
Abstract
Background
Adenosine deaminases acting on RNA (ADARs) are involved in adenosine-to-inosine (A-to-I) editing and implicated in tumorigenesis and prognosis. Emerging evidence has indicated that ADAR1, an ADAR family member, participates in the regulation of various cancers; however, its biological function in oral squamous cell carcinoma (OSCC) remains unclear. This study aimed to determine the role of ADAR1 in OSCC progression.
Methods
ADAR1 expression in both normal tissues and carcinoma tissues and in OSCC cell lines was examined by real-time PCR and western blotting. Gain-of-function and loss-of-function approaches were used to examine the effect of ADAR1 on the migration, invasion, epithelial-mesenchymal transition (EMT) and stemness of OSCC. Furthermore, the relationship between ADAR1 and Dicer was determined by co-immunoprecipitation, and the expression of OSCC-associated oncogenic miRNAs was evaluated by real-time PCR. For in vivo experiments, a xenograft model where OSCC cells stably expressing ADAR1 were implanted was used to investigate the effect of ADAR1 on tumor growth and progression, and the expression of ADAR1, PCNA, SOX2 and POU5F1 was further detected by immunohistochemistry. The impact of ADAR1 expression on the survival status of OSCC patients was determined by survival analysis.
Results
ADAR1 was overexpressed in OSCC and significantly associated with poor patient survival. There was a positive correlation between ADAR1 and the migration, invasion, EMT and stemness of OSCC. Mechanistically, ADAR1 was physically associated with Dicer, and six OSCC-associated oncogenic miRNAs were increased in OSCC cells with ADAR1 overexpression. In the mouse xenograft model of OSCC, ADAR1 overexpression promoted tumor growth and progression. Moreover, ADAR1 was highly expressed in OSCC patients with low survival rates.
Conclusions
Our findings demonstrated that ADAR1 may play a significant role in OSCC progression via combining with Dicer to regulate oncogenic miRNA maturation and further affect cell migration and invasion.
Similar content being viewed by others


Background
Oral squamous cell carcinoma (OSCC) is one of the most prevalent cancers and includes epithelial neoplasms of the oral cavity and oropharynx; OSCC is a serious and growing problem in many parts of the world [1, 2]. OSCC has a locoregional evolution, which is characterized by invasion of regional anatomical structures, and is prone to metastasis through lymphatic channels. Despite recent advancements in surgical techniques and other anticancer methods, the overall survival has improved little over the past few decades [3, 4]. A better understanding of the biology of OSCC is urgently needed for improving the poor outcome of this disease.
RNA editing, which was first discovered in trypanosome mitochondria, directly alters RNA sequences through post-transcriptional modifications [5, 6]. The most well known type of RNA editing is adenosine-to-inosine (A-to-I) editing, which is catalyzed by the enzyme family of adenosine deaminases acting on RNA (ADARs) targeting double-stranded RNA (dsRNA) [6,7,8]. Growing evidence supports the role of ADARs in various biological processes via RNA editing-independent mechanisms, including gene expression regulation [9], miRNA generation [3, 10, 11] and protein-protein complex formation [12, 13]. In humans, three ADAR proteins (ADAR1–3) have been identified, and ADAR1 has been verified to play important roles in a variety of diseases, such as cancer and infection [14, 15]. Recent results suggest that ADAR1 is upregulated in various solid cancers and associated with poor prognosis, but the biological significance of ADAR1 in OSCC remains largely unknown.
MicroRNAs (miRNAs) are approximately 22 nucleotides in length and post-transcriptionally regulate the expression of genes involved in multiple cellular functions [16, 17]. Some miRNAs (oncogenic miRNAs) play a vital role in tumor progression and therapeutic resistance [18]. Dicer, an RNase III gene family member, is an important component of the miRNA processing mechanism [19]. After primary transcripts of miRNA genes (pri-miRNAs) are cleaved into precursor transcripts of miRNA genes (pre-miRNAs) in the nucleus, pre-miRNAs are exported to the cytoplasm and cleaved into mature miRNAs by Dicer [3, 20]. ADARs are thought to participate in several steps during miRNA generation [21, 22]. Global suppression of miRNA synthesis in ADAR1-null mice led to an embryonic lethal phenotype [23]. Recent research has revealed that there is a robust interaction between Dicer and ADAR1 [3] and that ADAR1 increases the rate of miRNA processing by Dicer [6, 8]. In E11–12 embryos, upregulation of Dicer and ADAR1 p100 resulted in increased pre-miRNA processing capability [3].
In the present study, we found that ADAR1 was overexpressed in OSCC tissues and cell lines and was associated with poor prognosis. Additionally, we found that an abundance of ADAR1 enhanced the malignant phenotype, including stemness and EMT, of OSCC cell lines. Furthermore, we demonstrated that ADAR1 could interact with Dicer and promote the maturation of oncogenic miRNAs. Taken together, our findings showed that ADAR1 may be a key regulator of OSCC progression and a potential therapeutic target.
Methods
Patients and tissue specimens
Sixty-one OSCC tissue samples and matched noncancerous oral mucosa tissue samples (collected postoperatively from June 2014 to October 2015) used in this study were obtained from the Department of Oral Pathology, School of Stomatology, Nanjing Medical University. None of the patients had received radiotherapy or chemotherapy before their operation. Upon removal, the specimen was examined and divided into cancerous and normal tissues by pathology faculty. Each sample was snap-frozen in liquid nitrogen and stored at − 80 °C prior to RNA isolation. The tumor stage was determined according to the TNM classification system of the International Union Against Cancer. Tumor histological grade was determined according to Broder’s classification system. All surgical specimens were reviewed and analyzed after the study protocol was approved by the Ethics Committee of Nanjing Medical University. All procedures performed in studies involving human participants were in accordance with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent for tissue donation for this research was obtained before specimen collection.
Cell culture
Human OSCC cell lines (SCC9 and Cal27) were obtained from the American Type Culture Collection (ATCC). HOK cells were obtained from ScienCell, and HN4 and HN6 cells were from patients with head and neck squamous cell carcinoma (HNSCC). HN4, HN6, SCC9 and Cal27 cells were cultured in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12, Gibco, USA) containing 10% fetal calf serum (FBS, HyClone, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, USA). HOK cells were cultured in oral keratinocyte medium (OKM, ScienCell, USA) with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were incubated in a humidified atmosphere at 37 °C in the presence of 5% CO2. All cell lines were passaged for fewer than 6 months.
Real-time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, San Diego, CA, USA). RNA was reverse transcribed into cDNA using a Primer-Script™ one-step RT-PCR kit (TaKaRa, Dalian, China). The cDNA template was amplified by real-time PCR using a SYBR® Pre-mix Dimmer Eraser kit (TaKaRa, Dalian, China). GAPDH and U6 were used as internal controls, and mRNA and miRNA values were normalized to GAPDH and U6, respectively. The primers used were as follows: ADAR1, F: 5′ - TGCTGCTGAATTCAAGTTGG - 3′; R: 5′ - TCGTTCTCCCCAATCAAGAC- 3′, ADAR1-p110, F: 5′ -GGCAGCCTCCGGGTG- 3′; R: 5′ -CTGTCTGTGCTCATAGCCTTGA- 3′, ADAR1-p150, F: 5′ -CCACCTCCAGTGCGGAGTAGCG- 3′; R: 5′ -TGCCCCTTGAGAAATTCTATTTGC- 3′, Dicer, F: 5′ -TCCACGAGTCACAATCAACACGG- 3′; R: 5′ -GGGTTCTGCATTTAGGAGCTAGATGAG- 3′, GAPDH, F: 5′ - CCGGGAAACTGTGGCGTGATGG - 3′; R: 5′ - AGGTGGAGGAGTGGGTGTCGCTGTT- 3′. Real-time PCR was performed using an ABI7900 system (Applied Biosystems, CA). The relative expression fold change of mRNAs was calculated by the 2-ΔΔCt method. All miRNA and U6 primers were purchased from Genecoponeia, Guangzhou, China.
Western blot analysis
Harvested cells were lysed on ice for 30 min in RIPA Lysis Buffer (Beyotime Biotechnology, Shanghai, China) containing 100 mM PMSF (Beyotime Biotechnology) and centrifuged at 12000×g for 10 min to collect total protein samples. To isolate the cytoplasmic components from the nuclear components, the cells were mechanically homogenized and treated with a nuclear protein extraction kit (Beyotime Biotechnology). The protein lysate supernatants were mixed with loading buffer, separated on a 10% SDS-polyacrylamide gel and transferred to an Immobilon-PVDF membrane (Millipore Corporation, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk at 22 °C and incubated with primary antibodies at 4 °C overnight. Detailed information on the primary antibodies is provided in Additional file 5: Table S1. The membranes were incubated with secondary peroxidase-conjugated antibodies. Finally, the protein bands on the membranes were visualized using chemiluminescence reagents (WBKLS0100; Millipore Corporation, Billerica, MA, USA) according to the manufacturer’s instructions.
RNA interference
Small interfering RNA (siRNA) targeting ADAR1 and scrambled siRNA were designed and synthesized by GenePharma (Shanghai, China). Three sequences of siRNAs targeting ADAR1-p110 were used: siRNA1, 5′ - CCUUCUACAGUCAUGGCUUTT - 3′, 3′ - AAGCCAUGACUGUAGAAGGTT − 5′; siRNA2, 5′ - CCACUAUUCCACAGAGAAATT -3′, 3′ - UUUCUCUGUGGAAUAGUGGTT − 5′; siRNA3, 5′ - CCAUGAACCCAAGUUCCAATT -3′, 3′ - UUGGAACUUGGGUUCAUGGTT − 5′. The siRNA sequence used for targeting Dicer was 5′ - GCCAAGGAAAUCAGCUAAATT -3′, 3′ - UUUAGCUGAUUUCCUUGGCTT − 5′. According to the literature [24], the siRNA sequence used for targeting ADAR1-p150 was 5′ - GCCUCGCGGGCGCAAUGAATT -3′, 3′ - UUCAUUGCGCCCGCGAGGCAT − 5′. All of these siRNA duplexes (final concentration 50 nM) were transfected into cells using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA) according to the manufacturer’s instructions. Knockdown efficiency was determined after 48 h of culture.
Lentiviral vector construction and transfection
The ADAR1 p110 mRNA sequence (GenBank Accession NM_001025107.2) was synthesized and subcloned into the LV5 (EF-1aF/GFP &Puro) vector (LV-ADAR1), and the empty LV5 vector with the GFP gene (LV-GFP) served as a negative control (GenPharma, Shanghai, China). Cells were infected with LV-ADAR1 or LV-GFP and then cultured with DMEM containing 10% FBS in the presence of 0.5 μg/ml puromycin for 7 days. Stable clones of the cells were selected and used in the following experiments.
Wound healing assay and invasion assay
For wound healing assays, HN4 and Cal27 cells were seeded in a 6-well plate and then cultured in growth medium until they reached 80% confluency. The monolayer was then disrupted with a 1.2-mm cell scraper. Next, we used PBS to wash away the non-adherent cells and debris, and the cells were incubated with serum-free medium for 18 h. Lesion areas were imaged at 0 and 18 h under a phase-contrast microscope. The invasion assay was performed using Matrigel-coated transwell inserts. Briefly, 5 × 104 cells in 250 μl of serum-free medium were seeded into the upper chamber, and 750 μl of medium was added to the lower chamber. After incubation for 24 h, the chambers were first fixed in 4% paraformaldehyde for 30 min and then stained with a 0.05% crystal violet solution for 15 min. The numbers of cells at 100× magnification were counted using a positive microscope. Three random fields were recorded for each well.
Immunofluorescence
Cells were incubated for 24 h to reach approximately 60% confluence and then fixed with 4% paraformaldehyde and permeabilized in 0.5% Triton X100 for 10 min. Next, antigens were blocked for 1 h with 1% BSA, and the cells were incubated with primary antibodies at 4 °C overnight. The cells were washed with PBS, incubated for 1 h with secondary antibody and stained with DAPI. Images were acquired with a Laser scanning Confocal Microscopy and statistical analysis was performed by Image J software.
Subcutaneous nude mouse xenografts
Ten 4-week-old male nude mice (Institute of Zoology, China Academy of Sciences) were divided randomly into 2 groups (5 in each group). One million Cal27/Vector or Cal27/LV5-p110 cells in 100 μl of PBS were inoculated subcutaneously. Tumor nodules were measured every 7 days and calculated by the following formula: V = (Width2 × Length)/2. Xenografts were collected at the 5 th week for immunohistochemistry staining.
3D Colony formation assay
Briefly, 1000 cells per well were cultured in ultra-low attachment culture plastic ware (Corning Incorporated, catalog number: 3473 24-well plate, USA) with DMEM containing 10% Matrigel, 10 ng/ml EGF, 20 ng/ml FGF, 1× B27, and 5 μg/ml insulin. After 7 to 14 days, the cell colonies (diameter > 50 μm) were counted microscopically. Five fields per well were used to calculate the average value.
Co-immunoprecipitation
Cells were harvested and lysed using sonication in cell lysis buffer (50 mM Tris-HCl, pH 7.5, 130 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate and 1% protease inhibitor cocktail). Cell lysates were centrifuged at 5000 R/M, and the supernatants were incubated with the indicated antibodies and Protein A/G PLUS-Agarose beads (sc-2003, Santa Cruz, USA) at 4 °C overnight. The beads were washed three times with cell lysis buffer, and the precipitated proteins were further analyzed. For western blot details, please see the related assay section above.
Immunohistochemistry
Paraffin-embedded surgical tissue specimens were from 108 patients with OSCC who underwent surgery at the Affiliated Hospital of Stomatology, Nanjing Medical University. The specimens were sectioned at 5-μm thickness, and the sections were baked at 37 °C overnight. The sections were deparaffinized with xylene and hydrated with graded alcohol. Antigen retrieval was conducted by heating for 15 min in a pressure cooker with citrate buffer. Endogenous peroxidase was blocked by incubation with 3% H2O2 plus 1% Triton X100 in paraformaldehyde for 20 min at room temperature. The sections were then incubated overnight with primary antibodies for ADAR1 (1:50 dilution, Abcam, USA), PCNA (1:250 dilution, Abcam, USA), SOX2 (1:300 dilution, Cell Signaling Technology, USA) and POU5F1 (1:500 dilution, Proteintech, USA). Specimens were reacted with a secondary peroxidase-conjugated antibody, and the reaction products were visualized with a 3,3 -diaminobenzidine (DAB) solution. The samples were scored independently by two researchers who were blinded to the clinicopathological data at 200× magnification under a light microscope. The evaluation was based on the staining intensity and extent of staining. Staining intensity was scored as 0 (negative), 1 (weak) and 2 (strong). Staining extent was scored as 0 (0%), 1 (1–50%) and 2 (51–100%), depending on the percentage of positively stained cells. Positive staining was determined by the following formula: overall score = percentage score × intensity score. An overall score of ≤1 was defined as low expression, and an overall score of ≥2 was defined as high expression.
Flow cytometric analysis
Cells were seeded at a density of 1 × 106 cells/well in six-well plates. After 24 h, the cells were washed with PBS, fixed in ice-cold 70% ethanol for 1 h and then treated with 100 μL of 50 mg/L propidium iodide for 30 min at 4 °C in the dark. The cell cycle profiles were assayed using an Elite ESP flow cytometer at 488 nm, and the data were analyzed with the CELL Quest software (BD Biosciences, San Jose, CA, USA).
Cell growth assessment
Cell growth was assessed by the Cell counting Kit-8 (CCK-8; Medchem Express, New Jersey, USA) according to manufacturer instructions at 12, 24, 36, 48, 72 h. Cells were cultivated into 96-well culture plates with a density of 6 × 103 per well. Briefly, absorbance values were determined by the microplate reader at a wavelength of 450 nm. Each experiment was repeated three times and data are presented as the mean.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 and GraphPad Prism 7 software. The expression differences between high/low T category, N category, histological grades, high/low stages, cell lines, expression changes after transfection, protein expression, immunohistochemistry, cell migration and cell invasion assays were analyzed using the independent samples t-test. A chi-square test was used to evaluate the association between the expression of ADAR1 and the clinicopathological features and prognostic factors of OSCC. Kaplan-Meier analysis was used to assess the survival rate curves, and statistical significance was determined using the log-rank test. Univariate and multivariate Cox regression models were performed for the prognostic analyses. All data are presented as the mean ± standard error. A two-sided p value of less than 0.05 was considered statistically significant.
Results
ADAR1 is markedly upregulated in OSCC tissues and cell lines
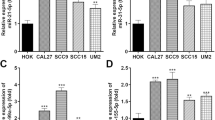
To determine the expression of ADAR1 in OSCC patients, the epicenter of the tumor tissues and matched distal normal tissues from 61 patients were examined by real-time PCR. The results revealed that tumor tissues had markedly higher levels of ADAR1 than matched distal normal tissues (Fig. 1a). Meanwhile, the expression of ADAR1-p110 and ADAR1-p150 in OSCC patients was detected and the result showed both ADAR1-p110 and ADAR1-p150 were enhanced expression in OSCC tissues (Additional file 1: Figure S1). The ADAR1 protein was similarly overexpressed in OSCC tissues that were harvested from 3 random patients (Fig. 1b). To better understand the underlying biological significance of ADAR1, we assessed the relationship between ADAR1 expression and the clinicopathological profiles of OSCC patients. Details of the clinical features are shown in Additional file 6: Table S2. ADAR1 expression was positively correlated with TNM staging (Fig. 1c) and lymph node metastasis (Fig. 1f). Although the data for different T categories and histology grades were not significantly different (Fig. 1d, e), ADAR1 tended to be higher in patients with a higher T stage and poor differentiation. We then focused on the expression of ADAR1 in OSCC cell lines. Consistent with the tissue results, ADAR1 expression was higher in OSCC cell lines than in human oral keratinocyte (HOK) cells (Fig. 1g, h). Together, these results revealed that ADAR1 was overexpressed in both OSCC tissues and cell lines and suggested that ADAR1 might play an important role in OSCC.

ADAR1 expression in OSCC and its clinical significance. a Differences in the expression of ADAR1 between OSCC tissue and matched non-tumor oral mucosa. The expression of ADAR1 was normalized to that of GAPDH. Significant differences between samples were analyzed with the paired Wilcoxon test and paired t test (n = 61, p < 0.01). b ADAR1 protein expression in tissue from three random patients. -, non-tumor oral mucosa; +, OSCC tissue. c Relationship between ADAR1 expression and TNM stage (p < 0.01). d Relationship between ADAR1 expression and primary tumor growth (p = 0.0958). e Relationship between ADAR1 expression and pathological grade (p = 0.0867). f Relationship between ADAR1 expression and lymph node metastasis (p = 0.05). g mRNA levels of ADAR1-p110 and ADAR1-p150 in HOK and OSCC cell lines. h ADAR1 protein expression in HOK and OSCC cell lines. The data were summarized from at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.00; #p < 0.05; ##p < 0.01
ADAR1 promotes migration, invasion, and proliferation in OSCC
To further learn the role of ADAR1 in OSCC, we chose HN4 and Cal27 as our model cell lines. A Conclusion Lentiviral vector carrying ADAR1 p110 gene was successfully constructed and used to induce high expression in both OSCC cell lines, and ADAR1 expression was decreased by siRNA transfection (Additional file 2: Figure S2a-f). In the invasion study, obviously more HN4 and Cal27 cells with ADAR1 overexpression (LV-ADAR1) penetrated the membrane of the chambers than control cells (LV-GFP) (Fig. 2a). Both HN4 and Cal27 cells with either ADAR1-p110 knockdown (si-ADAR1-p110) or ADAR1-p150 knockdown (si-ADAR1-p150) showed weaker invasion abilities than control cells (scrambled) (Fig. 2b). A wound-healing assay was then performed to detect the role of ADAR1 in the progression of OSCC. In the wound healing experiment, the LV-ADAR1 group was significantly faster than the LV-GFP group (Fig. 2c). Conversely, both si-ADAR1-p110 and si-ADAR1-p150 attenuated the migration of HN4 and Cal27 cells (Fig. 2d). Furthermore, the flow cytometry results revealed that upregulation of ADAR1 promoted cell proliferation by increasing the proportion of OSCC cells in S-phase (Additional file 2: Figure S2g). The CCK8 assay results also implied that LV-ADAR1 group exhibited enhanced cell proliferation ability comparing with LV-GFP at 72 h (Additional file 2: Figure S2h, i). These results may imply that ADAR1 promoted the migration, invasion and proliferation of OSCC cell lines.

Effect of ADAR1 on the invasion, migration of OSCC cell lines. a Representative images of the invasion assay using OSCC cell lines transfected with the ADAR1 overexpression vector (LV-ADAR1) and the respective vector control (LV-GFP). b Representative image of the invasion assay using OSCC cell lines transfected with ADAR1-p110, ADAR1-p150 siRNA (si-ADAR1-p110, si-ADAR1-p150) and the respective scrambled vector control (Scrambled). c A scratch-wound assay was used to study the migration ability of LV-ADAR1 and LV-GFP cells over a period of 18 h. d Migration assay of both si-ADAR1-p110 and si-ADAR1-p150 groups during a period of 18 h
ADAR1 participates in the process of TGF-β-induced EMT in OSCC
Previous studies have confirmed that EMT may play a fatal role in OSCC [25]. To examine whether ADAR1 enhanced the EMT of OSCC, the protein expression of CDH1 and VIM was examined by western blot. Both Cal27 and HN4 cells transfected with LV-ADAR1 had higher expression of VIM but lower expression of CDH1 than cells transfected with LV-GFP (Fig. 3a). Conversely, both si-ADAR1-p110 and si-ADAR1-p150 group showed less mesenchymal characteristics than the scrambled cells (Fig. 3c). Similar results were observed by immunofluorescence (Fig. 3b, d), thus validating that ADAR1 could promote EMT in OSCC. Cal27 and HN4 cells were exposed to 10 ng/ml TGF-β [26], which induced EMT, and the protein expression of VIM, CDH1 and ADAR1 was examined at 12 h and 24 h (Fig. 3e). Both Cal27 and HN4 cells showed mesenchymal characteristics at 12 h following increased ADAR1 expression. Higher CDH1 expression but lower VIM expression were detected in Cal27 and HN4 cells transfected with si-ADAR1-p110 under 24 h TGF-β treatment (Fig. 3f). Cal27 and HN4 cells with ADAR1-p110 overexpression were transfected by ADAR1-p150 siRNA and reduced VIM expression and enhanced CDH1 expression were detected by western blot assay (Additional file 3: Figure S3). As shown in Fig. 3g, ADAR1 expression was upregulated at 12 h. Similarly, immunofluorescence also revealed that ADAR1 expression was significantly increased in both the nucleus and cytoplasm (Fig. 3h) upon TGF-β treatment. Collectively, we suggest that ADAR1 is involved in the EMT process of OSCC.

Effect of ADAR1 on EMT of OSCC cell lines. a Western blot analysis of CDH1 and VIM in the LV-ADAR1 and LV-GFP groups. b CDH1 and VIM protein expression was detected by immunofluorescence analysis. c Western blot analysis of CDH1 and VIM in both si-ADAR1-p110 and si-ADAR1-p150 groups. d Representative image of immunofluorescence indicating that ADAR1-p110 knockdown attenuated EMT in OSCC cell lines. e Western blot analysis of CDH1, VIM and ADAR1 in OSCC cell lines treated with TGF-β for 0, 12, and 24 h. f Western blot analysis of CDH1 and VIM in the Scramble and si-ADAR1-p110 groups with 24 h TGF-β treatment. g The nuclear (ADAR1-N) and cytoplasm (ADAR1-C) expression levels of ADAR1 were measured in OSCC cell lines treated with TGF-β for 0, 12, and 24 h by western blot analysis. h Representative image of immunofluorescence showing ADAR1 expression in the process of TGF-β treatment. Scale bars: 20 μm. The data from at least three independent experiments were expressed as the mean ± SEM. *p < 0.05; **p < 0.01; #p > 0.05
ADAR1 maintains the stemness of OSCC in vitro
We then determined whether ADAR1 could promote the stemness of OSCC cell lines. The protein expression of SOX2 and POU5F1 was examined by western blot. The LV-ADAR1 group exhibited higher expression of SOX2 and POU5F1 than the LV-GFP group (Fig. 4a), whereas si-ADAR1-p110 and si-ADAR1-p150 groups attenuated the stemness of OSCC cell lines (Fig. 4b). To validate our observation, a suspended cell culture assay revealed more suspended cells in the LV-ADAR1 group than in the LV-GFP group (Fig. 4c). However, the same assay using ADAR1-p110 and ADAR1-p150 knockdown cells was unsuccessful because of the extremely small number of suspended cells. These findings demonstrated that ADAR1 could maintain and promote the stemness of OSCC.

ADAR1 promoted the stemness of OSCC cell lines in vitro. a Western blot analysis of SOX2 and POU5F1 in LV-ADAR1 and LV-GFP groups. b Western blot analysis of SOX2 and POU5F1 in si-ADAR1-p110, si-ADAR1-p150 and scrambled groups. c Representative image of suspended cell culture indicating the colony formation ability of the LV-ADAR1 and LV-GFP groups. The data were expressed as the mean ± SEM (n ≥ 3). *p < 0.05; **p < 0.01
ADAR1 physically interacts with Dicer and is involved in the maturation of oncogenic miRNAs
ADAR1 p110 has been confirmed to increase the rate of miRNA processing by Dicer [3], so co-immunoprecipitation was performed to further verify the interaction of ADAR1 and Dicer in Cal27 cells (Fig. 5a). The results suggested that ADAR1 could mainly interact with Dicer comparing with Drosha, Argonaute1, Argonaute2 and TRBP. Immunofluorescence was also performed to validate our conclusion and showed that ADAR1 and Dicer were co-localized in the cytoplasm (Fig. 5b). The Pearson’s coefficients of co-localization assay was 0.45 ± 0.11. MiR-21–3p, miR-18a-3p, miR-210-3p, miR-155-5p, miR-181a-5p and miR-19a-3p were found to be oncogenic miRNAs according to supporting experimental data that showed worse tumor prognosis for HNSCC [27,28,29,30,31]. Increased expression in Cal27 cells treated with 10 ng/ml TGF-β for 24 h implied that these miRNAs may be involved in the EMT process (Additional file 4: Figure S4). We further examined whether ADAR1 could promote the maturation of these oncogenic miRNAs. Six mature oncogenic miRNAs were remarkably increased in the LV-ADAR1 group (Fig. 5c). However, the precursors of these six oncogenic miRNAs were significantly lower in the LV-ADAR1 group than in the LV-GFP group (Fig. 5d, e). Conversely, both oncogenic miRNAs and tumor suppressor miRNAs were reduced expression in either si-ADAR1-p110 or si-Dicer group (Fig. 5f, g). These findings implied that ADAR1 could physically interact with Dicer and promote the formation of oncogenic miRNAs in OSCC.

ADAR1 could bind to Dicer and promote the maturation of oncogenic miRNAs. a Representative image of Co-IP. Cell lysates were incubated with normal rabbit IgG, anti-ADAR1, anti-Dicer, anti-Drosha, anti-Argonaute1, anti-Argonaute2, and anti-TRBP antibodies. b Representative image of immunofluorescence indicated that ADAR1 and Dicer were co-located in Cal27 cells. Scale bar: 20 μm. c ADAR1 overexpression upregulated oncogenic miRNAs. d ADAR1 overexpression decreased the precursor levels of six oncogenic miRNAs. e ADAR1 overexpression decreased the primary levels of six oncogenic miRNAs. f ADAR1-p110 knockdown reduced oncogenic miRNAs and tumor suppressor miRNAs expression. g Dicer knockdown inhibited oncogenic miRNAs and tumor suppressor miRNAs expression. The data were expressed as the mean ± SEM (n ≥ 3). *p < 0.05; **p < 0.01; ***p < 0.00; #p > 0.05
ADAR1 promotes tumor growth in vivo
Given the promising results found in vitro, we attempted to assess the effect of ADAR1 in murine models. The growth rate of xenografts with LV-ADAR1 cells was remarkably faster than that of xenografts with LV-GFP cells (Fig. 6a), and the paired tumor weights of the xenografts were also considerable (Fig. 6b). Tumors from the LV-ADAR1 group were larger than those from the LV-GFP group (Fig. 6c). ADAR1 expression in xenografts was detected by IHC (Fig. 6d) and was significantly higher in the LV-ADAR1 group than in the LV-GFP group. Notably, the expression of PCNA was also positively increased in the LV-ADAR1 group (Fig. 6e), indicating that ADAR1 may promote OSCC proliferation in vivo. As shown in Fig. 6f, the stemness markers were augmented correspondingly in the LV-ADAR1 group according to an immunohistochemistry analysis of SOX2 and POU5F1.

ADAR1 promoted tumor growth and stemness in the nude mouse xenograft model. a Image representing the volume of tumors after injection in ten nude mice (p < 0.05). Significant differences were analyzed using an independent t test. b Image representing the differences in the volume of tumors from nude mice injected with either LV-GFP or LV-ADAR1 cells. c Image of excised tumors from nude mice with LV-ADAR1 or LV-GFP cell injection. d and e ADAR1 and PCNA expression levels were examined by IHC. f SOX2 and POU5F1 expression levels were examined by IHC. Scale bars: 100 μm and 200 μm
ADAR1 is overexpressed in OSCC patients and associated with poor prognosis
Paraffin sections from 108 patients were examined by IHC to assess the expression of ADAR1. ADAR1 protein expression in the OSCC samples was categorized as high or low (Fig. 7a). The relationship between ADAR1 and the clinicopathological features is shown in Table 1. The abundance of ADAR1 was positively associated with primary tumor size, lymph node metastasis and TNM stage. However, there was no remarkable correlation between ADAR1 and other clinicopathological features, such as gender, age or histology grade of the patient.

The prognosis of patients correlated with ADAR1 expression and clinical features. a Representative image indicating high expression and low expression of ADAR1 in OSCC tissues. b Overall survival of OSCC patients with high and low expression levels. c Disease-free survival of OSCC patients with high and low expression levels. d Overall survival of OSCC patients with different histopathological grades. e Overall survival of OSCC patients with different lymph node metastasis levels. f Overall survival of OSCC patients with different TNM stages. g Overall survival of OSCC patients with different primary tumor sizes
Further indicating the impact of ADAR1 expression on the survival status of OSCC patients, ADAR1 upregulation was significantly associated with poor overall and disease-free survival rates (Fig. 7b, c). Moreover, high histopathological grade (Fig. 7d), lymph node metastasis (Fig. 7e) and late TNM stage (Fig. 7f) were also associated with a poor survival rate. Although large tumor size was not significantly different (Fig. 7g), a decrease in the survival rate with increasing tumor size was observed. We also performed univariate and multivariate Cox regression analyses in our research. Both univariate and multivariate Cox regression analyses revealed that patients with high ADAR1 expression, late T category, or lymph node metastasis had poor survival outcomes. Moreover, univariate Cox regression analysis results also showed that patients with a III + IV TNM stage had a poor prognosis (Table 2). Altogether, these data indicated that increased ADAR1 expression may have a significant relationship with poor prognosis in OSCC.
Discussion
As the sixth most common cancer worldwide, oral squamous cell carcinoma (OSCC) is a grievous health burden [32]. Finding the potential molecular mechanism underlying OSCC invasion and metastasis is urgently needed to improve the prognosis of patients. Recent studies investigating the role of ADAR1 in multiple tumors have shown that ADAR1 works as either an oncogene or a suppressive gene depending on the type of cancer [33]. Upregulation of ADAR1 was detected in some malignant tissues, such as esophageal squamous cell carcinoma [34] and hepatocellular carcinoma [35]. Moreover, brain cancer and metastatic melanoma tissues showed low expression of ADAR1 [36,37,38]. However, little is known about the effect of ADAR1 on OSCC. In the current study, we provided novel evidence showing that high levels of ADAR1 were detected in both OSCC tissues and cell lines and associated with the clinicopathological characteristics of patients.
ADAR1 has two isoforms: the shorter and constitutive ADAR1 p110 and the full-length interferon-inducible ADAR1 p150 [39]. In human cell lines, it has been proven that ADAR1 p150 is located in the cytoplasm and nucleus, while ADAR1 p110 is mainly expressed in the nucleus [40]. ADAR1 p150 can move between the nucleus and cytoplasm as a shuttling protein. Therefore, ADAR1 p150-mediated A-to-I editing occurs not only in the nucleus but also in the cytoplasm, whereas editing by ADAR1 p110 occurs in the nucleus [41, 42]. Interestingly, shuttling of ADAR1 p110 between the nucleus and cytoplasm was also reported [8]. In the present research, both real-time PCR and western blot results indicated that the ADAR1 p110 was the dominant isoform in OSCC tissues and cell lines. Both nuclear and cytoplasmic expression of ADAR1 p110 were detected in our research. To verify the role of ADAR1 in OSCC, we constructed a lentiviral vector carrying ADAR1 p110 and knocked down either ADAR1-p110 or ADAR1-p150 expression by siRNA transfection. Our data clearly showed that both ADAR1-p110 and ADAR1-p150 could promote the migration, invasion and proliferation of OSCC. However, considering the weaker ADAR1-p150 expression in OSCC tissues and cell lines, we speculate that ADAR1-p110 may play a more important role in OSCC progression.
Recently, heterogeneity among tumor cells is widely accepted. Documented data suggest that a high percentage of surgically resected OSCC patients eventually develop recurrence and distant tumor spread from residual tumors, possibly driven by OSCC-CSCs (oral squamous cell carcinoma-cancer stem cells) [43, 44]. The stemness of OSCC-CSCs is related to tumor initiation and self-renewal properties and further leads to poor prognosis [45, 46]. Many studies suggested that reducing cancer cell stemness could improve radio-sensitivity. By knocking out JARID1B, the cancer stem cell activity was reduced and the stemness radiotherapy sensitivity of OSCC was greatly improved [47]. The presence of CSC phenotype in clonospheres could increase DNA repair capacity [48]. SMC1A knockdown could limit CSC properties and enhance efficacy of radiation therapy [49]. By using western blot assays, we revealed that ADAR1 may play a potential role in promoting the stemness of OSCC cell lines. Overexpression of ADAR1 was conducive to promoting anti-anoikis and proliferation in tumor cells and negatively affecting the prognosis of OSCC. However, radio-resistance assays should be performed in future studies to validate the role of ADAR1.
OSCC cell lines retain epithelium characteristics and lack the ability to rebuild extracellular matrix [50,51,52]. The acquisition of EMT contributes to cancer cell matrix invasion and metastasis. High expression of VIM and low expression of CDH1 are features of EMT. In our EMT-induction model, the results implied that ADAR1 participated in the EMT process in OSCC cell lines. However, the mechanism of this characteristic remains unknown and needs further study. Nevertheless, the increased expression of six head-neck cancer-related oncogenic miRNAs, which were chosen by reviewing the literature of ADAR1 overexpression, implied that oncogenic miRNAs may be involved in the process. In previous studies, TGF-β was reported to regulate the expression of miRNA at either transcription or post-transcription level [53,54,55]. The smad4 as a key player in the TGF-β/smad signaling pathway cascades could cause downregulation or upregulation of the miRNAs [56]. Our studies demonstrated that ADAR1 regulated the expression of miRNAs at post-transcription levels while TGF-β regulated ADAR1 expression. With ADAR1-p110 knockdown, the EMT which induced by TGF-β was reduced, implying ADAR1 play a role in TGF-β induced EMT. However, there still need further experiments to answer whether the TGF-β regulates miRNAs via ADAR1.
MiRNAs could bind to RISC to degrade target mRNA, which may determine the biological characteristics of tumor cells by epigenetic modification [3]. Tumorigenesis and progression involve a series of cellular pathways that control cell biology, and the proteins associated with these cellular pathways may be potential oncogenes. It has been reported that ADAR1 regulates the generation of miRNAs via an editing-independent mechanism [6]. ADAR1 can not only suppress the processing of pri-miRNA to pre-miRNA by interacting with DGCR8 but also increase the processing of miRNAs by forming a heterodimer with Dicer [3, 33, 37]. In our study, we confirmed that ADAR1 could physically interact with Dicer, a key component of the miRNAs processing machinery in the cytoplasm, but weakly or not interact with Drosha, Argonaute1, Argonaute2 or TRBP. Upregulation of six oncogenic miRNAs following the reductions in both pre-miRNA and pri-miRNA by ADAR1 overexpression and downregulation of these miRNAs by ADAR1-p110 knockdown implied that ADAR1 may promote OSCC progression by promoting the maturation of oncogenic miRNAs. Furthermore, downregulation of both oncogenic miRNAs and tumor suppressor miRNAs by Dicer knockdown implied that ADAR1 may regulate miRNAs expression through Dicer. However, more oncogenic miRNAs and the regulation mechanism should be examined in OSCC cell lines in future research.
Conclusions
In summary, we demonstrate that upregulation of ADAR1 may promote OSCC progression through facilitating the maturation of oncogenic miRNAs and EMT. Our findings extend the current understanding of the molecular mechanisms of ADAR1 and suggest that ADAR1 may hold promise as a novel therapeutic target in OSCC.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADARs:
-
Adenosine deaminases acting on RNA
- CDH1:
-
Cadherin 1
- dsRNA:
-
Double-stranded RNA
- EMT:
-
Epithelial-mesenchymal transition
- HNSCC:
-
Head and neck squamous cell carcinoma
- HOK:
-
Human oral keratinocyte
- miRNA:
-
MicroRNA
- OSCC:
-
Oral squamous cell carcinoma
- OSCC-CSC:
-
Oral squamous cell carcinoma-cancer stem cell
- PCNA:
-
Proliferating cell nuclear antigen
- POU5F1:
-
POU class 5 homeobox1
- pre-miRNA:
-
Precursor transcript of miRNA gene
- pri-miRNA:
-
Primary transcript of miRNA gene
- SOX2:
-
SRY-box 2
- TGF:
-
Transforming growth factor
- VIM:
-
Vimentin
References
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32.
Kanemaru A, Yamamoto K, Kawaguchi M, Fukushima T, Lin CY, Johnson MD, et al. Deregulated matriptase activity in oral squamous cell carcinoma promotes the infiltration of cancer-associated fibroblasts by paracrine activation of protease-activated receptor 2. Int J Cancer. 2017;140(1):130–41.
Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, et al. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153(3):575–89.
Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002;52(4):195–215.
Keegan LP, Leroy A, Sproul D, O'Connell MA. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004;5(2):209.
Nishikura K, Sakurai M, Ariyoshi K, Ota H. Antagonistic and stimulative roles of ADAR1 in RNA silencing. RNA Biol. 2013;10(8):1240–7.
Czermak P, Amman F, Jantsch MF, Cimatti L. Organ-wide profiling in mouse reveals high editing levels of Filamin B mRNA in the musculoskeletal system. RNA Biol. 2018;15(7):1–9.
Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17(2):83–96.
Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013;5(3):849–60.
Heale BS, Eulalio A, Schulte L, Vogel J, O'Connell MA. Analysis of A to I editing of miRNA in macrophages exposed to Salmonella. RNA Biol. 2010;7(5):621–7.
Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550(7675):249–54.
Nemlich Y, Baruch EN, Besser MJ, Shoshan E, Bar-Eli M, Anafi L, et al. ADAR1-mediated regulation of melanoma invasion. Nat Commun. 2018;9(1):2154.
Nie Y, Ding L, Kao PN, Braun R, Yang JH. ADAR1 interacts with NF90 through double-stranded RNA and regulates NF90-mediated gene expression independently of RNA editing. Mol Cell Biol. 2005;25(16):6956–63.
Galeano F, Leroy A, Rossetti C, Gromova I, Gautier P, Keegan LP, et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int J Cancer. 2010;127(1):127–37.
Qiu W, Wang X, Buchanan M, He K, Sharma R, Zhang L, et al. ADAR1 is essential for intestinal homeostasis and stem cell maintenance. Cell Death Dis. 2013;4:e599.
Karnati HK, Panigrahi MK, Gutti RK, Greig NH, Tamargo IA. miRNAs: key players in neurodegenerative disorders and epilepsy. J Alzheimers Dis. 2015;48(3):563–80.
Makeyev EV, Maniatis T. Multilevel regulation of gene expression by microRNAs. Science. 2008;319(5871):1789–90.
Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, Joo C. A dynamic search process underlies MicroRNA targeting. Cell. 2015;162(1):96–107.
Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482(7385):347–55.
Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, et al. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151(4):900–11.
Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36(16):5270–80.
Kawamata T, Seitz H, Tomari Y. Structural determinants of miRNAs for RISC loading and slicer-independent unwinding. Nat Struct Mol Biol. 2009;16(9):953–60.
Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, et al. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279(6):4952–61.
Sakurai M, Shiromoto Y, Ota H, Song C, Kossenkov AV, Wickramasinghe J, et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat Struct Mol Biol. 2017;24(6):534–43.
Li W, Zhu D, Qin S. SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. J Exp Clin Cancer Res. 2018;37(1):148.
Sun X, Sit A, Feinberg MW. Role of miR-181 family in regulating vascular inflammation and immunity. Trends Cardiovasc Med. 2014;24(3):105–12.
Tagawa H, Ikeda S, Sawada K. Role of microRNA in the pathogenesis of malignant lymphoma. Cancer Sci. 2013;104(7):801–9.
Mirzaei H, Masoudifar A, Sahebkar A, Zare N, Sadri Nahand J, Rashidi B, et al. MicroRNA: a novel target of curcumin in cancer therapy. J Cell Physiol. 2018;233(4):3004–15.
Li Z, Wang Y, Zhu Y, Yuan C, Wang D, Zhang W, et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol Oncol. 2015;9(6):1091–105.
Shiiba M, Uzawa K, Tanzawa H. MicroRNAs in head and neck squamous cell carcinoma (HNSCC) and Oral squamous cell carcinoma (OSCC). Cancers (Basel). 2010;2(2):653–69.
John K, Wu J, Lee BW, Farah CS. MicroRNAs in head and neck cancer. Int J Dent. 2013;2013:650218.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
Nakano M, Nakajima M. Significance of A-to-I RNA editing of transcripts modulating pharmacokinetics and pharmacodynamics. Pharmacol Ther. 2018;181:13–21.
Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF, Liu H, et al. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014;74(3):840–51.
Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013;19(2):209–16.
Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17(11):1586–95.
Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Invest. 2013;123(6):2703–18.
Galore-Haskel G, Nemlich Y, Greenberg E, Ashkenazi S, Hakim M, Itzhaki O, et al. A novel immune resistance mechanism of melanoma cells controlled by the ADAR1 enzyme. Oncotarget. 2015;6(30):28999–9015.
Wang C, Zou J, Ma X, Wang E, Peng G. Mechanisms and implications of ADAR-mediated RNA editing in cancer. Cancer Lett. 2017;411:27–34.
Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15(10):5376–88.
Desterro JM, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116(Pt 9):1805–18.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168(4):670–91.
Kim JK, Jeon HY, Kim H. The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch Pharm Res. 2015;38(3):389–401.
Kaseb HO, Fohrer-Ting H, Lewis DW, Lagasse E, Gollin SM. Identification, expansion and characterization of cancer cells with stem cell properties from head and neck squamous cell carcinomas. Exp Cell Res. 2016;348(1):75–86.
Lin S, Peng C, Liao Y, Chou M, Hsieh P, Yu C. miR-1246 targets CCNG2 to enhance cancer stemness and chemoresistance in oral carcinomas. Cancers (Basel). 2018;10(8):272.
Krishnamurthy S, Nor JE. Head and neck cancer stem cells. J Dent Res. 2012;91(4):334–40.
Lin CS, Lin YC, Adebayo BO, Wu A, Chen JH, Peng YJ, et al. Silencing JARID1B suppresses oncogenicity, stemness and increases radiation sensitivity in human oral carcinoma. Cancer Lett. 2015;368(1):36–45.
Anuja K, Chowdhury AR, Saha A, Roy S, Rath AK, Kar M, et al. Radiation-induced DNA damage response and resistance in colorectal cancer stem-like cells. Int J Radiat Biol. 2019;95(6):667–79.
Yadav S, Kowolik CM, Lin M, Zuro D, Hui SK, Riggs AD, et al. SMC1A is associated with radioresistance in prostate cancer and acts by regulating epithelial-mesenchymal transition and cancer stem-like properties. Mol Carcinog. 2019;58(1):113–25.
Labernadie A, Kato T, Brugues A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19(3):224–37.
Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155(7):1639–51.
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–400.
Carrella S, Barbato S, D'Agostino Y, Salierno FG, Manfredi A, Banfi S, et al. TGF-beta controls miR-181/ERK regulatory network during retinal axon specification and growth. PLoS One. 2015;10(12):e0144129.
Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123(1):150–63.
Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, et al. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2010;29(12):1787–97.
Lashgarian PDHE. Effect of TGF-β/smad signaling pathway blocking on expression profiles of miR-335, miR-150, miR-194, miR-27a, and miR-199a of hepatic stellate cells (HSCs). J Gastroen Hepatol. 2017;10(2):112–7.
Acknowledgments
We thank Dr. Xiaoling Song and Dr. Bing Qi for technical assistance.
Funding
This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 2018–87) and the Jiangsu Provincial Medical Innovation Team (CXTDA2017036).
Author information
Authors and Affiliations
Contributions
XL, YF and HBJ conceived and designed the experiments. JDH and MW performed the experiments and drafted the manuscript. ZZZ, RYX, PZ, and SWZ participated in the experiments and analyzed the data. LKL participated in the histology experiment. HBJ oversaw of all aspects of the study. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Research Committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Differences in the expression of ADAR1-p110 and ADAR1-p150 between OSCC tissue and matched non-tumor oral mucosa. The expression of ADAR1-p110 and ADAR1-p150 were normalized to that of GAPDH. The data were expressed as the mean ± SEM (n ≥ 3). **p < 0.01. (TIF 1001 kb)
Additional file 2:
Figure S2 Real-time PCR (a) and western blot (b) assays were used to detect ADAR1 expression in Cal27 cells transfected with an ADAR1 overexpression lentiviral vector. (c) The interference efficiency of three si-ADAR1-p110 sequences was analyzed by real-time PCR. (d) Western blot assay was used to detect ADAR1-p110 protein expression in Cal27 and HN4 cells with ADAR1-p110 knockdown. (e) The interference efficiency of si-ADAR1-p150 sequences was detected by real-time PCR. (f) Western blot assay was used to detect ADAR1-p150 protein expression in Cal27 and HN4 cells with ADAR1-p150 knockdown. (g) Representative image indicated the cell cycle of LV-GFP and LV-ADAR1 groups. (h) Cell proliferation ability was showed in LV-GFP and LV-ADAR1 groups as detected by CCK-8 assay. (i) Cell proliferation ability was showed in both Cal27 and HN4 cells with ADAR1-p150 knockdown as detected by CCK-8 assay. The data were summarized for at least three independent experiments. *p < 0.05; **p < 0.01; #p > 0.05. (TIF 4844 kb)
Additional file 3:
Figure S3. Western blot analysis of CDH1 and VIM expression in ADAR1 overexpressed Cal27 and HN4 cells accompany with ADAR1-p150 knockdown. (TIF 599 kb)
Additional file 4:
Figure S4. Real-time PCR assay was used to detect miR-21–3p, miR-18a-3p, miR-210-3p, miR-155-5p, miR-181a-5p and miR-19a-3p expression. The data were summarized from at least three independent experiments. *p < 0.05; **p < 0.01; #p > 0.05. (TIF 902 kb)
Additional file 5:
Table S1. The primary antibodies used in western blot. (XLSX 34 kb)
Additional file 6:
Table S2. Clinicopathological profiles of 61 primary OSCC patients. OSCC, oral squamous cell carcinoma; P, P value; T, tumor size; N, lymph node; M, distant metastasis; TNM, tumor-node-metastases. (XLSX 26 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, X., Fu, Y., Huang, J. et al. ADAR1 promotes the epithelial-to-mesenchymal transition and stem-like cell phenotype of oral cancer by facilitating oncogenic microRNA maturation. J Exp Clin Cancer Res 38, 315 (2019). https://doi.org/10.1186/s13046-019-1300-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1300-2