
Abstract
Peripheral T-cell lymphoma (PTCL) is a rare and heterogeneous group of hematological malignancies. Compared to our knowledge of B-cell tumors, our understanding of T-cell leukemia and lymphoma remains less advanced, and a significant number of patients are diagnosed with advanced stages of the disease. Unfortunately, the development of drug resistance in tumors leads to relapsed or refractory peripheral T-Cell Lymphomas (r/r PTCL), resulting in highly unsatisfactory treatment outcomes for these patients. This review provides an overview of potential mechanisms contributing to PTCL treatment resistance, encompassing aspects such as tumor heterogeneity, tumor microenvironment, and abnormal signaling pathways in PTCL development. The existing drugs aimed at overcoming PTCL resistance and their potential resistance mechanisms are also discussed. Furthermore, a summary of ongoing clinical trials related to PTCL is presented, with the aim of aiding clinicians in making informed treatment decisions.
Similar content being viewed by others

Introduction
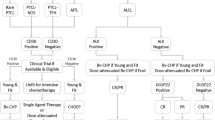
Peripheral T-cell lymphoma (PTCL) is a rare but heterogeneous group of hematological malignancies. This group of mature T-cell non-Hodgkin's lymphomas (NHL) is an aggressive disease associated with poor prognosis. PTCL accounts for 5–10% of all non-Hodgkin's lymphomas [1]. Our understandings on T-cell leukemia and lymphoma lag behind that of B-cell tumors, and a large proportion of patients have advanced disease at diagnosis. The history of our understanding and choices of PTCL is summarized in Fig. 1. The World Health Organization (WHO) categorizes PTCL into approximately 30 different types. Broadly speaking, nodal, extranodal, and leukemic PTCL typically manifest as aggressive diseases, with a five-year survival rate of about 30%. On the other hand, cutaneous PTCL tends to present as a more slow-growing disease [2]. The WHO classification of haematolymphoid tumors (WHO-HAEM5) presents new molecular and histopathological findings that facilitate the diagnostic classification of this type of tumor [3].

The incorporation of CD20-based combination chemotherapy has improved the long-term survival rate of diffuse large B-cell lymphoma, now reaching an impressive range of 60- 70%. However, regimens that have been successfully used in many aggressive B-cell lymphomas (i.e., anthracycline-based multiagent regimens, including CHOP) are not as effective in T-cell lymphomas [6, 7]. Approximately 70% of patients with PTCL develop relapsed or refractory disease after the first-line therapy. Attempts to improve outcomes by "adding" or "substituting" anthracyclines have consistently failed, and most patients with T-cell lymphoma will die from lymphoma- or treatment-related complications within a few years after diagnosis [8]. Undoubtedly, addressing most T-cell lymphomas has proven to be a difficult and unmet medical challenge. This article thoroughly examines the significant impact of tumor heterogeneity and the tumor microenvironment (TME) on treatment resistance in PTCL. Additionally, it presents an up-to-date analysis of the molecular mechanisms contributing to treatment resistance in PTCL. Finally, potential strategies to both prevent and overcome treatment resistance in PTCL are discussed.
Sources of treatment resistance in PTCL
The occurrence of relapsed or refractory disease in PTCL is highly prevalent due to the emergence of drug resistance during treatment. Despite the availability of various treatment options, none have been universally curative, and eventually, drug resistance may develop in response to available treatments [9]. Although these two phenomena can coexist, intrinsic treatment resistance is often distinguished from acquired resistance. Specifically, intrinsic resistance arises from the disease itself and easily leads to treatment refractoriness, whereas acquired resistance arises from the acquisition of resistance-mediated features through mutation or non-mutation during treatment and often leads to disease relapse [10, 11]. In PTCL, treatment resistance often arises from a cancer ecosystem composed of multiple sources, including the heterogeneous cancer cells themselves and their surrounding tumor microenvironment (TME).
Heterogeneity of tumors
PTCL represents a remarkably heterogeneous group of diseases, characterized by the absence of distinct molecular markers and morphological features [12]. T-cell lymphomas consist of a variety of rare diseases that can be classified as indolent or aggressive and account for 12% of all NHL. In 2016, the World Health Organization (WHO) released a revised classification of T cell/NK cell lymphoma, categorizing it into two main groups: precursor T cell tumors and mature T cell tumors. The mature subgroup was further sub-divided into leukemic, intranodal, extranodal, and cutaneous types. Additionally, lymphoma can be further classified into indolent (slow-growing) disease and aggressive forms [2]. Indolent lymphomas are characterized by a long disease course and are usually resistant to standard chemotherapy, whereas aggressive lymphomas usually have an acute presentation with B symptoms(such as weight loss, night sweats, and fever) and rapid progression. Cutaneous T-cell lymphoma (CTCL) is considered indolent, whereas peripheral mature lymphoma (PTCL), including other types, is considered aggressive. PTCL-nos (not otherwise specified) is the most common PTCL, followed by anaplastic large cell lymphoma (ALCL) and angioimmunoblastic T-cell lymphoma (AITL) [13, 14] (Fig. 2). The 2022 update of the WHO classification of lymphoid neoplasms of blood (WHO-HAEM5) includes new insights into pathogenesis and molecular genetics as well as new concepts underlying the classification [3].

Incidence of different Peripheral T-cell lymphoma subtypes according to IPTCLP [7]
T lymphoblastic leukemia/lymphoma (T-ALL/LBL) can present as lymphoma, often accompanied by rapid enlargement of mediastinal lymph nodes and pleural effusion, and can also be accompanied by leukemia of blood and/or bone marrow [15, 16]. In T-ALL /LBL, the most common genetic alteration in 50 to 70% of patients is NOTCH1 activating mutations. NOTCH1 signaling is required to induce maturation of immature lymphoid precursor cells into T cells, and through NOTCH1 activation, MYC is activated [17, 18]. In addition to NOTCH1 activation, TAL1 activation is another common driver of tumorigenesis, occurring in 25% to 35% of T-ALL/LBL cases [19]. CDKN2A/B gene deletion occurs in up to 70% of T-ALL/LBL patients [17]. NUP214 fusion, PTEN and WT1 deletion or mutation, PHF6 deletion and other genetic changes can occur in some T-ALL/LBL patients [20,21,22]. In contrast, early T-cell precursor lymphoblastic leukemia (ETP-ALL) harbors mutations commonly found in myeloid tumors, including alterations in FLT3, DNMT3A, NRAS, KRAS, IDH1, or IDH2, all of which are genetically rare in classical T-ALL/LBL [23].
Although all or even most cases of systemic EBV + T-cell lymphoma (sEBV + TNHL) do not have specific chromosomal abnormalities, there are some common features: cases with alterations usually have complex karyotypes (3 or more chromosomal alterations), and nearly 40% of cases in the literature have add(9)(p24) [24]. In addition, variants in chromosomes 1, 7, 11, 17, 20, 21, and X are present in more than 20% of reported cases, whereas DDX3X, BCOR/BCORL2, and TET2 are present in 20% of chronic active EBV disease [25, 26]. Chromosomal alterations associated with CAEBV in HV-LPD patients include 6q deletion or 6p gain. These molecular and chromosomal abnormalities are also seen in extranodal nasal-type NK/ T-cell lymphoma. A study of primary EBV-positive T/ NK-cell lymphoma showed that approximately 20% of the cases had recurrent copy-number aberrations, including deletions of chr14q11.2(100%), chr3q26.1(67%), and chr22q11.23(33%) [27, 28].
Extra-nodal NK/T-cell lymphoma (ENKTL) can be divided into three different subtypes with different molecular characteristics and treatment outcomes (3-year OS rates were 79.1%, 91.7% and 38.5%, respectively) [29]: TSIM (tumor-suppressor/immune-modulator) subtype is associated with JAK/ STAT pathway activation, NK-cell origin, TP53 mutations, genomic instability (including 6q21 deletion and 9p24.1 and/or 17q21.2 amplifications), and PD-L1/2 overexpression [30]; HEA (HDAC9-EP300-ARID1A) subtype is associated with epigenetic changes through HDAC9, EP300 and ARID1A mutations, NF-KB activation, T-cell origin, and T-cell receptor signaling activation; Finally, MB (MGA-BRDT)subtype is associated with MYC overexpression and poor prognosis [29, 31].
ATXN1 or CIC alterations are present in 53% of adult T-cell leukemia/lymphoma (ATLL) cases, and CCR4 (C–C chemokine receptor 4) mutations are common, and the majority of ATLL patients exhibit CCR4 overexpression, which is associated with skin involvement and worse prognosis [32]. 9p24 amplification with PDL1 amplification occurs in 10 to 20% of cases, and TP53 mutations occur in 16% of cases, both of which are associated with a poor prognosis [33].
The most common translocation in anaplastic large cell lymphoma(ALCL) is t(2; 5) (p23; q35), NPM1::ALK is found in approximately 85% of ALK + ALCL [12]. The prognosis of ALK + ALCL is better than that of ALK-ALCL, and the 5-year overall survival rate is 70–90% [34]. Alk-negative ALCL cases have considerable genetic heterogeneity, and DUSP22 gene rearrangement is found in approximately 30% of systemic ALK-negative ALCL patients [35]. Certain studies have proposed that dusp22 rearrangement in ALK-negative ALCL demonstrates a favorable outcome, similar to that of ALK-positive disease. However, other studies have indicated a more aggressive disease course compared to ALK-positive disease [35, 36]. TP63 rearrangement occurs in about 8% of Alk-negative ALCL cases and is associated with poor prognosis, with a 5-year overall survival rate of only 17%, which is lower than that of PTCL-NOS [37]. PTCL-NOS is a T-cell lymphoma that does not meet any of the specific subtype criteria. This tumor is also the most common type of T-cell lymphoma, accounting for 25–30% of all cases [38]. The prognosis of PTCL-NOS relies on its genetic heterogeneity, leading to its classification based on gene expression profiles: one with high expression of GATA3 (PTCLGATA3) and the other with high expression of TBX21 (PTCL-tbx1) [39]. PTCL-GATA3 is linked to PI3K/mTOR pathway activation, exhibiting a more aberrant genome, and thus, associated with a poor prognosis, with a 5-year overall survival rate of 19%. In contrast, PTCL-TBX21 is characterized by NFKB pathway activation, fewer genomic abnormalities, and a more favorable prognosis, with a 5-year overall survival rate of 38% [40]. PTCL-NOS cases with TP53/CDKN2A alterations show considerable chromosomal instability and a poor overall survival rate, exhibiting an inverse correlation with TFH marker expression. The majority of PTCL-NOS cases have a homozygous deletion of CDKN2A, which appears to be linked to an unfavorable prognosis [25].
In the latest WHO classification, Tfh (T follicular helper) PTCL encompasses AITL with Tfh phenotype, follicular PTCL, and nodal PTCL, which are tumors with similar clinical presentation, gene expression, DNA copy number, abnormalities, and mutational profiles [2]. 3, 5, 21, and X chromosome gain/trisomy are the most common cytogenetic abnormalities in AITL. Other visible chromosomal alterations include increases in 11q13, 19, or 22q and loss of 13q10. Mutations in genes observed in AITL overlap for the most part with those observed in medullary tumors [22].TET2 mutations are seen in 47% ~ 83% of AITL cases, and DNMT3A alterations are seen in 26% ~ 38% of cases. These genes are involved in epigenetic regulation, and their mutations lead to 5-hydroxymethylcytosine loss through a common mechanism in PTCL. There are also IDH2 mutations seen in 20–45% of AITL, and RHOA p.G17V mutations are seen in 70% of AITL [41,42,43]. The fact that RHOA and IDH2 mutations are confined to tumor cells suggests that they may be the second strike in a multistep oncogenic process [44]. These mutations are also present in Tfh-type PTCL, with the exception of the IDH2R172 mutation, which is strongly associated with AITL labeling and correlates with specific pathological manifestations, and the SYK fusion, which is more frequently detected in follicular PTCL [45, 46].
Tumor microenvironment
Lymphoma occurrence is not solely driven by tumor-autonomous processes; rather, it requires the intricate interaction of the tumor microenvironment (TME). The TME encompasses tumor cells, immune cells, stromal cells, blood vessels, and the extracellular matrix surrounding the tumor. Interactions between stroma cells and tumor cells within the TME, along with the secretion of soluble factors, have been identified as contributors to treatment resistance in various cancer types [47, 48]. As PTCL is relatively rare, our understanding of the PTCL TME is still in its early stages, with most knowledge derived from B-cell NHL experiences.
Programmed cell death ligand I (PD-L1) expression was more pronounced in the TME of PTCL, seen in 73% of CTCL cases and 39% of other PTCL cases. PD-L1 is also highly expressed in malignant cells such as nasal NK/ T cell lymphoma (NKTCL) and extranasal NKTCL. Almost all EBV-associated lymphomas are associated with high PD-L1 expression [27]. Serum PD-L1 level is associated with the prognosis of ENKTCL. For example, Nagato and colleagues reported that elevated levels of PD-L1 in tumor cells is correlated with elevated levels of PD-L1 in serum and worse OS, which is associated with immune escape [47]. As previously described, GATA3 expression in PTCL-NOS is associated with a poor prognosis [39] and is characterized by type 2 helper t cell (Th2) -related cytokines, including interleukin (IL)-4, IL-5, IL-10, and IL-13 [48], which promote macrophage polarization to alternative M2-type macrophages [49]. M2 macrophages promote angiogenesis by secreting pro-angiogenic cytokines such as VEGF. In addition, they secrete IL-10 and transforming growth factors, which up-regulate the expression of PD-L1 on macrophages in an autocrine manner. Binding of PD-L1 to PD-1 expressed on T cells results in suppression of T cell function and, consequently, immunosuppression [50, 51]. Clinic pathological correlation studies have provided evidence for the prognostic significance of tumor-associated macrophages in T-cell lymphoma [52]. Among 64 T-cell lymphomas, high tumor-associated macrophage content was associated with poor prognosis in multivariate analysis (high macrophage content vs low macrophage content, overall survival, OS: 28.1% vs 44.3%) [53]. The degree of macrophage infiltration is also inversely correlated with survival, with multiple studies showing that a high content of CD163-positive macrophages is associated with low survival [54, 55].
The infiltration of other non-neoplastic T cells in T-cell lymphomas may also regulate TME. Regulatory T cells (Tregs) are a subset of CD4-positive T cells that inhibit immune responses and maintain immune tolerance. Tregs are characterized by high levels of CD25 and forkhead transcription factor FOXP3 [56, 57]. Tregs in the lymphoma microenvironment may suppress immune-mediated antitumor responses, thereby enhancing tumor cell survival. However, Tregs may also down-regulate the inflammatory response in the microenvironment and promote tumor progression, thereby inhibiting tumor cell proliferation. The opposite regulatory effects of Tregs on tumor cells may explain the apparently contradictory prognostic effects of Tregs on different types of T-cell lymphomas. The number of Tregs remained an independent prognostic biomarker in multivariate analysis, and a large number of Tregs infiltration was associated with higher survival [58, 59].
Multiple drug resistance
Drug resistance is a common and difficult obstacle in the treatment of mature T/ NK-cell lymphoma, and multidrug resistance (MDR) phenotype is considered to be one of its mechanisms [60]. MDR refers to the acquired cross-resistance to a variety of structurally and functionally unrelated drugs. MDR is often associated with increased expression of drug efflux transporters of the ATP-binding cassette (ABC) protein family [61, 62]. The ABC transporter superfamily contains 48 ABC transporters, which are divided into seven subfamilies based on sequence homology and protein organization. Accumulating evidence suggests that ABC transporters play a key role in the physiological transport and export of drugs and toxic substances, which can export a variety of chemotherapeutic agents outside of cells [63]. T/ NK-cell lymphoma cases showed a high frequency of MDR protein expression [64]. In earlier reports, poor response to chemotherapy in ATL patients was partly due to high expression of P-gp or MDR1. In one study, the expression of P-glycoprotein (P-gp), multidrug resistance-related protein 1 (MRP-1), breast cancer resistance protein (BCRP), and lung resistance protein (LRP) in 45 cases of mature NK/ T-cell lymphoma was examined by immunohistochemistry. The positive rates of P-gp, MRP-1, BCRP and LRP were 31% (13/42), 74% (31/42), 78% (32/41) and 59%(26/44), respectively [65]. Jung et al., in a study of drug resistance in T—and NK-cell lymphomas, reported a statistically significant association with treatment failure and overall survival [66]. Yamaguchi et al. also reported high P-gp expression in their study of nasal NK-cell lymphoma patients and suggested that the poor prognosis of these patients may be related to P-gp expression [67]. Egashira et al. reported that P-gp expression in CD56-positive NK-cell tumors was associated with poor prognosis [68].
It has been confirmed that in B-cell lymphoma, the five drugs containing R-CHOP do not show synergistic effect, but cross-resistance is very low, indicating that the efficacy of RCHOP is produced by the combination of non-overlapping active drugs, and the reason for the poor efficacy of CHOP against ENKTL is the expression of the multidrug efflux pump P-glycoprotein /MDR1 [69].The high expression of P-glycoprotein in PTCL lymphoma cells, doxorubicin, vincristine, and prednisone, which are substrates of Pgp, easily induces intrinsic resistance to drugs due to the upregulation of P-gp expression [70]. ABCC4 and ABCG4 were significantly up-regulated in human NK/T cell lymphoma YTS and SNK-6 cells compared with normal NK cells [71]. Overall, it is important to highlight the role of ABCC4 in drug resistance. Based on gene expression regulation technology, overexpression of ABCC4 and ABCG4 can induce epirubicin (EPI) and cisplatin (DDP) resistance in human NK/ T cell lymphoma YTS cells and reduce cell apoptosis [72]. Meanwhile,IL-6, IL-10, and IL13 mediate ABCC4 resistance in T-cell lymphoma [64, 73, 74].
Signaling pathways in PTCL
The mechanism of drug resistance of lymphoma is closely related to the signaling pathways of lymphoma cells. Recent genetic analyses of PTCL have improved our understanding of the pathogenesis of this malignancy. The activation mutation of NF-κB, Notch, JAK/ STAT3, RHOA and PI3K/AKT signaling pathways play an important role in the pathogenesis of PTCL [29, 75]. The expression of JAK/STAT pathway genes is upregulated in ENKTL, and mutations in JAK3, STAT3, and STAT5B lead to constitutive activation of the JAK/STAT pathway, which occurs on the transcription factor jak3 in about 35% of cases, resulting in severe immunodeficiency characterized by a lack of T and NK cells [76, 77]. DDX3X, the RNA helicase gene, which mutated in 20% of PTCL, resulting in cell cycle arrest and loss of transcriptional activation of the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways. Clinically, the presence of DDX3X mutation indicates a poor prognosis [78, 79]. PTPRK is known to dephosphorylate phosphorylated stat3, resulting in its inactivation. Loss of PTPRK and low expression of PTPRK due to aberrant promoter hypermethylation can cause constitutive activation of STAT3, leading to proliferation and development of PTCL. Downregulation of PTPRK is associated with advanced disease and poor outcomes in patients treated with the steroids, methotrexate, ifosfamide, L-asparaginase, and etoposide (SMILE) regimen [80]. NF-κB is involved in pro-proliferative signal transduction in a variety of lymphoid malignancies [75, 81]. Although the mechanism needs to be further investigated, GEP studies suggest increased expression of NF-κB-related genes in PTCL and that NF-κb inhibitors induce apoptosis in PTCL cells [82], findings that support the hypothesis that this pathway plays an important role in PTCL. In addition, NF-κB is involved in the pathogenesis of hemophagocytosis, which is the main cause of death in PTCL patients [83].(Fig. 3). In addition, increasing data suggest that viral components are involved in multidrug chemotherapy resistance in lymphoma cells, and several mechanisms may be associated with oncogenic viral-mediated chemotherapy resistance, which is caused by changes in disease signaling pathways [84]. Under latent EBV infection, intracellular ROS production increases P-gp expression via the STAT1 pathway, and ROS scavenger NecroX-5 down-regulates ROS, effectively attenuating P-gp-associated chemotherapy resistance in EBV-positive NK/ T-cell lymphoma. LMP1 and/or other viral components are also involved in P-gp-dependent chemoresistance [28, 85].

Part of the pathways, such as NF-κB, JAK/ STAT3 and PI3K/AKT signaling pathways in the pathogenesis of PTCL
Epigenetics in PTCL
Epigenetics is the stable clonal inheritance of expression states that cannot be explained by DNA variants. Gene expression is typically maintained through DNA methylation and post-translational methylation, acetylation, phosphorylation, and ubiquitination of histone and non-histone proteins [86]. Mutations in genes encoding proteins involved in epigenetic regulation are seen in various malignancies including PTCL, particularly in those expressing follicular helper T cell (TFH) differentiation markers, such as angioimmunoblastic T-cell lymphoma (AITL) and some PTCL-NOS [87]. Disruption of DNA methylation and histone modifications has become a hallmark of these diseases and the basis for epigenetically targeted therapies [88]. As the understanding of epigenetic complexity continues to deepen, it has been found that mutations in these epigenetic regulators have a global impact on lymphoma development and drug sensitivity, and the ability to silence multiple genes at the same time through the regulation of a large number of genes leads to polygenic drug resistance [89, 90].
MicroRNAs in PTCL
MicroRNA is a type of RNA with length of 19 to 22 nucleotides, which is involved in important biological processes such as development, proliferation, differentiation and apoptosis [91]. Recent studies have shown that the differentiation of various T cell subsets is regulated by multiple miRNAs targeting different signaling pathway proteins/molecules, resulting in initiation or inhibition/termination of differentiation [92, 93]. Different microRNAs are uniquely expressed in lymphoid T cells and play a role in the development and differentiation of various subtypes by targeting their target genes. Aberrant expression of miRNAs may be involved in T-cell leukemia and lymphopoiegenesis and may function as tumor suppressor genes such as miR-451, miR-31, miR-150, and miR-29a or oncogenes such as miR-222, miR-223, miR-17–92, and miR-155. In T-cell leukemia and lymphoma, microRNA can be used as novel biomarkers for prognosis and diagnosis, or as indicators of disease severity [94, 95].
The increased expression of miR-122 is associated with poor prognosis in advanced mycosis fungoides (MF). The up-regulation of miR-21, miR-486 and miR-214 is involved in promoting SzS cell survival and is involved in the apoptotic resistance of CTCL cell lines, and may even become incurable CTCL [96]. The expression levels of miR-21 and miR-155 in NK cell lymphoma cell lines were significantly higher than those in normal NK cells [97]. Plasma miR-221 level may have diagnostic and prognostic significance in extranodal NK/ T cell lymphoma (ENKTCL) [98]. In fact, there is an indirect relationship between miR-221 expression and overall survival after treatment of this lymphoma. Evaluation of miR-146a expression in NKTL tissues showed that patients with low levels of miR-146a expression were associated with chemotherapy resistance and poor prognosis. This miRNA acts as a tumor suppressor and therefore can be used for prognosis of PTCL. Evaluation of the expression level of miR-16 in paraffin-embedded lymph node samples of T lymphoblastic lymphoma/leukemia (T-LBL/ALL) patients showed that the overall survival of patients with high levels of miR-16 was higher than that of patients with low levels of miR-16, and it can be used as a prognostic marker for T LBL/ALL108 patients [99]. Elevated exosomal miR-4454, miR-21-5p, and miR-320e levels were associated with poor overall survival. Elevated levels were also found in patients who relapsed after treatment. These three miRNAs were overexpressed in PTCL cell lines resistant to etoposide [100].
Approved and emerging therapies for R/R PTCL
The exploration of treatment for PTCL is very active, and the development of treatments has been driven primarily by advances in the understanding of the biology of the disease.
CD30 monoclonal antibody agents
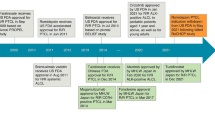
CD30 receptors are expressed in Hodgkin's disease, T-cell lymphoma subsets, and activated T cells [101]. In 376 PTCL patient samples, CD30 was expressed in 58% of PTCL-NOS, 63% of AITL, and almost 100% of ALCL. Similar to the therapeutic effect of CD20 monoclonal antibodies in B-cell lymphomas, there is an increasing interest in targeting CD30 as a potential therapeutic option for PTCL [102]. Brentuximab Vedotin (BV) is an antibody–drug conjugate in which an anti-CD30 monoclonal antibody is coupled to the tubulin toxin monomethyl lauristatin E (MMAE) [103]. Once bound to the CD30 receptor, it is internalized and causes disruption of microtubule polymerization and cell death. BV-CHP has shown clinical benefit in ALCL, making it the preferred option for the treatment of this disease, and it is strongly considered for other CD30 + PTCL [8]. In a large prospective phase II study and another large phase III randomized study (ECHELON-2), BV combined with other chemotherapy agents showed good therapeutic effect, and BV also achieved good efficacy in relapsed and refractory PTCL as a single agent [104,105,106]. At ASH 2022, the updated 5-year ECHELON-2 clinical trial showed that compared to CHOP, first-line treatment of PTCL patients with A + CHP continued to provide clinically meaningful improvements in PFS and OS with a manageable safety profile, with 5-year PFS rates at a median follow-up of 47.6 months in the A + CHP group and CHOP group of 51.4% and 43.0%, with 5-year overall survival rates of 70.1% and 61.0% and a 28% reduction in the risk of death (HR = 0.72; 95% CI: 0.53–0.99), respectively. Among patients treated with vibutuximab after relapse, the objective remission rate was 59% for re-treatment with BV after A + CHP and 50% for re-treatment with vibutuximab after CHOP. In ECHELON-2, patients over 65 years of age who received a + CHP had significantly improved outcomes. An ongoing phase 2 trial is providing additional data on BV retreatment in patients with classical Hodgkin's lymphoma, sALCL, or other cd30-positive PTCL (NCT03947255) [107]. Although BV has high activity in R/R HL, most patients eventually develop BV resistance. It was reported that antigen downregulation is a potential resistance mechanism to any antigen-targeted therapy [108], however, it seemed CD30 loss does not appear to be a common event in BV-refractory HL. The investigators found that CD30 expression was maintained in tumor samples from patients with BV-refractory HL, as well as in two independent BV-resistant cell lines, both of which were found to have upregulation of the multidrug resistance gene MDR1, suggesting that BV resistance may be mediated in part by increased MDR1 activity rather than CD30 loss [109]. Upregulation of NF-κB activity is responsible for increased MDR1 expression in drug resistant clones [110]. The addition of MDR1 inhibitors to BV treatment may be a potential treatment option for PTCL [109, 111].
Pralatrexate
Pralatrexate(PDX), a new antifolate agent that is more potent than methotrexate, is active against T-cell lymphomas. Pralatrexate is a highly selective antifolate drug with high affinity for reduced folate carrier (RFC). RFC is a protein that regulates natural folate uptake and, in tumor cells, is required for purine and pyrimidine biosynthesis [112]. Pralatrexate is the first drug to be approved for patients with relapsed/refractory PTCL based on the PROPEL study. The PROPEL trial study population included patients with all aggressive TCL subtypes, including challenging disease that was partially excluded by other studies, including blastic NTKCL, transformed MF and HTLV-1 ATLL, responses were observed for PTCL in all subtypes. The ORR was 29%(investigator-assessed 39%), with 18% of patients achieving PR and 11% achieving CR or unconfirmed CR (CRu) [113]. After pralatrexate's approval, a phase II trial explored alternating CEOP(cyclophosphamide, etoposide, vincristine, and prednisone) with first-line therapy (each cycle consisted of CEOP (A) with Pralatrexate (B) 30 mg/m2 IV days 15, 22 and 29). However, this study failed to show better results with CHOP than with historical data [114]. During the use of pralatrexate, the overall response rate (ORR) of treated patients was about 30%, which is not ideal for the treatment of T-cell lymphoma. In addition to intrinsic resistance, pralatrexate has cross-resistance with other conventional cytotoxic chemotherapy drugs, and it accumulates over time [113]. Moreover, toxicity of this agent can be significant, the most common grade 3/4 adverse events were thrombocytopenia (32%), mucositis (22%), neutropenia (22%), and anemia (18%) [113, 115], thus we need more research to improve the quality of life of these patients with acceptable comfort indices. The combination appears to overcome the inherent resistance to pratrexin to some extent, and the resistance mechanism of PDX is related to the reduced cellular uptake of PDX and/or the overexpression of DNMT3B. Epigenetic alterations are also thought to play a role in resistance mechanisms. DAC combined with PDX has a synergistic effect, which is expected to improve the clinical efficacy [116]. In addition, pratrexin also has significant synergistic effects with histone deacetylase inhibitors (such as romidispin [117]) and proteasome inhibitors (such as bortezomib [118]).
PD-1/PD-L1
Programmed death receptor 1 (PD-1) recognizes ligands such as PD-L1 on tumor cells to evade host immune responses. Inhibition of the programmed cell death ligand 1 (PD-L1) pathway has emerged as a promising strategy for the treatment of tumors [119]. PD-L1 expression ranged from 39 to 100% in NKTCL [30, 120, 121]. Nivolumab is a humanized immunoglobulin G4 monoclonal antibody that targets the programmed death (PD)-1 receptor on T cells and has shown marked antitumor activity, improving survival in a number of solid tumors and hematologic malignancies, including Hodgkin's lymphoma [122, 123]. In a phase Ib study that included R/R hematologic malignancies, the ORR was 40% in patients with PTCL, but no CR was observed [119]. Although Pembrolizumab has been used to treat various subtypes of NHL, there are limited real-world data on the efficacy of Pembrolizumab in patients with NKTCL. Several studies published some clinical trial data, but the sample sizes were small, and some studies showed that PD-1 inhibition with pembrolizumab was a favorable strategy for the treatment of refractory or relapsed NKTCL [124,125,126]. In addition, PD-L1 mutations and a diverse baseline T-cell receptor (TCR) repertoire have been shown to be potential biomarkers for better selection of NKTCL patients for anti-PD-1 therapy [14]. NKTCL patients can undergo PD-L1 mutation and TCR sequence analysis before receiving immune checkpoint inhibitor therapy to avoid excessive financial burden and reduce adverse events [127].
Histone deacetylase inhibitors
Histone deacetylation inhibitors are a class of drugs that can acetylate histone proteins, thereby regulating gene transcription, leading to cell cycle arrest, differentiation and apoptosis [128]. Histone deacetylase inhibitors (HDACis) have long been shown to have anti-tumor activity, and the mechanism is related to acetylation of histones and other proteins involved in tumor suppression, apoptosis, and cell cycle regulation [129]. Romidepsin, a cyclic peptide originally isolated from Chromobacterium violaceum, is a pan-HDAC inhibitor with potent inhibitory activity against selected class 1 HDAC isoforms, such as HDAC-1, -2, and -3, and is a selective and potent bicyclic histone deacetylase inhibitor. First approved by the FDA in 2009 for the treatment of patients with relapsed cutaneous T-cell lymphoma [130], Romidepsin was subsequently approved for the treatment of patients with relapsed/refractory PTCL based on two independent phase 2 clinical trials [131, 132]. The Ro-CHOP Phase III trial was designed to compare the efficacy and safety of standard CHOP versus Romidepsin-CHOP regimens for patients with first-line PTCL.As of December 13, 2019, with a median follow-up of 27.5 months, the Ro-CHOP combination regimen did not improve PFS, remission rates, or OS, nor did it increase the incidence of grade ≥ 3 treatment-related TEAEs in patients with PTCL compared to CHOP treatment. The 6-month, 1-year, and 2-year PFS rates were 67.4%, 67.4%, 67.3%, and 43.2%, respectively, in the Ro-CHOP group, 49.8% and 43.2% in the Ro-CHOP group and 65.9%, 44.3% and 36.3% in the CHOP group, respectively; the 1-year and 2-year OS rates were 78.2% and 63.6% in the Ro-CHOP group and 77.5% and 63.4% in the CHOP group, respectively. However, the high incidence of TEAE in Ro-CHOP treatment limited the intensity of the regimen measured in CHOP. Overall, Ro-CHOP did not demonstrate a satisfactory benefit in patients with previously untreated PTCL [133]. Based on this study, on May 6, 2022, the FDA formally announced that it was withdrawing approval of romidepsin for the indication of PTCL, but that it was still approved for the treatment of cutaneous T-cell lymphoma in patients who have received at least one systemic therapy.
Belinostat, a pan-class 1 and class 2 hydroxamic acid-based HDAC inhibitor, is currently approved for patients with relapsed or refractory PTCL who have received at least one line of previous therapy [134].
Chidamide is another HDACi that has shown monotherapy activity in R/R PTCL [135]. Despite the promising anti-lymphoma activity of histone deacetylase (HDAC) inhibitors, drug resistance is an important clinical problem. Belinostat-resistant cells showed significant cross-resistance to other HDAC inhibitors, including romidepsin, panobinostat, and vorinostat. Consistent with the insensitivity to HDAC inhibitors, resistant cells failed to induce an increase in acetylated histones. Resistance of tumor cells to HDAC inhibitors may involve both "intrinsic" and "acquired" mechanisms. Aberrant expression and modification of signaling molecules lead to the inherent resistance of cancer cells to HDAC inhibitors [128, 136, 137].
Inhibitors of the PI3K/Akt/mTOR pathway
In T lymphocytes, activation of the phosphatidylinositol 3-kinase (PI3K)/Akt/ mammalian target of rapamycin (mTOR) pathway in response to exogenous stimuli is known to drive cell survival and clonal proliferation, and PI3K activation is tightly controlled by signals transduced through the TCR complex [138, 139]. Thus, in the presence of stable TCR signaling, costimulatory signaling driven primarily by the PI3K/Akt/mTOR pathway leads to complete T-cell proliferation activation, making inhibition of this pathway and/or TCR signaling a reasonable approach for PTCL therapy [140, 141]. Duvelisib (IPI-145) is an oral inhibitor of the PI3K isoforms PI3Kδ and PI3Kγ, which are thought to be required for full TCR signaling. In an open-label phase 1 study, 16 patients with R/R PTCL and 19 patients with CTCL were treated with duvelisib, with an ORR of 50%, a CR rate of 19%, and a median PFS of 8.3 months [140]. Copanlisib (bay80-6946), a PI3Kα and δ inhibitor, is active in B-cell lymphomas and is FDA approved for the treatment of follicular (B-cell) lymphomas. The activity of copanlisib in T-cell lymphomas is currently being further investigated [142]. Aurora A kinase (AAK) has also recently been shown to play a key function in cell entry into mitosis, and its overexpression has been linked to the development of a number of tumors [143]. This prompted us to explore AAK inhibition as a potential therapeutic strategy for a variety of cancers, including PTCL. Alisertib is a selective AAK inhibitor that has shown promising activity in preclinical models of T—and B-cell lymphomas and in vivo lymphoma models [144, 145]. Despite promising early activity, enrollment was stopped early because of poor odds of superior PFS compared with other agents. The findings, although less encouraging, cannot completely rule out a future role for Alisertib combination therapy.
Other drugs
In addition to the above FDA- approved drugs, R/R PTCL can also be treated from other angles. Bendamustine, CCR4 inhibitors [146], ALK inhibitors [147], DNA methyltransferase (DNMT) inhibitors [8], CD138 monoclonal antibody, CD52 monoclonal antibody [148], antiviral drugs, immunomodulators, EZH2 and EZH1 dual inhibitors [149], are drugs that have achieved very encouraging clinical trials in relapsed and refractory PTCL. Furthermore, there is significant emphasis on conducting clinical trials targeting relapsed or refractory T-Cell Lymphomas. In this context, we present a list of ongoing r/r PTCL clinical trials, which encompass novel combinations of established drugs along with newly developed drugs that are not yet available in the market (refer to Table 1).
Conclusion
To date, most PTCL subtypes are aggressive and chemotherapy-resistant, and their prognosis remains poor. Multiple mechanisms, such as tumor heterogeneity, tumor microenvironment, and signaling pathways, contribute to PTCL resistance. Over the past few years, considerable efforts have been made to identify novel molecular targets and deregulated molecules in the oncogenic pathway. Many ongoing clinical trials are exploring other targeted drugs, novel cell therapies, and immunotherapies. As precision medicine catches up with this disease, we are likely to see new treatments that can overcome tumor resistance and thus improve the treatment efficacy of PTCL.
Availability of data and materials
The data and materials supporting the study are available upon request by writing to the corresponding authors.
Abbreviations
- PTCL:
-
Peripheral T-cell lymphoma
- r/r PTCL:
-
Relapsed or refractory peripheral T-Cell Lymphomas
- NHL:
-
Non-Hodgkin's lymphomas
- WHO:
-
World Health Organization
- WHO-HAEM5:
-
WHO classification of haematolymphoid tumors
- TME:
-
Tumor microenvironment
- CTCL:
-
Cutaneous T-cell lymphoma
- ALCL:
-
Anaplastic large cell lymphoma
- AITL:
-
Angioimmunoblastic T-cell lymphoma
- T-ALL/LBL:
-
T lymphoblastic leukemia/lymphoma
- ETP-ALL:
-
Early T-cell precursor lymphoblastic leukemia
- sEBV + TNHL:
-
Systemic EBV + T-cell lymphoma
- ENKTL:
-
Extra-nodal NK/T-cell lymphoma
- ATLL:
-
Adult T-cell leukemia/lymphoma
- CCR4:
-
C-C chemokine receptor 4
- ALCL:
-
Anaplastic large cell lymphoma
- PD-L1:
-
Programmed cell death ligand I
- OS:
-
Overall survival
- Tregs:
-
Regulatory T cells
- MDR:
-
Multidrug resistance
- ABC:
-
ATP-binding cassette
- P-gp:
-
P-glycoprotein
- MRP-1:
-
Multidrug resistance-related protein 1
- BCRP:
-
Breast cancer resistance protein
- LRP:
-
Lung resistance protein
- EPI:
-
Epirubicin
- NF-κB:
-
Nuclear factor κB
- MAPK:
-
Mitogen-activated protein kinase
- SMILE:
-
Steroids, methotrexate, ifosfamide, L-asparaginase, and etoposide
- TFH:
-
Follicular helper T cell
- MF:
-
Mycosis fungoides
- ENKTCL:
-
Extranodal NK/ t cell lymphoma
- T-LBL/ALL:
-
T lymphoblastic lymphoma/leukemia
- BV:
-
Brentuximab Vedotin
- MMAE:
-
Monomethyl lauristatin E
- NLG:
-
Nordic Lymphoma Group
- PDX:
-
Pralatrexate
- RFC:
-
Reduced folate carriers
- CRu:
-
Unconfirmed CR
- CEOP:
-
Cyclophosphamide,etoposide, vincristine, and prednisone
- ORR:
-
Overall response rate
- PD-1:
-
Programmed death receptor 1
- TCR:
-
T-cell receptor
- HDAC:
-
Histone Deacetylase Inhibitors
- PI3K:
-
Phosphatidylinositol 3-kinase
- AAK:
-
Aurora A kinase
- DNMT:
-
DNA methyltransferase
References
Bellei M, Chiattone CS, Luminari S, Pesce EA, Cabrera ME, Souza CAD, Gabús R, Zoppegno L, Milone J, Pavlovsky A, Connors JM, Foss FM, Horwitz SM, Liang R, Montoto S, Pileri SA, Polliack A, Vose JM, Zinzani PL, Zucca E, Federico M. T-Cell Lymphomas in South America and Europe. Rev Bras Hematol Hemoter. 2011;34(1):42–7. https://doi.org/10.5581/1516-8484.20120013.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood. 2016;127(20):2375–90. https://doi.org/10.1182/blood-2016-01-643569.
Murga-Zamalloa C, Inamdar K. Classification and Challenges in the Histopathological Diagnosis of Peripheral T-Cell Lymphomas, Emphasis on the WHO-HAEM5 Updates. Front Oncol. 2022;12:1099265. https://doi.org/10.3389/fonc.2022.1099265.
Aisenberg AC. Historical review of lymphomas: historical review. Br J Haematol. 2000;109(3):466–76. https://doi.org/10.1046/j.1365-2141.2000.01988.x.
Stuver R, Moskowitz AJ. Therapeutic advances in relapsed and refractory peripheral T-cell lymphoma. Cancers. 2023;15(3):589. https://doi.org/10.3390/cancers15030589.
O’Connor OA, Bhagat G, Ganapathi K, Pedersen MB, D’Amore F, Radeski D, Bates SE. Changing the paradigms of treatment in peripheral T-cell lymphoma: from biology to clinical practice. Clin Cancer Res. 2014;20(20):5240–54. https://doi.org/10.1158/1078-0432.CCR-14-2020.
Savage KJ, Chhanabhai M, Gascoyne RD, Connors JM. Characterization of Peripheral T-Cell Lymphomas in a Single North American Institution by the WHO Classification. Ann Oncol. 2004;15(10):1467–75. https://doi.org/10.1093/annonc/mdh392.
Bachow SH, O'Connor OA. Emerging therapies in relapsed and refractory peripheral T-cell lymphoma. Clin Adv Hematol Oncol. 2015;13(12):837–46.
Foley NC, Mehta-Shah N. Management of peripheral T-cell lymphomas and the role of transplant. Curr Oncol Rep. 2022;24(11):1489–99. https://doi.org/10.1007/s11912-022-01310-3.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309. https://doi.org/10.1038/s41586-019-1730-1.
Bukowski K, Kciuk M, Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. IJMS. 2020;21(9):3233. https://doi.org/10.3390/ijms21093233.
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBDO, Berti E, Bhagat G, Borges AM, Boyer D, Calaminici M, Chadburn A, Chan JKC, Cheuk W, Chng W-J, Choi JK, Chuang S-S, Coupland SE, Czader M, Dave SS, De Jong D, Du M-Q, Elenitoba-Johnson KS, Ferry J, Geyer J, Gratzinger D, Guitart J, Gujral S, Harris M, Harrison CJ, Hartmann S, Hochhaus A, Jansen PM, Karube K, Kempf W, Khoury J, Kimura H, Klapper W, Kovach AE, Kumar S, Lazar AJ, Lazzi S, Leoncini L, Leung N, Leventaki V, Li X-Q, Lim MS, Liu W-P, Louissaint A, Marcogliese A, Medeiros LJ, Michal M, Miranda RN, Mitteldorf C, Montes-Moreno S, Morice W, Nardi V, Naresh KN, Natkunam Y, Ng S-B, Oschlies I, Ott G, Parrens M, Pulitzer M, Rajkumar SV, Rawstron AC, Rech K, Rosenwald A, Said J, Sarkozy C, Sayed S, Saygin C, Schuh A, Sewell W, Siebert R, Sohani AR, Tooze R, Traverse-Glehen A, Vega F, Vergier B, Wechalekar AD, Wood B, Xerri L, Xiao W. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720–48. https://doi.org/10.1038/s41375-022-01620-2.
Pearse WB, Pro B. Diagnosis, risk stratification, and treatment of peripheral T-Cell Lymphomas: past and present. Cancer J. 2020;26(3):253–9. https://doi.org/10.1097/PPO.0000000000000452.
Tse E, Zhao W-L, Xiong J, Kwong Y-L. How we treat NK/T-Cell lymphomas. J Hematol Oncol. 2022;15(1):74. https://doi.org/10.1186/s13045-022-01293-5.
Wang, X.; Wang, W.; Vega, F.; Quesada, A. E. Aggressive Mediastinal Lymphomas. Semin Diagn Pathol 2021, S0740257021000496. https://doi.org/10.1053/j.semdp.2021.06.010.
You MJ, Medeiros LJ, Hsi ED. T-Lymphoblastic Leukemia/Lymphoma. Am J Clin Pathol. 2015;144(3):411–22. https://doi.org/10.1309/AJCPMF03LVSBLHPJ.
Shiraz P, Jehangir W, Agrawal V. T-cell acute lymphoblastic leukemia—current concepts in molecular biology and management. Biomedicines. 2021;9(11):1621. https://doi.org/10.3390/biomedicines9111621.
Fattizzo B, Rosa J, Giannotta JA, Baldini L, Fracchiolla NS. The Physiopathology of T- Cell acute lymphoblastic leukemia: focus on molecular aspects. Front Oncol. 2020;10:273. https://doi.org/10.3389/fonc.2020.00273.
Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16(8):494–507. https://doi.org/10.1038/nrc.2016.63.
Xu X, Zhai Q, Jin H, Yu Y, Han D, Zhang H, Fu K, Meng B. SET-NUP214 fusion gene involved early T-cell precursor acute lymphoblastic leukemia in adult with B marker expression. IJGM. 2021;14:659–64. https://doi.org/10.2147/IJGM.S294715.
Tesio M, Trinquand A, Macintyre E, Asnafi V. Oncogenic PTEN functions and models in T-cell malignancies. Oncogene. 2016;35(30):3887–96. https://doi.org/10.1038/onc.2015.462.
Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, Zhang W, Li Y, Dave BJ, Nairismägi M-L, Greiner TC, Vose J, Weisenburger DD, Lachel C, Wang C, Fu K, Stevens JM, Lim ST, Ong CK, Gascoyne RD, Missiaglia E, Lemonnier F, Haioun C, Hartmann S, Pedersen MB, Laginestra MA, Wilcox RA, Teh BT, Yoshida N, Ohshima K, Seto M, Rosenwald A, Ott G, Campo E, Rimsza LM, Jaffe ES, Braziel RM, d’Amore F, Inghirami G, Bertoni F, De Leval L, Gaulard P, Staudt LM, McKeithan TW, Pileri S, Chan WC, Iqbal J. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133(15):1664–76. https://doi.org/10.1182/blood-2018-09-872549.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen S-C, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui C-H, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63. https://doi.org/10.1038/nature10725.
Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. Role of NF-κB in cell survival and transcription of latent membrane protein 1-expressing or epstein-barr virus latency III-infected cells. J Virol. 2004;78(8):4108–19. https://doi.org/10.1128/JVI.78.8.4108-4119.2004.
Parilla M, Quesada AE, Medeiros LJ, Thakral B. An update on genetic aberrations in T-cell neoplasms. Pathology. 2023;55(3):287–301. https://doi.org/10.1016/j.pathol.2022.12.350.
Hue SS-S, Oon ML, Wang S, Tan S-Y, Ng S-B. Epstein-barr virus-associated T- and NK-cell lymphoproliferative diseases: an update and diagnostic approach. Pathology. 2020;52(1):111–27. https://doi.org/10.1016/j.pathol.2019.09.011.
Ng S-B, Chung T-H, Kato S, Nakamura S, Takahashi E, Ko Y-H, Khoury JD, Yin CC, Soong R, Jeyasekharan AD, Hoppe MM, Selvarajan V, Tan S-Y, Lim S-T, Ong C-K, Nairismägi M-L, Maheshwari P, Choo S-N, Fan S, Lee C-K, Chuang S-S, Chng W-J. Epstein-barr virus-associated primary nodal T/NK-cell lymphoma shows a distinct molecular signature and copy number changes. Haematologica. 2018;103(2):278–87. https://doi.org/10.3324/haematol.2017.180430.
Shafiee, A.; Shamsi, S.; Kohandel Gargari, O.; Beiky, M.; Allahkarami, M. M.; Miyanaji, A. B.; Aghajanian, S.; Mozhgani, S. EBV Associated T‐ and NK‐cell lymphoproliferative diseases: a comprehensive overview of clinical manifestations and novel therapeutic insights. Rev Med Virol. 2022, 32 (4). https://doi.org/10.1002/rmv.2328.
Xiong J, Cui B-W, Wang N, Dai Y-T, Zhang H, Wang C-F, Zhong H-J, Cheng S, Ou-Yang B-S, Hu Y, Zhang X, Xu B, Qian W-B, Tao R, Yan F, Hu J-D, Hou M, Ma X-J, Wang X, Liu Y-H, Zhu Z-M, Huang X-B, Liu L, Wu C-Y, Huang L, Shen Y-F, Huang R-B, Xu J-Y, Wang C, Wu D-P, Yu L, Li J-F, Xu P-P, Wang L, Huang J-Y, Chen S-J, Zhao W-L. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell. 2020;37(3):403-419.e6. https://doi.org/10.1016/j.ccell.2020.02.005.
Song TL, Nairismägi M-L, Laurensia Y, Lim J-Q, Tan J, Li Z-M, Pang W-L, Kizhakeyil A, Wijaya G-C, Huang D-C, Nagarajan S, Chia BK-H, Cheah D, Liu Y-H, Zhang F, Rao H-L, Tang T, Wong EK-Y, Bei J-X, Iqbal J, Grigoropoulos N-F, Ng S-B, Chng W-J, Teh B-T, Tan S-Y, Verma NK, Fan H, Lim S-T, Ong C-K. Oncogenic Activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132(11):1146–58. https://doi.org/10.1182/blood-2018-01-829424.
Kim H, Ko YH. The pathologic and genetic characteristics of extranodal NK/T-cell lymphoma. Life (Basel). 2022;12(1):73. https://doi.org/10.3390/life12010073.
Nasr, R.; Marçais, A.; Hermine, O.; Bazarbachi, A. Overview of Targeted Therapies for Adult T-Cell Leukemia/Lymphoma. In Human T-Lymphotropic Viruses; Casoli, C., Ed.; Methods in Molecular Biology; Springer New York: New York. 2017; 1582: 197–216. https://doi.org/10.1007/978-1-4939-6872-5_15.
Stengel A, Kern W, Zenger M, Perglerová K, Schnittger S, Haferlach T, Haferlach C. Genetic Characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker: comprehensive genetic characterization of T-PLL. Genes Chromosomes Cancer. 2016;55(1):82–94. https://doi.org/10.1002/gcc.22313.
Sibon D, Nguyen D-P, Schmitz N, Suzuki R, Feldman AL, Gressin R, Lamant L, Weisenburger DD, Rosenwald A, Nakamura S, Ziepert M, Maurer MJ, Bast M, Armitage JO, Vose JM, Tilly H, Jais J-P, Savage KJ. ALK-positive anaplastic large-cell lymphoma in adults: an individual patient data pooled analysis of 263 patients. Haematologica. 2019;104(12):e562–5. https://doi.org/10.3324/haematol.2018.213512.
Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Vasmatzis G, Gibson SE, Ondrejka S, Nicolae A, Grogg KL, Allmer C, Ristow KM, Wilson WH, Macon WR, Law ME, Cerhan JR, Habermann TM, Ansell SM, Dogan A, Maurer MJ, Feldman AL. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473–80. https://doi.org/10.1182/blood-2014-04-571091.
Qiu, L.; Tang, G.; Li, S.; Vega, F.; Lin, P.; Wang, S. A.; Wang, W.; Iyer, S. P.; Malpica, L.; Miranda, R. N.; Konoplev, S.; Tang, Z.; Fang, H.; Medeiros, L. J.; Xu, J. DUSP22 Rearrangement is associated with distinctive immunophenotype but not outcome in patients with systemic ALK-negative anaplastic large cell lymphoma. Haematol 2022. https://doi.org/10.3324/haematol.2022.281222.
Oishi N, Brody GS, Ketterling RP, Viswanatha DS, He R, Dasari S, Mai M, Benson HK, Sattler CA, Boddicker RL, McPhail ED, Bennani NN, Harless CA, Singh K, Clemens MW, Medeiros LJ, Miranda RN, Feldman AL. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood. 2018;132(5):544–7. https://doi.org/10.1182/blood-2017-12-821868.
Weiss J, Reneau J, Wilcox RA. PTCL, NOS: an update on classification, risk-stratification, and treatment. Front Oncol. 2023;13:1101441. https://doi.org/10.3389/fonc.2023.1101441.
Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, Greiner TC, Smith L, Guo S, Wilcox RA, Teh BT, Lim ST, Tan SY, Rimsza LM, Jaffe ES, Campo E, Martinez A, Delabie J, Braziel RM, Cook JR, Tubbs RR, Ott G, Geissinger E, Gaulard P, Piccaluga PP, Pileri SA, Au WY, Nakamura S, Seto M, Berger F, De Leval L, Connors JM, Armitage J, Vose J, Chan WC, Staudt LM. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915–23. https://doi.org/10.1182/blood-2013-11-536359.
Krug A, Tari G, Saidane A, Gaulard P, Ricci J-E, Lemonnier F, Verhoeyen E. Novel T follicular helper-like T-cell lymphoma therapies: from preclinical evaluation to clinical reality. Cancers (Basel). 2022;14(10):2392. https://doi.org/10.3390/cancers14102392.
Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129(9):1095–102. https://doi.org/10.1182/blood-2016-09-692541.
Palomero T, Couronné L, Khiabanian H, Kim M-Y, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, Nicolas C, Balbin M, Bastard C, Bhagat G, Piris MA, Campo E, Bernard OA, Rabadan R, Ferrando AA. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46(2):166–70. https://doi.org/10.1038/ng.2873.
Steinhilber J, Mederake M, Bonzheim I, Serinsöz-Linke E, Müller I, Fallier-Becker P, Lemonnier F, Gaulard P, Fend F, Quintanilla-Martinez L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2R172 mutations. Mod Pathol. 2019;32(8):1123–34. https://doi.org/10.1038/s41379-019-0254-4.
Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, Martin N, Fataccioli V, Pelletier R, Wakeham A, Snow BE, de Leval L, Pujals A, Haioun C, Paci A, Tobin ER, Narayanaswamy R, Yen K, Jin S, Gaulard P, Mak TW. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A. 2016;113(52):15084–9. https://doi.org/10.1073/pnas.1617929114.
Vallois D, Dupuy A, Lemonnier F, Allen G, Missiaglia E, Fataccioli V, Ortonne N, Clavert A, Delarue R, Rousselet M-C, Fabiani B, Llamas-Gutierrez F, Ogawa S, Thome M, Ko YH, Kataoka K, Gaulard P, de Leval L. RNA Fusions involving CD28 are rare in peripheral T-cell lymphomas and concentrate mainly in those derived from follicular helper T cells. Haematologica. 2018;103(8):e360–3. https://doi.org/10.3324/haematol.2017.186767.
Carbone A, Gloghini A. Subclassifying peripheral T-cell lymphoma NOS. Blood. 2019;134(24):2120–1. https://doi.org/10.1182/blood.2019003385.
Nagato T, Ohkuri T, Ohara K, Hirata Y, Kishibe K, Komabayashi Y, Ueda S, Takahara M, Kumai T, Ishibashi K, Kosaka A, Aoki N, Oikawa K, Uno Y, Akiyama N, Sado M, Takei H, Celis E, Harabuchi Y, Kobayashi H. Programmed death-Ligand 1 and its soluble form are highly expressed in nasal natural killer/T-cell lymphoma: a potential rationale for immunotherapy. Cancer Immunol Immunother. 2017;66(7):877–90. https://doi.org/10.1007/s00262-017-1987-x.
Wang T, Feldman AL, Wada DA, Lu Y, Polk A, Briski R, Ristow K, Habermann TM, Thomas D, Ziesmer SC, Wellik LE, Lanigan TM, Witzig TE, Pittelkow MR, Bailey NG, Hristov AC, Lim MS, Ansell SM, Wilcox RA. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014;123(19):3007–15. https://doi.org/10.1182/blood-2013-12-544809.
Boutilier AJ, Elsawa SF. Macrophage polarization States in the tumor microenvironment. IJMS. 2021;22(13):6995. https://doi.org/10.3390/ijms22136995.
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–55. https://doi.org/10.1016/j.semcancer.2008.03.004.
Tse E, Kwong Y-L. T-cell lymphoma: microenvironment-related biomarkers. Semin Cancer Biol. 2015;34:46–51. https://doi.org/10.1016/j.semcancer.2015.06.001.
Zhang W, Wang L, Zhou D, Cui Q, Zhao D, Wu Y. Expression of tumor-associated macrophages and vascular endothelial growth factor correlates with poor prognosis of peripheral T-cell lymphoma not otherwise specified. Leukemia Lymphoma. 2011;52(1):46–52. https://doi.org/10.3109/10428194.2010.529204.
Lin Z-X, Bai B, Cai Q-C, Cai Q-Q, Wang X-X, Wu X-Y, Huang H-Q. High numbers of tumor-associated macrophages correlate with poor prognosis in patients with mature T- and natural killer cell lymphomas. Med Oncol. 2012;29(5):3522–8. https://doi.org/10.1007/s12032-012-0244-6.
Niino D, Komohara Y, Murayama T, Aoki R, Kimura Y, Hashikawa K, Kiyasu J, Takeuchi M, Suefuji N, Sugita Y, Takeya M, Ohshima K. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-Cell Lymphoma (AITL). Pathol Int. 2010;60(4):278–83. https://doi.org/10.1111/j.1440-1827.2010.02514.x.
Komohara Y, Niino D, Saito Y, Ohnishi K, Horlad H, Ohshima K, Takeya M. Clinical Significance of CD 163 + tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. 2013;104(7):945–51. https://doi.org/10.1111/cas.12167.
Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. https://doi.org/10.1038/nri1806.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. https://doi.org/10.1126/science.1079490.
Bonzheim I, Geissinger E, Tinguely M, Roth S, Grieb T, Reimer P, Wilhelm M, Rosenwald A, Müller-Hermelink HK, Rüdiger T. Evaluation of FoxP3 expression in peripheral T-cell lymphoma. Am J Clin Pathol. 2008;130(4):613–9. https://doi.org/10.1309/L65GWEQ803PP6VX1.
Shareef MM, Elgarhy LH, Wasfy RE-S. Expression of granulysin and FOXP3 in cutaneous T cell lymphoma and sézary syndrome. Asian Pac J Cancer Prev. 2015;16(13):5359–64. https://doi.org/10.7314/APJCP.2015.16.13.5359.
Kant S, Kumar A, Singh SM. Tumor Growth Retardation and Chemosensitizing Action of Fatty Acid Synthase Inhibitor Orlistat on T Cell Lymphoma: Implication of Reconstituted Tumor Microenvironment and Multidrug Resistance Phenotype. Biochimica et Biophysica Acta (BBA) - General Subjects. 2014;1840(1):294–302. https://doi.org/10.1016/j.bbagen.2013.09.020.
Nobili S, Lapucci A, Landini I, Coronnello M, Roviello G, Mini E. Role of ATP-binding cassette transporters in cancer initiation and progression. Semin Cancer Biol. 2020;60:72–95. https://doi.org/10.1016/j.semcancer.2019.08.006.
Jacobo-Albavera L, Domínguez-Pérez M, Medina-Leyte DJ, González-Garrido A, Villarreal-Molina T. The role of the ATP-Binding Cassette A1 (ABCA1) in human disease. IJMS. 2021;22(4):1593. https://doi.org/10.3390/ijms22041593.
Dean M, Rzhetsky A, Allikmets R. The human ATP-Binding Cassette (ABC) transporter superfamily. Genome Res. 2001;11(7):1156–66. https://doi.org/10.1101/gr.184901.
Gao M, Liu L, Zhang X, Li Z, Zhang M. Interleukin-6 reverses Adriamycin resistance in nasal NK/T-cell lymphoma via downregulation of ABCC4 and inactivation of the JAK2/STAT3/NF-κB/P65 pathway. Environ Toxicol Pharmacol. 2021;85:103639. https://doi.org/10.1016/j.etap.2021.103639.
Saglam A, Hayran M, Uner AH. Immunohistochemical expression of multidrug resistance proteins in mature T/NK-cell lymphomas. APMIS. 2008;116(9):791–800. https://doi.org/10.1111/j.1600-0463.2008.00974.x.
Jung CK, Lee KY, Kim Y, Han K, Shim SI, Kim BK, Kang CS. Epstein-barr virus infection, drug resistance and prognosis in Korean T- and NK-cell lymphomas. Pathol Int. 2001;51(5):355–63. https://doi.org/10.1046/j.1440-1827.2001.01214.x.
Yamaguchi M, Kita K, Miwa H, Nishii K, Oka K, Ohno T, Shirakawa S, Fukumoto M. Frequent expression of P-Glycoprotein/MDR1 by nasal T-cell lymphoma cells. Cancer. 1995;76(11):2351–6. https://doi.org/10.1002/1097-0142(19951201)76:11%3c2351::AID-CNCR2820761125%3e3.0.CO;2-1.
Egashira M, Kawamata N, Sugimoto K, Kaneko T, Oshimi K. P-Glycoprotein expression on normal and abnormally expanded natural killer cells and inhibition of P-Glycoprotein function by Cyclosporin A and its analogue, PSC833. Blood. 1999;93(2):599–606.
Chen Q, Feng J, Wu J, Yu Z, Zhang W, Chen Y, Yao P, Zhang H. HKDC1 C-terminal based peptides inhibit extranodal natural Killer/T-cell lymphoma by modulation of mitochondrial function and EBV suppression. Leukemia. 2020;34(10):2736–48. https://doi.org/10.1038/s41375-020-0801-5.
Jillella AP, Murren JR, Hamid KK, Longley BJ, Edelson RL, Cooper DL. P-Glycoprotein expression and multidrug resistance in cutaneous T-cell lymphoma. Cancer Invest. 2000;18(7):609–13. https://doi.org/10.3109/07357900009032827.
Ali W, Spengler G, Kincses A, Nové M, Battistelli C, Latacz G, Starek M, Dąbrowska M, Honkisz-Orzechowska E, Romanelli A, Rasile MM, Szymańska E, Jacob C, Zwergel C, Handzlik J. Discovery of phenylselenoether-hydantoin hybrids as ABCB1 efflux pump modulating agents with cytotoxic and antiproliferative actions in resistant T-lymphoma. Eur J Med Chem. 2020;200:112435. https://doi.org/10.1016/j.ejmech.2020.112435.
Wu S, Zhang X, Dong M, Yang Z, Zhang M, Chen Q. sATP-binding Cassette subfamily G member 2 enhances the multidrug resistance properties of human nasal natural killer/T cell lymphoma side Population cells. Oncol Rep. 2020. https://doi.org/10.3892/or.2020.7722.
Ni M, Qin B, Xie L, Zhang X, Yang J, Lv H, Yang M, Zhang M. IL-13 contributes to drug resistance of NK/T-cell lymphoma cells by regulating ABCC4. Biomed Res Int. 2018;2018:1–9. https://doi.org/10.1155/2018/2606834.
Huo J, Fu L, Jin M, Li Z, Zhang M. IL-10 contributes to gemcitabine resistance in extranodal NK/T-cell lymphoma cells via ABCC4. Invest New Drugs. 2022;40(3):537–45. https://doi.org/10.1007/s10637-022-01224-8.
De Mel S, Hue SS-S, Jeyasekharan AD, Chng W-J, Ng S-B. Molecular Pathogenic Pathways in Extranodal NK/T Cell Lymphoma. J Hematol Oncol. 2019;12(1):33. https://doi.org/10.1186/s13045-019-0716-7.
Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh K-W, Yap SP, Lee KA, Lim LC, Tan D, Goh C, Cutcutache I, Yu W, Young Ng CC, Rajasegaran V, Heng HL, Gan A, Ong CK, Rozen S, Tan P, Teh BT, Lim ST. Janus kinase 3–activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–7. https://doi.org/10.1158/2159-8290.CD-12-0028.
Zhang Y, Li C, Xue W, Zhang M, Li Z. Frequent mutations in natural killer/T cell lymphoma. Cell Physiol Biochem. 2018;49(1):1–16. https://doi.org/10.1159/000492835.
Jiang L, Gu Z-H, Yan Z-X, Zhao X, Xie Y-Y, Zhang Z-G, Pan C-M, Hu Y, Cai C-P, Dong Y, Huang J-Y, Wang L, Shen Y, Meng G, Zhou J-F, Hu J-D, Wang J-F, Liu Y-H, Yang L-H, Zhang F, Wang J-M, Wang Z, Peng Z-G, Chen F-Y, Sun Z-M, Ding H, Shi J-M, Hou J, Yan J-S, Shi J-Y, Xu L, Li Y, Lu J, Zheng Z, Xue W, Zhao W-L, Chen Z, Chen S-J. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–6. https://doi.org/10.1038/ng.3358.
Küçük C, Hu X, Jiang B, Klinkebiel D, Geng H, Gong Q, Bouska A, Iqbal J, Gaulard P, McKeithan TW, Chan WC. Global promoter methylation analysis reveals novel candidate tumor suppressor genes in natural killer cell lymphoma. Clin Cancer Res. 2015;21(7):1699–711. https://doi.org/10.1158/1078-0432.CCR-14-1216.
Chen Y-W, Guo T, Shen L, Wong K-Y, Tao Q, Choi WWL, Au-Yeung RKH, Chan Y-P, Wong MLY, Tang JCO, Liu W-P, Li G-D, Shimizu N, Loong F, Tse E, Kwong Y-L, Srivastava G. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood. 2015;125(10):1589–600. https://doi.org/10.1182/blood-2014-07-588970.
Jost PJ, Ruland J. Aberrant NF-κB signaling in lymphoma: mechanisms, consequences, and therapeutic implications. Blood. 2007;109(7):2700–7. https://doi.org/10.1182/blood-2006-07-025809.
Lv K, Li X, Yu H, Chen X, Zhang M, Wu X. Selection of new immunotherapy targets for NK/T cell lymphoma. Am J Transl Res. 2020;12(11):7034–47.
Wi SM, Moon G, Kim J, Kim S-T, Shim J-H, Chun E, Lee K-Y. TAK1-ECSIT-TRAF6 complex plays a key role in the TLR4 signal to activate NF-κB. J Biol Chem. 2014;289(51):35205–14. https://doi.org/10.1074/jbc.M114.597187.
Kimura, H. EBV in T-/NK-Cell Tumorigenesis. In Human Herpesviruses; Kawaguchi, Y., Mori, Y., Kimura, H., Eds.; Advances in Experimental Medicine and Biology; Springer Singapore: ,2018; 1045: 459–475. https://doi.org/10.1007/978-981-10-7230-7_21.
Uno M, Tsuchiyama J, Moriwaki A, Noguchi T, Mizoguchi K, Ogino T, Yoshino T, Okada S, Harada M. In Vitro induction of apoptosis for nasal Angiocentric natural killer cell lymphoma-derived cell line, NK-YS, by etoposide and cyclosporine a: drug resistance and apoptosis of NK-YS cell line. Br J Haematol. 2001;113(4):1009–14. https://doi.org/10.1046/j.1365-2141.2001.02844.x.
Nirmaladevi R. Epigenetic Alterations in Cancer. Front Biosci. 2020;25(6):1058–109. https://doi.org/10.2741/4847.
Zhang P, Zhang M. Epigenetic Alterations and Advancement of Treatment in Peripheral T-Cell Lymphoma. Clin Epigenet. 2020;12(1):169. https://doi.org/10.1186/s13148-020-00962-x.
Chen I-C, Sethy B, Liou J-P. Recent update of HDAC inhibitors in lymphoma. Front Cell Dev Biol. 2020;8:576391. https://doi.org/10.3389/fcell.2020.576391.
Lee, J.-H.; Choy, M. L.; Marks, P. A. Mechanisms of Resistance to Histone Deacetylase Inhibitors. In Advances in Cancer Research; Elsevier, 2012;116: 39–86. https://doi.org/10.1016/B978-0-12-394387-3.00002-1.
Yamagishi M. The role of epigenetics in T-cell lymphoma. Int J Hematol. 2022;116(6):828–36. https://doi.org/10.1007/s12185-022-03470-1.
Mohr A, Mott J. Overview of MicroRNA biology. Semin Liver Dis. 2015;35(01):003–11. https://doi.org/10.1055/s-0034-1397344.
Lone W, Bouska A, Sharma S, Amador C, Saumyaranjan M, Herek TA, Heavican TB, Yu J, Lim ST, Ong CK, Slack GW, Savage KJ, Rosenwald A, Ott G, Cook JR, Feldman AL, Rimsza LM, McKeithan TW, Greiner TC, Weisenburger DD, Melle F, Motta G, Pileri S, Vose JM, Chan WC, Iqbal J. Genome-wide miRNA expression profiling of molecular subgroups of peripheral T-cell lymphoma. Clin Cancer Res. 2021;27(21):6039–53. https://doi.org/10.1158/1078-0432.CCR-21-0573.
Lin Y, Chen W-M, Wang C, Chen X-Y. MicroRNA profiling in peripheral T-cell lymphoma not otherwise specified. CBM. 2017;18(4):339–47. https://doi.org/10.3233/CBM-160126.
Mei M, Zhang M. Non-Coding RNAs in Natural Killer/T-cell lymphoma. Front Oncol. 2019;9:515. https://doi.org/10.3389/fonc.2019.00515.
Saki N, Abroun S, Soleimani M, Hajizamani S, Shahjahani M, Kast RE, Mortazavi Y. Involvement of MicroRNA in T-Cell Differentiation and Malignancy. Int J Hematol Oncol Stem Cell Res. 2015;9(1):33–49.
Manfè V, Biskup E, Rosbjerg A, Kamstrup M, Skov AG, Lerche CM, Lauenborg BT, Ødum N, Gniadecki R. miR-122 regulates P53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS One. 2012;7(1):e29541. https://doi.org/10.1371/journal.pone.0029541.
Yamanaka Y, Tagawa H, Takahashi N, Watanabe A, Guo Y-M, Iwamoto K, Yamashita J, Saitoh H, Kameoka Y, Shimizu N, Ichinohasama R, Sawada K. Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood. 2009;114(15):3265–75. https://doi.org/10.1182/blood-2009-06-222794.
Guo H-Q, Huang G-L, Guo C-C, Pu X-X, Lin T-Y. Diagnostic and prognostic value of circulating miR-221 for extranodal natural killer/T-cell lymphoma. Dis Markers. 2010;29(5):251–8. https://doi.org/10.1155/2010/474692.
Xi Y, Li J, Zan L, Wang J, Wang G, Ning Y. Micro-RNA-16 expression in paraffin-embedded specimen correlates with overall survival of T-lymphoblastic lymphoma/leukemia. Hum Pathol. 2013;44(6):1011–6. https://doi.org/10.1016/j.humpath.2012.08.023.
Ryu KJ, Lee JY, Choi ME, Yoon SE, Cho J, Ko YH, Shim JH, Kim WS, Park C, Kim SJ. Serum-derived exosomal MicroRNA profiles can predict poor survival outcomes in patients with extranodal natural killer/T-cell lymphoma. Cancers. 2020;12(12):3548. https://doi.org/10.3390/cancers12123548.
Viviani S, Guidetti A. Efficacy of antibody-drug conjugate brentuximab vedotin in treating hodgkin’s lymphoma. Expert Opin Biol Ther. 2018;18(8):841–9. https://doi.org/10.1080/14712598.2018.1499723.
Othman T, Herrera A, Mei M. Emerging therapies in relapsed and refractory Hodgkin lymphoma: what comes next after Brentuximab Vedotin and PD-1 inhibition? Curr Hematol Malig Rep. 2021;16(1):1–7. https://doi.org/10.1007/s11899-020-00603-3.
Chen R, Hou J, Newman E, Kim Y, Donohue C, Liu X, Thomas SH, Forman SJ, Kane SE. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to Brentuximab Vedotin. Mol Cancer Ther. 2015;14(6):1376–84. https://doi.org/10.1158/1535-7163.MCT-15-0036.
Richardson NC, Kasamon YL, Chen H, De Claro RA, Ye J, Blumenthal GM, Farrell AT, Pazdur R. FDA approval summary: Brentuximab Vedotin in first-line treatment of peripheral T-cell lymphoma. Oncologist. 2019;24(5):e180–7. https://doi.org/10.1634/theoncologist.2019-0098.
Horwitz S, O’Connor OA, Pro B, Illidge T, Fanale M, Advani R, Bartlett NL, Christensen JH, Morschhauser F, Domingo-Domenech E, Rossi G, Kim WS, Feldman T, Lennard A, Belada D, Illés Á, Tobinai K, Tsukasaki K, Yeh S-P, Shustov A, Hüttmann A, Savage KJ, Yuen S, Iyer S, Zinzani PL, Hua Z, Little M, Rao S, Woolery J, Manley T, Trümper L, Aboulafia D, Advani R, Alpdogan O, Ando K, Arcaini L, Baldini L, Bellam N, Bartlett N, Belada D, Yehuda DB, Benedetti F, Borchman P, Bordessoule D, Brice P, Briones J, Caballero D, Carella AM, Chang H, Cheong JW, Cho S-G, Choi I, Choquet S, Colita A, Congui AG, D’amore F, Dang N, Davison K, De Guibert S, Brown PDN, Delwail V, Demeter J, Di Raimondo F, Do YR, Domingo E, Douvas M, Dreyling M, Ernst T, Fanale M, Fay K, Feldman T, Ferrero SF, Flinn IW, Forero-Torres A, Fox C, Friedberg J, Fukuhara N, Garcia-Marco J, Cruz JG, Codina JG, Gressin R, Grigg A, Gurion R, Christensen JH, Haioun C, Hajek R, Hanel M, Hatake K, Hensen R, Horowitz N, Horwitz S, Huttmann A, Illes A, Illidge T, Ishizawa K, Islas-Ohlmayer M, Jacobsen E, Janakiram M, Jurczak W, Kaminski M, Kato K, Kim WS, Kirgner I, Iyer S, Kuo C-Y, Lazaroiu MC, Du KL, Lee J-S, LeGouill S, Lennard A, LaRosee P, Levi I, Link B, Maisonneuve H, Maruyama D, Mayer J, McCarty J, McKay P, Minami Y, Mocikova H, Morra E, Morschhauser F, Munoz J, Nagai H, O’Connor O, Opat S, Pettengell R, Pezzutto A, Pfreundschuh M, Pluta A, Porcu P, Pro B, Quach H, Rambaldi A, Renwick W, Reyes R, Izquierdo AR, Rossi G, Ruan J, Rusconi C, Salles G, Santoro A, Sarriera J, Savage K, Shibayama H, Shustov A, Suh C, Sureda A, Tanimoto M, Taniwaki M, Tilly H, Tobinai K, Trneny M, Trumper L, Tsukamoto N, Tsukasaki K, Vitolo U, Walewski J, Weidmann E, Wilhelm M, Witzens-Harig M, Yacoub A, Yamamoto K, Yeh S-P, Yoon S-S, Yuen S, Yun HJ, Zain J, Zinzani PL. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229–40. https://doi.org/10.1016/S0140-6736(18)32984-2.
Kim HK, Moon SM, Moon JH, Park JE, Byeon S, Kim WS. Complete remission in CD30-positive refractory extranodal NK/T-cell lymphoma with Brentuximab Vedotin. Blood Res. 2015;50(4):254. https://doi.org/10.5045/br.2015.50.4.254.
Horwitz S, O’Connor OA, Pro B, Trümper L, Iyer S, Advani R, Bartlett NL, Christensen JH, Morschhauser F, Domingo-Domenech E, Rossi G, Kim WS, Feldman T, Menne T, Belada D, Illés Á, Tobinai K, Tsukasaki K, Yeh S-P, Shustov A, Hüttmann A, Savage KJ, Yuen S, Zinzani PL, Miao H, Bunn V, Fenton K, Fanale M, Puhlmann M, Illidge T. The ECHELON-2 trial: 5-Year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol. 2022;33(3):288–98. https://doi.org/10.1016/j.annonc.2021.12.002.
Villamor N, Montserrat E, Colomer D. Mechanism of action and resistance to monoclonal antibody therapy. Semin Oncol. 2003;30(4):424–33. https://doi.org/10.1016/S0093-7754(03)00261-6.
Liu-Kreyche P, Shen H, Marino AM, Iyer RA, Humphreys WG, Lai Y. Lysosomal P-Gp-MDR1 confers drug resistance of Brentuximab Vedotin and its cytotoxic payload monomethyl Auristatin E in tumor cells. Front Pharmacol. 2019;10:749. https://doi.org/10.3389/fphar.2019.00749.
Mehra V, Pomplum S, Ireland R, Yallop D, Devereux S, Marcus R, Shah C, Patten P, Kassam S. ALK-positive large B-cell lymphoma with strong CD30 expression; a diagnostic pitfall and resistance to Brentuximab and Crizotinib. Histopathology. 2016;69(5):880–2. https://doi.org/10.1111/his.13002.
Chen R, Herrera AF, Hou J, Chen L, Wu J, Guo Y, Synold TW, Ngo VN, Puverel S, Mei M, Popplewell L, Yi S, Song JY, Tao S, Wu X, Chan WC, Forman SJ, Kwak LW, Rosen ST, Newman EM. Inhibition of MDR1 overcomes resistance to Brentuximab Vedotin in Hodgkin lymphoma. Clin Cancer Res. 2020;26(5):1034–44. https://doi.org/10.1158/1078-0432.CCR-19-1768.
New Drug. Pralatrexate for Lymphoma. Aust Prescr. 2019;42(2):68. https://doi.org/10.18773/austprescr.2019.020.
O’Connor OA, Pro B, Pinter-Brown L, Bartlett N, Popplewell L, Coiffier B, Jo Lechowicz M, Savage KJ, Shustov AR, Gisselbrecht C, Jacobsen E, Zinzani PL, Furman R, Goy A, Haioun C, Crump M, Zain JM, Hsi E, Boyd A, Horwitz S. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. JCO. 2011;29(9):1182–9. https://doi.org/10.1200/JCO.2010.29.9024.
Advani, R. H.; Ansell, S. M.; Lechowicz, M. J.; Beaven, A. W.; Loberiza, F.; Carson, K. R.; Evens, A. M.; Foss, F.; Horwitz, S.; Pro, B.; Pinter-Brown, L. C.; Smith, S. M.; Shustov, A. R.; Savage, K. J.; M. Vose, J. A Phase II Study of Cyclophosphamide, Etoposide, Vincristine and Prednisone (CEOP) Alternating with Pralatrexate (P) as Front Line Therapy for Patients with Peripheral T-Cell Lymphoma (PTCL): Final Results from the T- Cell Consortium Trial. Br J Haematol 2016, 172 (4), 535–544. https://doi.org/10.1111/bjh.13855.
Shimanovsky A, Dasanu CA. Pralatrexate : evaluation of clinical efficacy and toxicity in T-cell lymphoma. Expert Opin Pharmacother. 2013;14(4):515–23. https://doi.org/10.1517/14656566.2013.770474.
Oiwa K, Hosono N, Nishi R, Scotto L, O’Connor OA, Yamauchi T. Characterization of newly established Pralatrexate-resistant cell lines and the mechanisms of resistance. BMC Cancer. 2021;21(1):879. https://doi.org/10.1186/s12885-021-08607-9.
Jain S, Jirau-Serrano X, Zullo KM, Scotto L, Palermo CF, Sastra SA, Olive KP, Cremers S, Thomas T, Wei Y, Zhang Y, Bhagat G, Amengual JE, Deng C, Karan C, Realubit R, Bates SE, O’Connor OA. Preclinical pharmacologic evaluation of pralatrexate and romidepsin confirms potent synergy of the combination in a murine model of human T-cell lymphoma. Clin Cancer Res. 2015;21(9):2096–106. https://doi.org/10.1158/1078-0432.CCR-14-2249.
Marchi E, Paoluzzi L, Scotto L, Seshan VE, Zain JM, Zinzani PL, O’Connor OA. Pralatrexate is synergistic with the proteasome inhibitor Bortezomib in In Vitro and In Vivo models of T-cell lymphoid malignancies. Clin Cancer Res. 2010;16(14):3648–58. https://doi.org/10.1158/1078-0432.CCR-10-0671.
Rossi C, Casasnovas R-O. PD-1 Inhibitors in patients with hodgkin lymphoma. Eur J Cancer. 2022;164:114–6. https://doi.org/10.1016/j.ejca.2021.06.059.
Xie M, Huang X, Ye X, Qian W. Prognostic and clinicopathological significance of PD-1/PD-L1 expression in the tumor microenvironment and neoplastic cells for lymphoma. Int Immunopharmacol. 2019;77:105999. https://doi.org/10.1016/j.intimp.2019.105999.
Jalili-Nik M, Soltani A, Mashkani B, Rafatpanah H, Hashemy SI. PD-1 and PD-L1 inhibitors foster the progression of adult T-cell leukemia/lymphoma. Int Immunopharmacol. 2021;98:107870. https://doi.org/10.1016/j.intimp.2021.107870.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P. PD-1 Blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9. https://doi.org/10.1056/NEJMoa1411087.
Houot R, Poeschel V, Altmann B, Angel S, Thurner L, Illmer T, Andre M, Dreyling M, Maisonneuve H, Tilly H, Mayer S, Casasnovas O, Le Gouill S, Offner F, Cartron G, Kerkhoff A, Weber T, Hoffmann J, Ziepert M, Klapper W, Itti E, Hellwig D, Natchkebia G, De Leval L, Rosenwald A, Haioun C, Dercle L, Gaulard P, Held G. Prolonged remissions after nivolumab plus gemcitabine/Oxaliplatin in relapsed/refractory T-cell lymphoma. HemaSphere. 2022;6(2):e672. https://doi.org/10.1097/HS9.0000000000000672.
Kwong Y-L, Chan TSY, Tan D, Kim SJ, Poon L-M, Mow B, Khong P-L, Loong F, Au-Yeung R, Iqbal J, Phipps C, Tse E. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-Asparaginase. Blood. 2017;129(17):2437–42. https://doi.org/10.1182/blood-2016-12-756841.
Li X, Cheng Y, Zhang M, Yan J, Li L, Fu X, Zhang X, Chang Y, Sun Z, Yu H, Zhang L, Wang X, Wu J, Li Z, Nan F, Tian L, Li W, Young KH. Activity of pembrolizumab in relapsed/refractory NK/T-cell lymphoma. J Hematol Oncol. 2018;11(1):15. https://doi.org/10.1186/s13045-018-0559-7.
Kwok G, Yau TCC, Chiu JW, Tse E, Kwong Y-L. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016;12(11):2777–89. https://doi.org/10.1080/21645515.2016.1199310.
Chen X, Wu W, Wei W, Zou L. Immune checkpoint inhibitors in peripheral T-cell lymphoma. Front Pharmacol. 2022;13:869488. https://doi.org/10.3389/fphar.2022.869488.
Von Knethen A, Heinicke U, Weigert A, Zacharowski K, Brüne B. Histone deacetylation inhibitors as modulators of regulatory T cells. IJMS. 2020;21(7):2356. https://doi.org/10.3390/ijms21072356.
Zahnow, C. A.; Topper, M.; Stone, M.; Murray-Stewart, T.; Li, H.; Baylin, S. B.; Casero, R. A. Inhibitors of DNA Methylation, Histone Deacetylation, and Histone Demethylation. In Advances in Cancer Research; Elsevier, 2016; 130: 55–111. https://doi.org/10.1016/bs.acr.2016.01.007.
McGraw AL. Romidepsin for the treatment of T-cell lymphomas. Am J Health Syst Pharm. 2013;70(13):1115–22. https://doi.org/10.2146/ajhp120163.
Falchi L, Ma H, Klein S, Lue JK, Montanari F, Marchi E, Deng C, Kim HA, Rada A, Jacob AT, Kinahan C, Francescone MM, Soderquist CR, Park DC, Bhagat G, Nandakumar R, Menezes D, Scotto L, Sokol L, Shustov AR, O’Connor OA. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood. 2021;137(16):2161–70. https://doi.org/10.1182/blood.2020009004.
Iyer SP, Foss FF. Romidepsin for the treatment of peripheral T-cell lymphoma. Oncologist. 2015;20(9):1084–91. https://doi.org/10.1634/theoncologist.2015-0043.
Bachy E, Camus V, Thieblemont C, Sibon D, Casasnovas R-O, Ysebaert L, Damaj G, Guidez S, Pica GM, Kim WS, Lim ST, André M, García-Sancho AM, Penarrubia MJ, Staber PB, Trotman J, Hüttmann A, Stefoni V, Re A, Gaulard P, Delfau-Larue M-H, de Leval L, Meignan M, Li J, Morschhauser F, Delarue R. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral T-cell lymphoma: results of the Ro-CHOP phase III study (Conducted by LYSA). J Clin Oncol. 2022;40(3):242–51. https://doi.org/10.1200/JCO.21.01815.
Campbell P, Thomas CM. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J Oncol Pharm Pract. 2017;23(2):143–7. https://doi.org/10.1177/1078155216634178.
Shi Y, Jia B, Xu W, Li W, Liu T, Liu P, Zhao W, Zhang H, Sun X, Yang H, Zhang X, Jin J, Jin Z, Li Z, Qiu L, Dong M, Huang X, Luo Y, Wang X, Wang X, Wu J, Xu J, Yi P, Zhou J, He H, Liu L, Shen J, Tang X, Wang J, Yang J, Zeng Q, Zhang Z, Cai Z, Chen X, Ding K, Hou M, Huang H, Li X, Liang R, Liu Q, Song Y, Su H, Gao Y, Liu L, Luo J, Su L, Sun Z, Tan H, Wang H, Wang J, Wang S, Zhang H, Zhang X, Zhou D, Bai O, Wu G, Zhang L, Zhang Y. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017;10(1):69. https://doi.org/10.1186/s13045-017-0439-6.
Ni X, Li L, Pan G. HDAC inhibitor-induced drug resistance involving ATP-binding cassette transporters (Review). Oncol Lett. 2015;9(2):515–21. https://doi.org/10.3892/ol.2014.2714.
Bi J, Zhang Y, Malmrose PK, Losh HA, Newtson AM, Devor EJ, Thiel KW, Leslie KK. Blocking autophagy overcomes resistance to dual histone deacetylase and proteasome inhibition in gynecologic cancer. Cell Death Dis. 2022;13(1):59. https://doi.org/10.1038/s41419-022-04508-2.
Peng Z, Xiong J, Dong H. Valproic acid inhibits peripheral T cell lymphoma cells behaviors via restraining PI3K/AKT pathway. Evid Based Complementa Altern Med. 2022;2022:1–9. https://doi.org/10.1155/2022/7350489.
Wu SJ, Chen J, Wu B, Wang YJ, Guo KY. MicroRNA-150 enhances radiosensitivity by inhibiting the AKT pathway in NK/T cell lymphoma. J Exp Clin Cancer Res. 2018;37(1):18. https://doi.org/10.1186/s13046-017-0639-5.
Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, Myskowski P, Officer A, Jaffe JD, Morrow SN, Allen K, Douglas M, Stern H, Sweeney J, Kelly P, Kelly V, Aster JC, Weaver D, Foss FM, Weinstock DM. Activity of the PI3K-δ, γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood. 2018;131(8):888–98. https://doi.org/10.1182/blood-2017-08-802470.
Huang D, Song TL, Nairismägi M, Laurensia Y, Pang W, Zhe DCM, Wong EKY, Wijaya GG, Tan J, Tan SH, Lim J, Chia BKH, Chan JY, Tang TPL, Somasundaram N, Cheng CL, Politz O, Liu N, Lim ST, Ong CK. Evaluation of the PIK3 pathway in peripheral T-cell lymphoma and NK/T-cell lymphoma. Br J Haematol. 2020;189(4):731–44. https://doi.org/10.1111/bjh.16435.
Markham A. Copanlisib: first global approval. Drugs. 2017;77(18):2057–62. https://doi.org/10.1007/s40265-017-0838-6.
Alrifai D, Pettengell R. MLN8237 (Alisertib) and its role in peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(12):1731–6. https://doi.org/10.1517/13543784.2014.972501.
Zullo KM, Guo Y, Cooke L, Jirau-Serrano X, Mangone M, Scotto L, Amengual JE, Mao Y, Nandakumar R, Cremers S, Duong J, Mahadevan D, O’Connor OA. Aurora A kinase inhibition selectively synergizes with histone deacetylase inhibitor through cytokinesis failure in T-cell lymphoma. Clin Cancer Res. 2015;21(18):4097–109. https://doi.org/10.1158/1078-0432.CCR-15-0033.
Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Roselló S, Andreu J, Jung J, Sanchis-Garcia JM, Piera A, Blasco I, Maños L, Pérez-Fidalgo J-A, Fingert H, Baselga J, Tabernero J. Phase I Pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective Aurora A kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(17):4764–74. https://doi.org/10.1158/1078-0432.CCR-12-0571.
Ishitsuka K. T cell lymphoma: time to make discoveries and advance treatment. Int J Hematol. 2023;117(4):473–4. https://doi.org/10.1007/s12185-023-03573-3.
Zain JM, Hanona P. Aggressive T-cell Lymphomas: 2021 updates on diagnosis, risk stratification and management. Am J Hematol. 2021;96(8):1027–46. https://doi.org/10.1002/ajh.26270.
Chamuleau ED, Van De Loosdrecht MA, Huijgens PC. Monoclonal antibody therapy in haematological malignancies. CCP. 2010;5(3);148–59. https://doi.org/10.2174/157488410791498752.
Mina A, Pro B. T time: emerging and new therapies for peripheral T-cell lymphoma. Blood Rev. 2022;52:100889. https://doi.org/10.1016/j.blre.2021.100889.
Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221:107753. https://doi.org/10.1016/j.pharmthera.2020.107753.
Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updates. 2020;53:100715. https://doi.org/10.1016/j.drup.2020.100715.
Funding
This work supported by The Reserve Talents Project for Young and Middle-aged Academic and technical Leaders of Department of Science and Technology of Yunnan Province (202105AC160047).
Author information
Authors and Affiliations
Contributions
Yunpeng Luan and Dong-Hua Yang conceived and designed the study. Yunpeng Luan and Xiang Li and Yunqi Luan were responsible for manuscript writing. Junyu Luo, Qinzuo Dong, and Shili Ye, were responsible for the collection and assembly of data. Yuejin Li, Yanmei Li, Lu Jia and Jun Yang were responsible for data analysis and interpretation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luan, Y., Li, X., Luan, Y. et al. Therapeutic challenges in peripheral T-cell lymphoma. Mol Cancer 23, 2 (2024). https://doi.org/10.1186/s12943-023-01904-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01904-w