
Abstract
Background
Apple replant disease (ARD) has been reported from all major fruit-growing regions of the world, and is often caused by biotic factors (pathogen fungi) and abiotic factors (phenolic compounds). In order to clarify the proteomic differences of Fusarium moniliforme under the action of phloridzin, and to explore the potential mechanism of F. moniliforme as the pathogen of ARD, the role of Fusarium spp. in ARD was further clarified.
Methods
In this paper, the quantitative proteomics method iTRAQ analysis technology was used to analyze the proteomic differences of F. moniliforme before and after phloridzin treatment. The differentially expressed protein was validated by qRT-PCR analysis.
Results
A total of 4535 proteins were detected, and 293 proteins were found with more than 1.2 times (P< 0.05) differences. In-depth data analysis revealed that 59 proteins were found with more than 1.5 times (P< 0.05) differences, and most proteins were consistent with the result of qRT-PCR. Differentially expressed proteins were influenced a variety of cellular processes, particularly metabolic processes. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium. Functional pathway analysis indicated that up-regulated proteins were mainly distributed in amino sugar, nucleotide sugar metabolism, glycolysis/ gluconeogenesis and phagosome pathways.
Conclusions
This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in F. moniliforme under phloridzin conditions. The results confirmed that F. moniliforme presented a unique protein profile that indicated the adaptive mechanisms of this species to phloridzin environments. The results deepened our understanding of the proteome in F. moniliforme in response to phloridzin inducers and provide a basis for further exploration for improving the efficiency of the fungi as biocontrol agents to control ARD.
Similar content being viewed by others


Background
Apple is one of the most important fruit trees worldwide, and China has the largest area of apple tree cultivation. Apple replant disease (ARD) is widespread in China because repeated production in the same field is a common practice owing land scarcity and the replant of aged apple orchards. ARD causes the inhibition of root system development, stunts tree growth, and reduces yield and quality in replanted apple orchards [1,2,3], and is common in all of the major apple growing regions of the world [1]. The etiology of ARD is complex, many research results suggest that ARD is not attributed to only one factor, but rather to a combined effect of biotic factors and abiotic factors [4, 5]. Biotic factors including nematodes, bacteria, actinomycete, oomycetes and fungi species [6, 7], and most researches demonstrated fungal and oomycete genera were the main reason for ARD, i.e. fungal genera: Fusarium [5, 6, 8], Rhizoctonia [5, 6], Cylindrocarpon [3, 6, 9]; oomycete genera: Phytophthora [5, 6], Pythium [5, 6]. Abiotic factors such as soil structure, nutrition, and the release of allelochemicals through leaching, root exudation, volatilization, and/or decomposition of residues may also play roles in replant problems [10,11,12].
The harmful fungi were different in replant soil of different area’s orchards. Manici et al. [13] investigated three orchards in Germany, Austria and Italy, found that Cylindrocarpon spp. (Ilyonectria spp. and Thelonectria spp) is the main pathogenic fungus in three replanted orchards, while pythium spp. is just a pathogenic fungus in German. Van Schoor et al. [7] showed that pathogenic species of Fusarium, Cylindrocarpon, and Pythium were the primary obstacles to continuous cropping in replanted South African orchards. Kelderer et al. [14] showed that Fusarium solani, F. oxysporum, binucleate Rhizoctonia spp. were the main soil-borne pathogens for ARD in Italy. So for, F. moniliforme also have been shown to be pathogenic on forest nurseries in Spain [15], Sorghum in India, Maize in Iowa State [16], Scots pine seedlings in Palencia [17], Peach in California [18] and apple in China [19]. F. moniliforme (F. verticillioides), F. oxysporum, and F. proliferatum isolated from replanted apple soil near the Bohai Bay region in China were highly pathogenic to seedlings of Malus hupehensis in our laboratory research cooperation studies [20, 21]. Phloridzin is a characteristic dihydrochalcone of the phenolic compounds produced by apple seedlings [11, 22,23,24], and phloridzin in the root exudates of Malus x domestica Borkh. and Malus hupehensis Rehd. might have consequences for research on the etiology of ARD [11, 12]. But how interaction of phloridzin (a phenolic compound) and F. moniliforme (a soil-borne pathogen) in the ARD is not yet fully understood.
In our study, we observed the effect of phloridzin on F. moniliforme on potato dextrose agar medium and inorganic salt culture medium, respectively. The effect of phloridzin on the proteome of F. moniliforme was further studied using iTRAQ technology. To the best of our knowledge, this is the first application of iTRAQ coupled nanoUHPLC-MS/MS technique to investigate the mechanism of F. moniliforme hyphae growth in response to phloridzin inducers. The primary aims of this research were to study: 1) Does phloridzin provide a carbon source for F. moniliforme? 2) How does the mycelium proteome of F. moniliforme change with phloridzin? 3) Which metabolic pathways of F. moniliforme have been changed by phloridzin?
Methods
Strains and culture conditions
The experimental processing settings were as follows: F. moniliforme+PDA(as CK), PDA medium + F. moniliforme + 1 ml phloridzin solution(as T). Stock cultures of the stains were prepared on potato dextrose agar (PDA) medium (20% potato extract, 2% glucose, 2% agar, pH 7.0). After being incubated for 7 days at 28 °C, mycelia were collected and stored at − 80 °C for protein preparation, respectively. Three biological replicates were used for both control and treatment in this study.
Our previous study found that phloridzin promoted the growth and division of F. moniliforme with the help of fluorescence labeling with SiC quantum dots (Fig. S1) [20]. The details of the experiment were as follows: The F. moniliforme was inoculated into Potato Dextrose (PD: 200 g potato and 20 g glucosein 1 L distilled water) liquid culture media and incubated in a thermostatic orbital shaker at 28 °C and 200 rpm for 48 h, and PD liquid culture media containing F. moniliforme was prepared. Three treatments were arranged: no phloridzin (T1: 45 mL PD liquid culture media containing F. moniliforme and 15 mL sterilized SiC quantum dots solution and 5 mL sterilized water). 0.5 mM phloridzin (T2: 45 mL PD liquid culture media containing F. moniliforme and 15 mL sterilized SiC quantum dots solution and 5 mL 0.5 mM phloridzin). 1.0 mM phloridzin (T3: 45 mL PD liquid culture media containing F. moniliforme and 15 mL sterilized SiC quantum dots solution and 5 mL 1.0 mM phloridzin), each treatment with 3 replicates. All samples of three treatments were incubated in a thermostatic shaker at 28 °C and 200 rpm for 3-40d. The growth and conidia division of F. moniliforme were observed at different time points under a fluorescence microscope. Living cell morphologies were observed and photographed using an Olympus IX-71 type fluorescence microscope (Japan).
In order to further verify whether phloridzin provides carbon source for F. moniliforme, inorganic salt culture medium (1.0 g of ammonium sulfate, 0.5 g of sodium chloride, 0.5 g of potassium dihydrogen phosphate, 1.5 g of dipotassium hydrogen phosphate, 0.2 g of magnesium sulfate, 20 g of agar, make up to 1000 ml with distilled water, and autoclave at 121 °C for 20 min) and phloridzin inorganic salt culture medium (1.0 g of ammonium sulfate, 0.5 g of sodium chloride, 0.5 g of potassium dihydrogen phosphate, 1.5 g of dipotassium hydrogen phosphate, 0.2 g of magnesium sulfate, 20 g of agar, 0.044 g of phloridzin, make up to 1000 ml with distilled water, and autoclave at 121 °C for 20 min) were set in this study.
Protein preparation and sample preparation
Took a 2 g sample, added liquid extraction buffer and protease inhibitor in a certain ratio after grinding in liquid nitrogen in a frozen state, vortexed for 10 min. Added the same volume of Tris-saturated phenol (pH 8.0) and vortexed for 10 min; centrifuged at 12000 g at 4 °C for 20 min to obtain a phenol phase; took the phenol phase into a new centrifuge tube, added an equal volume of extraction buffer, vortexed Shake for 10 min; centrifuged at 12,000 g for 20 min at 4 °C to obtain a phenol phase; took the phenol phase into a new centrifuge tube, added the pre-chilled ammonium acetate methanol solution in proportion, and precipitated the protein overnight at − 20 °C; centrifuged at 12000 g for 20 min at 4 °C; discarded Supernatant, added 90% acetone and vortexed to mix and washed twice. Suspended the pellet with an appropriate volume of lysate (8 M urea + 1% SDS, protease inhibitor has been added) to fully dissolved the sample protein. Centrifuged at 4 °C, 12000 g for 20 min. Collected the supernatant. The quantitative results of BCA were shown in Fig. S2 and Table S1.
Lysis buffer (1% SDS, 8 M urea, 1x Protease Inhibitor Cocktail (Roche Ltd. Basel, Switzerland) was added into the samples. The lysis was performed by sonication on ice for 2 min and kept on ice for 30 min. After centrifugation at 15000 rpm for 15 min at 4 °C, the supernatant was collected and transferred to a new Eppendorf tube.
Protein digestion and iTRAQ labeling
The protein concentration was determined by using the BCA protein assay, and then 100 μg of protein per condition was transferred into a new Eppendorf tube and the final volume was adjusted to 100 μL with 8 M urea. 2 μL of 0.5 M TCEP was added and the sample was incubated at 37 °C for 1 h, and then 4 μL of 1 M iodoacetamide was added to the sample and the incubation was last for 40 min protected from light at room temperature. After that, five volumes of − 20 °C pre-chilled acetone was added to precipitate the proteins overnight at − 20 °C. The precipitates were washed by pre-chilled 90% acetone aqueous solution for twice and then re-dissolved in 100 μL 100 mM TEAB. Sequence grade modified trypsin (Promega, Madison, WI) was added at the ratio of 1:50 (enzyme: protein, weight: weight) to digest the proteins at 37 °C overnight. The peptide mixture was desalted by C18 ZipTip, quantified by Pierce™ Quantitative Colorimetric Peptide Assay (23275) and then lyophilized by SpeedVac.
The resultant peptide mixture was labeled with iTRAQ 8Plex labeling kit (Sciex) following the manufacturer’s instructions. The labeled peptide samples were then pooled and lyophilized in a vacuum concentrator.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
To validate differentially proteins, qRT-PCR was performed in triplicate using the same RNA samples as were used for the iTRAQ construction. An Fungal RNA Kit (Omega Bio-Tek, Norcross, Georgia, USA) was used to extract RNA. The concentration (ng/μL) and quality (A260/A280) of the total RNA were determined using a Nanodrop 2000 spectrophotometer (ThermoScientific, USA), and the integrity of the RNA was tested on an Agilent Technologies 2100 Bioanalyzer. First-strand cDNA was synthesized from 1 μg of total RNA using RevertAid First Strand cDNA Synthesis Kit (TransGEN, Beijing, China). SYBR® Green PCR Master Mix (TransGen) was then used for qRT-PCR. The β-actin was used as an internal control [25] and the relative quantification of specific mRNA levels was performed using the cycle threshold (Ct) 2-ΔΔCt method (SoftwareIQ5 2.0) [26].
High pH reverse phase separation
The peptide mixture was re-dissovled in the buffer A (buffer A: 20 mM ammonium formate in water, pH 10.0, adjusted with ammonium hydroxide), and then fractionated by high pH separation using Ultimate 3000 system (ThermoFisher scientific, MA, USA) connected to a reverse phase column (XBridge C18 column, 4.6 mm × 250 mm, 5 μm, (Waters Corporation, MA, USA). High pH separation was performed using a linear gradient, starting from 5% B to 45% B in 40 min (B: 20 mM ammonium formate in 80% ACN, pH 10.0, adjusted with ammonium hydroxide). The column was re-equilibrated at the initial condition for 15 min. The column flow rate was maintained at 1 mL/min and the column temperature was maintained at 30 °C. Twelve fractions were collected; each fraction was dried in a vacuum concentrator for the next step.
nanoUHPLC-MS/MS analysis
The peptides were re-dissolved in solvent A containing 0.1% formic acid and analyzed by on-line nanospray LC-MS/MS on Q Exactive™ (Thermo Fisher Scientific, MA, USA) coupled to EASY-nano-LC 1200 system (Thermo Fisher Scientific, MA, USA). 2.5 μL peptide sample was loaded, and separated with 90 min-gradient, from 5 to 35% B (B: 0.1% formic acid in ACN). The column flow rate was maintained at 300 nL/min. The electrospray voltage of 2 kV versus the inlet of the mass spectrometer was used.
The mass spectrometer was run under data dependent acquisition mode, and automatically switched between MS and MS/MS mode. The parameters was: (1) MS: scan range (m/z)=300–1800; resolution=70,000; AGC target=3e6; maximum injection time=60 ms; include charge states=2–7; dynamic exclusion=20s; (2) HCD-MS/MS: resolution=17,500; isolation window=2.2; AGC target=5e4; maximum injection time=80 ms; collision energy=30.
Proteomic data analysis
Tandem mass spectra were processed by PEAKS Studio version 8.5 (Bioinformatics Solutions Inc., Waterloo, Canada). PEAKS DB was set up to search the uniprot-proteome_UP000009096 databases (17,877 entries) assuming trypsin as the digestion enzyme. PEAKS DB were searched with a fragment ion mass tolerance of 0.05 Da and a parent ion tolerance of 10 ppm. Carbamidomethylation (C) and Itraq 8plex (K, N-term) were specified as the fixed modification. Oxidation (M), Deamidation (NQ) were specified as the variable modifications. Peptides were filter by 1% FDR and 1 unique. ANOVA was used for peptide and protein abundance calculation. Normalization was performed on averaging the abundance of all peptides. Medians were used for averaging. Different expressed proteins were filtered if their fold change were over 1.2 and contained at least 1 unique peptides with significance over 13 (p< 0.05).
Bioinformatics analysis
The volcano plot, which plots significance versus fold-change on the y and x axes respectively, is a type of scatter-plot that is used to quickly identify changes in large data sets composed of replicate data. It is drawn by using ggplot2 package (http://ggplot2.org). Blast2GO version 5 was used for functional annotation, and GOATOOLS was used for functional enrichment of differential proteins. Blast2GO version 4 was used for functional annotation. Whole protein sequence database was analyzed by BlastP using whole database and mapped, annotated with gene ontology database. Statistically altered functions of different expressed proteins was calculated by Fisher’s exact test in BLAST2GO [27, 28]. KEGG Pathway analysis was processed by KOBAS (http://kobas.cbi.pku.edu.cn/) [29].
Results
Phloridzin provides carbon source for F. moniliforme
Our previous study found that phloridzin promoted the growth and division of F. moniliforme, The mycelial growth and conidia division of F. moniliforme were fast with the help of phloridzin, and division were faster in the 1.0 mM phloridzin than in the 0.5 mM phloridzin (Fig. S1) [20]. But how does phloridzin promote the growth of F. moniliforme? Provide carbon sources? In this study, the effects of phloridzin on F. moniliforme were further determined by plate culture and ITRAQ-based proteomics.
In order to further verify whether phloridzin provides carbon source for F. moniliforme, inorganic salt culture medium (without any carbon source) and phloridzin inorganic salt culture medium (phloridzin is the only carbon source) were set in this study. After 1–7 days of culture (Fig. 1a, b), it was found that F. moniliforme grew faster on the phloridzin inorganic salt medium than on the inorganic salt medium, which indicated that phloridzin provided carbon source for F. moniliforme. The color of F. moniliforme hyphae also changed under the action of phloridzin on PDA medium (Fig. 1c, d), which indicated that phloridzin had various effects on F. moniliforme. In order to further verify the effect of phloridzin on F. moniliforme, the cultured hyphae were sent to Jiachen Hengye (Beijing) Technology Limited company to determine the effect of phloridzin on the proteome of F. moniliforme.

Growth of F. moniliforme on different media for 7 days. a inorganic salt culture medium; b phloridzin+inorganic salt culture medium; c Potato Dextrose Agar medium; d phloridzin+Potato Dextrose Agar medium
Overview of protein identification on ITRAQ technology
The volcano map is a scatter plot with the log2 value of the Fold Change as the horizontal axis and the -log10 value of the P value as the vertical axis. According to the threshold of the significant change, the data is divided into three lines. The red dot is up, the blue dot is down, and the gray dot is unchanged. It can be seen in Fig. 2 that there are significant differentially expressed proteins in the T.vs.CK group, and there are more significant differentially expressed proteins with a more uniform distribution. In this project experiment, a total of 4535 Protein Groups were identified (Card value standard: Peptide Threshold 1.0% FDR, 1 Unique Peptide), and 293 proteins were found with more than 1.2 times (P< 0.05) differences.

Analysis of differentially expressed proteins. a Numbers of up- and down-regulated proteins between the T.vs.CK inoculated at the collection. b Volcano Plot. Each point in the figure represents a protein, the abscissa represents the log2 (fold change) of the difference in the expression of the protein in the two groups, and the ordinate represents the negative logarithm of the P value of the change in the amount of protein expression. The blue dots represent the proteins down-regulate significantly, red dots represent the proteins up-regulate significantly, and gray dots are the proteins with no difference
Table 1 and Table 2 summarize the partial differential proteins by taking the difference fold value change more than 1.5 times as a significant upward adjustment, and using the difference fold value change less than − 1.5 as a significant downward adjustment change standard. It can be seen from Table 2 that there were 9 significantly up-regulated differential proteins, including Thiamine thiazole synthase(W7MF57), Protein NMT1(W7N1I1), and 7 proteins with unknown functional properties in the Uniport database. Among the above differential proteins, Thiamine thiazole synthase(W7MF57) protein closely related to F. moniliforme after phloridzin action.
It can be seen from Table 2 that there were 50 significantly down-regulated differential proteins, including Beta-xylanase (W7MUY6), Serine/threonine protein kinase (W7MT22), Sphingosine N-acyltransferase-like protein FUM18 (FUM18), Carboxylesterase 1 (W7N9Q1), Endo-1 4-beta-xylanase (W7MHE4), Mannan polymerase II complex ANP1 subunit (W7LYG9), Chitinase (W7MNY9, W7NGN0), CPA1 family monovalent cation:H+antiporter (W7N377), Serine/threonine protein kinase (W7M0R4), Murein transglycosylase (W7LU13), 5-oxoprolinase (ATP-hydrolysing, W7LQM7), D-fructose-6-phosphate amidotransferase (W7LZB4), Eburicol 14-alpha-demethylase (W7LY87), DNA primase large subunit (W7M6R7), 40S ribosomal protein S24 (W7MX74), 12 proteins with domain-containing protein and 22 proteins with unknown functional properties in the Uniport database. Among the above differential proteins, Beta-xylanase (W7MUY6), Serine/threonine protein kinase (W7MT22), Sphingosine N-acyltransferase-like protein FUM18 (FUM18), Carboxylesterase 1 (W7N9Q1), Endo-1 4-beta-xylanase (W7MHE4), annan polymerase II complex ANP1 subunit (W7LYG9), Chitinase (W7MNY9, W7NGN0), Murein transglycosylase (W7LU13), 5-oxoprolinase (ATP-hydrolysing, W7LQM7), D-fructose-6-phosphate amidotransferase (W7LZB4), Eburicol 14-alpha-demethylase (W7LY87) protein closely related to F. moniliforme after phloridzin action.
Based on methods of quantitative proteomics iTRAQ analysis, qRT-PCR identified 9 up-regulated proteins and 9 down-regulated proteins. Among the 9 up-regulated proteins, most proteins were basically consistent with the result of qRT-PCR. Although there were some quantitative differences in expression level, the trend of expression level was similar in the data between iTRAQ and qRT-PCR. We found that most of the 9 down-regulated proteins were highly consistent with the expression levels obtained by qRT-PCR. Primers for qRT-PCR and protein reference number are listed (Table S2, Fig. 3).

Comparison of qRT-PCR analysis for eighteen selected proteins with different abundance identified by iTRAQ data. Changes in the transcript (qRT-PCR analysis) and protein abundance (iTRAQ analysis) of T and CK were compared. For iTRAQ data analysis and qRT-PCR data analysis, details can be found in the Materials and methods section. The qRT-PCR data represent the mean ± S.D. (standard deviation) of three biological replicates. All primers and gene abbreviations are listed in Table S2
GO enrichment analysis of differential proteins
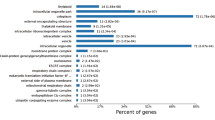
GO functional annotation is mainly divided into three categories: Biological Process (BP), Molecular Function (MF) and Cellular Component (CC). The summarized GO mapping and annotation data of differential proteins at GO level3 and level4 were shown in Fig. 4a, b, respectively. Figure 4 showed that differential proteins were mainly enrich in cell cortex part (GO:0044448), intrracellular organelle (GO:0043229), cytoplasm (GO:0005737), intrracellular organelle part (GO:0044446), organelle envelope (GO:0031967), ribosomal subunit (GO:0044391) of cellular component. In biological process, differential proteins were mainly distributed in carbohydrate derivative metabolic process (GO:1901135), sulfur compound metabolic process(GO:0006790), endoplasmic reticulum membrane organization(GO:0090158), vitamin biosynthetic process(GO:0009110), small molecule biosynthetic process(GO:0044283), organonitrogen compound metabolic process(GO:1901564), nuclear membrane biogenesis(GO:0101025), lipid biosynthetic process(GO:0008610), organic hydroxy compound biosynthetic process(GO:1901617) and water-soluble vitamin biosynthetic process(GO:0042364). While under the category of molecular function, differential proteins were mostly related to transferase activity, transferring phosphorus-containing groups(GO:0016772), heme binding(GO:0020037), purine nucleoside binding(GO:0001883), tetrapyrrole binding(GO:0046906), intramolecular transferase activity(GO:0016866), nucleosomal histone binding(GO:0031493), transferase activity, transferring acyl groups(GO:0016746) and RNA binding(GO:0003723).

Gene ontology enrichment analysis of differentially expressed proteins. a The proportion of protein number enriched in the first 20 items with the lowest P-value among the three classification enrichment results of Biological process, Molecular function and Cellular component at Level 3; b The proportion of protein number enriched in the first 20 items with the lowest P-value among the three classification enrichment results of Biological process, Molecular function and Cellular component at Level 4
KEGG pathway enrichment analysis of differential proteins
To identify the biological pathway information of F. moniliforme in response to Phloridzin inducers, these 293 proteins were further mapped to the corresponding pathways included in the KEGG database. These 293 proteins could be mapped to 64 pathways. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium (Table 3) when the p-value was set at 0.05. Ribosome and Biosynthesis of antibiotics were the first and second significantly enriched pathways, respectively. Other representative pathways included Amino sugar and nucleotide sugar metabolism, Glycolysis / Gluconeogenesis and Phagosome were also significantly enriched pathways.
As shown in Fig. 5, the expression levels of glycolysis / gluconeogenesis-related proteins in F. moniliforme were 2 down-regulated and 4 up-regulated. Moreover, it was found that the differentially expressed proteins W7N5X1, W7MA30, W7M3M6, and W7MPI9, which collectively show significantly up-regulation, participate in 19 metabolic pathways in total, and the differentially expressed differentially-formed proteins W7LNK6, W7M651, participate in 13 metabolic pathways.

KEGG pathway enriched by differentially expressed proteins. Blue box nodes indicate down regulation and red box nodes indicate up regulation
Ribosome proteins can affect and regulate biological processes such as gene transcription, translation, cell proliferation and apoptosis. In this omics experiment, we used the Kanehisa database to analyze the differential proteins detected by the ITRAQ quantitative proteomics method. A total of 32 ribosome-associated proteins were found to have significant differential expression. These ribosomal proteins are structurally distributed in large ribosomal subunits and small ribosomal subunits (Fig. 6). Among them, the expression levels of ribosomal-related proteins in F. moniliforme are all down-regulated, showing a huge change in expression levels.

KEGG ribosome pathway enriched by differentially expressed proteins. Blue box nodes indicate differentially expressed proteins
Discussion
Primary fungal and oomycete genera reported as containing species that were pathogenic toward apple trees, include the fungal genera Cylindrocarpon, Fusarium and Rhizoctonia, and the oomycete genera Phytophthora and Pythium [3, 30]. Fusarium sp. was a significant plant pathogen, which had the diversity of affected hosts, numbers of pathogen taxa, and types of habitats in plant pathology [31]. Fusarium isolates, such as F. tricinctum (Corda) Sacc, F. graminearum, F. solani (Mart.) Sacc. and F. avenaceum (Fr.) Sacc. have been shown to be pathogenic [3, 6, 32,33,34,35,36,37,38,39]. And F. moniliforme have also been shown to be pathogenic on forest nurseries in Spain [15], Sorghum in India, Maize in Iowa State [16], Scots pine seedlings in Palencia [17], Peach in California [18] and apple in China [19, 21]. After Fusarium contacts the host plant, it initiates physiological and biochemical reactions inside the strain through processes such as surface molecular interaction and signal transmission, and produces metabolites that act on the plant body to cause pathogenic effects. It mainly includes cell wall degrading enzymes, toxins, growth regulators and their analogues.
Thiamine has a relatively important role in the body, and it has an effect on the growth of bacteria and amino acid metabolism. Thiamine is an important enzyme co-factor in all organisms. It has four forms in the body: thiamine triphosphate (TTP), bisamine (pyrophosphate) thiamine (TPP / TDP), single Thiamine Phosphate (TMP), Free Thiamine (FT). TPP plays a key role in the metabolism of amino acids and carbohydrates in the body (glycolytic pathway, tricarboxylic acid cycle, pentose phosphate cycle, etc.). Thiazole synthase is a rate-limiting enzyme for thiamine synthesis, and is involved in the synthesis of thiamine, and its function is related to the growth of bacteria and spore formation. In this test, a large amount of thiamine biosynthetic enzyme expression in F. moniliforme under phloridzin environment is beneficial to the synthesis of thiamine in F. moniliforme, and then promotes the growth of F. moniliforme and spore formation. It is consistent with the previous research results of this research group [20]. Phloridzin was utilized directly as a sugar alternative and a toxin accelerator by Valsa mali [40]. Legumes can co-exist with rhizobium to form a nodule, and the cell group containing the rhizobium in the nodule forms a bacterial-containing tissue. Recent studies have reported that exogenous thiamine can increase the diameter of root nodules, which indicates that leguminous plants and rhizobium treated with exogenous thiamine have more active division and differentiation of nodule-forming cells [41].
To identify the biological pathway information of F. moniliforme in response to Phloridzin inducers, these 293 proteins were further mapped to the corresponding pathways included in the KEGG database. These 293 proteins could be mapped to 64 pathways. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium when the p-value was set at 0.05. Ribosome and Biosynthesis of antibiotics were the first and second significantly enriched pathways, respectively. Other representative pathways included Amino sugar and nucleotide sugar metabolism, Glycolysis / Gluconeogenesis and Phagosome were also significantly enriched pathways. Phosphoenolpyruvate carboxykinase is widely present in animals, plants, microorganisms, and cells. It catalyzes the conversion of oxaloacetate to phosphoenolpyruvate and is a key enzyme that regulates the gluconeogenesis pathway. Gluconate 5-dehydrogenase is a coenzyme-dependent reductase that belongs to the short-chain dehydrogenase SDR family. It can catalyze D-gluconic acid and 5-keto-D-gluconic acid (5 -KGA) reversible redox conversion, so as to regulate the conversion of carbon source and reducing power in the body to maintain metabolic balance in the body, has an important role in glucose metabolism [42, 43]. According to GO annotation classification, most proteins are related to carbon metabolism. In biological process, differential proteins were mainly distributed in carbohydrate derivative metabolic process (GO:1901135), sulfur compound metabolic process(GO:0006790), vitamin biosynthetic process(GO:0009110), organonitrogen compound metabolic process(GO:1901564). While under the category of molecular function, differential proteins were mostly related to transferase activity, transferring phosphorus-containing groups(GO:0016772), heme binding(GO:0020037), transferase activity, transferring acyl groups(GO:0016746). In this study, the large expression of phosphoenolpyruvate carboxykinase and 5-gluconate dehydrogenase in F. moniliforme under the phloridzin environment is helpful to promote the gluconeogenesis pathway in F. moniliforme Amino acid metabolism and carbohydrate metabolism. In addition, 3-methyl-2-oxobutanoate hydroxymethyltransferase, phosphopantothenoylcysteine decarboxylase, acyl-CoA dehydrogenase, triosephosphate isomerase, related to beta-1,4-mannosyltransferase, related to gamma-glutamyltransferase, alcohol dehydrogenase 1, L-lactate dehydrogenase also up-regulated, L-lactate dehydrogenase is a key enzyme that produces lactic and phenyllactate [44].
β-1,4-xylanase is a key enzyme in the process of xylan degradation. Caleronieto et al. [45] inactivated xylR gene and xylR gene in Fusarium oxysporum tomato specialization, and found that the knockout of xlnR can make the expression of XYL3 and XYL genes Decreased, xylanase activity was correspondingly reduced, but the virulence was not weakened. Analysis may be because the knockout of xlnR did not completely eliminate the xylanase activity, and the remaining enzyme activity was still sufficient to produce a △ xlnR mutant strain. Virulence. Garcia-Maceira et al. [46] used an inactive allele to replace the target gene and found that the pathogenicity of the XYL3 mutant variant of the xylanase gene is similar to that of the wild-type strain, indicating that the XYL3 gene has Pathogenicity does not play a key role. Therefore, xylanase and cellulase may not play a key role in the pathogenicity of pathogenic bacteria. In this test, the expression of endo-1,4-beta-xylanase C and endo-1,4-beta-xylanase in F. moniliforme under phloridzin environment was down-regulated, which may be caused by xylanase degradation the accumulation of xylose in the culture medium, the accumulation of xylose inhibited the secretion of xylanase. Many regulatory genes in F. moniliforme infection may exist in multiple metabolic pathways at the same time, function in multiple biological processes, and constitute a complex biological system involving pathogens and hosts, proteins within pathogens, and proteins and Other complex inter-molecular networks of interactions [47]. Therefore, protein-based interaction networks based on pathogens will help to further understand the mechanism of pathogen infection.
Conclusions
This study is the first to perform quantitative proteomic investigation by iTRAQ labeling and LC-MS/MS to identify differentially expressed proteins in F. moniliforme under phloridzin conditions. The results confirmed that F. moniliforme presented a unique protein profile that indicated the adaptive mechanisms of this species to phloridzin environments. Differentially expressed proteins were influenced a variety of cellular processes, particularly metabolic processes. Among these metabolic pathways, a total of 8 significantly enriched KEGG pathways were identified with at least 2 affiliated proteins with different abundance in conidia and mycelium. Functional pathway analysis indicated that up-regulated proteins were mainly distributed in amino sugar, nucleotide sugar metabolism, glycolysis/ gluconeogenesis and phagosome pathways. The results deepened our understanding of the proteome in F. moniliforme in response to phloridzin inducers and provide a basis for further exploration for improving the efficiency of the fungi as biocontrol agents to control ARD.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
References
Mazzola M, Manici LM. Apple Replant Disease: role of microbial ecology in cause and control. Annu Rev Phytopathol. 2012;50:45–65.
Laurent AS, Merwin IA, Thies JE. Long-term orchard groundcover management systems affect soil microbial communities and apple replant disease severity. Plant Soil. 2008;304:209–25.
Manici L, Ciavatta C, Kelderer M, Erschbaumer G. Replant problems in South Tyrol: role of fungal pathogens and microbial population in conventional and organic apple orchards. Plant Soil. 2003;256:315–24.
Bai R, Ma FW, Liang D, Zhao X. Phthalic acid induces oxidative stress and alters the activity of some antioxidant enzymes in roots of Malus prunifolia. J Chem Ecol. 2009;35:488–94.
Tewoldemedhin YT, Mazzola M, Labuschagne I, McLeod A. A multi-phasic approach reveals that apple replant disease is caused by multiple biological agents, with some agents acting synergistically. Soil Biol Biochem. 2011;43:1917–27.
Tewoldemedhin YT, Mazzola M, Botha WJ, Spies CFJ, McLeod A. Characterization of fungi (Fusarium and Rhizoctonia) and oomycetes (Phytophthora and Pythium) associated with apple orchards in South Africa. Eur J Plant Pathol. 2011;130:215–29.
Schoor LV, Denman S, Cook NC. Characterisation of apple replant disease under South African conditions and potential biological management strategies. Sci Hortic. 2009;119:153–62.
Franke-Whittle IH, Manici LM, Insam H, Stres B. Rhizosphere bacteria and fungiassociated with plant growth in soils of three replanted apple orchards. Plant Soil. 2015;395:317–33.
Tewoldemedhin YT, Mazzola M, Mostert L, McLeod A. Cylindrocarpon species associated with apple tree roots in South Africa and their quantification using real-time PCR. Eur J Plant Pathol. 2011;129:637–51.
Gao XB, Zhao FX, Shen X, Hu YL, Hao YH, Yang SQ, et al. Effects of cinnamon acid on respiratory rate and its related enzymes activity in roots of seedlings of Malus hupehensis Rehd. Agri Sci China. 2010;9:833–9.
Hofmann A, Wittenmayer L, Arnold G, Schieber A, Merbach W. Root exudation of phloridzin by apple seedlings (Malus x domestica Borkh.) with symptoms of apple replant disease. J Appl Bot Food Qual. 2009;82:193–8.
Zhang JH, Mao ZQ, Wang LQ, Shu HR. Bioassay and identification of root exudates of three fruit tree species. J Integr Plant Biol. 2007;49:257–61.
Manici L, Kelderer M, Franke-Whittle I, Rühmer T, Baab G, Nicoletti F, Caputo F, Topp A, Insam H, Naef A. Relationship between root-endophytic microbial communities and replant disease in specialized apple growing areas in Europe. App Soil Ecol. 2013;72:207–14.
Kelderer M, Manici LM, Caputo F, Thalheimer M. Planting in the‘inter-row’to overcome replant disease in apple orchards: a study on the effectiveness of the practice based on microbial indicators. Plant Soil. 2012;357:381–93.
Pinto PM, Alonso JAP, Fernandez VP, Casero JJD. Fungi isolated from diseased nursery seedlings in Spain. New Forest. 2006;31:41–56.
Da Silva MP. Interactions between lesion nematodes and corn pathogens. Ames: Iowa State University;; 2010.
Yang JI, Ruegger PM, McKenry MV, Becker JO, Borneman J. Correlations between root-associated microorganisms and peach replant disease symptoms in a California soil. Plos One. 2012;7:e46420.
Machón P, Pajares JA, Diez JJ, Alves-Santos FM. Influence of the ectomycorrhizal fungus Laccaria laccata on pre-emergence, post-emergence and late damping-off by Fusarium oxysporum and F. verticillioides on Stone pine seedlings. Symbiosis. 2006;42:153–60.
Ju RC, Zhao YH, Li JY, Jiang HX, Liu P, Yang T, et al. Identification and evaluation of a potential biocontrol agent, Bacillus subtilis, against Fusarium sp. in apple seedlings. Ann. Microbiol. 2014;64:377–83.
Yin CM, Xiang L, Wang GS, Wang YF, Shen X, Chen XS, Mao ZQ. Phloridzin promotes the growth of Fusarium moniliforme (Fusarium verticillioides). Sci Hortic. 2017;214:187–94.
Wang GS, Yin CM, Pan FB, Wang XB, Xing L, Wang YF, Wang JZ, Tian CP, Chen J, Mao ZQ. Analysis of the Fungal Community in Apple Replanted Soil Around Bohai Gulf. Horticult Plant J. 2018;4(05):175–81.
Gosch C, Halbwirth H, Stich K. Phloridzin: biosynthesis, distribution and physiological relevance in plants. Phytochemistry. 2010;71:838–43.
Petkovsek MM, Slatnar A, Stampar F, Veberic R. Phenolic compounds in apple leaves after infection with apple scab. Biol Plant. 2011;55:725–30.
Petkovsek MM, Stampar F, Veberic R. Parameters of inner quality of the apple scab resistant and susceptible apple cultivars ( Malus domestica Borkh.). Sci Hortic. 2007;114(1):37–44.
Lanubile A, Bernardi J, Battilani P, Logrieco A, Marocco A. Resistant and susceptible maize genotypes activate different transcriptional responses against Fusarium verticillioides. Physiol Mol Plant. 2012;77(1):52–9.
Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔ CT Method. Methods. 2001;25:402–8.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Klopfenstein DV, Liangsheng Z, Pedersen BS, Ramírez F, Vesztrocy AW, Naldi A, et al. Goatools: a python library for gene ontology analyses. Sci Rep. 2018;8(1):10872.
Chen X, Mao XZ, Huang JJ, Dang Y, Wu JM, Dong S, Kong L, Gao G, Li CY, Wei LP. Kobas 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucl Acids Res. 2011;39:316–22.
Mazzola M. Elucidation of the microbial complex having a causal role in the development of apple replant disease in Washington. Phytopathology. 1998;88:930–8.
Wang X, Zhu X, Tooley P, Zhang X. Cloning and functional analysis of three genes encoding polygalacturonase-inhibiting proteins fromcapsicum annuumand transgenic capgip1 in tobacco in relation to increased resistance to two fungal pathogens. Plant Mol Biol. 2013;81(4–5):379–400.
Celar F. Cucurbit diseases. Sodobno Kmetijstvo. 2000;33:162–5.
Ding M, Zhu Q, Liang Y, Li J, Fan X, Yu X, He F, Xu H, Liang Y, Yu JF. Differential roles of three FgPLD genes in regulating development and pathogenicity in Fusarium graminearum. Fungal Genet Biol. 2017;109:46–52.
Duan X, Bi HG, Li T, Wu GX, Li QM, Ai XZ. Root characteristics of grafted peppers and their resistance to Fusarium solani. Biol Plantarum. 2017;61(3):579–86.
Hibar K, Daami-Remadi M, Hamada W, EI-Mahjoub M. Bio-fungicides as an alternative for tomato Fusarium crown and root rot control. Tunis J Plant Prot. 2006;1:19–29.
Ramamoorthy V, Raguchander T, Samiyappan R. Enhancing resistance of tomato and hot pepper to Pythium diseases by seed treatment with fluorescent pseudomonads. Eur J Plant Pathol. 2002;108:429–41.
Steinkellner S, Mammerler R, Vierheilig H. Germination of Fusarium oxysporum in root exudates from tomato plants challenged with different Fusarium oxysporum strains. Eur J Plant Pathol. 2008;122:395–401.
Yan K, Han G, Bian T. Fusarium solani Infection Depressed Photosystem Performance by Inducing Foliage Wilting in Apple Seedlings. Front Plant Sci. 2018;9:497.
Zhu Q, Sun L, Lian J, Gao X, Zhao L, Ding M, Li J, Liang Y. The phospholipase c (fgplc1) is involved in regulation of development, pathogenicity, and stress responses in Fusarium graminearum. Fungal Genet Biol. 2016;97:1–9.
Zhou K, Hu LY, Li YTS, Chen XF, Zhang ZJ, Liu BB, Li PM, Gong XQ, Ma FW. Md UGT88F1-mediated phloridzin biosynthesis regulating apple development and Valsa canker resistance. Plant Physiol. 2019;180:2290–305.
Nagae M, Parniske M, Kawaguchi M, Takeda N. The thiamine biosynthesis gene THI1 promotes nodule growth and seed maturation. Plant Physiol. 2016;172(3):2033–43.
Hanke T, Noh K, Noack S, Polen T, Bringer S, Sahm H, et al. Combined fluxomics and transcriptomics analysis of glucose catabolism via a partially cyclic pentose phosphate pathway in gluconobacter oxydans 621h. Appl Environ Microb. 2013;79(7):2336–48.
Silberbach M, Maier B, Zimmermann M, Büchs J. Glucose oxidation bygluconobacter oxydans: characterization in shaking-flasks, scale-up and optimization of the ph profile. Appl Microbiol Biot. 2003;62(1):92–8.
Hou W, Mu J, Li A, Wang H, Kong L. Identification of a wheat polygalacturonase-inhibiting protein involved in Fusarium head blight resistance. Eur J Plant Pathol. 2015;141(4):731–45.
Caleronieto F, Pietro AD, Roncero MIG, Hera C. Role of the transcriptional activator xlnr of fusarium oxysporum in regulation of xylanase genes and virulence. Mol Plant Microbe Interact. 2007;20(8):977–85.
Garcia-Maceira FI, Di Pietro A, Huertas-Gonzalez MD, Ruiz-Roldan MC, Roncero MIG. Molecular characterization of an endopolygalacturonase from Fusarium oxysporum expressed during early stages of infection. Appl Environ Microbiol. 2001;67(5):2191–6.
Göhre V, Robatzek S. Breaking the barriers: microbial effector molecules subvert plant immunity. Annu Rev Phytopathol. 2008;46(1):189–215.
Acknowledgements
Not applicable.
Funding
The research was supported by the National Natural Science Foundation of China (32072510, 31501720, 31672104), earmarked fund for National Modern Agro-industry Technology Research System of China (CARS-27), Qingchuang Science and Technology Support Project of Shandong Colleges and Universities(2019KJF020); Shandong Agricultural Major Applied Technology Innovation Project (SD2019ZZ008); Taishan Scholar Funded Project (NO.ts20190923); the Taishan Scholar Assistance Program from Shandong Provincial Government; the National Key Research and Development Program of China (2016YFD0201114), Fruit innovation team in Shandong Province, China (SDAIT-06-07).
Author information
Authors and Affiliations
Contributions
CMY and ZQM designed the experiments, RZ, WTJ and XL wrote and improved the manuscript, YFW and YND executed the experiments, XS and YMJ and XSC helped in polishing the language. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in strict accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals of the State Council of the People’s Republic of China. The protocol was approved by the Committee on Experimental Animal Management of Shandong Agricultural University, China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. F. moniliforme labeled with the SiC quantum dots in different concentrations of the phloridzin and observed with fluorescence microscopy (200 ×) on the 40 th day. (a) No phloridzin, T1. (b) 0.5 mM phloridzin, T2. (c) 1.0 mM phloridzin, T3. Fig. S2. Protein standard curve. Dilute protein standards with ultrapure water into samples of different concentrations (0.0 mg·mL-1, 0.1 mg·mL-1, 0.2 mg·mL-1, 0.3 mg·mL-1, 0.4 mg·mL-1, 0.5 mg·mL-1, 0.6 mg·mL-1). According to the BCA kit instructions, draw the standard curve with protein content as the abscissa and absorbance as the ordinate. The standard equation was y=1.1872x-0.1164, and the correlation coefficient R2=0.9989. Table S1. Sample protein quantification results. Table S2. Primer sequencesused for the qRT-PCR validation of selected differentially expressed proteins and β-actin.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, R., Jiang, W., Liu, X. et al. ITRAQ-based quantitative proteomic analysis of Fusarium moniliforme (Fusarium verticillioides) in response to Phloridzin inducers. Proteome Sci 19, 2 (2021). https://doi.org/10.1186/s12953-021-00170-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12953-021-00170-2