
Abstract
Adenosine-to-inosine (A-to-I) editing of RNA, catalyzed by adenosine deaminase acting on RNA (ADAR) enzymes, is a prevalent RNA modification in mammals. It has been shown that A-to-I editing plays a critical role in multiple diseases, such as cardiovascular disease, neurological disorder, and particularly cancer. ADARs are the family of enzymes, including ADAR1, ADAR2, and ADAR3, that catalyze the occurrence of A-to-I editing. Notably, A-to-I editing is mainly catalyzed by ADAR1. Given the significance of A-to-I editing in disease development, it is important to unravel the complex roles of ADAR1 in cancer for the development of novel therapeutic interventions.
In this review, we briefly describe the progress of research on A-to-I editing and ADARs in cancer, mainly focusing on the role of ADAR1 in cancer from both editing-dependent and independent perspectives. In addition, we also summarized the factors affecting the expression and editing activity of ADAR1 in cancer.
Similar content being viewed by others


Background
Adenosine-to-inosine (A-to-I) RNA editing is a biological process that converts adenosine to inosine in double-stranded RNA (dsRNA) molecules. Inosine is recognized by the cell as guanosine and paired with cytosine. Therefore, the base conversion may affect the amino acid sequence of the protein. ADARs, a family of enzymes, act on double-stranded RNA (dsRNA) to catalyze this conversion. Among them, ADAR1 has been extensively studied in cancer, while ADAR2 and ADAR3 have received less attention, and ADAR3 has a total lack of activity [1]. ADAR1 is widely expressed throughout mammalian tissues and has been shown to modify substrates within both nuclear and cytoplasmic compartments [2]. Exploring the biological mechanisms of ADAR1 in cancer may provide important implications for clinical diagnosis and treatment.
In this review, we first provide information on the characters of ADAR1, and then we describe the roles that ADAR1 can promote or suppress cancer in RNA editing-dependent and independent manners. Finally, we summarize the modulation of the level and editing activity of ADAR1 by multiple factors.
A-to-I RNA editing
Deamination of adenosine-to-inosine (A-to-I) is a widespread post-transcriptional modification of RNA and is catalyzed by Adenosine deaminase acting on RNA (ADAR) enzymes in dsRNA [3]. As early as 2004, it was reported that ADAR1 plays an important role in embryonic development by editing dsRNA to keep embryos alive and prevent stress-induced apoptosis [4]. A large number of A-to-I editing sites exist in the human genome which lays the foundation for transcriptome diversity and most editing occurs in the Alus sequence [5, 6]. With the development of high-throughput sequencing analysis, RNA editing has been seen in many diseases, especially in cancer. The RNA modification of A-to-I editing is significantly altered in many cancer types, most of which show increased editing and are associated with patient survival, and also affect drug sensitivity [7, 8].
Given the important roles of A-to-I editing in cancer, this RNA modification process is thought to be potentially involved in carcinogenesis as an epigenetic mechanism [9]. To more intuitively and accurately understand the activity of ADAR enzymes and the changes in A-to-I editing in organisms, scientists have developed and designed a series of tools to objectively reflect the level of A-to-I editing (Table 1). Many of them are bioinformatics algorithms, by which A-to-I editing sites can be identified based on transcriptome sequencing [10, 11]. To quantify ADAR activity, Roth et al. present the Alu editing index (AEI), a robust and simple-to-use computational tool, based on the fact that almost all human editing events occur in Alu repeats. They defined the AEI as the ratio of the number of A-G to the number of A-G and A-A at the RNA-DNA mismatch sites within human Alu repeats. Unlike previous approaches, the AEI offers a reliable, unbiased estimate of the editing level that averages over millions of sites, enables direct comparison of several samples, and is simple to use for any kind of organism. It can also enable us to investigate the factors influencing the global editing activity [12]. To circumvent the errors in RNA-seq data and make the detection of editing sites more accurate, researchers exploited EndoVIPER-seq to improve such problems [13]. Recently, Zhu et al. developed a network interactive server to facilitate users to study the relationship between A-to-I editing sites of interest and cancer [14]. Alternatively, a sensitive ADAR editing reporter can be stably introduced into the cell to determine the editing activity according to the fluorescent signal quantitatively [15].
ADARs isoforms and domain structures
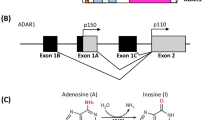
The three members of the ADAR family, ADAR1, ADAR2, and ADAR3, all contain highly evolutionarily conserved catalytic deaminase domain (DM) and three or two double-stranded RNA-binding domains (dsRBDs). ADAR1 is more specific because it has a part of the structure that is different from ADAR2 and ADAR3 (Fig. 1). As early as 1996, it was reported that ADAR1 contains two major isoforms, the interferon-inducible ADAR1 p150, and the constitutively expressed ADAR1 p110. ADAR1 p150 is localized both in the cytoplasm and nucleus, whereas ADAR1 p110 is mainly in the nucleus [31]. In the three dsRBD (RI, RII, and RIII) domains of ADAR1, scientists mutated each dsRBD separately to explore their effects on the binding activity of dsRNA and adenosine deaminase activity. The results showed that the binding activity of RNA would be greatly reduced after the mutation of RIII, so it is the most important of the three dsRBDs [32]. Additionally, RIII as an RNA-sensitive nucleocytoplasmic transport signal in ADAR1 can mediate nuclear import [33]. Nuclear localization signal (NLS) that overlaps the RIII domain, mediates nuclear import by interacting with the import receptor transportin 1 (Trn1). And the RIII domain can serve as a bridge between the two sides of the NLS structure, facilitating the localization of the NLS and the interaction with Trn1 [34, 35]. In addition, ADAR1 contains the Z-DNA/RNA binding domain (ZBD), also known as Zalpha (Zα) or Zbeta (Zβ), which is a 78-amino acid protein-fold structure that binds exclusively to Z-DNA and Z-RNA but not to B-DNA. ZBDs may play an important role in the innate antiviral immune response. Both ADAR1 isoforms contain a Zβ domain, but besides the Zβ domain, ADAR1 p150 contains a Zα domain [36,37,38]. There is a nuclear export signal (NES) in the Zα domain, which regulates ADAR1 p150 penetration out of the nucleus [39]. This also clarifies why the majority of ADAR1 p110, ADAR2, and ADAR3 are found in the nucleus rather than the cytoplasm.

The domain structures of ADARs. All ADARs contain catalytically active deaminase domains and nuclear localization signal (NLS). There are two isoforms of ADAR1, p150 and p110, both of them contain a zβ domain, three dsRNA binding domains. ADAR1 p150 also contains a zα domain and a nuclear export signal (NES) at the N-terminal sequence. Compared to ADAR1, ADAR2 and ADAR3 have one fewer dsRNA binding domain. ADAR3 has an R-domain, which enables it to bind not only double-stranded RNA but also single-stranded RNA.
In contrast to ADAR1, ADAR2 and ADAR3 have only two dsRBD structural domains and their NLS domains are localized to the N terminus. In addition, ADAR3 contains a unique R domain, which enables it to bind not only double-stranded RNA but also single-stranded RNA [1].
RNA editing-dependent ADAR1 functions in tumors
RNA editing in coding regions
Recently, the sequencing technologies advances rapidly, which enable the detection of millions of A-to-I editing sites in human [11, 20, 22]. At these sites, base changes may alter amino acid sequences during translation, largely affecting the translation of transcripts or even altering the localization and function of encoded proteins (Table 2).
In cancer, antizyme inhibitor 1 (AZIN1) is one of the most studied proteins for A-to-I editing in the coding region. Initially, studies on the role played by over-edited AZIN1 were performed in liver cancer. They found that A-to-I editing occurs at position 367, which results in a serine-to-glycine substitution. The edited form of AZIN1 gains higher protein stability, and stronger affinity to antizyme, and undergoes cytoplasmic-to-nuclear translocation, which promotes cell proliferation [44]. Then, Ghalali et al. reported that the nuclear translocation of edited AZIN1 is dependent on binding to the actin/myosin9 complex, which can enhance cellular aggressiveness and is associated with worse outcomes of prostate cancer [47]. Recently, studies on RNA editing of AZIN1 have gradually increased and expanded to other cancer types, such as colorectal cancer, esophageal cancer, endometrial cancer, gastric cancer, lung cancer, etc. (Table 2).
By analyzing RNA and DNA data, Hu et al. found that Bladder cancer-associated protein (BLCAP) was also over-edited in liver cancer tissues, and over-edited BLCAP showed a cell proliferation promoting phenotype both in vivo and in vitro, which may be achieved by activating the AKT/mTOR signaling pathway [52]. Notably, over-edited GABAA receptor alpha3 (Gabra3) can inhibit AKT activation, thereby suppressing breast cancer progression [60]. In addition, similar findings regarding the phenomenon that over-edited BLCAP promotes cancer progression were obtained in another study on cervical cancer. It was shown that wild-type BLCAP could inhibit the phosphorylation of signal transducer and activator of transcription 3 (STAT3) by directly interacting with it, whereas over-edited BLCAP lost its ability to inhibit STAT3 activation, which in turn promoted cancer progression [53].
There are still some unmentioned studies on the action of ADAR1-mediated RNA editing in the coding region. Here, we compile recent studies on the impact of over-editing proteins on cancer progression (Table 2).
RNA editing in non-coding regions (3’UTR, Alus, miRNA, etc.)
ADAR1-mediated A-to-I editing is also important for the regulation of non-coding RNAs. Although editing of non-coding RNAs cannot affect protein translation by directly changing codons, it can indirectly regulate protein expression through other pathways.
In 3’ untranslated region
It is well established that the transmission of genetic information from mRNA to protein comes from the correct translation of the coding sequence on mRNA, and the untranslated regions (UTRs) at the 5’ and 3’ ends of mRNA have a complex role in regulating its stable translation. Currently, a large number of studies have shown the existence of multiple RNA modification types within the 3’ UTR, including A-to-I editing [62]. A double-stranded structure formed by the inverted-repeat Alus (IR-Alus) are often found in 3’UTR, which is more likely to recruit ADAR1 and undergo an A-to-I transition. This editing can regulate the localization, translation, and stability of mRNA [63]. In addition, A-to-I editing occurring on IR-Alus can also neutralize its immunogenicity, thereby functioning as a specific negative regulator of the MDA5-MAVS pathway, which prevents the production of type I interferon (IFN-I) [64,65,66].
By analyzing the transcriptomic data from the tumor and its paired “normal” tissues, researchers found that the number of editing sites and editing levels of some known sites in 3’UTR are both significantly increased [67]. We have compiled current studies of A-to-I editing on 3’UTR (Table 3).
In Alus
Among transposable elements (TES), Alus is the most abundant repetitive element, accounting for about 10% of the human genome. The dsRNA structure of Alus facilitates the recruitment of ADAR1 for A-to-I editing [5, 87]. It was shown that A-to-I editing occurs predominantly in Alus sequence (> 90%) and is mainly catalyzed by ADAR1. It had been demonstrated that the endogenous “self” Alus sequence can be converted to “nonself” sequence by forming stem-loop structures, the latter can be sensed by melanoma differentiation-associated protein 5 (MDA5), which lead to the production of IFN-I. ADAR1-mediated A-to-I editing can neutralize the immunogenicity of Alus by disrupting the formation of stem-loop, converting the aberrantly expressed “nonself” Alus sequence into a “self” sequence, which avoids its stimulation of MDA5 and inhibits the expression of IFN-I regulated by the MDA5/MAVS/IRF7 signaling pathway [66, 88, 89]. Meanwhile, researchers found that IFN-I activated by the immunogenicity of Alus plays a key role in tumor immune surveillance, which could make the tumor more vulnerable to inhibition of ADAR1. This suggests that ADAR1 can serve as a new target in tumor therapy [90,91,92].
In MicroRNA
MicroRNAs (miRNAs) are known to be non-coding RNAs whose main function is to target the 3’UTR of mRNAs to degrade them and/or inhibit their translation [93]. The binding of miRNA to 3’UTR of mRNAs can be intervened by A-to-I RNA editing, which occurs on 3’UTR of mRNAs or miRNA (Table 3). For example, miR-25-3p and miR-125a-3p can bind to unedited Dihydrofolate reductase (DHFR) but not edited-DHFR, leading to upregulation of DHFR, which can increase tumor resistance to methotrexate and promote tumor proliferation [68]. Besides, miRNAs themselves undergo A-to-I editing and alter their binding preference. For example, in metastatic melanoma, edited but not unedited miR-378a-3p was found to target the 3’UTR of the oncogene PARVA to suppress its expression [83]. ADAR1, an enzyme that specifically recognizes dsRNA, can regulate miRNA maturation at various critical nodes of miRNA processing maturation. It can target the dsRNA structure of certain pri-miRNAs, causing them to undergo editing, thereby inhibiting the processing of pri-miRNA by RNase III DROSHA and reducing miRNA levels [94]. In addition, ADAR1 can also regulate miRNA biological processes by directly interacting with DROSHA and/or DGCR8 [95].
In CircRNA and LncRNA
In recent years, the study of ADAR1 in cancer has gradually expanded to circular RNA (circRNA) and Long noncoding RNA (lncRNA). CircRNA is considered a unique epigenetic regulatory molecule involved in the cancer process. After the booming development of high-throughput sequencing technology and bioinformatics, the biological significance of circRNA has gradually been elucidated [78]. The most important function of circRNA is to act as miRNA sponges, which can enhance gene expression levels by derepressing miRNAs on their target genes [96]. Recently, studies have reported that ADAR1 can reduce circRNA biogenesis by interacting with RNA helicase DHX9 and disrupting the formation of double-stranded RNA or by altering the secondary structure of the precursor of circRNA [97, 98]. Additionally, there is also research that hsa_circ_0004872 can inhibit ADAR1 expression through hsa_circ_0004872/miR-224/Smad4/ADAR1 negative feedback loop and exert oncogenic effects in gastric cancer (GC) [78].
Long noncoding RNAs (LncRNAs) are non-coding RNAs with more than 200 nucleotides that are involved in the regulation of a variety of biological processes in cancer [99, 100]. Some lncRNA also undergo ADAR1-mediated A-to-I editing in cancer. For instance, in prostate cancer, Prostate cancer antigen 3 (PCA3), a lncRNA, can form a double-stranded RNA with PRUNE2 (a human homolog of the Drosophila prune gene) to attract ADAR1 binding and exert an editing effect to regulate PRUNE2’s level [86].
RNA editing-independent ADAR1 functions in tumors
Acting in a protein-protein interaction manner
Although most of the studies on ADAR1 have been conducted on its editing function, the editing-independent role of ADAR1 has been successively confirmed by studies [101]. Currently, most of the studies on ADAR1’s editing-independent role in cancer are related to the processing of miRNA.
miRNA maturation requires a series of processes. In the nucleus, primary miRNA (pri-miRNA) is transcribed by RNA polymerase II and is processed by the DROSHA-DGCR8 complex, through which precursor miRNA (Pre-miRNA) with about 70 nucleotides is generated. Pre-miRNA is then exported to the cytoplasm where it can be further processed by the DICER enzyme into a double-stranded miRNA. Subsequently, one miRNA strand of the double-strand is degraded and the other is the mature miRNA. By binding to 3’UTR of the target mRNA, the mature miRNA can mediate its degradation [102]. During this process, the function of the three critical proteins, DROSHA, DGCR8, and DICER, are influenced by ADAR1 in an editing-independent manner. For example, ADAR1 can form a complex with DGCR8 interfering with the formation of the DGCR8-DROSHA complex [103] (Fig. 2A). ADAR1 can also promote the degradation of DROSHA by enhancing its ubiquitination [104]. In addition, ADAR1 can form a heterodimer with DICER, which accelerates the cleavage of pre-miRNA and promotes the loading of miRNA-induced silencing complex (miRISC) onto 3’UTR of the target mRNA, thereby regulating the expression of target genes [105,106,107] (Fig. 2B). Notably, ADAR1 can also regulate the expression level of DICER by regulating the level of let-7 [103].

Editing-independent functions of ADAR1. A ADAR1 forms a complex with DGCR8 and interferes with DGCR8-DROSHA complex formation. B ADAR1 binds to DICER, accelerating the cleavage of pre-miRNAs to promote miRNA maturation, the later targets mRNAs, and downregulates protein levels. C ADAR1 competes with 3’UTR-binding factors for binding to the 3’UTR. When ADAR1 is downregulated, these proteins may have greater access to the 3'UTR, which could result in 3'UTR lengthening. D ADAR1 facilitates the binding of HUR to its target transcripts and increases transcript stability, which in turn enhances the translation efficiency of its target mRNAs
Acting as RNA binding protein
ADAR1, as an RNA-binding protein, acts directly on RNA to perform some editing-independent functions. Besides the aforementioned interactions of ADAR1 with some key proteins in miRNA processing, ADAR1 can also directly bind to pri-miRNA, which regulates the maturation of miRNA [108]. Furthermore, ADAR1 competes with some 3’UTR processing factors for “occupancy” of the 3’UTR to be involved in the processing [95] (Fig. 2C).
ADAR1 and other RNA-binding proteins can jointly regulate gene expression by binding to the same RNA substrates. For instance, the interaction of ADAR1 with nuclear factor 90 (NF90) family proteins rely on the bridge of dsRNA and mediates gene expression [109]. Human antigen R (HuR) is another RNA binding protein, it can specifically bind the ARE element (AU-rich element) of target mRNA and upregulates its expression by increasing the stability and translation efficiency of target mRNA [110]. During this process, ADAR1 can exert its function by cooperating with HuR [111] (Fig. 2D). Similarly, it has also been shown that ADAR1 can bind to cyclin-dependent kinases 2 (CDK2) mRNA to exert a pro-oncogenic effect, even though the deeper mechanisms behind this are not clear. However, it is certain that none of these processes is dependent on the editing activity of ADAR1 [108, 112].
These findings highlight the versatility of ADAR1 and suggest that it may have multiple roles beyond its well-characterized A-to-I RNA editing function. In summary, ADAR1 possesses both editing-dependent and independent functions in tumors, which exert diverse effects on various biological processes. Further studies are required to elucidate the precise molecular mechanisms underlying these functions and their implications for disease pathogenesis and therapeutic development.
Influencing factors of ADAR1 expression and activity
Regulation of ADAR1 through post-transcriptional or post-translational modification
As the coding gene, ADAR1 modifications that occur at the mRNA level or protein level can have an impact on its expression, localization, function, etc. In a recent study on glioma, researchers found that RNA methyltransferase-like 3 (METTL3) can target the m6A site near the termination codon of the ADAR1 transcript to regulate ADAR1 protein expression thereby affecting the proliferation of glioma cells [112].
The impact of post-translational modifications (PTMs) on the function of the protein itself and the interaction with other molecules cannot be ignored. Common types of modifications include phosphorylation, ubiquitination, glycosylation, and palmitoylation [113]. Among them, phosphorylation is one of the most studied modifications in PTMs, which can phosphorylate some proteins to regulate their localization, conformation, and activity [114]. It has been shown that MKK6–p38–MSK1/2 mitogen-activated protein (MAP) kinases phosphorylate two threonine sites and three serine sites (T808, T811, S814, S823, and S825) of ADAR1, and tethers it to the nuclear export protein Exportin-5 to facilitate its nuclear exportation. Then, ADAR1-p110 can compete with Staufen1 for the 3’UTR of some antiapoptotic transcripts to participate in the stress response and protect cells from apoptosis under stress conditions. Notably, the aforementioned function is independent of its RNA editing activity [115] (Fig. 3A). Akt (Protein Kinase B) activity has been reported to be associated with several cancer processes [116]. Recently, Alberto Bavelloni et al. showed that ADAR1 p110 can act as a substrate for Akt kinase, and phosphorylation at T738 of ADAR1 p110 can significantly reduce its editing activity, which in turn influences the progression of editing-related diseases [117]. According to the information from the Uniprot website (https://www.uniprot.org/uniprotkb/P55265/entry), we note that the phosphorylation sites of ADAR1 are not evenly distributed. Among them, most of the sites tend to be distributed on unknown functional domains and are densely distributed in the region where the RI and RII structural domains join. This suggests that it may be a phosphorylation-dependent functional regulatory region that influences protein structure, activity, and function. So far, there are only four phosphorylation sites distributed in the known functional domains of ADAR1. Three of these phosphorylation sites are located within the second dsRNA binding domain (RII), and one is in the deaminase domain. Therefore, it suggests that modification of these sites affects the binding of ADAR1 to some double-stranded RNAs and may also affect their activity and function (Fig. 3B). In addition, modification of ubiquitination has also been reported. The E3 ubiquitin ligase SMURF2 binds directly to ADAR1 p110, which ubiquitinates the lysine at position 744, increases its stability, and promotes A-to-I editing activity [118]. Sumoylation is a post-translational modification of proteins similar to ubiquitination. SUMO-1, a ubiquitin-like protein modification molecule, can modify the lysine residue at position 418 of ADAR1 and be found to reduce the editing activity of ADAR1 [119].

The phosphorylation of ADAR1. A Phosphorylation of ADAR1 by MKK6–p38–MSK1/2 mitogen-activated protein (MAP) kinase promotes its binding to Exportin-5 (Xpo5), which exits the nucleus and competes with Staufen1 for the 3'-UTR of antiapoptotic genes. B Phosphorylation modification sites of ADAR1 (from UniProt: https://www.uniprot.org/uniprotkb/P55265/entry, positions source: UniProt and PRIDE S: phosphoserine, T: phosphothreonine, Y: phosphotyrosine)
Impact of protein-protein interaction on the catalytic activity of ADAR1
Both homodimerization and heterodimerization can be formed between ADAR1 and ADAR2, which is extremely important for the activity of the ADAR family [120]. Other proteins in the cell can also act on ADARs, thereby affecting their activity. For example, death-associated protein 3 (DAP3) is considered as an editing repressor, it can inhibit A-to-I editing in cancer cells by interfering with the homodimerization of ADAR1 and inhibiting the binding of ADAR2 to dsRNA [121]. In addition, cytoplasmic polyadenylation element-binding protein 3 (CPEB3) inhibits ADAR1-mediated RNA editing by localizing ADAR1 mRNA to the processing body (P-body), inhibiting the translation of ADAR1, thereby suppressing gastric cancer progression [122]. It has been shown that in the nucleus, DDX6 interacts with ADAR1 to regulate A-to-I editing, and the C-terminal domain or nuclear entry features of DDX6 were found to repress ADAR1 activity [123].
It is noteworthy that recent research has shown that Long non-coding RNAs (LINC00624) can also affect the stability of ADAR1 by binding to it [79].
Impact of ADAR1 by other factors
It has been reported that a high level of editing can be triggered by the presence of an Edit Inducing Element (EIE) in the vicinity of the editing sites, which can form long double-stranded structures that are easily hyper-editing, recruiting ADAR1, thereby increasing editing efficiency [124].
While the secondary structure (dsRNA) is more important for ADAR1 to bind, the base pair of the duplex RNA adjacent to editing sites also influence the editing efficiency. For example, study had shown that a non-Watson–Crick pairing of guanosine (G: A or G: G) 5′ nearest neighbor of editing sites in duplex RNA can facilitate ADAR editing [125, 126].
It has also been reported that intracellular acidification enhances RNA editing by increasing ADAR1 base flipping and deamination rates [127].
Conclusions and prospects
A-to-I editing is one of the most studied types of RNA modifications to date, especially today with the rapid development of high-throughput sequencing technologies and bioinformatics. A series of tools have been developed to detect editing sites and to predict the relevance of editing to cancer [13, 14].
In this review, we briefly summarize studies related to the involvement of ADAR1 in cancer progression from both editing-dependent and editing-independent perspectives. We also describe a number of factors that affect ADAR1’s expression and activity, including ADAR1’s own modifications (post-transcriptional and post-translational modifications), interactions of other proteins with ADAR1, the primary sequence and secondary structure of the substrates (RNAs) of ADAR1, and the effect of the microenvironment on its function.
Currently, the role of ADAR1 in tumor immunotherapy is receiving attention. For example, the editing of Alus by ADAR1 reduces its immunogenicity, which in turn reduces the body’s tumor immune surveillance and tumor immunotherapy effects [64]. The Warburg effect is widely present in cancerous tissues. Currently, a large number of abnormalities and mutations in metabolic enzymes have been identified in many cancer types, most of which may directly or indirectly lead to an increase of lactate that acidifies the extracellular environment of the tumor [128]. In addition, it has been shown that the acidic tumor microenvironment can increase A-to-I editing levels [127, 129]. Thus, it suggests that lactic acid may be involved in cancer development as a potent editing inducer to reduce the immunogenicity of Alus thereby suppressing tumor immune surveillance. This provides a new clue for immunotherapy by targeting ADAR1, and further studies on the factors affecting ADAR1 activity are expected to provide new strategies for tumor immunotherapy.
Additionally, aberrant A-to-I editing is commonly found in cancers, where over-editing of some sites is accompanied by under-editing of others. For example, high editing of the FLNB and low editing of the COPA are present in hepatocellular carcinoma (HCC). Both of them can drive the development of HCC [45]. Currently, several site-directed RNA editing (SDRE) systems utilizing endogenous or exogenous ADARs have been developed and have attracted the attention of scientists due to their biological tolerance and greater biosafety [130,131,132]. It has been shown that SDRE can restore the activity of TP53 by correcting its pathogenic mutations [133]. Thus, the SDRE system may be a promising candidate in tumor therapy. However, the strategies of targeted RNA A-to-I editing by using endogenous or exogenous ADARs are both limited. For example, endogenous ADARs expression levels are insufficient, and delivered exogenous ADARs reduce their substrate activity [134]. Therefore, detailed understanding the factors that influence ADARs editing activity is essential to enhance their activity. In addition, in tumors, there is usually a large of number aberrant editing sites, many of which have been demonstrated to be involved in the progression of the disease. Therefore, correcting single-site editing abnormalities is often not sufficient for therapeutic needs, and the development of tools for multisite editing has still to be explored to provide new strategies for the treatment of tumors.
Availability of data and materials
Not applicable.
References
Chen C-X, Cho D-SC, Wang Q, Lai F, Carter KC, Nishikura K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6:755–67.
Caponio VCA, Troiano G, Botti G, Pedicillo MC, Lo Russo L, Mastrangelo F, et al. Overexpression of ADAR1 into the cytoplasm correlates with a better prognosis of patients with oral squamous cells carcinoma. J Oral Pathol Med. 2019;48:108–14.
Bass BL. RNA editing by Adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–46.
Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, et al. Stress-induced apoptosis Associated with null mutation of ADAR1 RNA editing Deaminase Gene. J Biol Chem. 2004;279:4952–61.
Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-Containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:15.
Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–76.
Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, et al. Elevated RNA editing activity is a major contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015;13:267–76.
Han L, Diao L, Yu S, Xu X, Li J, Zhang R, et al. The genomic Landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–28.
Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17:1586–95.
Wang H, Chen S, Wei J, Song G, Zhao Y. A-to-I RNA editing in Cancer: from evaluating the editing level to exploring the Editing effects. Front Oncol. 2021;10:632187.
Mansi L, Tangaro MA, Lo Giudice C, Flati T, Kopel E, Schaffer AA, et al. REDIportal: millions of novel A-to-I RNA editing events from thousands of RNAseq experiments. Nucleic Acids Res. 2021;49:D1012–9.
Roth SH, Levanon EY, Eisenberg E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat Methods. 2019;16:1131–8.
Knutson SD, Heemstra JM. EndoVIPER-seq for Improved Detection of A‐to‐I Editing Sites in Cellular RNA. Current Protocols in Chemical Biology [Internet]. 2020 [cited 2022 Sep 25];12. Available from: https://onlinelibrary.wiley.com/doi/https://doi.org/10.1002/cpch.82.
Zhu H, Huang L, Liu S, Dai Z, Songyang Z, Weng Z, et al. REIA: a database for cancer A-to-I RNA editing with interactive analysis. Int J Biol Sci. 2022;18:2472–83.
Fritzell K, Xu L-D, Otrocka M, Andréasson C, Öhman M. Sensitive ADAR editing reporter in cancer cells enables high-throughput screening of small molecule libraries. Nucleic Acids Res. 2019;47:e22–2.
Zhang F, Lu Y, Yan S, Xing Q, Tian W. SPRINT: an SNP-free toolkit for identifying RNA editing sites. Bioinformatics. 2017;33:3538–48.
Xiong H, Liu D, Li Q, Lei M, Xu L, Wu L et al. RED-ML: a novel, effective RNA editing detection method based on machine learning. GigaScience [Internet]. 2017 [cited 2023 Dec 20];6. Available from: https://academic.oup.com/gigascience/article/doi/https://doi.org/10.1093/gigascience/gix012/3059653.
Tang AD, Hrabeta-Robinson E, Volden R, Vollmers C, Brooks AN. Detecting haplotype-specific transcript variation in long reads with FLAIR2 [Internet]. Bioinformatics; 2023 Jun. https://doi.org/10.1101/2023.06.09.544396.
Tamazian G, Dobrynin P, Krasheninnikova K, Komissarov A, Koepfli K-P, O’Brien SJ. Chromosomer: a reference-based genome arrangement tool for producing draft chromosome sequences. GigaSci. 2016;5:38.
Ramaswami G, Li JB. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucl Acids Res. 2014;42:D109–13.
Piechotta M, Wyler E, Ohler U, Landthaler M, Dieterich C. JACUSA: site-specific identification of RNA editing events from replicate sequencing data. BMC Bioinformatics. 2017;18:7.
Picardi E, D’Erchia AM, Lo Giudice C, Pesole G. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017;45:D750–7.
Marceca GP, Distefano R, Tomasello L, Lagana A, Russo F, Calore F, et al. MiREDiBase, a manually curated database of validated and putative editing events in microRNAs. Sci Data. 2021;8:199.
Kofman E, Yee B, Medina-Munoz HC, Yeo GW. FLARE: a fast and flexible workflow for identifying RNA editing foci. BMC Bioinformatics. 2023;24:370.
Kluesner MG, Tasakis RN, Lerner T, Arnold A, Wüst S, Binder M, et al. MultiEditR: the first tool for the detection and quantification of RNA editing from Sanger sequencing demonstrates comparable fidelity to RNA-seq. Mol Therapy - Nucleic Acids. 2021;25:515–23.
Kim M, Hur B, Kim S. RDDpred: a condition-specific RNA-editing prediction model from RNA-seq data. BMC Genomics. 2016;17:5.
John D, Weirick T, Dimmeler S, Uchida S. RNAEditor: easy detection of RNA editing events and the introduction of editing islands. Brief Bioinform. 2017;18:993–1001.
Chen W, Feng P, Yang H, Ding H, Lin H, Chou K-C. iRNA-3typeA: identifying three types of modification at RNA’s Adenosine sites. Mol Therapy - Nucleic Acids. 2018;11:468–74.
Chen R, Li F, Guo X, Bi Y, Li C, Pan S, et al. ATTIC is an integrated approach for predicting A-to-I RNA editing sites in three species. Brief Bioinform. 2023;24:bbad170.
Chen L, Ou L, Jing X, Kong Y, Xie B, Zhang N, et al. DeepEdit: single-molecule detection and phasing of A-to-I RNA editing events using nanopore direct RNA sequencing. Genome Biol. 2023;24:75.
Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–88.
Liu Y, Samuel CE. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J Virol. 1996;70:1961–8.
Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch MF. RNA-Regulated Interaction of Transportin-1 and Exportin-5 with the double-stranded RNA-Binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol. 2009;29:1487–97.
Barraud P, Banerjee S, Mohamed WI, Jantsch MF, Allain FH-T. A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc Natl Acad Sci USA [Internet]. 2014 [cited 2022 Nov 26];111. https://doi.org/10.1073/pnas.1323698111.
Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. The human but not the Xenopus RNA-editing enzyme ADAR1 has an Atypical Nuclear Localization Signal and displays the characteristics of a shuttling protein. Mol Biol Cell. 2001;12:1911–24.
Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Zα Domain of the human editing enzyme ADAR1 bound to Left-handed Z-DNA. Science. 1999;284:1841–5.
Schade M, Turner CJ, Kühne R, Schmieder P, Lowenhaupt K, Herbert A, et al. The solution structure of the Zα domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc Natl Acad Sci USA. 1999;96:12465–70.
Ha SC, Lowenhaupt K, Rich A, Kim Y-G, Kim KK. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature. 2005;437:1183–6.
Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. CRM1 mediates the export of ADAR1 through a Nuclear Export Signal within the Z-DNA binding domain. Mol Cell Biol. 2001;21:7862–71.
Qin Y-R, Qiao J-J, Chan THM, Zhu Y-H, Li F-F, Liu H, et al. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014;74:840–51.
Shigeyasu K, Okugawa Y, Toden S, Miyoshi J, Toiyama Y, Nagasaka T, et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018;3:e99976.
Takeda S, Shigeyasu K, Okugawa Y, Yoshida K, Mori Y, Yano S, et al. Activation of AZIN1 RNA editing is a novel mechanism that promotes invasive potential of cancer-associated fibroblasts in colorectal cancer. Cancer Lett. 2019;444:127–35.
Takahashi K, Shigeyasu K, Kondo Y, Gotoh K, Yano S, Umeda Y, et al. RNA editing is a Valuable Biomarker for Predicting Carcinogenesis in Ulcerative Colitis. J Crohns Colitis. 2023;17:754–66.
Chen L, Li Y, Lin CH, Chan THM, Chow RKK, Song Y, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19:209–16.
Chan THM, Lin CH, Qi L, Fei J, Li Y, Yong KJ, et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut. 2014;63:832–43.
Nakamura K, Shigeyasu K, Okamoto K, Matsuoka H, Masuyama H. ADAR1 and AZIN1 RNA editing function as an oncogene and contributes to immortalization in endometrial cancer. Gynecol Oncol. 2022;166:326–33.
Ghalali A, Wang L, Stopsack KH, Rice JM, Wu S, Wu C-L, et al. AZIN1 RNA editing alters protein interactions, leading to nuclear translocation and worse outcomes in prostate cancer. Exp Mol Med. 2022;54:1713–26.
Okugawa Y, Toiyama Y, Shigeyasu K, Yamamoto A, Shigemori T, Yin C, et al. Enhanced AZIN1 RNA editing and overexpression of its regulatory enzyme ADAR1 are important prognostic biomarkers in gastric cancer. J Transl Med. 2018;16:366.
Hu X, Chen J, Shi X, Feng F, Lau KW, Chen Y, et al. RNA editing of AZIN1 induces the malignant progression of non-small-cell lung cancers. Tumour Biol. 2017;39:1010428317700001.
Ramírez-Moya J, Miliotis C, Baker AR, Gregory RI, Slack FJ, Santisteban P. An ADAR1-dependent RNA editing event in the cyclin-dependent kinase CDK13 promotes thyroid cancer hallmarks. Mol Cancer. 2021;20:115.
Dong X, Chen G, Cai Z, Li Z, Qiu L, Xu H, et al. CDK13 RNA over-editing mediated by ADAR1 Associates with Poor Prognosis of Hepatocellular Carcinoma patients. Cell Physiol Biochem. 2018;47:2602–12.
Hu X, Wan S, Ou Y, Zhou B, Zhu J, Yi X, et al. RNA over-editing of BLCAP contributes to hepatocarcinogenesis identified by whole-genome and transcriptome sequencing. Cancer Lett. 2015;357:510–9.
Chen W, He W, Cai H, Hu B, Zheng C, Ke X, et al. A-to-I RNA editing of BLCAP lost the inhibition to STAT3 activation in cervical cancer. Oncotarget. 2017;8:39417–29.
Baker AR, Miliotis C, Ramírez-Moya J, Marc T, Vlachos IS, Santisteban P, et al. Transcriptome profiling of ADAR1 targets in Triple-negative breast Cancer cells reveals mechanisms for regulating Growth and Invasion. Mol Cancer Res. 2022;20:960–71.
Gao C, Zhou G, Shi J, Shi P, Jin L, Li Y, et al. The A-to-I editing of KPC1 promotes intrahepatic cholangiocarcinoma by attenuating proteasomal processing of NF-κB1 p105 to p50. J Exp Clin Cancer Res. 2022;41:338.
Teoh PJ, An O, Chung T-H, Chooi JY, Toh SHM, Fan S, et al. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood. 2018;132:1304–17.
Luo J, Gong L, Yang Y, Zhang Y, Liu Q, Bai L et al. Enhanced mitophagy driven by ADAR1-GLI1 editing supports the self-renewal of cancer stem cells in hepatocellular carcinoma. Hepatology [Internet]. 2023 [cited 2023 May 1];Publish Ahead of Print. Available from: https://journals.lww.com/https://doi.org/10.1097/HEP.0000000000000299.
Amin EM, Liu Y, Deng S, Tan KS, Chudgar N, Mayo MW, et al. The RNA-editing enzyme ADAR promotes lung adenocarcinoma migration and invasion by stabilizing FAK. Sci Signal. 2017;10:eaah3941.
Amweg A, Tusup M, Cheng P, Picardi E, Dummer R, Levesque MP, et al. The A to I editing landscape in melanoma and its relation to clinical outcome. RNA Biol. 2022;19:996–1006.
Gumireddy K, Li A, Kossenkov AV, Sakurai M, Yan J, Li Y, et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated akt activation and breast cancer metastasis. Nat Commun. 2016;7:10715.
Shimokawa T, Rahman MF-U, Tostar U, Sonkoly E, Ståhle M, Pivarcsi A, et al. RNA editing of the GLI1 transcription factor modulates the output of hedgehog signaling. RNA Biol. 2013;10:321–33.
Chan JJ, Tabatabaeian H, Tay Y. 3′UTR heterogeneity and cancer progression. Trends in Cell Biology. 2023;33:568-82.
Chen L-L, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO J. 2008;27:1694–705.
Mehdipour P, Marhon SA, Ettayebi I, Chakravarthy A, Hosseini A, Wang Y, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. 2020;588:169–73.
Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing enzyme ADAR1 independently Control Nucleic Acid Sensor MDA5-Driven autoimmunity and Multi-organ Development. Immunity. 2015;43:933–44.
Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–20.
Zhang L, Yang C-S, Varelas X, Monti S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci Rep. 2016;6:23226.
Nakano M, Fukami T, Gotoh S, Nakajima M. A-to-I RNA editing Up-regulates human dihydrofolate reductase in breast Cancer. J Biol Chem. 2017;292:4873–84.
Li Y, Wang N-X, Yin C, Jiang S-S, Li J-C, Yang S-Y. RNA editing enzyme ADAR1 regulates METTL3 in an editing dependent manner to promote breast Cancer progression via METTL3/ARHGAP5/YTHDF1 Axis. IJMS. 2022;23:9656.
Yang C-C, Chen Y-T, Chang Y-F, Liu H, Kuo Y-P, Shih C-T, et al. ADAR1-mediated 3’ UTR editing and expression control of antiapoptosis genes fine-tunes cellular apoptosis response. Cell Death Dis. 2017;8:e2833.
Cho CJ, Jung J, Jiang L, Lee EJ, Kim D-S, Kim BS, et al. Combinatory RNA-Sequencing analyses reveal a Dual Mode of Gene Regulation by ADAR1 in gastric Cancer. Dig Dis Sci. 2018;63:1835–50.
Sagredo EA, Blanco A, Sagredo AI, Pérez P, Sepúlveda-Hermosilla G, Morales F, et al. ADAR1-mediated RNA-editing of 3’UTRs in breast cancer. Biol Res. 2018;51:36.
Wang Q, Hui H, Guo Z, Zhang W, Hu Y, He T, et al. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA. 2013;19:1525–36.
Jiang L, Hao Y, Shao C, Wu Q, Prager BC, Gimple RC, et al. ADAR1-mediated RNA editing links ganglioside catabolism to glioblastoma stem cell maintenance. J Clin Invest. 2022;132:e143397.
Wang Y, Xu X, Yu S, Jeong KJ, Zhou Z, Han L, et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017;27:1112–25.
Kim HS, Na MJ, Son KH, Yang HD, Kim SY, Shin E, et al. ADAR1-dependent mir-3144-3p editing simultaneously induces MSI2 expression and suppresses SLC38A4 expression in liver cancer. Exp Mol Med. 2023;55:95–107.
Shen H, An O, Ren X, Song Y, Tang SJ, Ke X-Y, et al. ADARs act as potent regulators of circular transcriptome in cancer. Nat Commun. 2022;13:1508.
Ma C, Wang X, Yang F, Zang Y, Liu J, Wang X, et al. Circular RNA hsa_circ_0004872 inhibits gastric cancer progression via the miR-224/Smad4/ADAR1 successive regulatory circuit. Mol Cancer. 2020;19:157.
Zhang Q, Xiu B, Zhang L, Chen M, Chi W, Li L, et al. Immunosuppressive lncRNA LINC00624 promotes tumor progression and therapy resistance through ADAR1 stabilization. J Immunother Cancer. 2022;10:e004666.
Shiromoto Y, Sakurai M, Minakuchi M, Ariyoshi K, Nishikura K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat Commun. 2021;12:1654.
Roberts JT, Patterson DG, King VM, Amin SV, Polska CJ, Houserova D, et al. ADAR mediated RNA editing modulates MicroRNA targeting in human breast Cancer. Processes (Basel). 2018;6:42.
Park MJ, Jeong E, Lee EJ, Choi HJ, Moon BH, Kang K et al. RNA Editing Enzyme ADAR1 Suppresses the Mobility of Cancer Cells via ARPIN. MolCells [Internet]. 2023 [cited 2023 Apr 30]; Available from: http://molcells.org/journal/view.html?doi=10.14348/molcells.2023.2174.
Velazquez-Torres G, Shoshan E, Ivan C, Huang L, Fuentes-Mattei E, Paret H, et al. A-to-I miR-378a-3p editing can prevent melanoma progression via regulation of PARVA expression. Nat Commun. 2018;9:461.
Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, et al. Reduced adenosine-to-inosine mir-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;17:311–21.
Choudhury Y, Tay FC, Lam DH, Sandanaraj E, Tang C, Ang B-T, et al. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;122:4059–76.
Salameh A, Lee AK, Cardó-Vila M, Nunes DN, Efstathiou E, Staquicini FI, et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc Natl Acad Sci USA. 2015;112:8403–8.
International Human Genome Sequencing Consortium, Whitehead Institute for Biomedical Research, Center for Genome Research:, Lander ES, Linton LM, Birren B, Nusbaum C, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
Eisenberg E, Levanon EY. A-to-I RNA editing — immune protector and transcriptome diversifier. Nat Rev Genet. 2018;19:473–90.
Jain M, Jantsch MF, Licht K. The editor’s I on Disease Development. Trends Genet. 2019;35:903–13.
Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med. 2019;25:95–102.
Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565:43–8.
Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun. 2018;9:5450.
Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, Ghaffari SH. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J Cell Physiol. 2019;234:5451–65.
Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21.
Bahn JH. Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat Commun. 2015;13.
Panda AC. Circular RNAs Act as miRNA Sponges. In: Xiao J, editor. Circular RNAs [Internet]. Singapore: Springer Singapore; 2018 [cited 2023 Jul 14]. p. 67–79. Available from: http://link.springer.com/https://doi.org/10.1007/978-981-13-1426-1_6.
Kristensen LS, Jakobsen T, Hager H, Kjems J. The emerging roles of circRNAs in cancer and oncology. Nat Rev Clin Oncol. 2022;19:188–206.
Omata Y, Okawa M, Haraguchi M, Tsuruta A, Matsunaga N, Koyanagi S, et al. RNA editing enzyme ADAR1 controls mir-381-3p-mediated expression of multidrug resistance protein MRP4 via regulation of circRNA in human renal cells. J Biol Chem. 2022;298:102184.
Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21:446–60.
Ulitsky I, Bartel DP, lincRNAs. Genomics, Evolution, and mechanisms. Cell. 2013;154:26–46.
Licht K, Jantsch MF. The other Face of an editor: ADAR1 functions in editing-independent ways. BioEssays. 2017;39:1700129.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest. 2013;123:2703–18.
Cai D, Sun C, Murashita T, Que X. ADAR1 non-editing function in macrophage activation and abdominal aortic aneurysm. Circulation Res. 2023;132.
Liu X, Fu Y, Huang J, Wu M, Zhang Z, Xu R, et al. ADAR1 promotes the epithelial-to-mesenchymal transition and stem-like cell phenotype of oral cancer by facilitating oncogenic microRNA maturation. J Exp Clin Cancer Res. 2019;38:315.
Qi L, Song Y, Chan THM, Yang H, Lin CH, Tay DJT, et al. An RNA editing/dsRNA binding-independent gene regulatory mechanism of ADARs and its clinical implication in cancer. Nucleic Acids Res. 2017;45:10436–51.
Ota H, Sakurai M, Gupta R, Valente L, Wulff B-E, Ariyoshi K, et al. ADAR1 forms a complex with dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–89.
Chen T, Xiang J-F, Zhu S, Chen S, Yin Q-F, Zhang X-O, et al. ADAR1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res. 2015;25:459–76.
Nie Y, Ding L, Kao PN, Braun R, Yang J-H. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol. 2005;25:6956–63.
Majumder M, Chakraborty P, Mohan S, Mehrotra S, Palanisamy V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Adv Drug Deliv Rev. 2022;188:114442.
Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. ADAR regulates RNA editing, transcript Stability, and Gene Expression. Cell Rep. 2013;5:849–60.
Tassinari V, Cesarini V, Tomaselli S, Ianniello Z, Silvestris DA, Ginistrelli LC, et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021;22:51.
Czuba LC, Hillgren KM, Swaan PW. Post-translational modifications of transporters. Pharmacol Ther. 2018;192:88–99.
Watanabe N, Osada H. Phosphorylation-dependent protein-protein Interaction modules as potential molecular targets for Cancer Therapy. CDT. 2012;13:1654–8.
Sakurai M, Shiromoto Y, Ota H, Song C, Kossenkov AV, Wickramasinghe J, et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat Struct Mol Biol. 2017;24:534–43.
Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B. The relation between PI3K/AKT signalling pathway and cancer. Gene. 2019;698:120–8.
Bavelloni A, Focaccia E, Piazzi M, Raffini M, Cesarini V, Tomaselli S, et al. AKT-dependent phosphorylation of the adenosine deaminases ADAR-1 and – 2 inhibits deaminase activity. FASEB J. 2019;33:9044–61.
Koganti P, Kadali VN, Manikoth Ayyathan D, Emanuelli A, Paolini B, Levy-Cohen G, et al. The E3 ubiquitin ligase SMURF2 stabilizes RNA editase ADAR1p110 and promotes its adenosine-to-inosine (A-to-I) editing function. Cell Mol Life Sci. 2022;79:237.
Desterro JMP, Keegan LP, Jaffray E, Hay RT, O’Connell MA, Carmo-Fonseca M. SUMO-1 modification alters ADAR1 editing activity. MBoC. 2005;16:5115–26.
Chilibeck KA, Wu T, Liang C, Schellenberg MJ, Gesner EM, Lynch JM, et al. FRET analysis of in vivo dimerization by RNA-editing enzymes. J Biol Chem. 2006;281:16530–5.
Han J, An O, Hong H, Chan THM, Song Y, Shen H, et al. Suppression of adenosine-to-inosine (A-to-I) RNA editome by death associated protein 3 (DAP3) promotes cancer progression. Sci Adv. 2020;6:eaba5136.
Chen J, Li L, Liu T-Y, Fu H-F, Lai Y-H, Lei X, et al. CPEB3 suppresses gastric cancer progression by inhibiting ADAR1-mediated RNA editing via localizing ADAR1 mRNA to P bodies. Oncogene. 2022;41:4591–605.
Shih C-Y, Chen Y-C, Lin H-Y, Chu C-Y. RNA helicase DDX6 regulates A-to-I editing and neuronal differentiation in human cells. IJMS. 2023;24:3197.
Daniel C, Widmark A, Rigardt D, Öhman M. Editing inducer elements increases A-to-I editing efficiency in the mammalian transcriptome. Genome Biol. 2017;18:195.
Feng C, Cao X, Du Y, Chen Y, Xin K, Zou J, et al. Uncovering Cis-Regulatory Elements Important for A-to-I RNA Editing in Fusarium graminearum. Lin X, editor. mBio. 2022;13:e01872-22.
Doherty EE, Karki A, Wilcox XE, Mendoza HG, Manjunath A, Matos VJ, et al. ADAR activation by inducing a synconformation at guanosine adjacent to an editing site. Nucleic Acids Res. 2022;50:10857–68.
Malik TN, Doherty EE, Gaded VM, Hill TM, Beal PA, Emeson RB. Regulation of RNA editing by intracellular acidification. Nucleic Acids Res. 2021;49:4020–36.
Spencer NY, Stanton RC. The Warburg Effect, Lactate, and nearly a century of trying to Cure Cancer. Semin Nephrol. 2019;39:380–93.
Certo M, Tsai C-H, Pucino V, Ho P-C, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21:151–61.
Li M, Yan C, Jiao Y, Xu Y, Bai C, Miao R, et al. Site-directed RNA editing by harnessing ADARs: advances and challenges. Funct Integr Genomics. 2022;22:1089–103.
Casati B, Stamkopoulou D, Tasakis RN, Pecori R, ADAR-Mediated RNA. Editing and Its Therapeutic Potentials. In: Jurga S, Barciszewski J, editors. Epitranscriptomics [Internet]. Cham: Springer International Publishing; 2021 [cited 2023 Dec 14]. p. 471–503. https://doi.org/10.1007/978-3-030-71612-7_18.
Booth BJ, Nourreddine S, Katrekar D, Savva Y, Bose D, Long TJ, et al. RNA editing: expanding the potential of RNA therapeutics. Mol Ther. 2023;31:1533–49.
Qu L, Yi Z, Zhu S, Wang C, Cao Z, Zhou Z, et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat Biotechnol. 2019;37:1059–69.
Diaz Quiroz JF, Siskel LD, Rosenthal JJC. Site-directed A → I RNA editing as a therapeutic tool: moving beyond genetic mutations. RNA. 2023;29:498–505.
Funding
This work was funded by the Natural Science Foundation of Shandong Province, China (ZR202111200086).
Author information
Authors and Affiliations
Contributions
Yue Jiao, Yuqin Xu, and Chengbin Liu reviewed the literature and wrote the article. Rui Miao, Chunyan Liu, and Yilong Wang designed the figures and tables. Jiao Liu critically revised the manuscript. All authors revised the manuscript and approved its final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethical approval is not required for this study.
The authors declare that their participation in writing this review as well as its publication is completely voluntary without afecting their actual research work.
Consent for publication
The authors give Molecular Cancer permission to publish this work.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiao, Y., Xu, Y., Liu, C. et al. The role of ADAR1 through and beyond its editing activity in cancer. Cell Commun Signal 22, 42 (2024). https://doi.org/10.1186/s12964-023-01465-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01465-x