
Abstract
Background
Besides being a risk factor for multiple metabolic disorders, obesity could affect female reproduction. While increased adiposity is associated with hormonal changes that could disrupt the function of the hypothalamus and the pituitary, compelling data suggest that obesity-related hormonal and inflammatory changes could directly impact ovarian function.
Objective
To review the available data related to the mechanisms by which obesity, and its associated hormonal and inflammatory changes, could affect the female reproductive function with a focus on the hypothalamic-pituitary-ovarian (HPO) axis.
Methods
PubMed database search for publications in English language until October 2017 pertaining to obesity and female reproductive function was performed.
Results
The obesity-related changes in hormone levels, in particular leptin, adiponectin, ghrelin, neuropeptide Y and agouti-related protein, are associated with reproductive dysfunction at both the hypothalamic-pituitary and the ovarian levels. The pro-inflammatory molecules advanced glycation end products (AGEs) and monocyte chemotactic protein-1 (MCP-1) are emerging as relatively new players in the pathophysiology of obesity-related ovarian dysfunction.
Conclusion
There is an intricate crosstalk between the adipose tissue and the inflammatory system with the HPO axis function. Understanding the mechanisms behind this crosstalk could lead to potential therapies for the common obesity-related reproductive dysfunction.
Similar content being viewed by others

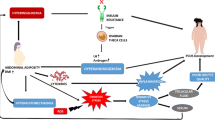
Background
According to a recent population study, approximately 39% of the population over the age of 20 and 18% of children between the ages of 2–19 are obese [1]. Obesity causes a huge economic burden where it is estimated that obesity will add 48–66 billion dollars in related health care expenditures by the year 2030 [2]. This is due to obesity-related comorbidities such as diabetes mellitus, hypertension, dyslipidemia, and cardiovascular disease [3]. Besides these chronic disorders, obesity is also associated with reproductive and obstetric complications such as menstrual irregularities, subfertility, endometrial hyperplasia and cancer, as well as poor obstetrical and perinatal outcomes [4,5,6,7]. This review will focus on the relationship between obesity and female reproductive function with a focus on alterations in the hypothalamic pituitary ovarian (HPO) axis and the direct effect of obesity-related inflammatory processes on ovarian function.
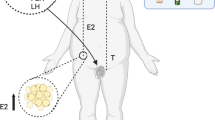
Normal HPO Axis
The female reproductive physiology is a complex interaction between neuroendocrine and endocrine signaling affecting the hypothalamus, the pituitary gland and the ovaries. At the level of the hypothalamus, gonadotropin releasing hormone (GnRH) pulses activate the pituitary release of the two gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) [8, 9]. FSH and LH in turn act on the ovaries to stimulate follicular growth and result in the production of estradiol and, following ovulation, progesterone [10]. Estradiol, together with ovarian inhibin B, acts primarily in a negative feedback loop on the hypothalamus suppressing the release of FSH. When estradiol reaches a sustained threshold in concentration and duration, for at least 24–48 h [11,12,13], it provides a positive feedback that increases the frequency and decreases the amplitude of GnRH pulses thereby activating increased pituitary release of LH surge for ovulation to occur [14]. There are multiple regulators of this cycle and our review is limited to those regulators, which are altered, directly or indirectly, in overweight and obese women.
The HPO axis plays a crucial role in pubertal transition. Normal pubertal development is under two main controls, adrenal and hypothalamic [15]. The adrenal glands participate in adrenarche, which often precedes the remainder of pubertal events. Adrenarche is independent of the hypothalamic pubertal changes as observed in several disorders of sexual differentiation [16]. The hypothalamus, with its GnRH pulse generator responsible for the initiation of puberty, is quiescent in childhood, likely under GABAergic (gamma-aminobutyric acid) inhibitory control [17, 18]. Normal central activation of the hypothalamus via kisspeptin and glutamate neurons in the arcuate nucleus results in nocturnal increases in low frequency LH pulses [19]. These pulses become more prevalent throughout the day until the GnRH pulse generator achieves the frequency and amplitude sufficient for cyclic ovarian hormonal function and ultimately menarche. During normal puberty, there is a physiologic insulin resistance that helps achieve the anticipated pubertal weight gain and growth [20,21,22]. The insulin resistance leads to decreased hepatic derived circulating sex hormone-binding globulin (SHBG), which in turn increases circulating free estradiol levels. This may also contribute to a parallel adrenal activation resulting in more androgen production and ultimately adrenarche [17, 18]. Increasing estradiol levels, in turn, are significantly associated with growth velocity, [23] possibly through the stimulation of the growth hormone – Insulin like growth factor 1 (IGF-1) axis [24, 25].
Obesity and the HPO axis
It has been thought that obesity changes the expected time course of puberty and leads to earlier thelarche, adrenarche and menarche [26,27,28]. Adiposity, and specifically the distribution of the adipose tissue, contributes to fluctuations in peripheral steroid hormone secretion, thereby impacting pubertal development [29]. For instance, the adipocytes contain aromatase, that both assists in the production [30] and the conversion of steroid hormones, mainly androgens to estrogens. Population studies have demonstrated a trend for earlier menarche, due to the obesity epidemic, over the past 30 years [31], highlighting the potential for significant increase in health care risks and cost for these young girls. Increased adipose tissue quantity rather than increased sensitization or increased aromatase activity during aging [32] and therefore increased peripheral production of estrogens leads to increased rate of serious hormone dependent cancers, such as endometrial and breast cancer [33, 34].
Obesity decreases pituitary LH pulse amplitude and mean LH release without changing its frequency, leading to impaired luteal phase [35]. Additionally, obesity may affect various components of the HPO axis, and may have direct effect on ovarian function independent of hypothalamic pituitary function. We will elucidate in the following sections the changes that occur in the metabolic and inflammatory systems that are related to ovarian dysfunction.
Leptin
Leptin is a 16-kDa adipokine secreted primarily by white adipose tissue, although it can be secreted from other tissues, such as gastric mucosa [36]. Serum leptin levels positively correlate with the amount of adipose tissue in the body [37]. Leptin has several functions, which in concert reflect body energy homeostasis. It acts to stimulate energy expenditure and suppresses appetite via its signaling at the level of the hypothalamus [18, 38]. These findings were confirmed with mouse knockout model where leptin receptor knockout mice become obese with a rapid and persistent weight gain [39]. Because leptin suppresses appetite, it was studied as a potential target for weight loss drug development [40]. However, it has been shown that increased exogenous leptin does not necessarily induce weight loss [41, 42] suggesting that there is a resistance state to the action of leptin at the level of the hypothalamus. Indeed, high circulating leptin levels observed in obese women supports the concept of leptin resistance [37]. This desensitization to leptin is not a complete blockage of leptin signaling and can possibly be reversed with weight loss [43].
Leptin’s energy homeostatic function in part contributes to its effect on puberty. Puberty requires a certain energy balance to proceed [22, 44]. Leptin has a permissive action for pubertal progression, but it is not the initiator or the sole mechanism by which puberty occurs [45]. Conversely, hyperleptinemia may initially cause earlier reproductive maturation, and prolonged high leptin levels could lead to ovulatory dysfunction [46]. On the other hand, low leptin levels observed in energy-deficient states are associated with delayed or lack of puberty, when puberty is not physiologically desired [47].
Leptin affects GnRH pulse neurons indirectly though GABA or kisspeptin [39, 48]. Both GABA and kisspeptin neurons have a focus in the arcuate nucleus, where leptin receptors are readily available [39, 49, 50]. Knockout models for the leptin receptor that is found on GABA specific neurons display delayed puberty and decreased fecundability in animal models [39]. Similarly, loss of function gene mutation in the kisspeptin gene results in delayed or absent puberty [51]. Hypothalamic kisspeptin levels often correlate with leptin levels [50] in line with low energy states, making this an alternate pathway for leptin signaling [52]. Both low serum, and therefore hypothalamic, leptin levels and low hypothalamic kisspeptin levels decrease LH secretion by the pituitary, ultimately affecting ovulation [52].
Studies on the effect of leptin on ovarian folliculogenesis and ovulation have shown conflicting results. Leptin has been isolated in both mural and cumulus granulosa cells and within the follicular fluid of pre-ovulatory follicles in women undergoing in vitro fertilization (IVF) [53]. Serum leptin levels increase after ovulation and have peak levels in the mid-secretory phase, possibly contributing to the implantation window within the endometrium [54]. Some studies suggested an inhibitory effect of leptin, especially on early follicular development, while others suggested that leptin could induce ovulation, independent of LH [55] since there seems to be an interaction between estradiol and leptin [55, 56]. In a mouse model, leptin administration increased LH levels and follicular development corresponding to an increase in ovarian tissue weight [57]. In addition, IVF outcome studies pointed to leptin as negatively correlated to reproductive outcome. In one study of serum leptin levels in patients undergoing IVF, higher leptin:BMI ratio was associated with a decreased number of good quality embryos and lower implantation and pregnancy rates [58]. At the ovarian level [46], supraphysiologic levels of leptin inhibits androstenedione and progesterone production [59, 60]. Additionally, human granulosa and cumulus cells exposed to leptin in vitro lead to a downregulation in anti-Müllerian hormone (AMH) gene expression via the JAK/STAT pathway [61] potentially leading to ovulatory dysfunction. In summary, normal leptin homeostasis is required for normal physiologic functions at the level of the hypothalamus and the ovaries. While low levels of leptin could disrupt the GnRH pulsatility, supraphysiologic levels of leptin could disrupt ovarian folliculogenesis [46, 55, 62].
Ghrelin
Ghrelin is an orexigenic enterokine composed of 28 amino acids related to the oxyntomodulin family of intestinal peptides [63]. It is predominantly produced by the stomach, but is also detectable in many other tissues, like bowel, pancreas, hypothalamus and pituitary [64]. Ghrelin is increased in fasting states and stimulates appetite to compensate for decreased nutritional intake [65]. Mouse models confirm that ghrelin increases food consumption as an immediate and short-term effect [66] by acting on the arcuate nucleus in the hypothalamus and stimulates appetite via neuropeptide Y (NPY) and agouti related protein (AgRP) neurons [67]. One of ghrelin’s targets is the hypothalamic arcuate nucleus where it increases the expression of NPY/AgRP mRNA [68] resulting in increased body weight and inhibiting proopiomelanocortin (POMC) neurons [69]. Double knockout (NPY-/AgRP-) mouse models, or other NPY/AgRP deficient models demonstrated suppression of ghrelin-induced appetite stimulation as compared to wild type control animals. Deficiency of both NPY and AgRP is necessary to abolish the effect of ghrelin while knockout of only one of these mediators is not sufficient to suppress ghrelin’s effect [66].
In the hypothalamus, ghrelin can decrease both GnRH secretion and pulsatility [70,71,72,73] possibly through NPY- and AgRP- mediated mechanisms [74]. Vulliémoz et al. suggested that this effect is part of the homeostatic control of reproductive function in response to nutritional changes. In low energy states, such as fasting, ghrelin levels are increased, thereby altering the HPO axis cyclic activity [73]. Ghrelin also stimulates other pituitary hormone release, such as adrenocorticotropic hormone and prolactin [75]. In low energy states where ghrelin levels are elevated, prolactin levels may be increased potentially leading to disruption of ovarian cyclicity. Ghrelin has not clearly been shown to have a direct effect on pubertal transition. However, ghrelin levels decrease throughout childhood as puberty approaches [75]. Given its inverse relationship to adiposity, decreasing ghrelin levels may be a reflection of the increasing weight gain observed in the pre-pubertal stage [76].
Studies on the effect of ghrelin on ovarian steroidogenesis have shown mixed results [74, 77]. Animal studies suggest that ghrelin could induce estradiol and/or progesterone production, [78] or could inhibit estradiol release via inhibiting CYP19A1 (aromatase enzyme) expression [74, 79, 80]. These effects may be dose-dependent [81] or related to the activated portion of the ghrelin molecule [79]. The full-length ghrelin molecule stimulates estradiol secretion while certain amino acid fragments of ghrelin are inhibitory for estradiol secretion [79]. Ghrelin could affect folliculogenesis by increasing cell proliferation and decreasing apoptosis and follicular atresia [82, 83]. Similarly, chronic ghrelin infusion leads to increase in follicle number and decrease in corpus luteum number in a rat model [84]. Additionally, ghrelin, with its connection to obesity, is related to insulin regulation and insulin resistance where it decreases insulin secretion and sensitivity [64, 85, 86] ultimately leading to insulin resistance [87].
Thus, these findings suggest that ghrelin may indirectly contribute to puberty through general energy homeostasis such as insulin action, and it acts at the level of the hypothalamus, pituitary and ovary leading to alterations in normal reproductive function.
Neuropeptide Y and Agouti-related protein: (NPY/AgRP)
The connection between peripherally circulating adipokines, enterokines and the HPO axis centers on a collection of neurons within the arcuate nucleus, which secrete the neuropeptides NPY and AgRP [88]. NPY is a 36 amino acid neurotransmitter peptide predominantly expressed in sympathetic neurons. It stimulates fat angiogenesis and proliferation via both a central hypothalamic and peripheral mechanism [49, 89]. AgRP is a 112 amino acid neuropeptide expressed in the arcuate nucleus [90, 91] and stimulates appetite [91]. Both NPY and AgRP are orexigenic neurons and interact with ghrelin to promote appetite [66].
Under normal physiologic conditions, NPY concentration increases in the portal capillary blood during the ovulatory surge to potentiate the action of GnRH on pituitary gonadotropin secretion [92, 93]. Similarly, AgRP engages the GnRH pulse generator neurons within the hypothalamus and regulates gonadotropin release. Both NPY and AgRP have tonic inhibitory effects where they could be stimulated by ghrelin in order to decrease GnRH pulse frequency and amplitude [94]. Infusion of NPY, independent of ghrelin, also decreases pituitary LH secretion [95]. NPY could have a negative regulatory impact on ovarian folliculogenesis where it could have a pro-apoptotic and an anti-proliferative effect [96]. NPY has been shown to have no effect on progesterone secretion [97], but it recently has been found to stimulate estradiol and testosterone secretion in catfish [98]. Data pertaining to the direct effect of NPY and AgRP on ovarian steroidogenesis and folliculogenesis are still limited and further studies are required.
Adiponectin
Adiponectin is a 30-kDa adipokine secreted by the adipose tissue [99]. Opposite to leptin, adiponectin levels increase with starvation [99]. Adiponectin acts in the brain by binding to the adiponectin receptor 1 (AdipoR1) in the arcuate nucleus. This binding leads to activation of AMPK resulting in increased food intake and reduced energy expenditure [100]. Adiponectin production increases insulin sensitivity and is inversely correlated with adiposity [101]. Knockout studies demonstrated that the absence of adiponectin causes severe insulin resistance that is reversible with administration of exogenous adiponectin [102]. Low adiponectin levels, as seen in certain genetic polymorphisms, have been linked to insulin resistance, metabolic syndrome and type 2 diabetes mellitus [103,104,105]. Interestingly, the adiponectin gene is located at 3q27, near the diabetes susceptibility gene locus [103, 106]. Adiponectin is structurally similar to the pro-inflammatory TNF- α family, however it functions as an anti-inflammatory adipokine, inhibiting the production of TNF-α within the adipocyte [102]. Conversely, pro-inflammatory agents such as TNF-α, IL-6 and IL-8 are implicated in the development of insulin resistance via hypothalamic inflammation [107]. This inflammation in turn contributes to both insulin and leptin resistance, promoting further obesity and subsequently diabetes [107]. In fact, in obese women adiponectin levels are low and pro-inflammatory markers, such as TNF-α, IL-6, and CRP are increased [46].
Adiponectin and its receptors are found in many organs including the ovaries [108,109,110,111]. Adiponectin acts in concert with insulin and IGF-1 to mediate changes within the granulosa cells during the periovulatory phase. Through IGF-1, it increases ovarian production of estradiol and progesterone in rat ovaries [108] possibly by up-regulating StAR gene [109]. It also causes vasodilation with the upregulation of VEGF and COX-2 expression in the periovulatory ovary [109]. Adiponectin knockout mice show ovarian dysfunction reflected by fewer oocytes, more atretic follicles, prolonged diestrus cycles and decreased LH receptor activity [112]. In a retrospective case-controlled analysis, adiponectin levels were higher in women who conceived after IVF and positively correlated with the number of oocytes retrieved, independent of BMI [113]. Similarly, while adiponectin levels are low or minimal in human and mouse granulosa cells, its presence enhances fertilization rates and embryo development [114, 115].
These findings suggest that adiponectin is notable for its action in mediating insulin sensitivity, with its receptors found at every level of the reproductive axis making it a great therapeutic target for ovulatory dysfunction.
Insulin
Insulin is a 51 amino acid protein synthesized in the beta islet cells of the pancreas. Its release is stimulated by glucose in the gastrointestinal tract from ingestion, as well as various amino acids directly [116]. Insulin levels rise and its sensitivity decreases with obesity [116]. Insulin resistance, in conjunction with obesity, impacts reproduction. While not a direct adipokine, adipose tissue stimulates pancreatic beta islet cells to release insulin. Hyperinsulinemia acts on the liver to cause a decrease in SHBG production [117]. This in turn leads to increased free circulating steroid hormone levels, such as estrogens and androgens. Insulin increases androgen production by two independent pathways; first by up-regulating CYP17A1 enzymes, which increase androgen production in both the adrenal gland and the ovary [118]. Second, insulin augments LH action on the ovary to increase androgen production and secretion [118, 119]. Insulin resistance is associated with higher leptin levels [38]. As noted previously, higher circulating leptin levels lead to leptin resistance which in turn leads to greater insulin resistance.
Insulin acts on the pituitary to modulate the GnRH receptor and increases LH secretion after GnRH stimulation [120]. Insulin augments FSH activity by increasing ovarian steroidogenesis and inducing LH responsiveness [121]. Hyperinsulinemia is consequently associated with elevated basal LH levels and hyperandrogenism [119]. Insulin alone does not have an effect on ovarian response to gonadotropic hyperstimulation during IVF in women without underlying insulin resistance [122]. Rather, the changes seen during IVF stimulation are due to insulin resistance [122]. Syndromic severe insulin resistance, as seen in some genetic disorders, is associated with enlarged ovaries and hyperandrogenism independent of gonadotropin levels. Elevated insulin levels over a long period of time can lead to increased autophosphorylation of its receptor, which can inactivate one of its downstream transducers, GSK3. This inactivation can lead to spindle disruption within growing oocytes [123, 124]. Additionally high insulin levels during oocyte development disrupt chromatin remodeling within mouse oocytes, thereby contributing to poorer oocyte quality [123]. Mouse models show that the pituitary is still sensitive to changes in insulin levels despite peripheral insulin resistance and basal hyperinsulinemia [120].
Disruption of insulin signaling in diet-induced obesity improves reproductive cyclicity in mice, suggesting that insulin represents a mediator for pituitary LH dysregulation in obesity [120]. Further study of insulin at the level of the ovary, specifically in insulin receptor knockout mice in theca cells, also demonstrates improved cyclic reproduction in mice, showing a coordinated effect of insulin along the HPO axis to disrupt cyclicity but not pubertal onset [118]. In summary, insulin’s action is known to be necessary for changes in food intake and body weight. Its well-studied actions on the HPO axis and its relationship to adipokines such as leptin and adiponectin makes it a major player in female reproduction.
AGEs and MCP-1
Obesity is a state of chronic inflammation with macrophage infiltration into various tissues. Macrophage infiltration into adipose tissue is directly correlated to both the degree of adiposity as well as chemokine/adipokine production, such as MCP-1 [125]. The pattern of macrophage infiltration is similar to that found in disorders associated with chronic inflammation such as rheumatoid arthritis [125]. In addition to elevated circulating inflammatory markers, such as TNF-α, IL-6, and CRP [46], obese women have elevated levels of circulating AGEs [126] and MCP-1 [127]. In animal studies, obese mice have higher MCP-1 levels, which correlate with insulin resistance [128].
The pro-inflammatory AGEs may be part of the link between diet-induced obesity and inflammation, with AGEs inducing MCP-1 gene expression [129] thus forming the AGEs/MCP-1 axis. AGEs are highly reactive molecules formed by non-enzymatic cross-linking of proteins, lipids and nucleic acids with glucose [130, 131]. They may be formed endogenously or exogenously ingested as part of diet or through smoking [132]. AGE levels are elevated in chronic diseases such as type 2 Diabetes Mellitus, metabolic syndrome, cardiovascular disease, and neurodegenerative disorders [133,134,135]. AGEs have also been studied for their effect on reproduction [135,136,137].
Kandaraki et al. has demonstrated in human immortalized granulosa cells (KGN cell line) that AGEs attenuate LH- and FSH-induced ERK signaling needed for cell proliferation and proper follicular growth [138], one possible mechanism for AGE-induced ovulatory dysfunction. Similarly, AGEs interfere with glucose transport within granulosa cells [136]. Our recent data have shown that high-AGE diet could induce ovulatory dysfunction in a mouse model, as reflected by prolonged diestrus phase (unpublished data). We have also shown that high-fat diet induced obesity leads to ovulatory dysfunction in mice, as demonstrated by fewer oocytes ovulated following superovulation compared to mice on normal chow diet (controls) [139]. This ovarian dysfunction was not observed in MCP-1 knockout mice that became obese following ingestion of a high-fat diet, suggesting that lack of MCP-1 may be protective against high-fat- and obesity-induced ovarian dysfunction. Further supporting this hypothesis, we showed that elevated serum MCP-1 levels were associated with poorer outcome in women undergoing IVF [140], an effect that is pronounced in women with already diminished ovarian reserve. Additionally, AGE levels in follicular fluid were negatively correlated with IVF outcome parameters: fewer oocytes retrieved and fertilized, fewer embryos and lower ongoing pregnancy rate [137]. Taken together, these observations suggest that obesity may have direct deleterious effects on the ovaries partly through activation of inflammatory AGE/MCP-1 axis.
Relationship between obesity and assisted reproductive technology (ART) outcome
Population studies on the clinical sequelae of obesity provide a connection between obesity and subfertility. Several studies indicated that obesity is a risk factor for ovulatory dysfunction [141,142,143]. For women who ovulate regularly, obesity increases the time to conception; for instance, a high waist- height ratio decreases fecundity by 30% [144]. Van der Steeg et al. found that in ovulatory infertile women (who underwent fertility evaluation but did not yet receive treatment), for every BMI unit over 29, there was a 5% decrease in the probability of a conception [145]. This increased time to conception is not only in the infertile population. Even in obese fertile women there was an increased time to conception from 3 to 5 months [146]. Other population studies have shown that 33% of obese women at age 23 did not conceive spontaneously when trying to conceive for 12 months [147]. This increases the number of couples meeting criteria for infertility and therefore for potential ART interventions [147]. Obesity could also impact ovarian reserve markers. Studies have shown that obesity is negatively correlated with serum AMH [148, 149], FSH, LH and inhibin B levels [150]. These markers provide added support for a clinician’s analysis of a couple’s fertility potential and may guide treatment options [151].
Obesity does not only affect spontaneous pregnancy rates in fertile patients, but it also confers a risk for poorer ART outcome. While earlier data could not demonstrate adequate convincing evidence of poorer ART outcome parameters, recent data supports this correlation. Large national cohort studies as well as systemic reviews and meta-analyses suggest that increasing BMI is negatively correlated with implantation, clinical pregnancy, and live birth rates [152,153,154]. Obese patients require higher doses of gonadotropins but achieve lower serum estradiol levels and lower number of oocytes retrieved [152, 155]. The oocytes of obese women tend to be smaller with decreased fertilization potential, leading to a decreased blastocyst formation and decreased trophectoderm cell number [156]. Additionally, obese women have higher cycle cancellation rates, possibly due to changes in pharmacodynamics of GnRH antagonists (clinically used to inhibit ovulation) leading to early LH surge and premature ovulation [157]. While not all studies identified these specific adverse ART outcome measures (i.e. smaller oocytes, decreased embryo quantity and quality), they still demonstrated a lower clinical pregnancy and live birth rates-- up to 50% decrease compared to control women with normal BMI [152, 158].
The poorer IVF outcomes observed in obese patients are quite intriguing. These observations suggest that the effect of obesity on ovarian function is not solely dependent on the HPO axis, since gonadotropins are supplied exogenously during IVF cycles, thus bypassing the HPO axis. Obesity adversely impacts ART outcome differently in different ethnic populations [159,160,161]. However, these findings were inconsistent in the literature [162,163,164] and may actually be in part due to ethnic variations in BMI rather than difference in ethnicity itself [159, 161].
Obesity could disrupt endometrial receptivity leading to poorer implantation rates [158, 165, 166], arguably due to endometrial inflammation. Inflammatory markers such as IL-6 and TNF-α have been implicated in lower implantation rates [46]. Similar to macrophage infiltration in the adipose tissue [125], we have shown that there is increased expression of macrophage markers in the ovaries of obese mice following the ingestion of a high-fat diet [167]. Moreover, mice given high-fat diet ovulated fewer oocytes following superovulation, further supporting the notion that obesity may have direct effect on ovaries, independent of HPO axis. Clearly the data to date demonstrates that obesity affects ART outcome in women undergoing IVF possibly via actions on all aspects: oocyte, embryo and endometrium.
Conclusion
With the uncurbed obesity epidemic, more reproductive-aged women will face metabolic and reproductive complications. Body energy hemostasis is closely linked to reproductive function through many hormones, adipokines, cytokines, and growth factors that act at the level of the brain and the ovaries. Obesity is also a state of chronic inflammation, which may directly affect ovarian function possibly by increased macrophage infiltration in the ovaries through MCP-1 mediated pathways. The elevation of AGEs in the serum and tissues of obese women may exacerbate the reproductive dysfunction associated with adiposity and may provide, along with MCP-1, a crucial link between obesity and ovarian macrophage infiltration. Each of these molecules and their prospective pathways may represent potential therapeutic targets in order to improve the overall reproductive health of overweight/obese women. Obesity, with its alterations in the AGEs/MCP-1 axis, could disrupt the ovarian microenvironment potentially compromising oocyte competence, formation of healthy embryos and ultimately conception.
Finally, this review underscores a critical need to uncover the mechanistic actions of molecules that affect almost every level of the HPO axis. Obesity, a significant and growing public health problem, is an overwhelming condition that causes reproductive disturbances in women in part due to ovarian dysfunction. Losing weight is commonly challenging and often not sustainable. Thus there is a need to establish therapies for ovarian dysfunction and to improve ovarian health in the obese patient population.
References
Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity among adults and youth: United States, 2015-2016. NCHS Data Brief. 2017;(288):1–8.
Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378(9793):815–25.
Tzeng CR, Chang YC, Chang YC, Wang CW, Chen CH, Hsu MI. Cluster analysis of cardiovascular and metabolic risk factors in women of reproductive age. Fertil Steril. 2014;101(5):1404–10.
Wei S, Schmidt MD, Dwyer T, Norman RJ, Venn AJ. Obesity and menstrual irregularity: associations with SHBG, testosterone, and insulin. Obesity (Silver Spring). 2009;17(5):1070–6.
Wise MR, Jordan V, Lagas A, Showell M, Wong N, Lensen S, Farquhar CM. Obesity and endometrial hyperplasia and cancer in premenopausal women: a systematic review. Am J Obstet Gynecol. 2016;214(6):689.e681–17.
Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113(10):1126–33.
Massetti GM, Dietz WH, Richardson LC. Excessive weight gain, obesity, and Cancer: opportunities for clinical intervention. JAMA. 2017;318(20):1975–6.
Belchetz PE, Plant TM, Nakai Y, Keogh EJ, Knobil E. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science (New York, NY). 1978;202(4368):631–3.
Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128(1):509–17.
Adashi EY. Endocrinology of the ovary. Hum Reprod. 1994;9(5):815–27.
Evans NP, Dahl GE, Mauger DT, Padmanabhan V, Thrun LA, Karsch FJ. Does estradiol induce the preovulatory gonadotropin-releasing hormone (GnRH) surge in the ewe by inducing a progressive change in the mode of operation of the GnRH neurosecretory system. Endocrinology. 1995;136(12):5511–9.
Xia L, Van Vugt D, Alston EJ, Luckhaus J, Ferin M. A surge of gonadotropin-releasing hormone accompanies the estradiol-induced gonadotropin surge in the rhesus monkey. Endocrinology. 1992;131(6):2812–20.
Knobil E. The neuroendocrine control of ovulation. Hum Reprod. 1988;3(4):469–72.
Marshall JC, Griffin ML. The role of changing pulse frequency in the regulation of ovulation. Hum Reprod. 1993;8(Suppl 2):57–61.
Baker ER. Body weight and the initiation of puberty. Clin Obstet Gynecol. 1985;28(3):573–9.
Counts DR, Pescovitz OH, Barnes KM, Hench KD, Chrousos GP, Sherins RJ, Comite F, Loriaux DL, Cutler GB Jr. Dissociation of adrenarche and gonadarche in precocious puberty and in isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1987;64(6):1174–8.
Jasik CB, Lustig RH. Adolescent obesity and puberty: the “perfect storm”. Ann N Y Acad Sci. 2008;1135:265–79.
Burt Solorzano CM, McCartney CR. Obesity and the pubertal transition in girls and boys. Reproduction. 2010;140(3):399–410.
Cortés ME, Carrera B, Rioseco H, Pablo del Río J, Vigil P. The role of Kisspeptin in the onset of puberty and in the ovulatory mechanism: a mini-review. J Pediatr Adolesc Gynecol. 2015;28(5):286–91.
Bloch CA, Clemons P, Sperling MA. Puberty decreases insulin sensitivity. J Pediatr. 1987;110(3):481–7.
Kelsey MM, Zeitler PS. Insulin resistance of puberty. Curr Diab Rep. 2016;16(7):64.
Frisch RE, Revelle R. Height and weight at menarche and a hypothesis of critical body weights and adolescent events. Science (New York, NY). 1970;169(3943):397–9.
Goji K. Twenty-four-hour concentration profiles of gonadotropin and estradiol (E2) in prepubertal and early pubertal girls: the diurnal rise of E2 is opposite the nocturnal rise of gonadotropin. J Clin Endocrinol Metab. 1993;77(6):1629–35.
Decensi A, Robertson C, Ballardini B, Paggi D, Guerrieri-Gonzaga A, Bonanni B, Manetti L, Johansson H, Barreca A, Bettega D, et al. Effect of tamoxifen on lipoprotein(a) and insulin-like growth factor-I (IGF-I) in healthy women. Eur J Cancer. 1999;35(4):596–600.
van den Berg G, Veldhuis JD, Frölich M, Roelfsema F. An amplitude-specific divergence in the pulsatile mode of growth hormone (GH) secretion underlies the gender difference in mean GH concentrations in men and premenopausal women. J Clin Endocrinol Metab. 1996;81(7):2460–7.
Chen C, Zhang Y, Sun W, Chen Y, Jiang Y, Song Y, Lin Q, Zhu L, Zhu Q, Wang X, et al. Investigating the relationship between precocious puberty and obesity: a cross-sectional study in shanghai, China. BMJ Open. 2017;7(4):e014004.
Kaplowitz PB, Slora EJ, Wasserman RC, Pedlow SE, Herman-Giddens ME. Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics. 2001;108(2):347–53.
Rosenfield RL, Lipton RB, Drum ML. Thelarche, pubarche, and menarche attainment in children with normal and elevated body mass index. Pediatrics. 2009;123(1):84–8.
de Ridder CM, Bruning PF, Zonderland ML, Thijssen JH, Bonfrer JM, Blankenstein MA, Huisveld IA, Erich WB. Body fat mass, body fat distribution, and plasma hormones in early puberty in females. J Clin Endocrinol Metab. 1990;70(4):888–93.
Li J, Daly E, Campioli E, Wabitsch M, Papadopoulos V. De novo synthesis of steroids and oxysterols in adipocytes. J Biol Chem. 2014;289(2):747–64.
Freedman DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR, Berenson GS. Relation of age at menarche to race, time period, and anthropometric dimensions: the Bogalusa heart study. Pediatrics. 2002;110(4):e43.
Cleland WH, Mendelson CR, Simpson ER. Effects of aging and obesity on aromatase activity of human adipose cells. J Clin Endocrinol Metab. 1985;60(1):174–7.
Zhao H, Zhou L, Shangguan AJ, Bulun SE. Aromatase expression and regulation in breast and endometrial cancer. J Mol Endocrinol. 2016;57(1):R19–33.
Nelson LR, Bulun SE. Estrogen production and action. J Am Acad Dermatol. 2001;45(3 Suppl):S116–24.
Jain A, Polotsky AJ, Rochester D, Berga SL, Loucks T, Zeitlian G, Gibbs K, Polotsky HN, Feng S, Isaac B, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92(7):2468–73.
Cammisotto PG, Bendayan M. Leptin secretion by white adipose tissue and gastric mucosa. Histol Histopathol. 2007;22(2):199–210.
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334(5):292–5.
Bozkurt L, Gobl CS, Rami-Merhar B, Winhofer Y, Baumgartner-Parzer S, Schober E, Kautzky-Willer A. The cross-link between Adipokines, insulin resistance and obesity in offspring of diabetic pregnancies. Horm Res Paediatr. 2016;86(5):300–8.
Zuure WA, Roberts AL, Quennell JH, Anderson GM. Leptin signaling in GABA neurons, but not glutamate neurons, is required for reproductive function. J Neurosci. 2013;33(45):17874–83.
Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282(16):1568–75.
Liu AG, Smith SR, Fujioka K, Greenway FL. The effect of leptin, caffeine/ephedrine, and their combination upon visceral fat mass and weight loss. Obesity (Silver Spring). 2013;21(10):1991–6.
Zelissen PM, Stenlof K, Lean ME, Fogteloo J, Keulen ET, Wilding J, Finer N, Rossner S, Lawrence E, Fletcher C, et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: a randomized, placebo-controlled trial. Diabetes Obes Metab. 2005;7(6):755–61.
Baver SB, Hope K, Guyot S, Bjorbaek C, Kaczorowski C, O'Connell KM. Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J Neurosci. 2014;34(16):5486–96.
Frisch RE. Body fat, menarche, fitness and fertility. Hum Reprod. 1987;2(6):521–33.
Cheung CC, Thornton JE, Kuijper JL, Weigle DS, Clifton DK, Steiner RA. Leptin is a metabolic gate for the onset of puberty in the female rat. Endocrinology. 1997;138(2):855–8.
Gosman GG, Katcher HI, Legro RS. Obesity and the role of gut and adipose hormones in female reproduction. Hum Reprod Update. 2006;12(5):585–601.
Egan OK, Inglis MA, Anderson GM. Leptin signaling in AgRP neurons modulates puberty onset and adult fertility in mice. J Neurosci. 2017;37(14):3875–86.
Sanchez-Garrido MA, Tena-Sempere M. Metabolic control of puberty: roles of leptin and kisspeptins. Horm Behav. 2013;64(2):187–94.
Backholer K, Smith JT, Rao A, Pereira A, Iqbal J, Ogawa S, Li Q, Clarke IJ. Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology. 2010;151(5):2233–43.
Smith JT, Acohido BV, Clifton DK, Steiner RA. KiSS-1 neurones are direct targets for leptin in the Ob/Ob mouse. J Neuroendocrinol. 2006;18(4):298–303.
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100(19):10972–6.
Castellano JM, Bentsen AH, Mikkelsen JD, Tena-Sempere M. Kisspeptins: bridging energy homeostasis and reproduction. Brain Res. 2010;1364:129–38.
Cioffi JA, Van Blerkom J, Antczak M, Shafer A, Wittmer S, Snodgrass HR. The expression of leptin and its receptors in pre-ovulatory human follicles. Mol Hum Reprod. 1997;3(6):467–72.
Hardie L, Trayhurn P, Abramovich D, Fowler P. Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol. 1997;47(1):101–6.
Tena-Sempere M. Roles of ghrelin and leptin in the control of reproductive function. Neuroendocrinology. 2007;86(3):229–41.
Brannian JD, Hansen KA. Leptin and ovarian folliculogenesis: implications for ovulation induction and ART outcomes. Semin Reprod Med. 2002;20(2):103–12.
Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, Clifton DK, Steiner RA. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137(7):3144–7.
Brannian JD, Schmidt SM, Kreger DO, Hansen KA. Baseline non-fasting serum leptin concentration to body mass index ratio is predictive of IVF outcomes. Hum Reprod. 2001;16(9):1819–26.
Spicer LJ, Chamberlain CS, Francisco CC. Ovarian action of leptin: effects on insulin-like growth factor-I-stimulated function of granulosa and thecal cells. Endocrine. 2000;12(1):53–9.
Spicer LJ, Francisco CC. Adipose obese gene product, leptin, inhibits bovine ovarian thecal cell steroidogenesis. Biol Reprod. 1998;58(1):207–12.
Merhi Z, Buyuk E, Berger DS, Zapantis A, Israel DD, Chua S Jr, Jindal S. Leptin suppresses anti-Mullerian hormone gene expression through the JAK2/STAT3 pathway in luteinized granulosa cells of women undergoing IVF. Hum Reprod. 2013;28(6):1661–9.
Carro E, Pinilla L, Seoane LM, Considine RV, Aguilar E, Casanueva FF, Dieguez C. Influence of endogenous leptin tone on the estrous cycle and luteinizing hormone pulsatility in female rats. Neuroendocrinology. 1997;66(6):375–7.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–60.
Dezaki K, Sone H, Yada T. Ghrelin is a physiological regulator of insulin release in pancreatic islets and glucose homeostasis. Pharmacol Ther. 2008;118(2):239–49.
Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, Makino S, Fujimiya M, Niijima A, Fujino MA, et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology. 2001;120(2):337–45.
Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, Shen Z, Marsh DJ, Feighner SD, Guan XM, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and Agouti-related protein. Endocrinology. 2004;145(6):2607–12.
Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85(2):495–522.
Kamegai J, Tamura H, Shimizu T, Ishii S, Sugihara H, Wakabayashi I. Chronic central infusion of ghrelin increases hypothalamic neuropeptide Y and Agouti-related protein mRNA levels and body weight in rats. Diabetes. 2001;50(11):2438–43.
Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15(10):1350–5.
Fernandez-Fernandez R, Tena-Sempere M, Navarro VM, Barreiro ML, Castellano JM, Aguilar E, Pinilla L. Effects of ghrelin upon gonadotropin-releasing hormone and gonadotropin secretion in adult female rats: in vivo and in vitro studies. Neuroendocrinology. 2005;82(5–6):245–55.
Furuta M, Funabashi T, Kimura F. Intracerebroventricular administration of ghrelin rapidly suppresses pulsatile luteinizing hormone secretion in ovariectomized rats. Biochem Biophys Res Commun. 2001;288(4):780–5.
Kluge M, Schussler P, Schmidt D, Uhr M, Steiger A. Ghrelin suppresses secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in women. J Clin Endocrinol Metab. 2012;97(3):E448–51.
Vulliémoz NR, Xiao E, Xia-Zhang L, Germond M, Rivier J, Ferin M. Decrease in luteinizing hormone pulse frequency during a five-hour peripheral ghrelin infusion in the Ovariectomized rhesus monkey. J Clin Endocrinol Metab. 2004;89(11):5718–23.
Rak-Mardyla A. Ghrelin role in hypothalamus-pituitary-ovarian axis. J Physiol Pharmacol. 2013;64(6):695–704.
Broglio F, Benso A, Castiglioni C, Gottero C, Prodam F, Destefanis S, Gauna C, van der Lely AJ, Deghenghi R, Bo M, et al. The endocrine response to ghrelin as a function of gender in humans in young and elderly subjects. J Clin Endocrinol Metab. 2003;88(4):1537–42.
Whatmore AJ, Hall CM, Jones J, Westwood M, Clayton PE. Ghrelin concentrations in healthy children and adolescents. Clin Endocrinol. 2003;59(5):649–54.
Gaytan F, Barreiro ML, Chopin LK, Herington AC, Morales C, Pinilla L, Casanueva FF, Aguilar E, Dieguez C, Tena-Sempere M. Immunolocalization of ghrelin and its functional receptor, the type 1a growth hormone secretagogue receptor, in the cyclic human ovary. J Clin Endocrinol Metab. 2003;88(2):879–87.
Sirotkin AV, Grossmann R, Maria-Peon MT, Roa J, Tena-Sempere M, Klein S. Novel expression and functional role of ghrelin in chicken ovary. Mol Cell Endocrinol. 2006;257-258:15–25.
Sirotkin AV, Grossmann R. Effects of ghrelin and its analogues on chicken ovarian granulosa cells. Domest Anim Endocrinol. 2008;34(2):125–34.
Tropea A, Tiberi F, Minici F, Orlando M, Gangale MF, Romani F, Miceli F, Catino S, Mancuso S, Sanguinetti M, et al. Ghrelin affects the release of luteolytic and luteotropic factors in human luteal cells. J Clin Endocrinol Metab. 2007;92(8):3239–45.
Viani I, Vottero A, Tassi F, Cremonini G, Sartori C, Bernasconi S, Ferrari B, Ghizzoni L. Ghrelin inhibits steroid biosynthesis by cultured granulosa-lutein cells. J Clin Endocrinol Metab. 2008;93(4):1476–81.
Rak A, Gregoraszczuk EL. Modulatory effect of ghrelin in prepubertal porcine ovarian follicles. J Physiol Pharmacol. 2008;59(4):781–93.
Tilly JL, Kowalski KI, Johnson AL, Hsueh AJ. Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology. 1991;129(5):2799–801.
Kheradmand A, Roshangar L, Taati M, Sirotkin AV. Morphometrical and intracellular changes in rat ovaries following chronic administration of ghrelin. Tissue Cell. 2009;41(5):311–7.
Muller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, Batterham RL, Benoit SC, Bowers CY, Broglio F, et al. Ghrelin. Mol Metab. 2015;4(6):437–60.
Sun Y, Asnicar M, Saha PK, Chan L, Smith RG. Ablation of ghrelin improves the diabetic but not obese phenotype of Ob/Ob mice. Cell Metab. 2006;3(5):379–86.
Ikezaki A, Hosoda H, Ito K, Iwama S, Miura N, Matsuoka H, Kondo C, Kojima M, Kangawa K, Sugihara S. Fasting plasma ghrelin levels are negatively correlated with insulin resistance and PAI-1, but not with leptin, in obese children and adolescents. Diabetes. 2002;51(12):3408–11.
Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1(4):271–2.
Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, Lee EW, Burnett MS, Fricke ST, Kvetnansky R, et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 2007;13(7):803–11.
Bures EJ, Hui JO, Young Y, Chow DT, Katta V, Rohde MF, Zeni L, Rosenfeld RD, Stark KL, Haniu M. Determination of disulfide structure in agouti-related protein (AGRP) by stepwise reduction and alkylation. Biochemistry. 1998;37(35):12172–7.
Cansell C, Denis RG, Joly-Amado A, Castel J, Luquet S. Arcuate AgRP neurons and the regulation of energy balance. Front Endocrinol. 2012;3:169.
Sutton SW, Toyama TT, Otto S, Plotsky PM. Evidence that neuropeptide Y (NPY) released into the hypophysial-portal circulation participates in priming gonadotropes to the effects of gonadotropin releasing hormone (GnRH). Endocrinology. 1988;123(2):1208–10.
Sahu A, Crowley WR, Kalra SP. Hypothalamic neuropeptide-Y gene expression increases before the onset of the ovarian steroid-induced luteinizing hormone surge. Endocrinology. 1994;134(3):1018–22.
Vulliémoz NR, Xiao E, Xia-Zhang L, Wardlaw SL, Ferin M. Central infusion of Agouti-related peptide suppresses pulsatile luteinizing hormone release in the Ovariectomized rhesus monkey. Endocrinology. 2005;146(2):784–9.
McDonald JK. Role of neuropeptide Y in reproductive function. Ann N Y Acad Sci. 1990;611:258–72.
Sirotkin AV, Kardosova D, Alwasel SH, Harrath AH. Neuropeptide Y directly affects ovarian cell proliferation and apoptosis. Reprod Biol. 2015;15(4):257–60.
Keator CS, Custer EE, Hoagland TA, Schreiber DT, Mah K, Lawson AM, Slayden OD, McCracken JA. Evidence for a potential role of neuropeptide Y in ovine corpus luteum function. Domest Anim Endocrinol. 2010;38(2):103–14.
Priyadarshini, Lal B. Seasonal ovarian immunolocalization of neuropeptide Y and its role in steriodogenesis in Asian catfish, Clarias batrachus. Gen Comp Endocrinol. 2018;255:32–9.
Lee B, Shao J. Adiponectin and energy homeostasis. Rev Endocr Metab Disord. 2014;15(2):149–56.
Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6(1):55–68.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83.
Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8(7):731–7.
Kondo H, Shimomura I, Matsukawa Y, Kumada M, Takahashi M, Matsuda M, Ouchi N, Kihara S, Kawamoto T, Sumitsuji S, et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes. 2002;51(7):2325–8.
Ohashi K, Ouchi N, Kihara S, Funahashi T, Nakamura T, Sumitsuji S, Kawamoto T, Matsumoto S, Nagaretani H, Kumada M, et al. Adiponectin I164T mutation is associated with the metabolic syndrome and coronary artery disease. J Am Coll Cardiol. 2004;43(7):1195–200.
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116(7):1784–92.
Vionnet N, Hani EH, Dupont S, Gallina S, Francke S, Dotte S, De Matos F, Durand E, Lepretre F, Lecoeur C, et al. Genomewide search for type 2 diabetes-susceptibility genes in French whites: evidence for a novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet. 2000;67(6):1470–80.
Thaler JP, Schwartz MW. Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151(9):4109–15.
Chabrolle C, Tosca L, Dupont J. Regulation of adiponectin and its receptors in rat ovary by human chorionic gonadotrophin treatment and potential involvement of adiponectin in granulosa cell steroidogenesis. Reproduction. 2007;133(4):719–31.
Ledoux S, Campos DB, Lopes FL, Dobias-Goff M, Palin M-F, Murphy BD. Adiponectin induces Periovulatory changes in ovarian follicular cells. Endocrinology. 2006;147(11):5178–86.
Chabrolle C, Tosca L, Crochet S, Tesseraud S, Dupont J. Expression of adiponectin and its receptors (AdipoR1 and AdipoR2) in chicken ovary: potential role in ovarian steroidogenesis. Domest Anim Endocrinol. 2007;33(4):480–7.
Campos DB, Palin MF, Bordignon V, Murphy BD. The ‘beneficial’ adipokines in reproduction and fertility. Int J Obes (Lond). 2008;32(2):223–31.
Cheng L, Shi H, Jin Y, Li X, Pan J, Lai Y, Lin Y, Jin Y, Roy G, Zhao A, et al. Adiponectin deficiency leads to female subfertility and ovarian dysfunctions in mice. Endocrinology. 2016;157(12):4875–87.
Liu YH, Tsai EM, Wu LC, Chen SY, Chang YH, Jong SB, Chan TF. Higher basal adiponectin levels are associated with better ovarian response to gonadotropin stimulation during in vitro fertilization. Gynecol Obstet Investig. 2005;60(3):167–70.
Richards JS, Liu Z, Kawai T, Tabata K, Watanabe H, Suresh D, Kuo FT, Pisarska MD, Shimada M. Adiponectin and its receptors modulate granulosa cell and cumulus cell functions, fertility, and early embryo development in the mouse and human. Fertil Steril. 2012;98(2):471–9. e471
Chang HJ, Lee JH, Lee JR, Jee BC, Suh CS, Kim SH. Relationship between follicular fluid adipocytokines and the quality of the oocyte and corresponding embryo development from a single dominant follicle in in vitro fertilization/intracytoplasmic sperm injection cycles. Clin Exp Reprod Med. 2014;41(1):21–8.
Lundon JR. Insulin-some current concepts. Can Fam Physician. 1970;16(7):58–61.
Preziosi P, Barrett-Connor E, Papoz L, Roger M, Saint-Paul M, Nahoul K, Simon D. Interrelation between plasma sex hormone-binding globulin and plasma insulin in healthy adult women: the telecom study. J Clin Endocrinol Metab. 1993;76(2):283–7.
Wu S, Divall S, Nwaopara A, Radovick S, Wondisford F, Ko C, Wolfe A. Obesity-induced infertility and hyperandrogenism are corrected by deletion of the insulin receptor in the ovarian theca cell. Diabetes. 2014;63(4):1270–82.
Barbieri RL, Makris A, Ryan KJ. Effects of insulin on steroidogenesis in cultured porcine ovarian theca. Fertil Steril. 1983;40(2):237–41.
Brothers KJ, Wu S, DiVall SA, Messmer MR, Kahn CR, Miller RS, Radovick S, Wondisford FE, Wolfe A. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab. 2010;12(3):295–305.
Willis D, Mason H, Gilling-Smith C, Franks S. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab. 1996;81(1):302–9.
La Marca A, Pati M, Giulini S, Levratti P, Caretto S, Volpe A. Does plasma insulin level affect ovarian response to exogenous administration of follicle-stimulating hormone in women without polycystic ovary syndrome? Gynecol Endocrinol. 2005;21(5):292–4.
Acevedo N, Ding J, Smith GD. Insulin signaling in mouse Oocytes1. Biol Reprod. 2007;77(5):872–9.
Ou XH, Li S, Wang ZB, Li M, Quan S, Xing F, Guo L, Chao SB, Chen Z, Liang XW, et al. Maternal insulin resistance causes oxidative stress and mitochondrial dysfunction in mouse oocytes. Hum Reprod. 2012;27(7):2130–45.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Van Puyvelde K, Mets T, Njemini R, Beyer I, Bautmans I. Effect of advanced glycation end product intake on inflammation and aging: a systematic review. Nutr Rev. 2014;72(10):638–50.
Vazzana N, Guagnano MT, Cuccurullo C, Ferrante E, Lattanzio S, Liani R, Romano M, Davi G. Endogenous secretory RAGE in obese women: association with platelet activation and oxidative stress. J Clin Endocrinol Metab. 2012;97(9):E1726–30.
Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2003;100(12):7265–70.
Gu L, Hagiwara S, Fan Q, Tanimoto M, Kobata M, Yamashita M, Nishitani T, Gohda T, Ni Z, Qian J, et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrol Dial Transplant. 2006;21(2):299–313.
Grandhee SK, Monnier VM. Mechanism of formation of the Maillard protein cross-link pentosidine. Glucose, fructose, and ascorbate as pentosidine precursors. J Biol Chem. 1991;266(18):11649–53.
Monnier VM, Sell DR, Nagaraj RH, Miyata S, Grandhee S, Odetti P, Ibrahim SA. Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes. 1992;41(Suppl 2):36–41.
Horiuchi S, Araki N, Morino Y. Immunochemical approach to characterize advanced glycation end products of the Maillard reaction. Evidence for the presence of a common structure. J Biol Chem. 1991;266(12):7329–32.
Yamagishi S, Nakamura K, Imaizumi T. Advanced glycation end products (AGEs) and diabetic vascular complications. Curr Diabetes Rev. 2005;1(1):93–106.
Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. Cellular and molecular aspects of ovarian follicle ageing. Hum Reprod Update. 2008;14(2):131–42.
Diamanti-Kandarakis E, Piperi C, Patsouris E, Korkolopoulou P, Panidis D, Pawelczyk L, Papavassiliou AG, Duleba AJ. Immunohistochemical localization of advanced glycation end-products (AGEs) and their receptor (RAGE) in polycystic and normal ovaries. Histochem Cell Biol. 2007;127(6):581–9.
Diamanti-Kandarakis E, Chatzigeorgiou A, Papageorgiou E, Koundouras D, Koutsilieris M. Advanced glycation end-products and insulin signaling in granulosa cells. Exp Biol Med. 2016;241(13):1438–45.
Jinno M, Takeuchi M, Watanabe A, Teruya K, Hirohama J, Eguchi N, Miyazaki A. Advanced glycation end-products accumulation compromises embryonic development and achievement of pregnancy by assisted reproductive technology. Hum Reprod. 2011;26(3):604–10.
Kandaraki EA, Chatzigeorgiou A, Papageorgiou E, Piperi C, Adamopoulos C, Papavassiliou AG, Koutsilieris M, Diamanti-Kandarakis E. Advanced glycation end products interfere in luteinizing hormone and follicle stimulating hormone signaling in human granulosa KGN cells. Exp Biol Med. 2018;243(1):29–33. 1535370217731288.
Asemota OA, Berger DS, Seki Y, Jindal SK, Charron MJ, Buyuk E. MCP-1, a central mediator of obesity and diet-induced ovarian dysfunction. Fertil Steril. 2014;102(3):e259.
Buyuk E, Asemota OA, Merhi Z, Charron MJ, Berger DS, Zapantis A, Jindal SK. Serum and follicular fluid monocyte chemotactic protein-1 levels are elevated in obese women and are associated with poorer clinical pregnancy rate after in vitro fertilization: a pilot study. Fertil Steril. 2017;107(3):632–40. e633
Grodstein F, Goldman MB, Cramer DW. Body mass index and ovulatory infertility. Epidemiology. 1994;5(2):247–50.
Rich-Edwards JW, Spiegelman D, Garland M, Hertzmark E, Hunter DJ, Colditz GA, Willett WC, Wand H, Manson JE. Physical activity, body mass index, and ovulatory disorder infertility. Epidemiology. 2002;13(2):184–90.
Green BB, Weiss NS, Daling JR. Risk of ovulatory infertility in relation to body weight. Fertil Steril. 1988;50(5):721–6.
Zaadstra BM, Seidell JC, Van Noord PA, te Velde ER, Habbema JD, Vrieswijk B, Karbaat J. Fat and female fecundity: prospective study of effect of body fat distribution on conception rates. BMJ. 1993;306(6876):484–7.
van der Steeg JW, Steures P, Eijkemans MJ, Habbema JD, Hompes PG, Burggraaff JM, Oosterhuis GJ, Bossuyt PM, van der Veen F, Mol BW. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod. 2008;23(2):324–8.
Gesink Law DC, Maclehose RF, Longnecker MP. Obesity and time to pregnancy. Hum Reprod. 2007;22(2):414–20.
Lake JK, Power C, Cole TJ. Women's reproductive health: the role of body mass index in early and adult life. Int J Obes Relat Metab Disord. 1997;21(6):432–8.
Moy V, Jindal S, Lieman H, Buyuk E. Obesity adversely affects serum anti-mullerian hormone (AMH) levels in Caucasian women. J Assist Reprod Genet. 2015;32(9):1305–11.
Freeman EW, Gracia CR, Sammel MD, Lin H, Lim LC, Strauss JF 3rd. Association of anti-mullerian hormone levels with obesity in late reproductive-age women. Fertil Steril. 2007;87(1):101–6.
De Pergola G, Maldera S, Tartagni M, Pannacciulli N, Loverro G, Giorgino R. Inhibitory effect of obesity on gonadotropin, estradiol, and inhibin B levels in fertile women. Obesity (Silver Spring). 2006;14(11):1954–60.
Practice Committee of the American Society for Reproductive Medicine. Testing and interpreting measures of ovarian reserve: a committee opinion. Fertil Steril. 2015;103(3):e9–e17.
Shah DK, Missmer SA, Berry KF, Racowsky C, Ginsburg ES. Effect of obesity on oocyte and embryo quality in women undergoing in vitro fertilization. Obstet Gynecol. 2011;118(1):63–70.
Provost MP, Acharya KS, Acharya CR, Yeh JS, Steward RG, Eaton JL, Goldfarb JM, Muasher SJ. Pregnancy outcomes decline with increasing body mass index: analysis of 239,127 fresh autologous in vitro fertilization cycles from the 2008–2010 Society for Assisted Reproductive Technology registry. Fertil Steril. 2016;105(3):663–9.
Maheshwari A, Stofberg L, Bhattacharya S. Effect of overweight and obesity on assisted reproductive technology--a systematic review. Hum Reprod Update. 2007;13(5):433–44.
Zhang D, Zhu Y, Gao H, Zhou B, Zhang R, Wang T, Ding G, Qu F, Huang H, Lu X. Overweight and obesity negatively affect the outcomes of ovarian stimulation and in vitro fertilisation: a cohort study of 2628 Chinese women. Gynecol Endocrinol. 2010;26(5):325–32.
Leary C, Leese HJ, Sturmey RG. Human embryos from overweight and obese women display phenotypic and metabolic abnormalities. Hum Reprod. 2015;30(1):122–32.
Roth LW, Bradshaw-Pierce EL, Allshouse AA, Lesh J, Chosich J, Bradford AP, Polotsky AJ, Santoro N. Evidence of GnRH antagonist escape in obese women. J Clin Endocrinol Metab. 2014;99(5):E871–5.
Bellver J, Ayllon Y, Ferrando M, Melo M, Goyri E, Pellicer A, Remohi J, Meseguer M. Female obesity impairs in vitro fertilization outcome without affecting embryo quality. Fertil Steril. 2010;93(2):447–54.
Sharara FI, McClamrock HD. Differences in in vitro fertilization (IVF) outcome between white and black women in an inner-city, university-based IVF program. Fertil Steril. 2000;73(6):1170–3.
Mitwally MF, Leduc MM, Ogunleye O, Albuarki H, Diamond MP, Abuzeid M. The effect of body mass index (BMI) on the outcome of IVF and embryo transfer in women of different ethnic backgrounds. Fertility Sterility. 2006;86:S68–9.
Patel AP, Patel JA, Cruz M, Gupte-Shah A, Garcia Velasco JA, Banker MR. Ethnicity is an independent predictor of IVF-ICSI outcome: a study of 5,549 cycles in Spain and India. Gynecol Endocrinol. 2016;32(10):819–22.
Dayal MB, Gindoff P, Dubey A, Spitzer TL, Bergin A, Peak D, Frankfurter D. Does ethnicity influence in vitro fertilization (IVF) birth outcomes? Fertil Steril. 2009;91(6):2414–8.
Lashen H, Afnan M, Sharif K. A controlled comparison of ovarian response to controlled stimulation in first generation Asian women compared with white Caucasians undergoing in vitro fertilisation. Br J Obstet Gynaecol. 1999;106(5):407–9.
Nichols JE Jr, Higdon HL 3rd, Crane MM, Boone WR. Comparison of implantation and pregnancy rates in African American and white women in an assisted reproductive technology practice. Fertil Steril. 2001;76(1):80–4.
Bellver J, Melo MA, Bosch E, Serra V, Remohi J, Pellicer A. Obesity and poor reproductive outcome: the potential role of the endometrium. Fertil Steril. 2007;88(2):446–51.
Loveland JB, McClamrock HD, Malinow AM, Sharara FI. Increased body mass index has a deleterious effect on in vitro fertilization outcome. J Assist Reprod Genet. 2001;18(7):382–6.
Thornton KAO, Jindal S, Charron M, Buyuk E. High fat diet and aging are associated with macrophage infiltration in the ovaries. Fertil Steril. 2015;104(3):e104–5.
Author information
Authors and Affiliations
Contributions
All authors contributed to the collection of literature data and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
N/A
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Goldsammler, M., Merhi, Z. & Buyuk, E. Role of hormonal and inflammatory alterations in obesity-related reproductive dysfunction at the level of the hypothalamic-pituitary-ovarian axis. Reprod Biol Endocrinol 16, 45 (2018). https://doi.org/10.1186/s12958-018-0366-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-018-0366-6