu L, Ke K, Su X, Cai Z, et al. Personalized neoantigen-based immunotherapy for advanced collecting duct carcinoma: case report. J Immunother Cancer. 2020;8(1):e000217.
Sheng X, Cao D, Yuan J, Zhou F, Wei Q, Xie X, Cui C, Chi Z, Si L, Li S, et al. Sorafenib in combination with gemcitabine plus cisplatin chemotherapy in metastatic renal collecting duct carcinoma: a prospective, multicentre, single-arm, phase 2 study. Eur J Cancer. 2018;100:1–7.
Pinto A, Garrido M, Aguado C, Alonso T, Gajate P, Maximiano C, García-Carbonero I, Martín A, Gallegos I, Arranz JA, et al. Collecting duct carcinoma of the kidney: analysis of our experience at the SPANISH ‘Grupo Centro’ of genitourinary tumors. Kidney Cancer. 2019;3:1–6.
Bratslavsky G, Gleicher S, Jacob JM, Sanford TH, Shapiro O, Bourboulia D, Gay LM, Andrea Elvin J, Vergilio JA, Suh J, et al. Comprehensive genomic profiling of metastatic collecting duct carcinoma, renal medullary carcinoma, and clear cell renal cell carcinoma. Urol Oncol. 2021;39(6):367.e1-5.
Motzer RJ, Bacik J, Mariani T, Russo P, Mazumdar M, Reuter V. Treatment outcome and survival associated with metastatic renal cell carcinoma of non-clear-cell histology. J Clin Oncol. 2002;20(9):2376–81.
Pal SK, Choueiri TK, Wang K, Khaira D, Karam JA, Van Allen E, Palma NA, Stein MN, Johnson A, Squillace R, et al. Characterization of clinical cases of collecting duct carcinoma of the kidney assessed by comprehensive genomic profiling. Eur Urol. 2016;70(3):516–21.
Becker F, Junker K, Parr M, Hartmann A, Füssel S, Toma M, Grobholz R, Pflugmann T, Wullich B, Strauss A, et al. Collecting duct carcinomas represent a unique tumor entity based on genetic alterations. PLoS ONE. 2013;8(10):e78137.
Wang J, Papanicolau-Sengos A, Chintala S, Wei L, Liu B, Hu Q, Miles KM, Conroy JM, Glenn ST, Costantini M, et al. Collecting duct carcinoma of the kidney is associated with CDKN2A deletion and SLC family gene up-regulation. Oncotarget. 2016;7(21):29901–15.
Gargiuli C, Sepe P, Tessari A, Sheetz T, Colecchia M, de Braud FGM, Procopio G, Sensi M, Verzoni E, Dugo M. Integrative transcriptomic analysis reveals distinctive molecular traits and novel subtypes of collecting duct carcinoma. Cancers (Basel). 2021;13(12):2903.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17(1):31.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2018;47(D1):D941–7.
Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, Arpino G, De Laurentiis M, Puglisi F, De Placido S, et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. 2018;8:608.
Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28(3):423–5.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Zhang H, Meltzer P, Davis S. RCircos: an R package for Circos 2D track plots. BMC Bioinform. 2013;14(1):244.
Hodge RG, Schaefer A, Howard SV, Der CJ. RAS and RHO family GTPase mutations in cancer: twin sons of different mothers? Crit Rev Biochem Mol Biol. 2020;55(4):386–407.
Li J, Xu C, Lee HJ, Ren S, Zi X, Zhang Z, Wang H, Yu Y, Yang C, Gao X, et al. A genomic and epigenomic atlas of prostate cancer in Asian populations. Nature. 2020;580(7801):93–9.
Kai M. Roles of RNA-binding proteins in DNA damage response. Int J Mol Sci. 2016;17(3):310.
Li J, Wang Y, Rao X, Wang Y, Feng W, Liang H, Liu Y. Roles of alternative splicing in modulating transcriptional regulation. BMC Syst Biol. 2017;11(Suppl 5):89.
Bozgeyik I, Yumrutas O, Bozgeyik E. MTUS1, a gene encoding angiotensin-II type 2 (AT2) receptor-interacting proteins, in health and disease, with special emphasis on its role in carcinogenesis. Gene. 2017;626:54–63.
Lv Q, Dong F, Zhou Y, Cai Z, Wang G. RNA-binding protein SORBS2 suppresses clear cell renal cell carcinoma metastasis by enhancing MTUS1 mRNA stability. Cell Death Dis. 2020;11(12):1056.
Ray MR, Wafa LA, Cheng H, Snoek R, Fazli L, Gleave M, Rennie PS. Cyclin G-associated kinase: a novel androgen receptor-interacting transcriptional coactivator that is overexpressed in hormone refractory prostate cancer. Int J Cancer. 2006;118(5):1108–19.
Jain PB, Guerreiro PS, Canato S, Janody F. The spectraplakin Dystonin antagonizes YAP activity and suppresses tumourigenesis. Sci Rep. 2019;9(1):19843.
Banh RS, Iorio C, Marcotte R, Xu Y, Cojocari D, Rahman AA, Pawling J, Zhang W, Sinha A, Rose CM, et al. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat Cell Biol. 2016;18(7):803–13.
Paul MR, Pan TC, Pant DK, Shih NN, Chen Y, Harvey KL, Solomon A, Lieberman D, Morrissette JJ, Soucier-Ernst D, et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J Clin Invest. 2020;130(8):4252–65.
Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–12.
Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Robertson AG, Chu A, Beroukhim R, Cibulskis K, Signoretti S, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9.
Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, Vocke CD, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374(2):135–45.
Weinstein JN, Akbani R, Broom BW, Wang WY, Verhaak RG, McConkey D, Lerner S, Morgan M, Creighton CJ, Smith C, et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22.
Kim K, Hu W, Audenet F, Almassi N, Hanrahan AJ, Murray K, Bagrodia A, Wong N, Clinton TN, Dason S, Mohan V, et al. Modeling biological and genetic diversity in upper tract urothelial carcinoma with patient derived xenografts. Nat Commun. 2020;11(1):1975.
Girgis AH. Clear cell renal cell carcinoma with biallelic inactivation of CDKN2A/B on 9p21 have distinct gene expression signature and are associated with poor prognosis. bioRxiv. 2017:143180.
Rose TL, Chism DD, Alva AS, Deal AM, Maygarden SJ, Whang YE, Kardos J, Drier A, Basch E, Godley PA, et al. Phase II trial of palbociclib in patients with metastatic urothelial cancer after failure of first-line chemotherapy. Br J Cancer. 2018;119(7):801–7.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35.
Logan JE, Mostofizadeh N, Desai AJ, Vone Euw E, Conklin D, Konkankit V, Hamidi H, Eckardt M, Anderson L, Chen HW, et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer Res. 2013;33(8):2997–3004.
Kancherla P, Schardein J, Woodford MR, Backe SJ, Smith G, Sager R, Bourboulia D, Shapiro O, Ross J, Bratslavsky G, et al. PD64-01 the status of the tumor suppressors VHL and CDKN2A impacts clear cell renal cell carcinoma sensitivity to CDK 4/6 inhibitors. J Urol. 2020;203(Suppl 4):e1298.
Pal SK, Ali SM, Ross J, Choueiri TK, Chung JH. Exceptional response to palbociclib in metastatic collecting duct carcinoma bearing a CDKN2A Homozygous deletion. JCO Precis Oncol. 2017;1:1–5.
Acknowledgements
Not applicable.
Funding
This work was supported by the Shanghai Rising-Star Program (20QA1411800) and the Youth National Science Foundation of Jiangxi Province (20192BAB215014).
Author information
Authors and Affiliations
Contributions
GSY, QY and JL conceived of the study and interpreted the data. XJL, GH and MMH collected and provided samples. WHZ, TW, ZWW and QC collected the clinical information and downloaded the public data. GH and HRZ carried out experiments and analysed the data. HRZ wrote the manuscript. All authors had final approval of the submitted and published versions.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All research participants or their legal representatives signed informed consent forms for participation in the research. Ethics approval was obtained from the institutional review board of Changhai Hospital (CHEC2021-064). The study was conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983, and the Good Clinical Practice guidelines.
Consent for publication
Not applicable.
In addition, the accessible links for SNV data in this paper are as follows: https://www.cbioportal.org/study/summary?id=kirc_tcga; https://www.cbioportal.org/study/summary?id=kirp_tcga; and https://www.cbioportal.org/study/summary?id=blca_tcga. The accessible links for SNV, CNV and survival data of CDKN2A in this manuscript are as follows: https://www.cbioportal.org/results/comparison?cancer_study_list=kirc_tcga&Z_SCORE_THRESHOLD=2.0&RPPA_SCORE_THRESHOLD=2.0&profileFilter=mutations%2Cgistic&case_set_id=kirc_tcga_cnaseq&gene_list=CDKN2A&geneset_list=%20&tab_index=tab_visualize&Action=Submit; https://www.cbioportal.org/results/oncoprint?cancer_study_list=kirp_tcga&Z_SCORE_THRESHOLD=2.0&RPPA_SCORE_THRESHOLD=2.0&profileFilter=mutations%2Cgistic&case_set_id=kirp_tcga_cnaseq&gene_list=CDKN2A&geneset_list=%20&tab_index=tab_visualize&Action=Submit; https://www.cbioportal.org/results/oncoprint?cancer_study_list=blca_tcga&Z_SCORE_THRESHOLD=2.0&RPPA_SCORE_THRESHOLD=2.0&profileFilter=mutations%2Cgistic&case_set_id=blca_tcga_cnaseq&gene_list=CDKN2A&geneset_list=%20&tab_index=tab_visualize&Action=Submit; https://www.cbioportal.org/results/oncoprint?cancer_study_list=brca_tcga&Z_SCORE_THRESHOLD=2.0&RPPA_SCORE_THRESHOLD=2.0&profileFilter=mutations%2Cgistic&case_set_id=brca_tcga_cnaseq&gene_list=CDKN2A&geneset_list=%20&tab_index=tab_visualize&Action=Submit.
Competing interests
The authors declared no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The summary of the clinicopathological features of the patients included.
Additional file 2: Table S2.
The summary of the whole exosome sequencing data in patients with CDC.
Additional file 3: Table S3.
The summary of the filtered somatic short variant discovery.
Additional file 4: Figure S1.
The Sanger validations of RBM14, GAK, DST and XIRP2.
Additional file 5: Figure S2.
The Sanger validations of SETD2, MTUS1, RNF213, ASPM and CDC27.
Additional file 6: Figure S3.
The mutation signature of collecting duct carcinoma patients in our study.
Additional file 7: Table S4.
GISTIC2.0 results of deleted genes in CDC, BLCA, KIRC and KIRP cohorts.
Additional file 8: Figure S4.
Somatic copy number variants (CNV) detection.
Additional file 9: Table S5.
GISTIC2.0 results of amplified genes in CDC, BLCA, KIRC and KIRP cohorts.
Additional file 10: Table S6.
Details of pathway-level alterations across the CDC, BLCA, KIRC and KIRP cohorts considering the single nucleotide variants (SNV).
Additional file 11: Table S7.
Details of pathway-level alterations across CDC, BLCA, KIRC and KIRP cohorts considering the copy number variants (CNV).
Additional file 12: Figure S5.
The mutation landscape on the KIRC specific pathway.
Additional file 13: Figure S6.
The mutation landscape on the KIRP specific pathway.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Lu, X., Huang, G. et al. A genomic mutation spectrum of collecting duct carcinoma in the Chinese population. BMC Med Genomics 15, 1 (2022). https://doi.org/10.1186/s12920-021-01143-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01143-2