
Abstract
Aim
Gastric cancer (GC) is one of the most diagnosed cancers worldwide. GC is a heterogeneous disease whose pathogenesis has not been entirely understood. Besides, the GC prognosis for patients remains poor. Hence, finding reliable biomarkers and therapeutic targets for GC patients is urgently needed.
Methods
GSE54129 and GSE26942 datasets were downloaded from Gene Expression Omnibus (GEO) database to detect differentially expressed genes (DEGs). Then, gene set enrichment analyses and protein-protein interactions were investigated. Afterward, ten hub genes were identified from the constructed network of DEGs. Then, the expression of hub genes in GC was validated. Performing survival analysis, the prognostic value of each hub gene in GC samples was investigated. Finally, the databases were used to predict microRNAs that could regulate the hub genes. Eventually, top miRNAs with more interactions with the list of hub genes were introduced.
Results
In total, 203 overlapping DEGs were identified between both datasets. The main enriched KEGG pathway was “Protein digestion and absorption.” The most significant identified GO terms included “primary alcohol metabolic process,” “basal part of cell,” and “extracellular matrix structural constituent conferring tensile strength.” Identified hub modules were COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, MUC6, CXCL8, and BGN. The overexpression of seven hub genes was associated with overall survival. Moreover, among the list of selected miRNAs, hsa-miR-27a-3, hsa-miR-941, hsa-miR-129-2-3p, and hsa-miR-1-3p, were introduced as top miRNAs targeting more than five hub genes.
Conclusions
The present study identified ten genes associated with GC, which may help discover novel prognostic and diagnostic biomarkers as well as therapeutic targets for GC. Our results may advance the understanding of GC occurrence and progression.
Similar content being viewed by others


Introduction
Gastric cancer (GC) has been reported as the fifth most diagnosed cancer worldwide, with more than 1 million newly diagnosed cases annually. Also, GC is considered one of the leading causes of death by cancer all over the world [1]. Despite all improvements in treating GC, survival rates for GC patients remain unsatisfying and depend on the disease diagnosed stage [1, 2]. While the five-year survival rate is about less than 30% in most GC cases with advanced stages [3, 4], it decreases to less than 5% in patients diagnosed with the distant disease [1]. However, since most patients are diagnosed at an advanced stage, they lose the chance of the most effective surgical intervention, the gold standard for GC therapy [5, 6]. Hence, screening, early diagnosis, and targeted therapies are essential to boost the survival rate of its patients [5]. The main reason for late diagnosis in GC patients is the lack of sensitive and precise predictive markers for diagnosis [7, 8]. Since diagnosing GC cases relies on invasive approaches such as endoscopy and biopsy, finding sufficient non-invasive tests and biomarkers for GC screening or diagnosis is necessary [5].
GC is a heterogeneous disease with phenotypic diversity [9]. A variety of genetic and epigenetic alterations have been reported associated with gastric precancerous lesions and GC [10]. A growing body of literature focused on the potential of microRNAs (miRNAs or miRs) as novel biomarkers and therapeutic targets for GC patients [2, 11, 12]. MiRNAs are a subclass of small non-coding RNAs regulating gene expression post-transcriptionally [13, 14]. Aberrantly expressed miRNAs are associated with the onset and progression of various cancers, like GC [15]. Therefore, miRNAs are studied as attractive biomarker candidates for diagnosis and prognosis, as well as predictors of drug responses.
Nowadays, microarray and sequencing-based technologies have facilitated the discovery of genes and underlying mechanisms of tumors, as well as the exploration of biomarkers, prognostic factors, and therapeutic targets for a variety of cancers [16, 17]. Moreover, in recent years, a growing body of literature has focused on investigating new therapeutic targets and diagnostic markers for diseases such as cancers through employing bioinformatics analysis [18, 19]. However, although several studies have focused on identifying genes, miRNAs, and their interactions in GC through bioinformatics methods [20,21,22,23], it is still far from enough to reveal and understand the underlying pathogenesis of the disease.
Hence, the present study aims to find the essential genes and miRNAs participating in GC by employing bioinformatics methods and public database resources. In this regard, we first identified the differentially expressed genes (DEGs) in GC from microarray datasets derived from the GEO database. We comprehensively analyzed the identified DEGs. First, we performed Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. Then, after constructing a protein-protein interaction (PPI) network, we searched for hub modules among the DEGs. Afterward, we explored the expression distribution and prognostic significance of the selected hub genes. Eventually, after predicting miRNAs targeting the identified hub genes, we constructed the network of miRNA-hub genes.
We hope the results of our study provide a theoretical basis for the discovery of promising biomarkers and therapeutic targets to improve the clinical diagnosis and treatment of GC.
Materials and methods
Data collection from GEO repository
The GEO is a public repository containing high-throughput functional genomic data [24]. The GEO database (http://www.ncbi.nlm.nih.gov/geo) was investigated to find suitable gene expression datasets, employing the following customized criteria: ‘Gastric cancer’ and ‘Healthy control’ as keywords, ‘Homo sapiens’ as the organism, ‘Expression profiling by array’ as the study type, and studies with sample count ‘Higher than 20’. Eventually, two datasets, GSE54129 and GSE26942, were selected for further examinations. Microarray data of GSE54129 was obtained from the GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) and collected from 111 gastric cancer tissues and 21 controls, comprised of biopsy normal gastric mucosa obtained from 21 volunteers underwent gastroscopy for health examinations. The GSE26942 data was based on the GPL6947 platform (Illumina HumanHT-12 V3.0 expression beadchip) and came from 206 GC patients and 12 normal samples.
Screening DEGs in GC
R software version 4.01 was used for screening and finding DEGs in the selected datasets. R packages employed to accomplish this purpose were Limma 3.48.3, data.Table 1.14.2, plyr 1.8.6, BiocGenerics 0.40.0, BioBase 2.54, and ggplot 3.3.5. Moreover, the “EnhancedVolcano” package was utilized to draw volcano plots. Significant DEGs were defined as upregulated DEGs with logFC ≥ 1 and downregulated DEGs with logFC ≤ − 1, with an adjusted p-value < 0.001.Then, the overlapping DEGs in the selected datasets were identified via a Venn diagram created by an online Venn diagram maker available at https://bioinformatics.psb.ugent.be/webtools/Venn.
Gene set enrichment analysis
To determine the biological implication of the overlapping DEGs, gene set enrichment analysis was performed using R software and employing several packages, including DOSE 3.20, org.Hs.eg.db 3.14.0, clusterProfiler 4.2.0. Results with a p.value < 0.05 were accepted as significant data.
Protein-protein interaction analysis
STRING (http://string.embl.de/) is a database designed for constructing PPI networks and analyzing the functional interactions among proteins [25, 26]. The PPI network of the identified DEGs was obtained from the STRING biological database and then visualized through Cytoscape software (version 3.7.2 [27]). Furthermore, the plugin of the CytoHubba 0.1 of Cytoscape software was applied to the obtained results to explore the top hub genes in the constructed network of DEGs based on their score calculated via the degree method. Eventually, the PPI network of top hub modules was constructed.
Validating the expression of the hub genes in GC
GEPIA 2.0 (available at http://gepia2.cancer-pku.cn/) is a cancer-specific database designed to analyze data based on TCGA and the Genotype-Tissue Expression (GTEx) databases [28]. Here, the GEPIA2 database was used to examine and validate the expression levels of the identified hub genes between GC and normal samples through the “Expression DIY” page of the database.
UALCAN (available at http://ualcan.path.uab.edu/analysis.html) is a web portal for conducting in-depth analyses of TCGA gene expression data [29, 30]. Here, the UALCAN database was employed to explore the differences in expression levels of each identified hub gene at various GC stages. A P-value < 0.05 were chosen as the p-value threshold for significant data.
Survival analysis
The Kaplan–Meier plotter is a web-based tool available at (http://kmplot.com/analysis/). This database is suitable for investigating the prognostic values of genes in samples from various tumor types, including GC [31]. The Kaplan–Meier plotter was applied to examine the correlation between the effect of the key genes and overall survival in GC patients. The database can calculate the hazard ratio (HR) with a 95% confidence interval (95% CI) and log-rank p.value. In this step, a p.value < 0.05 were set as a threshold to distinguish statistically significant results.
Identifying gene–miRNA interaction
Two online databases, including DIANA-TarBase v8 [32] and miRTarBase [33], were used to investigate the miRNAs regulating the hub genes. Both databases contain the experimentally validated associations between miRNAs and mRNAs [32, 33]. The lists of identified gene–miRNA interactions were merged for each hub gene to detect a single list of all valid interactions for the individual gene. Then, an online Venn diagram maker (https://bioinformatics.psb.ugent.be/webtools/Venn/) was used to find overlapping miRNAs. The miRNAs targeting more than four genes of the hub list were selected. Eventually, the final gene–miRNA interactions were visualized using Cytoscape software.
Results
Identification of DEGs in GC samples
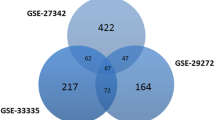
Two expression profiles (GSE54129 and GSE26942) were selected from the GEO database. A total of 317 GC tumors and 33 normal samples were obtained in this study. Employing the R software determined 3580 and 340 DEGs from GSE54129, and GSE26942, respectively. The Venn diagram identified 203 overlapping DEGs between selected datasets (Fig. 1). The lists of DEGs were presented in the supplementary file. Also, the volcano plots of the obtained DEGs from each dataset were drawn and illustrated in Fig. 2.

Venn diagram of the overlapping DEGs 203 common DEGs were detected between the two datasets (GSE54129 and GSE26942) using Venn diagram software (http://bioinformatics.psb.ugent.be/webtools/Venn/). DEGs = Differentially Expressed Genes

Volcano plots of DEGs in each GC dataset. Volcano Plot depicting DEGs between GC and control samples from GSE54129 and GSE26942 discriminated based on p-value and log2 fold-change. Colored dots represented genes with significant expression differences based on p-value (blue dots), only logFC (green dots), both p-value and log2 FC (red dots), or not significant in both terms (grey dots)
GO and pathway analysis of overlapping DEGs
In this step, GO, and KEGG pathway enrichment analyses of the overlapping DEGs were performed through R language. Based on the pathway analysis results, DEGs were particularly enriched in “Protein digestion and absorption” (P.value: 5.45E-07 and Gene count: 10), “Gastric acid secretion” (P.value: 4.28E-06 and Gene count: 8), “Metabolism of xenobiotics by cytochrome P450” (P.value: 5.21E-06 and Gene count: 8). Top identified pathways were presented in Table 1; Fig. 3. The results of GO enrichment analysis of 203 DEGs determined the most significant GO terms in biological process (BP), cellular component (CC), and molecular function (MF) as “primary alcohol metabolic process” (P.value: 3.54E-10 and Gene count: 12), “basal part of cell” (P.value: 2.27E-09 and Gene count: 17), and “extracellular matrix structural constituent conferring tensile strength” (P.value: 7.43E-09 and Gene count: 8), respectively. Other BP, CC, and MF were obtained in this study. The most significant terms of CC, BP, and MF were presented in Tables 2, 3 and 4; Fig. 3. Other results of GO and KEGG pathway enrichment analyses were presented in the supplementary file.

Gene Ontology and Pathway Analysis of overlapping DEGs. The most significant terms of BP, CC, MF, and KEGG pathways for overlapping DEGs were discovered and visualized using R software. The terms with a p.value and adjusted p.value < 0.05 were considered significant
PPI networks
At this step, we used 203 DEGs to construct a PPI network utilizing the STRING database and Cytoscape. The obtained network with 150 nodes and 416 edges is presented in Fig. 4. Subsequently, using the CytoHubba plugin, the ten hub genes, including COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, MUC6, CXCL8, and BGN, were identified. A network of hub genes was built with 52 nodes and 220 edges (Fig. 5; Table 5).

PPI Network of overlapping DEGs. The PPI network with 150 nodes and 416 edges was constructed via Cytoscape

PPI Network of top 10 hub genes. The PPI network with 52 nodes and 220 edges was constructed via Cytoscape
Validation of the gene expression
In this step, GEPIA was employed to investigate the expression levels of selected key genes in GC patients and healthy samples. The GEPIA results confirmed our data and reflected that all the selected hub genes except MUC6 were over-expressed in GC samples compared to normal samples. MUC6 was expressed at lower levels in GC compared with normal gastric tissues. All obtained results were significant (P < 0.05; Fig. 6).

mRNA expression of identified hub genes. Comparison of expression levels of 10 identified hub genes, including COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, MUC6, CXCL8, and BGN, in gastric cancer tissue (red; n = 408) and normal tissues (black; n = 211) using the GEPIA2 database. * P value < 0.05
Furthermore, the UALCAN results discerned that the expression pattern of the hub genes, including COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, CXCL8, and BGN, were significantly higher in GC stages I–IV than normal samples, whereas MUC6 was significantly downregulated in different stages of GC (Fig. 7). The obtained results were consistent with the finding of the selected microarray datasets, indicating that the expression of all hub genes except MUC6 was increased in GC.

Correlation between the expression levels of each identified hub gene and various stages of gastric cancer. Box-whisker plots made by UALCAN showed the expression of hub genes, including COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, CXCL8, and BGN, elevated in different stages of gastric cancer (stages 1, 2, 3, and 4) in TCGA samples. MUC6 is downregulated in the GC stages
Survival analysis of the hub genes
Overall survival curves were drawn to investigate the prognostic values of the identified hub genes in 875 GC patients by using the Kaplan-Meier plotter. Based on the median expression of the candidate genes, patients were divided into two groups [1] those with a high expression level of the desired gene and [2] others with a low expression level of it. The analysis determined that eight of ten key genes were significantly associated with the prognosis of GC patients. Among them, high expressions of COL1A1 (P.value: 8.9E-5), COL1A2 (P.value: 0.0015), TIMP1 (P.value: 1.5E-10), THBS2 (P.value: 1.2E-6), COL4A1 (P.value: 5.5E-7), MUC6 (P.value: 0.0151), and BGN (P.value: 1.5E-10) were significantly correlated with poor overall survival probability for GC patients (Fig. 8). High expression of CXCL8 (aka MDNCF) was detected to be associated with favorable overall survival (P.value: 1.5E-5). However, COL5A2 (P.value: 0.1769) and SPP1 (P.value: 0.2713) had nonsignificant log-rank p values and were independent of the prognosis of GC patients (Fig. 8).

Kaplan–Meier overall survival analysis for the top 10 hub genes. High expression of COL1A1, COL1A2, TIMP1, THBS2, COL4A1, MUC6, and BGN was associated with poor overall survival of GC patients. On the other hand, high expression of CXCL8 (aka MDNCF) was found to be associated with favorable overall survival. The expression of COL5A2 and SPP1 was not related to the overall survival of GC patients. GC: gastric cancer
Gene–miRNA interaction network
DIANA-TarBase v8 and miRTarBase were investigated for the miRNAs regulating the hub genes. A total of 101, 109, 32, 30, 58, 134, 117, 15, 75, and 33 miRNA-gene interactions were found for COL1A1, COL1A2, TIMP1, SPP1, COL5A2, THBS2, COL4A1, MUC6, CXCL8, and BGN, respectively. After merging the lists of identified gene–miRNA interactions, a single list containing ten miRNAs targeting more than four hub genes was chosen, including hsa-miR-27a-3, hsa-miR-941, hsa-miR-129-2-3p, hsa-miR-1-3p, hsa-miR-145-5p, hsa-let-7b-5p, hsa-miR-29a-3p, hsa-miR-124-3p, hsa-miR-16-5p, and hsa-miR-7-5p (Table 6) (supplementary file). Four miRNAs, including hsa-miR-27a-3, hsa-miR-941, hsa-miR-129-2-3p, and hsa-miR-1-3p, were selected as top miRNAs since they interact with more than half of the identified hub genes. Moreover, Cytoscape was employed to visualize the miRNA-gene interactions in a network with 27 nodes and 56 edges (Fig. 9).

miRNA-gene interaction network. The miRNA-gene interaction network with 27 nodes and 56 edges was constructed via Cytoscape
Discussion
In spite of a gradual decrease in the incidence and mortality rate, GC is still considered a leading cause of cancer mortality worldwide [1]. Besides, the early diagnosis and survival rate are still unfavorable for this cancer [34]. Therefore, it is critical to understand the underlying mechanisms and to determine biomarkers for developing strategies for screening, early diagnosis, and novel therapies for GC [35]. Hence, finding novel prognostic factors and/or biomarkers for early detection is required to improve patient outcomes.
In this study, we identified 203 DEGs in two GEO datasets of GC, GSE54129 and GSE26942. GO, and KEGG pathway enrichment analyses determined “Protein digestion and absorption,” “primary alcohol metabolic process,” “basal part of cell,” and “extracellular matrix structural constituent conferring tensile strength” as the most significant cancer-related pathways, BP, CC, and MF terms in which DEGs were enriched. The STRING and Cytoscape were employed to construct the PPI network. The plugin of CytoHubba introduced the top 10 hub genes, including COL1A1, COL1A2, COL5A2, COL4A1, TIMP1, SPP1, THBS2, MUC6, CXCL8, and BGN. All identified hub genes, except COL5A2 and SPP1, were significantly correlated with the overall survival of GC patients. After verifying the expression of all hub genes in GC, the miRNA-mRNA interactions were predicted for them. Among identified miRNAs, hsa-miR-27a-3, hsa-miR-941, hsa-miR-129-2-3p, and hsa-miR-1-3p, selected as top miRNAs interacting with more than half of the hub gene list.
Our results revealed the implication of different collagen family members, including COL1A1, COL4A1, COL5A2, and COL4A1, in GC samples. As critical parts of the ECM component, the members of this family are closely related to tumor prognosis, proliferation, invasion, and drug resistance [36]. COL1A1 and COL1A2 are overexpressed in GC and promote cell proliferation, invasion, and migration [37,38,39]. Li et al. shed light on the potential of COL1A1 as a monitoring factor for screening early GC. Besides, their results revealed a link between the overexpression of COL1A1 and COL1A2 with a poor overall survival rate of GC [40]. Previously, other studies proposed COL1A1, COL1A2, and COL4A1 as candidate diagnostic markers for this cancer [41, 42]. COL4A1 has been suggested as a potential biomarker and inflammation-related target for GC [43]. In silico studies have shed light on the potential of COL4A1 in conferring trastuzumab resistance and promoting gastric carcinoma recurrence [44, 45]. Additionally, COL4A1 has been implicated in trastuzumab resistance in gastric cancer, potentially conferring resistance to this targeted therapy [46]. Biglycan (BGN) is another critical component of ECM proteins involved in the development and aggressiveness of GC [47, 48]. This gene may implicate GC progression and development through the chronic activating of tumor angiogenesis [48]. A link was detected between BGN overexpression and worse clinical and prognostic parameters of GC [47, 49]. The mechanism of BGN-induced gastric cancer involves the induction of epithelial to mesenchymal transition (EMT) and upregulation of chromatin reprogramming factors [50]. The tissue inhibitor of metalloproteinases 1 (TIMP1) is an important player in ECM remodeling [51]. Preoperative TIMP1 expression level in peripheral blood may link to the GC stage, suggesting its potential application as a marker for tumor invasion and metastasis [52]. Hence, the expression level of TIMP1 has been suggested as a clinical biomarker for the screening, diagnosis, and prognostic of GC [52,53,54]. An in-silico study identified a correlation between overexpression of COL4A1, TIMP1, and COL1A2 with worse overall survival in GC [55]. Secreted phosphoprotein 1 (SPP1), an acidic glycoprotein known as osteopontin (OPN), participated in EMT and tumor metastasis [56]. Research showed that SPP1 suppresses the Wnt/β-catenin pathway, thereby enhancing the radiosensitivity of GAC through inhibition of invasion and acceleration of DNA damage, G2/M phase arrest, and apoptosis [57]. Lu et al. using bioinformatics analysis identified that SPP1 and FN1 were upregulated in GC than normal samples in their selected datasets [58]. Another study demonstrated that type I collagen promoted TIC-like phenotypes and chemoresistance through ITGB1/YBX1/SPP1/NF-κB pathway [59]. Thrombospondin 2 (THBS2) a member of the thrombospondin family, has been suggested as an early diagnostic marker for patients with GC [60]. Wang et al. showed that while SPP1 had no significant association with GC overall survival, high expression level of THBS2 in GC patients was correlated with shorter survival time [61] [60].Bioinformatics reanalysis suggested the prognostic value of COL1A1, COL1A2, and THBS2 in GC. KEGG reanalysis determined that these genes, together with COL2A1 and COL11A1, were enriched in the ECM-receptor interaction pathways [62].MUC6, Musin 6, is a marker of gastric foveolar and antral mucous glandular cells that shows gastric phenotypes. literatures indicated that the expression of MUC6 was regulated by promoter methylation which lead to the downregulation of MUC6 in GC and induce the progression of GC [63]. Zheng et al. reported a link between the downregulation of MUC6 with progression, poor prognosis, and metastasis of GC [64]. Considering transcriptomics and single-cell sequencing, another study suggested BGN and COL5A2 as GC diagnostic and prognostic biomarkers applicable for predicting drug sensitivity in GC [49]. Despite controversial evidence regarding the link between the expression level of COL5A2 and the survival of GC patients [49, 65,66,67,68], Zhang et al. and Cao et al. confirmed our results by reporting no significant correlation between COL5A2 and the overall survival of GC patients [66, 69]. Studies have also demonstrated that serum CXCL8 levels can significantly predict GC risk and reveal a role of the CXCL8/CXCR2 axis and inflammation in the pathogenesis of this malignancy [70]. CXCL8, which is primarily secreted by macrophages in gastric cancer, is linked to poor clinical outcomes and tumor progression. CXCL8 promotes an immunosuppressive environment by increasing PD-L1 expression on macrophages, hindering CD8 + T-cell function, and limiting infiltration [71]. Several studies identified positive impacts of higher levels of CXCL8 expression on the overall survival of GC patients [72,73,74,75].
Several studies highlighted the role of hsa-miR-27a-3p as oncomiR upregulated in GC [76,77,78]. Moreover, miR-27a-3p/BTG2 axis was proposed as not only a promising diagnostic biomarker but also a potential therapeutic target for GC patients [77]. Previously, the link between overexpression of miR-27a and significant up-regulation of COL1A2 was reported in hepatic stellate cells [79]. An inverse correlation between miR-27a-3p and CXCL8 was reported [80]. MicroRNA-27a-3p negatively regulates SPP1 to inhibit lung and skin fibrosis of systemic sclerosis [81]. MiR-129-2-3p regulates cell proliferation in GC cells [82]. Gastric juice miR-129-2-3p has been suggested as a potential biomarker for the screening GC [83]. MiR-129-5p regulates GC invasion through interacting with IL-8 and COL1A1 [84, 85]. The interaction between COL1A1 and MiR-129-5p has been proposed as a potential therapeutic target for GC [86]. MiR-1-3p is another miRNA suppressing proliferation and invasion of GC cells [87]. Although the role of miR-941 has been investigated in several cancers, such as breast and prostate cancer [88, 89], its association has not been reported with GC. Previous studies supported our results and revealed the link between selected miRNAs and GC. However, at the time of this study, most of the miRNA-gene interactions identified in this study have not been investigated in GC.
The results of this study provide a more comprehensive understanding of the underlying mechanisms of GC and suggest novel biomarkers for prognosis and diagnosis, as well as therapeutic targets for GC patients. The most important limitation of our study is the lack of experimental work to validate the results obtained from bioinformatics methods. Therefore, further studies with larger sample sizes, animal models, and clinical tissue verification are required to confirm our results.
Conclusion
In conclusion, using two datasets obtained from the GEO database and integrated bioinformatics analysis, ten GC-associated hub genes were found. Except for MUC6, the expression of other hub genes was revealed to be upregulated in GC. The overexpression of seven hub genes was associated with GC’s poor overall survival. Then, the miRNA-mRNA interactions were predicted for each hub gene. Although more experimental investigations with larger sample sizes are required to validate the findings of the present study, we hope that our results will assist in the discovery of novel biomarkers and therapeutic targets for GC and advance the understanding of its pathogenesis.
Data Availability
All databases (including NCBI GEO, Veen Diagram maker, etc.) are Freely available on the web. The raw data of this study are obtained from the GEO database (available at https://www.ncbi.nlm.nih.gov/geo/).
References
Thrift AP, El-Serag HB. Burden of gastric Cancer. Clin Gastroenterol Hepatol. 2020;18(3):534–42.
Zheng TH, Zhao JL, Guleng B. Advances in molecular biomarkers for gastric Cancer. Crit Rev Eukaryot Gene Expr. 2015;25(4):299–305.
Shi XJ, Wei Y, Ji B. Systems Biology of Gastric Cancer: perspectives on the Omics-based diagnosis and treatment. Front Mol Biosci. 2020;7:203.
Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2(9):533–43.
Liu L, Pang H, He Q, Pan B, Sun X, Shan J, et al. A novel strategy to identify candidate diagnostic and prognostic biomarkers for gastric cancer. Cancer Cell Int. 2021;21(1):335.
Choi YY, Noh SH, Cheong JH. Evolution of gastric Cancer treatment: from the Golden Age of Surgery to an era of Precision Medicine. Yonsei Med J. 2015;56(5):1177–85.
Hamashima C. Current issues and future perspectives of gastric cancer screening. World J Gastroenterol. 2014;20(38):13767–74.
Hu Y, Li J, Luo H, Song W, Yang J. Differential expression of COL1A1, COL1A2, COL6A3, and SULF1 as prognostic biomarkers in gastric Cancer. Int J Gen Med. 2021;14:5835–43.
Yu C, Chen J, Ma J, Zang L, Dong F, Sun J, et al. Identification of key genes and Signaling pathways Associated with the progression of gastric Cancer. Pathol Oncol Res. 2020;26(3):1903–19.
Battista S, Ambrosio MR, Limarzi F, Gallo G, Saragoni L. Molecular alterations in gastric preneoplastic lesions and early gastric Cancer. Int J Mol Sci. 2021;22(13).
Liu Y, Ao X, Ji G, Zhang Y, Yu W, Wang J. Mechanisms of action and clinical implications of MicroRNAs in the Drug Resistance of Gastric Cancer. Front Oncol. 2021;11:768918.
Kipkeeva F, Muzaffarova T, Korotaeva A, Nikulin M, Grishina K, Mansorunov D et al. MicroRNA in Gastric Cancer Development: Mechanisms and Biomarkers Diagnostics (Basel). 2020;10(11).
Bartel DP. Metazoan MicroRNAs. Cell. 2018;173(1):20–51.
Mortazavi SS, Bahmanpour Z, Daneshmandpour Y, Roudbari F, Sheervalilou R, Kazeminasab S, et al. An updated overview and classification of bioinformatics tools for MicroRNA analysis, which one to choose? Comput Biol Med. 2021;134:104544.
So JBY, Kapoor R, Zhu F, Koh C, Zhou L, Zou R, et al. Development and validation of a serum microRNA biomarker panel for detecting gastric cancer in a high-risk population. Gut. 2021;70(5):829–37.
Wu M, Li Q, Wang H. Identification of novel biomarkers Associated with the prognosis and potential pathogenesis of Breast Cancer via Integrated Bioinformatics Analysis. Technol Cancer Res Treat. 2021;20:1533033821992081.
Wang D, Liu Y, Cheng S, Liu G. Identification of novel genes and Associated Drugs in Cervical Cancer by Bioinformatics methods. Med Sci Monit. 2022;28:e934799.
Jiang P, Liu XS. Big data mining yields novel insights on cancer. Nat Genet. 2015;47(2):103–4.
Ding X, Duan H, Luo H. Identification of Core Gene Expression Signature and key pathways in Colorectal Cancer. Front Genet. 2020;11:45.
Li M, Wang X, Liu J, Mao X, Li D, Wang Z, et al. Identification of core prognosis-related candidate genes in Chinese gastric Cancer Population based on Integrated Bioinformatics. Biomed Res Int. 2020;2020:8859826.
Liu S, Zhao Y, Duan R, Wu Y, Chen X, Li N. Identification of core genes associated with type 2 Diabetes Mellitus and gastric cancer by bioinformatics analysis. Ann Transl Med. 2022;10(5):247.
Yang G, Zhang Y, Yang J. Identification of Potentially Functional CircRNA-miRNA-mRNA Regulatory Network in Gastric Carcinoma using Bioinformatics Analysis. Med Sci Monit. 2019;25:8777–96.
Wang Y, Zhang J, Liu M, Zhang S, Wang W, Cheng S. Clinical values and potential pathways of mir-183-5p in gastric cancer: a study based on integrational bioinformatics analysis. J Gastrointest Oncol. 2021;12(5):2123–31.
Clough E, Barrett T. The Gene expression Omnibus Database. Methods Mol Biol. 2016;1418:93–110.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49:D1.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D1.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11).
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W1.
Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M et al. UALCAN: an update to the integrated cancer data analysis platform. Neoplasia (United States). 2022;25.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK et al. UALCAN: a portal for facilitating Tumor Subgroup Gene expression and survival analyses. Neoplasia (United States). 2017;19(8).
Győrffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in Breast cancer. Comput Struct Biotechnol J. 2021;19.
Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018;46:D1.
Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–302.
Rohatgi PR, Yao JC, Hess K, Schnirer I, Rashid A, Mansfield PF, et al. Outcome of gastric cancer patients after successful gastrectomy: influence of the type of recurrence and histology on survival. Cancer. 2006;107(11):2576–80.
Sun H. Identification of key genes associated with gastric cancer based on DNA microarray data. Oncol Lett. 2016;11(1):525–30.
Li X, Sun X, Kan C, Chen B, Qu N, Hou N, et al. COL1A1: a novel oncogenic gene and therapeutic target in malignancies. Pathol Res Pract. 2022;236:154013.
Shi Y, Duan Z, Zhang X, Zhang X, Wang G, Li F. Down-regulation of the let-7i facilitates gastric cancer invasion and Metastasis by targeting COL1A1. Vol. 10, Protein & cell. Germany; 2019. p. 143–8.
Guo Y, Lu G, Mao H, Zhou S, Tong X, Wu J, et al. miR-133b suppresses Invasion and Migration of Gastric Cancer cells via the COL1A1/TGF-β Axis. Onco Targets Ther. 2020;13:7985–95.
Yasui W, Oue N, Ito R, Kuraoka K, Nakayama H. Search for new biomarkers of gastric cancer through serial analysis of gene expression and its clinical implications. Cancer Sci. 2004;95(5):385–92.
Li J, Ding Y, Li A. Identification of COL1A1 and COL1A2 as candidate prognostic factors in gastric cancer. World J Surg Oncol. 2016;14(1):297.
Zhao Q, Xie J, Xie J, Zhao R, Song C, Wang H, et al. Weighted correlation network analysis identifies FN1, COL1A1 and SERPINE1 associated with the progression and prognosis of gastric cancer. Cancer Biomark. 2021;31(1):59–75.
Wang F, Xue Q, Xu D, Jiang Y, Tang C, Liu X. Identifying the hub gene in gastric cancer by bioinformatics analysis and in vitro experiments. Cell Cycle. 2020;19(11):1326–37.
Liu Y, Xu Y, Xiao F, Zhang J, Wang Y, Yao Y et al. Comprehensive Analysis of a circRNA-miRNA-mRNA Network to Reveal Potential Inflammation-Related Targets for Gastric Adenocarcinoma. Mediators Inflamm. 2020; 2020:9435608.
Huang R, Gu W, Sun B, Gao L. Identification of COL4A1 as a potential gene conferring trastuzumab resistance in gastric cancer based on bioinformatics analysis. Mol Med Rep. 2018;17(5):6387–96.
Li DF, Wang NN, Chang X, Wang SL, Wang LS, Yao J, et al. Bioinformatics analysis suggests that COL4A1 may play an important role in gastric carcinoma recurrence. J Dig Dis. 2019;20(8):391–400.
Li Y, Sun R, Zhao X, Sun B. RUNX2 promotes malignant progression in gastric cancer by regulating COL1A1. Cancer Biomarkers. 2021;31:227–38.
Wang B, Li GX, Zhang SG, Wang Q, Wen YG, Tang HM, et al. Biglycan expression correlates with aggressiveness and poor prognosis of gastric cancer. Exp Biol Med (Maywood). 2011;236(11):1247–53.
Hu L, de Zang M, Wang HX, Li JF, Su LP, Yan M, et al. Biglycan stimulates VEGF expression in endothelial cells by activating the TLR signaling pathway. Mol Oncol. 2016;10(9):1473–84.
Zhao X, Wu S, Jing J. Identifying diagnostic and prognostic biomarkers and candidate therapeutic Drugs of gastric Cancer based on Transcriptomics and single-cell sequencing. Pathol Oncol Res. 2021;27:1609955.
Han RF, Ji X, Dong XG, Xiao RJ, Liu YP, Xiong J, et al. An epigenetic mechanism underlying Doxorubicin Induced EMT in the human BGC-823 gastric Cancer cell. Asian Pac J Cancer Prev. 2014;15(10):4271–4.
Ries C. Cytokine functions of TIMP-1. Cell Mol Life Sci. 2014;71(4):659–72.
Wang YY, Li L, Zhao ZS, Wang HJ. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J Surg Oncol. 2013;11:81.
Zhang T, Wang BF, Wang XY, Xiang L, Zheng P, Li HY, et al. Key genes Associated with Prognosis and Tumor infiltrating Immune cells in gastric Cancer patients identified by Cross-database Analysis. Cancer Biother Radiopharm. 2020;35(9):696–710.
Li Z, Jing Q, Wu L, Chen J, Huang M, Qin Y, et al. The prognostic and diagnostic value of tissue inhibitor of metalloproteinases gene family and potential function in gastric cancer. J Cancer. 2021;12(13):4086–98.
Li T, Gao X, Han L, Yu J, Li H. Identification of hub genes with prognostic values in gastric cancer by bioinformatics analysis. World J Surg Oncol. 2018;16(1):114.
Jia R, Liang Y, Chen R, Liu G, Wang H, Tang M, et al. Osteopontin facilitates Tumor Metastasis by regulating epithelial–mesenchymal plasticity. Cell Death Dis. 2016;7(12):e2564–4.
Sun G, Shang Z, Liu W. SPP1 Regulates Radiotherapy Sensitivity of Gastric Adenocarcinoma via the Wnt/Beta-Catenin Pathway. J Oncol. 2021; 2021:1–10.
Lu XQ, Zhang JQ, Zhang SX, Qiao J, Qiu MT, Liu XR, et al. Identification of novel hub genes associated with gastric cancer using integrated bioinformatics analysis. BMC Cancer. 2021;21(1):697.
Deng S, Li L, Xu S, Wang X, Han T. Promotion of gastric tumor initiating cells in a 3D collagen gel culture model via YBX1/SPP1/NF-κB signaling. Cancer Cell Int [Internet]. 2021;21(1):599. https://doi.org/10.1186/s12935-021-02307-x.
Li L, Dong J, Fu L, Xia X, Pan F, Ning Y. Clinical Value of Serum Thrombospondin-2 Combined with CA19-9 in Early Diagnosis of Gastric Cancer. J Oncol. 2021; 2021:2483964.
Wang L, Feng L, Liu L, Han J, Zhang X, Li D, et al. Joint effect of THBS2 and VCAN accelerating the poor prognosis of gastric cancer. Aging. 2023;15(5):1343–57.
Li D, Yin Y, He M, Wang J. Identification of potential biomarkers Associated with Prognosis in Gastric Cancer via Bioinformatics Analysis. Med Sci Monit. 2021;27:e929104.
Shi D, Xi X. xia. Regulation of MUC6 Methylation Correlates with Progression of Gastric Cancer. Yonsei Med J. 2021;62(11):1005.
Zheng H, Takahashi H, Nakajima T, Murai Y, Cui Z, Nomoto K, et al. MUC6 down-regulation correlates with gastric carcinoma progression and a poor prognosis: an immunohistochemical study with tissue microarrays. J Cancer Res Clin Oncol. 2006;132(12):817–23.
Hao S, Lv J, Yang Q, Wang A, Li Z, Guo Y, et al. Identification of key genes and circular RNAs in human gastric Cancer. Med Sci Monit. 2019;25:2488–504.
Cao L, Chen Y, Zhang M, Xu DQ, Liu Y, Liu T, et al. Identification of hub genes and potential molecular mechanisms in gastric cancer by integrated bioinformatics analysis. PeerJ. 2018;6:e5180.
Tan Y, Chen Q, Xing Y, Zhang C, Pan S, An W et al. High expression of COL5A2, a member of COL5 family, indicates the poor survival and facilitates cell migration in gastric cancer. Biosci Rep. 2021;41(4).
Niu X, Ren L, Hu A, Zhang S, Qi H. Identification of potential diagnostic and prognostic biomarkers for gastric Cancer based on bioinformatic analysis. Front Genet. 2022;13:862105.
Zhang S, Xiang X, Liu L, Yang H, Cen D, Tang G. Bioinformatics Analysis of Hub Genes and potential therapeutic agents Associated with gastric Cancer. Cancer Manag Res. 2021;13:8929–51.
Pawluczuk E, Łukaszewicz-Zając M, Gryko M, Kulczyńska-Przybik A, Mroczko B. Serum CXCL8 and its specific receptor (CXCR2) in gastric Cancer. Cancers (Basel). 2021;13(20):5186.
Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68(10):1764–73.
Wang Y, Zheng K, Chen X, Chen R, Zou Y. Bioinformatics analysis identifies COL1A1, THBS2 and SPP1 as potential predictors of patient prognosis and immunotherapy response in gastric cancer. Biosci Rep. 2021;41(1).
Yan P, He Y, Xie K, Kong S, Zhao W. In silico analyses for potential key genes associated with gastric cancer. PeerJ. 2018;6:e6092.
Tian Y, Xing Y, Zhang Z, Peng R, Zhang L, Sun Y. Bioinformatics Analysis of Key Genes and circRNA-miRNA-mRNA Regulatory Network in Gastric Cancer. Biomed Res Int. 2020; 2020:1–16.
Shao C, Wang R, Kong D, Gao Q, Xu C. Identification of potential core genes in gastric cancer using bioinformatics analysis. J Gastrointest Oncol. 2021;12(5):2109–22.
Liang Y, Zhao Y, Li L, Wei H, Huang T, Zhang H, et al. MicroRNA profiles in five pairs of early gastric cancer tissues and adjacent non-cancerous tissues. Oncol Lett. 2021;22(2):595.
Zhou L, Liang X, Zhang L, Yang L, Nagao N, Wu H, et al. MiR-27a-3p functions as an oncogene in gastric cancer by targeting BTG2. Oncotarget. 2016;7(32):51943–54.
Liu T, Tang H, Lang Y, Liu M, Li X. MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett. 2009;273(2):233–42.
Zhang H, Yan XL, Guo XX, Shi MJ, Lu YY, Zhou QM, et al. MiR-27a as a predictor for the activation of hepatic stellate cells and Hepatitis B virus-induced liver Cirrhosis. Oncotarget. 2018;9(1):1075–90.
Kinget L, Roussel E, Lambrechts D, Boeckx B, Vanginderhuysen L, Albersen M et al. MicroRNAs possibly involved in the development of bone Metastasis in Clear-Cell Renal Cell Carcinoma. Cancers (Basel). 2021;13(7).
Cheng Q, Chen M, Wang H, Chen X, Wu H, Du Y, et al. MicroRNA-27a-3p inhibits lung and skin fibrosis of systemic sclerosis by negatively regulating SPP1. Genomics. 2022;114(4):110391.
Yu X, Song H, Xia T, Han S, Xiao B, Luo L, et al. Growth inhibitory effects of three miR-129 family members on gastric cancer. Gene. 2013;532(1):87–93.
Yu X, Luo L, Wu Y, Yu X, Liu Y, Yu X, et al. Gastric juice miR-129 as a potential biomarker for screening gastric cancer. Med Oncol. 2013;30(1):365.
Wang Q, Yu J. MiR-129-5p suppresses gastric cancer cell invasion and proliferation by inhibiting COL1A1. Biochem Cell Biol. 2018;96(1):19–25.
Jiang Z, Wang H, Li Y, Hou Z, Ma N, Chen W, et al. MiR-129-5p is down-regulated and involved in migration and invasion of gastric cancer cells by targeting interleukin-8. Neoplasma. 2016;63(5):673–80.
Yang W, Pan Y, Guan P, Li X, You C. [Bioinformatics analysis of COL1A1 regulated by mir-129-5p as a potential therapeutic target for gastric cancer]. Nan Fang Yi Ke Da Xue Xue Bao. 2019;39(5):540–6.
Ke J, Zhang BH, Li YY, Zhong M, Ma W, Xue H, et al. MiR-1-3p suppresses cell proliferation and invasion and targets STC2 in gastric cancer. Eur Rev Med Pharmacol Sci. 2019;23(20):8870–7.
Surapaneni SK, Bhat ZR, Tikoo K. MicroRNA-941 regulates the proliferation of Breast cancer cells by altering histone H3 ser 10 phosphorylation. Sci Rep. 2020;10(1):17954.
Larson NB, McDonnell SK, Fogarty Z, Liu Y, French AJ, Tillmans LS, et al. A microRNA transcriptome-wide Association study of Prostate Cancer risk. Front Genet. 2022;13:836841.
Acknowledgements
Not applicable.
Funding
No funding or sponsorship was received for this study or publication of this article.
Author information
Authors and Affiliations
Contributions
All authors contributed to this project. Dr Shahraki and Dr Sheervalilou designed the study. Abdolahi conducted research, data collection, and part of the initial analysis. Mortazavi performed analysis, drafted the work, and helped in the revision. Also, Dr Sheervalilou were responsible for the final edit and revision. Eventually, all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent to Publish
All authors are agreed.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Abdolahi, F., Shahraki, A., Sheervalilou, R. et al. Identification of differentially expressed genes associated with the pathogenesis of gastric cancer by bioinformatics analysis. BMC Med Genomics 16, 311 (2023). https://doi.org/10.1186/s12920-023-01720-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01720-7