
Abstract
Background
Gastric cancer (GC) is a prevalent malignant cancer of digestive system. To identify key genes in GC, mRNA microarray GSE27342, GSE29272, and GSE33335 were downloaded from GEO database.
Methods
Differentially expressed genes (DEGs) were obtained using GEO2R. DAVID database was used to analyze function and pathways enrichment of DEGs. Protein-protein interaction (PPI) network was established by STRING and visualized by Cytoscape software. Then, the influence of hub genes on overall survival (OS) was performed by the Kaplan-Meier plotter online tool. Module analysis of the PPI network was performed using MCODE. Additionally, potential stem loop miRNAs of hub genes were predicted by miRecords and screened by TCGA dataset. Transcription factors (TFs) of hub genes were detected by NetworkAnalyst.
Results
In total, 67 DEGs were identified; upregulated DEGs were mainly enriched in biological process (BP) related to angiogenesis and extracellular matrix organization and the downregulated DEGs were mainly enriched in BP related to ion transport and response to bacterium. KEGG pathways analysis showed that the upregulated DEGs were enriched in ECM-receptor interaction and the downregulated DEGs were enriched in gastric acid secretion. A PPI network of DEGs was constructed, consisting of 43 nodes and 87 edges. Twelve genes were considered as hub genes owing to high degrees in the network. Hsa-miR-29c, hsa-miR-30c, hsa-miR-335, hsa-miR-33b, and hsa-miR-101 might play a crucial role in hub genes regulation. In addition, the transcription factors-hub genes pairs were displayed with 182 edges and 102 nodes. The high expression of 7 out of 12 hub genes was associated with worse OS, including COL4A1, VCAN, THBS2, TIMP1, COL1A2, SERPINH1, and COL6A3.
Conclusions
The miRNA and TFs regulation network of hub genes in GC may promote understanding of the molecular mechanisms underlying the development of gastric cancer and provide potential targets for GC diagnosis and treatment.
Similar content being viewed by others


Background
Gastric cancer is the fourth common cancer worldwide, with an estimated 951,600 new cases of gastric cancer (GC) and estimated 723,100 deaths from it in 2012 [1]. In China, GC is one of the most common malignancies and is the third leading cause of cancer death in 2010 [2]. Despite recent improvements in multimodal therapy including surgery, chemotherapy, radiotherapy, and targeted therapy, its overall 5-year survival rate remains below 20% [3]. The abnormal intracellular signaling molecules contribute a part of poor prognosis due to tumor invasion and metastasis [4]. Such signaling molecules, like matrix metalloproteinase (MMP)-2, MMP-9, and vascular endothelial growth factor (VEGF), have been shown to promote tumor metastasis [5]. Moreover, the abnormal expression of non-coding RNA is also an important factor according to recent studies, especially microRNAs (miRNAs) [6]. MiRNAs are a class of evolutionary conserved small RNA that regulates gene expression by targeting mRNAs to translation repression or triggering mRNA degradation. Various biological processes were involved in the regulation network between miRNAs and target mRNAs, including differentiation, proliferation, survival, stress response, and oncogenesis [7,8,9,10]. Like miRNAs, transcription factors (TFs) regulate diverse cellular pathways and are widely believed to regulate most biological processes, including cancer. TFs are sequence-specific DNA-binding proteins which act as transcriptional activators and repressors.
In recent years, the application of high-throughput platforms in gene expression (GE) is becoming more valuable in clinical research, like molecular classification, prognosis prediction, and new targeted drug discovery [11,12,13]. Hundreds of DEGs were shown in many gene expression profiling studies on GC carcinogenesis, which involved in different pathways, biological process, molecular function. However, the interaction network of DEGs remains to be clarified. In this work, three mRNA microarray datasets were analyzed to obtain DEGs between GC tissues and normal tissues. We further explored GC development by a way of DEGs functional enrichment and interaction network analysis, combined with survival analysis and mRNA-miRNA interaction analysis, also constructed gene–transcription factor interaction network to identify key genes in GC.
Methods
Identification of DEGs
Three gene expression profiles (GSE27342, GSE29272, and GSE33335) were acquired from GEO database. The array data of GSE27342 consisted of 80 paired GC tissues and adjacent tissues [14]. GSE33335 contained 25 paired GC tissues and adjacent tissues [15]. GSE29272 included 134 paired GC tissues, and adjacent tissues were submitted by Wang et al. [16]. DEG was obtained from GEO database by a way of GEO2R analysis (http://www.ncbi.nlm.nih.gov/geo/geo2r/). The adj. P < 0.05 and |logFC| > 1.5 were set as DEGs cutoff criterion.
Gene ontology and pathway enrichment analysis of DEGs
The Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) has facilitated the transition from data collection to biological analysis [17]. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using DAVID online tool. P < 0.01 was set as the cutoff criterion.
Integration of protein-protein interaction network and modules selection
To explore the interaction of DEGs, we submitted the DEGs to the Search Tool for the Retrieval of Interacting Genes (STRING, http://string.embl.de/) database, and only validated interactions with combined score > 0.4 were selected as significant. Then, integration of protein-protein interaction (PPI) networks were visualized using Cytoscape software. The Molecular Complex Detection (MCODE) was applied to screen modules of PPI network with degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max. depth = 100 [18]. The functional enrichment and pathway enrichment analysis in the module was performed by DAVID.
Prediction of stem loop miRNAs for hub genes
miRecords, which is an integrated resource of 11 established miRNA target prediction programs [19], was used to identify the stem loop miRNAs of hub genes. The miRNAs predicted by at least four programs were regarded as the stem loop miRNAs of hub genes.
Negatively correlated stem loop miRNA of hub genes in gastric cancer
The raw counts of miRNA expression data of 380 gastric cancer tissues were obtained from the TCGA dataset (Illumina HiSeq Systems). MiRNA expression data was normalized by the R/Bioconductor package edgeR [20]. The correlation between hub genes and miRNAs was characterized by the |logFC|. MiRNAs with negative correlation (P < 0.05, FDR adjusted P < 0.05) were considered as negatively correlated miRNAs.
Prediction of transcription factor for DEGs
TFs of hub genes were explored combined with the human TF information (NetworkAnalyst, http://www.networkanalyst.ca), recorded using ChIP Enrichment Analysis (ChEA), and visualized using the Cytoscape software [21, 22].
Survival analysis of DEGs
Kaplan-Meier plotter (KM plotter, http://kmplot.com/analysis/) was capable to assess the survival of 10,188 cancer samples, including 5143 breast, 1648 ovarian, 2437 lung, and 1065 gastric cancer patients with a mean follow-up of 69/40/49/33 months [23]. According to the median expression of a particular gene, the patients with GC were split into high and low expression groups. The overall survival (OS) of GC patients was evaluated using a KM plot. The hazard ratio (HR) with 95% confidence intervals and log rank P values were shown on the webpage; then multiple hypothesis testing was calculated.
Results
Identification of DEGs
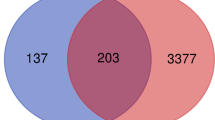
A total of 598, 350, and 418 DEGs were identified from GSE27342, GSE29272, and GSE33335 datasets. Sixty-seven genes were screened out in all three datasets and were selected for further analysis (Fig. 1). There are 36 upregulated genes and 31 downregulated genes in GC tissues compared to adjacent tissues.

Identification of DEGs in mRNA expression profiling datasets GSE27342, GSE29272, and GSE33335
GO term and KEGG pathway enrichment analysis of DEGs
To gain further insight into the function of identified DEGs, GO term and KEGG pathway enrichment analysis were performed using DAVID. The upregulated genes were mainly enriched in angiogenesis and extracellular matrix organization, while downregulated genes were mainly involved in ion transport and response to bacterium in biological processes (Table 1). Top ten terms of upregulation or downregulation genes were selected and are displayed in Table 1 according to P value. Moreover, seven KEGG pathways were overrepresented in upregulated genes, including ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathway, and two pathways were identified in downregulation (Table 2).
PPI network construction and modules selection
The PPI network of DEGs is consisted of 43 nodes and 87 edges, including 26 upregulated genes and 17 downregulated genes (Fig. 2a). There are 12 genes selected as hub genes, such as SPP1, TIMP1, MMP7, and COL1A1, enriched in a module when degrees ≥ 8 were set as the cutoff criterion.

PPI network and a significant module. a PPI network of DEGs, red means upregulated genes and green means downregulated genes. b A significant module selected from PPI network, all of them were upregulated genes
A significant module was obtained from PPI network of DEGs using MCODE, including 12 nodes and 45 edges (Fig. 2b). KEGG pathway enrichment analysis revealed that genes in this module were mainly involved in ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathway (Table 3).
MiRNA-hub genes pairs
To investigate the molecular mechanisms underlying the dysregulated hub genes, potential stem loop miRNAs were searched by bioinformatics methods. The stem loop miRNAs of hub genes were predicted by miRecords database. As shown in Table 4, top five predicted stem loop miRNAs of hub genes were displayed according to the prediction program numbers. To validate the accuracy of the predicted stem loop miRNAs for hub genes, we screened the negatively correlated stem loop miRNAs of hub genes in a TCGA dataset composed of 380 gastric cancer tissues (Additional file 1). Top five negatively correlated stem loop miRNAs of hub genes are shown in Table 5 in consideration of logFC. As shown in Table 6, COL1A1, COL1A2, COL4A1, COL6A3, SPARC, and THBS2 might be the targets of hsa-miR-29c. Moreover, COL1A1, COL1A2, COL3A1, COL6A3, SERPINH1, SPARC, and THBS2 were potential targets of hsa-miR-30c. Hsa-miR-335 may regulate COL1A1, COL1A2, COL3A1, COL4A1, COL6A3, SPARC, and THBS2. Hsa-miR-33b possibly bound to 3′ UTR of COL1A1, COL3A1, COL4A1, COL6A3, SPARC, THBS2, and VCAN. But COL1A1, COL3A1, SPARC, SPP1, and THBS2 would be targets of hsa-miR-101 in some ways.
Transcription factors-hub genes pairs
To further understand the regulatory network between TFs and hub genes, TFs with adj. P < 0.05 in ChEA through NetworkAnalyst were constructed by Cytoscape. As shown in Fig. 3, the transcription-regulated network with 182 edges and 102 nodes was obtained for hub genes. Different hub genes regulated by TFs are shown in Table 7, which androgen receptor (AR) had been predicted to regulate SPARC, COL6A3, SERPINH1, COL4A1, and VCAN, while COL4A1, VCAN, and COL6A3 could be regulated by EZH2.

The network of transcription factors and hub genes
The prognostic value of hub genes
The prognostic value of 12 hub genes in PPI network was obtained from KM plotter (http://kmplot.com/analysis/). In consideration of the semblable histopathological feature and adequate quantity, the below two datasets were used to assess survival time. In GSE15459, it was found that high mRNA expression of SERPINH1 was associated with worse OS for GC patients [24, 25], as well as COL1A1, THBS2, COL4A1, COL6A3, MMP7, COL1A2, TIMP1, SPP1, and VCAN (Fig. 4). To validate the prognostic significance of these genes, the GSE62254 was analyzed [26]. In Fig. 5, seven out of ten genes had statistical significance, including COL4A1, VCAN, THBS2, TIMP1, COL1A2, SERPINH1, and COL6A3.

Prognostic value of 12 genes in GSE15459. Prognostic value in GSE15459 of THBS2(a), TIMP1(b), VCAN(c), MMP7(d), COL4A1(e), COL1A2(f), SPP1(g), COL6A3(h), COL1A1(i), SERINH1(j), SPARC(k), and COL3A1(l) were obtained in www.kmplot.com. The desired Affymetrix IDs are valid: 203083_at (THBS2), 201666_at (TIMP1), 221731_x_at (VCAN), 204259_at (MMP7), 211980_at (COL4A1), 202403_s_at (COL1A2), 48580_at (SPP1), 201438_at (COL6A3), 202311_s_at (COL1A1), 207714_s_at (SERPINH1), 212667_at (SPARC), and 201852_x_at (COL3A1). HR, hazard ratio; CI, confidence interval; adj. p, adjusted p value

The validation of prognostic value of ten genes in GSE62254. Prognostic value in GSE62254 of COL4A1 (a), VCAN (b), THBS2 (c), TIMP1 (d), COL1A2 (e), SERINH1 (f), COL6A3 (g), COL1A1 (h), MMP7 (i), and SPP1 (j) were obtained in www.kmplot.com. The desired Affymetrix IDs are valid: 211980_at (COL4A1), 221731_x_at (VCAN), 203083_at (THBS2), 201666_at (TIMP1), 202403_s_at (COL1A2), 207714_s_at (SERPINH1), 201438_at (COL6A3), 202311_s_at (COL1A1), 204259_at (MMP7), and 48580_at (SPP1). HR, hazard ratio; CI, confidence interval; adj. p, adjusted p value
Discussion
Due to the high heterogeneity of GC, GC was still a disease with high rates of prevalence and mortality. With surgery as the main, the other treatments including radiotherapy, chemotherapy, targeted therapy, and gene therapy as a supplement to the limited treatment measures of GC, the 5-year survival rate was still less than 30% [27]. Therefore, it is essential to explore the mechanisms of GC progression to prevent its occurrence, guide pharmacy, indicate the prognosis, or improve survival rate. The high-throughput platforms for detection of gene expression have been developing rapidly in diseases progression, which provides the basis of new targets discovery for diagnosis, therapy, and prognosis of cancers.
In this study, a total of 67 DEGs were screened, consisting of 36 upregulated genes and 31 downregulated genes. These upregulated genes were mainly enriched in angiogenesis and extracellular matrix organization, while downregulated genes were mainly involved in ion transport and response to bacterium in biological processes. Among these DEGs, 12 genes had high degrees in the PPI network. Following survival analysis of these genes revealed that 7 of these 12 upregulated genes were significantly correlated with worse overall survival of GC patients, including COL4A1, VCAN, THBS2, TIMP1, COL1A2, SERPINH1, and COL6A3.
The pathogenesis of cancer is multifactorial, with genetic, environmental, and lifestyle factors interacting to produce a given pathological characteristic. SERPINH1, also known as Hsp47, is a collagen-specific molecule that is essential for collagen synthesis [28]. The overexpression of SERPINH1 was found in many different cancers, including lung cancer, cervical squamous cancer, and glioma [29,30,31]. SERPINH1 may play an important role in tumor metastasis because of promoting maturation of various types of procollagens [32, 33]. Knockdown of SERPINH1 has been shown to significantly inhibit cell proliferation, migration, and invasion [31].
Type I collagen, including COL1A1 and COL1A2, is a major structural component of the ECM, and epithelial tumorigenesis is often accompanied by abnormal expression of ECM [34]. Overexpression of type I collagen was correlated with staging and poor disease-free survival of CRC patients [35]. It also found that type I collagen is required for maintaining lung cancer cell growth in 3D culture [36]. COL3A1 was a member of type III collagen and was found in extensible connective tissues. In epithelial ovarian cancers, the increase of COL3A1 was prognostic markers of poor prognosis [37]. High COL4A1 was revealed to be associated with advanced tumor stage as well as with bad overall and disease-free survival in HCC patients [38]. And COL4A1 knockdown led to cell viability reduction and cell cycle arrest in breast cancer cells [39]. COL6A3 has been observed to be frequently overexpressed in the GC tissues and also in five GC cell lines, including AGS, HGC-27, BGC-823, SGC-7901, and MGC-803 [40].
The other three hub genes are SPARC, SPP1, and VCAN. SPARC was overexpressed in highly metastatic tumors such as melanoma, breast cancer, and prostate cancer and acted as an anti-tumor factor in anti-angiogenesis, pro-apoptosis, cell proliferation inhibition, and cell cycle arrest in less metastatic tumors such as ovarian cancer, pancreatic cancer, colorectal cancer, and gastric cancer [41]. The high expression level of SPARC in GC tissues is controversial to its role in GC cells that it inhibited VEGF-induced proliferation and arrested cell cycle by reducing the activation of VEGFR2, ERK1/2, and AKT signaling pathways [42]. Therefore, SPARC may play different roles in different cancers and in different development stages of the same cancer. Elevated SPP1 levels have been detected in a variety of human cancers [43]; it may serve as a potential prognostic factor in GC [44]. The expression of SPP1 is related to the invasion and metastases of GC, and its mechanism may be to upregulate the expression of MMP-9 by activating NF-kappaB pathway [45]. VCAN was known to favor the homeostasis of the ECM [46]. And abolition of VCAN could reverse the increased migration effect which was induced by exogenous IL-11 in GC [47]. However, the oncogenic role and clinical significance of VCAN for GC were rarely explored. As the hub genes are mainly related to ECM, invasion and migration of GC seem to play a more important role in the development of cancer.
MiRNAs are known to regulate protein translation inhibition or targeted mRNA cleavage [48]. Increasing evidence suggested that miRNAs are involved in cancer development and progression, including gastric cancer [49]. In this study, we identified that hsa-miR-29c, hsa-miR-30c, hsa-miR-335, hsa-miR-33b, and hsa-miR-101 possibly affect the development and prognosis of gastric cancer through regulating the hub genes. MiR-29c expression was significantly decreased in GC [50]. MiR-29c downregulation was required to develop lung metastasis for the premetastatic CRC cells [51]. Recent study showed that hsa-miR-30c-5p was downregulated in GC tissues and remarkably related with lymphatic metastasis [52], and it suppressed the invasion ability of cancer by targeting metastasis-associated protein 1(MTA1) [52, 53]. MiR-30c was shown to be downregulated both in colon cancer specimens and prostate cancer [54, 55]. Aberrant expression of miR-335 is a noticeable factor in the cancer development [56, 57]. Furthermore, miR-335 suppressed the motibility of gastric cancer through regulating Bcl-w and specificity protein 1 [58]. MiR-33b was reported as a protective factor in multiple cancers [59,60,61]. In 2016, the methylation that resulted in downregulation of miR-33b was significantly detected in GC metastasis patients [62]. The genomic loss of miR-101 led to overexpression of EZH2, resulting in cancer progression [63, 64]. Therefore, this indicated that miRNAs play a noticeable role in the development and prognosis of GC through regulating disease-associated genes.
TF-hub genes regulatory network was constructed to explore the molecular mechanism of gastric cancer. In this study, we found AR, SOX2, EZH2, GATA2, RUNX1, SMAD4, and SUZ12 were meaningful for the expression of hub genes. It was reported that AR-negative patients had a significantly better survival than AR-positive GC patients. The SOX2 (sex-determining region Y-box 2) was a highly conserved transcription regulator and played a vital role in the cancer progression [65]. SOX2 was controversial in GC tissues; it was reported as a tumor promoter to activate AKT signaling on the one hand, and SOX2 was also detected as a protective factor to inhibit proliferation and metastasis [66, 67]. Overexpression of EZH2 was associated with poor prognosis and distant metastases in GC [68, 69]. As a potential metastasis-driving gene in prostate cancer [70], GATA2 was lack of research in GC. RUNX1 was reported as a target of AR, and its promoter was bound by EZH2 in prostate cancer [71]. Interestingly, RUNX1 played a cancer suppressor role in GC [72]. SMAD4 was frequently altered as common as TP53 in human gastric cancer [73]. SUZ12 is an essential component of Polycomb Repressive Complex2 (PRC2), affecting transcription by methylating histone and DNA [74]. Increased expression of SUZ12 was detected in GC and associated with pathological stage, metastasis, and poor prognosis [75]. The intricate interaction between TFs and hub genes made great contribution to the development of cancer.
Conclusion
In summary, we intend to identify DEGs by bioinformatics analysis to find the potential biomarkers which may be involved in the progress of GC. The study provides a set of useful DEGs for future investigation into molecular mechanisms and biomarkers of GC. And the application of data mining and integration is available for prediction of GC progression. Nevertheless, further molecular biological explorations are required to verify the function of the DEGs in GC.
Abbreviations
- BP:
-
Biological process
- DEGs:
-
Differentially expressed genes
- GC:
-
Gastric cancer
- OS:
-
Overall survival
- PPI:
-
Protein-protein interaction
- TFs:
-
Transcription factors
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Chen W, Zheng R, Zhang S, Zhao P, Zeng H, Zou X. Report of cancer incidence and mortality in China, 2010. Ann Transl Med. 2014;2:61.
Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Cancer Netw. 2010;8:437–47.
Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–84.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Zhang Z, Dou M, Yao X, Tang H, Li Z, Zhao X. Potential biomarkers in diagnosis of human gastric cancer. Cancer Investig. 2016;34:115–22.
Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6.
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43.
Marsit CJ, Eddy K, Kelsey KT. MicroRNA responses to cellular stress. Cancer Res. 2006;66:10843–8.
Schmittgen TD. Regulation of microRNA processing in development, differentiation and cancer. J Cell Mol Med. 2008;12:1811–9.
Bustin SA, Dorudi S. Gene expression profiling for molecular staging and prognosis prediction in colorectal cancer. Expert Rev Mol Diagn. 2004;4:599–607.
Nannini M, Pantaleo MA, Maleddu A, Astolfi A, Formica S, Biasco G. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer Treat Rev. 2009;35:201–9.
Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5:588–99.
Cui J, Chen Y, Chou WC, Sun L, Chen L, Suo J, Ni Z, Zhang M, Kong X, Hoffman LL, et al. An integrated transcriptomic and computational analysis for biomarker identification in gastric cancer. Nucleic Acids Res. 2011;39:1197–207.
Cheng L, Wang P, Yang S, Yang Y, Zhang Q, Zhang W, Xiao H, Gao H, Zhang Q. Identification of genes with a correlation between copy number and expression in gastric cancer. BMC Med Genet. 2012;5:14.
Wang G, Hu N, Yang HH, Wang L, Su H, Wang C, Clifford R, Dawsey EM, Li JM, Ding T, et al. Comparison of global gene expression of gastric cardia and noncardia cancers from a high-risk population in China. PLoS One. 2013;8:e63826.
Dennis GJ, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. Bmc Bioinformatics. 2003;4:2.
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–10.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’Ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–44.
Xia J, Gill EE, Hancock RE. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc. 2015;10:823–44.
Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19:197–208.
Zhang X, Yang JJ, Kim YS, Kim KY, Ahn WS, Yang S. An 8-gene signature, including methylated and down-regulated glutathione peroxidase 3, of gastric cancer. Int J Oncol. 2010;36:405–14.
Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, Ward L, Koo JH, Gopalakrishnan V, Zhu Y, et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009;5:e1000676.
Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, Liu J, Yue YG, Wang J, Yu K, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–56.
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Ito S, Nagata K. Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone. Semin Cell Dev Biol. 2016;62:142–51.
Wu ZB, Cai L, Lin SJ, Leng ZG, Guo YH, Yang WL, Chu YW, Yang SH, Zhao WG. Heat shock protein 47 promotes glioma angiogenesis. Brain Pathol. 2016;26:31–42.
Kamikawaji K, Seki N, Watanabe M, Mataki H, Kumamoto T, Takagi K, Mizuno K, Inoue H. Regulation of LOXL2 and SERPINH1 by antitumor microRNA-29a in lung cancer with idiopathic pulmonary fibrosis. J Hum Genet. 2016;61:985.
Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, Mitsuhashi A, Usui H, Enokida H, Nakagawa M, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. 2013;43:1855–63.
Sauk JJ, Nikitakis N, Siavash H. Hsp47 a novel collagen binding serpin chaperone, autoantigen and therapeutic target. Front Biosci. 2005;10:107–18.
Morino M, Tsuzuki T, Ishikawa Y, Shirakami T, Yoshimura M, Kiyosuke Y, Matsunaga K, Yoshikumi C, Saijo N. Specific expression of HSP47 in human tumor cell lines in vitro. In Vivo. 1997;11:17–21.
Aitken KJ, Bagli DJ. The bladder extracellular matrix. Part I: architecture, development and disease. Nat Rev Urol. 2009;6:596–611.
Zou X, Feng B, Dong T, Yan G, Tan B, Shen H, Huang A, Zhang X, Zhang M, Yang P, et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J Proteome. 2013;94:473–85.
Li J, Li X, Lan T, Qi C, He X, Yang H, Li Y, Wang L, Guan X. Type I collagen secreted by lung cancer cells promotes cancer cell growth in a three- dimensional culture system. Nan Fang Yi Ke Da Xue Xue Bao. 2014;34:1129–34.
Santala M, Simojoki M, Risteli J, Risteli L, Kauppila A. Type I and III collagen metabolites as predictors of clinical outcome in epithelial ovarian cancer. Clin Cancer Res. 1999;5:4091–6.
Desert R, Mebarki S, Desille M, Sicard M, Lavergne E, Renaud S, Bergeat D, Sulpice L, Perret C, Turlin B, et al. “Fibrous nests” in human hepatocellular carcinoma express a Wnt-induced gene signature associated with poor clinical outcome. Int J Biochem Cell Biol. 2016;81:195.
Salem O, Erdem N, Jung J, Munstermann E, Worner A, Wilhelm H, Wiemann S, Korner C. The highly expressed 5′isomiR of hsa-miR-140-3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC Genomics. 2016;17:566.
Xie X, Liu X, Zhang Q, Yu J. Overexpression of collagen VI alpha3 in gastric cancer. Oncol Lett. 2014;7:1537–43.
Feng J, Tang L. SPARC in tumor pathophysiology and as a potential therapeutic target. Curr Pharm Des. 2014;20:6182–90.
Zhang J, Wang P, Zhu J, Wang W, Yin J, Zhang C, Chen Z, Sun L, Wan Y, Wang X, et al. SPARC expression is negatively correlated with clinicopathological factors of gastric cancer and inhibits malignancy of gastric cancer cells. Oncol Rep. 2014;31:2312–20.
Furger KA, Menon RK, Tuck AB, Bramwell VH, Chambers AF. The functional and clinical roles of osteopontin in cancer and metastasis. Curr Mol Med. 2001;1:621–32.
Raja UM, Gopal G, Shirley S, Ramakrishnan AS, Rajkumar T. Immunohistochemical expression and localization of cytokines/chemokines/growth factors in gastric cancer. Cytokine. 2016;89:82–90.
Zhang DT, Yuan J, Yang L, Guo XN, Hao ZM, Han ZY, Wu KC, Fan DM. Osteopontin expression and its relation to invasion and metastases in gastric cancer. Zhonghua Zhong Liu Za Zhi. 2005;27:167–9.
Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev. 2009;28:233–45.
Zhang Z, Zhang J, Miao L, Liu K, Yang S, Pan C, Jiao B. Interleukin-11 promotes the progress of gastric carcinoma via abnormally expressed versican. Int J Biol Sci. 2012;8:383–93.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61.
Han TS, Hur K, Xu G, Choi B, Okugawa Y, Toiyama Y, Oshima H, Oshima M, Lee HJ, Kim VN, et al. MicroRNA-29c mediates initiation of gastric carcinogenesis by directly targeting ITGB1. Gut. 2015;64:203–14.
Cristobal I, Madoz-Gurpide J, Manso R, Rojo F, Garcia-Foncillas J. MiR-29c downregulation contributes to metastatic progression in colorectal cancer. Ann Oncol. 2015;26:2199–200.
Cao JM, Li GZ, Han M, Xu HL, Huang KM. MiR-30c-5p suppresses migration, invasion and epithelial to mesenchymal transition of gastric cancer via targeting MTA1. Biomed Pharmacother. 2017;93:554–60.
Xia Y, Chen Q, Zhong Z, Xu C, Wu C, Liu B, Chen Y. Down-regulation of miR-30c promotes the invasion of non-small cell lung cancer by targeting MTA1. Cell Physiol Biochem. 2013;32:476–85.
Zhang Q, Yu L, Qin D, Huang R, Jiang X, Zou C, Tang Q, Chen Y, Wang G, Wang X, Gao X. Role of microRNA-30c targeting ADAM19 in colorectal cancer. PLoS One. 2015;10:e120698.
Ling XH, Han ZD, Xia D, He HC, Jiang FN, Lin ZY, Fu X, Deng YH, Dai QS, Cai C, et al. MicroRNA-30c serves as an independent biochemical recurrence predictor and potential tumor suppressor for prostate cancer. Mol Biol Rep. 2014;41:2779–88.
Wang YX, Zhang XY, Zhang BF, Yang CQ, Chen XM, Gao HJ. Initial study of microRNA expression profiles of colonic cancer without lymph node metastasis. J Dig Dis. 2010;11:50–4.
Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008;111:478–86.
Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang X, Jiang L, Sun Z, Miao Z, Xu H. MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene. 2012;31:1398–407.
Liao W, Gu C, Huang A, Yao J, Sun R. MicroRNA-33b inhibits tumor cell growth and is associated with prognosis in colorectal cancer patients. Clin Transl Oncol. 2016;18:449–56.
Qu J, Li M, An J, Zhao B, Zhong W, Gu Q, Cao L, Yang H, Hu C. MicroRNA-33b inhibits lung adenocarcinoma cell growth, invasion, and epithelial-mesenchymal transition by suppressing Wnt/beta-catenin/ZEB1 signaling. Int J Oncol. 2015;47:2141–52.
Tian Q, Xiao Y, Wu Y, Liu Y, Song Z, Gao W, Zhang J, Yang J, Zhang Y, Guo T, et al. MicroRNA-33b suppresses the proliferation and metastasis of hepatocellular carcinoma cells through the inhibition of Sal-like protein 4 expression. Int J Mol Med. 2016;38:1587–95.
Yin H, Song P, Su R, Yang G, Dong L, Luo M, Wang B, Gong B, Liu C, Song W, et al. DNA Methylation mediated down-regulating of MicroRNA-33b and its role in gastric cancer. Sci Rep. 2016;6:18824.
Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–9.
Carvalho J, van Grieken NC, Pereira PM, Sousa S, Tijssen M, Buffart TE, Diosdado B, Grabsch H, Santos MA, Meijer G, et al. Lack of microRNA-101 causes E-cadherin functional deregulation through EZH2 up-regulation in intestinal gastric cancer. J Pathol. 2012;228:31–44.
Tam WL, Ng HH. Sox2: masterminding the root of cancer. Cancer Cell. 2014;26:3–5.
Tian Y, Jia X, Wang S, Li Y, Zhao P, Cai D, Zhou Z, Wang J, Luo Y, Dong M. SOX2 oncogenes amplified and operate to activate AKT signaling in gastric cancer and predict immunotherapy responsiveness. J Cancer Res Clin Oncol. 2014;140:1117–24.
Wang S, Tie J, Wang R, Hu F, Gao L, Wang W, Wang L, Li Z, Hu S, Tang S, et al. SOX2, a predictor of survival in gastric cancer, inhibits cell proliferation and metastasis by regulating PTEN. Cancer Lett. 2015;358:210–9.
Zhou Y, Du WD, Wu Q, Liu Y, Chen G, Ruan J, Xu S, Yang F, Zhou FS, Tang XF, et al. EZH2 genetic variants affect risk of gastric cancer in the Chinese Han population. Mol Carcinog. 2014;53:589–97.
Choi JH, Song YS, Yoon JS, Song KW, Lee YY. Enhancer of zeste homolog 2 expression is associated with tumor cell proliferation and metastasis in gastric cancer. Apmis. 2010;118:196–202.
Chiang YT, Wang K, Fazli L, Qi RZ, Gleave ME, Collins CC, Gout PW, Wang Y. GATA2 as a potential metastasis-driving gene in prostate cancer. Oncotarget. 2014;5:451–61.
Takayama K, Suzuki T, Tsutsumi S, Fujimura T, Urano T, Takahashi S, Homma Y, Aburatani H, Inoue S. RUNX1, an androgen- and EZH2-regulated gene, has differential roles in AR-dependent and -independent prostate cancer. Oncotarget. 2015;6:2263–76.
Li N, Zhang QY, Zou JL, Li ZW, Tian TT, Dong B, Liu XJ, Ge S, Zhu Y, Gao J, Shen L. miR-215 promotes malignant progression of gastric cancer by targeting RUNX1. Oncotarget. 2016;7:4817–28.
Park JW, Park DM, Choi BK, Kwon BS, Seong JK, Green JE, Kim DY, Kim HK. Establishment and characterization of metastatic gastric cancer cell lines from murine gastric adenocarcinoma lacking Smad4, p53, and E-cadherin. Mol Carcinog. 2015;54:1521–7.
Pasini D, Bracken AP, Jensen MR, Lazzerini DE, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–71.
Xia R, Jin FY, Lu K, Wan L, Xie M, Xu TP, De W, Wang ZX. SUZ12 promotes gastric cancer cell proliferation and metastasis by regulating KLF2 and E-cadherin. Tumour Biol. 2015;36:5341–51.
Availability of data and materials
The datasets supporting the conclusion of this article are included within the article.
Author information
Authors and Affiliations
Contributions
TL and XG participated in the design of this study, and they both performed the statistical analysis and drafted the manuscript. LH, JY, and HL helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Negatively correlated stem loop miRNAs of hub genes in a TCGA dataset composed of 380 gastric cancer tissues. (DOCX 30 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, T., Gao, X., Han, L. et al. Identification of hub genes with prognostic values in gastric cancer by bioinformatics analysis. World J Surg Onc 16, 114 (2018). https://doi.org/10.1186/s12957-018-1409-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-018-1409-3