
Abstract
Tumor immunotherapy has transformed neoplastic disease management, yet low response rates and immune complications persist as major challenges. Extracellular vesicles including exosomes have emerged as therapeutic agents actively involved in a diverse range of pathological conditions. Mounting evidence suggests that alterations in the quantity and composition of extracellular vesicles (EVs) contribute to the remodeling of the immune-suppressive tumor microenvironment (TME), thereby influencing the efficacy of immunotherapy. This revelation has sparked clinical interest in utilizing EVs for immune sensitization. In this perspective article, we present a comprehensive overview of the origins, generation, and interplay among various components of EVs within the TME. Furthermore, we discuss the pivotal role of EVs in reshaping the TME during tumorigenesis and their specific cargo, such as PD-1 and non-coding RNA, which influence the phenotypes of critical immune cells within the TME. Additionally, we summarize the applications of EVs in different anti-tumor therapies, the latest advancements in engineering EVs for cancer immunotherapy, and the challenges encountered in clinical translation. In light of these findings, we advocate for a broader understanding of the impact of EVs on the TME, as this will unveil overlooked therapeutic vulnerabilities and potentially enhance the efficacy of existing cancer immunotherapies.
Similar content being viewed by others

Introduction
Immunotherapy was developed based on tumor escape, by manipulating the body's immune system to reactivate the antitumor effect [1]. Tumor immunotherapies include immune checkpoint inhibitors (ICI), pericyte therapies, lysing viruses, and cancer vaccines. Immunotherapy, represented by immune checkpoint blockade, has transformed the treatment of many solid and hematologic malignancies [2,3,4]. However, only 10–30% of solid tumors are treated clinically, and the treatment sensitivity is low. TME is a complex environment for tumor survival. Tumor cells and their microenvironment interact and co-evolve to promote tumor generation, development and metastasis. During the process of tumor development, the TME dynamically changes, generating an immunosuppressive microenvironment that leads to tumor immune escape. Similarly, the heterogeneity of TME allows for individual differences in tumors, which may be related to individual differences in tumor immunotherapy [5].
Extracellular vesicles (EVs) released by various cell types and play an important role in intercellular communication. As an important component of tumor-host interactions, EVs are increasingly recognized as a key molecular entity in constructing the immunosuppressive microenvironment in TME. Tumor cells actively release EVs into the surrounding microenvironment, and these vesicles have pleiotropic abilities in regulating tumor growth progression, neovascularization, immune escape, and promoting tumor invasion and metastasis [6, 7]. Thus, EVs regulate intercellular communication not only between cancer cells, but also between cells in TME [8].
Tumor-induced immune cell exhaustion represents a pervasive stratagem employed to elude vigilant immune surveillance [9]. Notably, the wellspring of malignancy expels a copious profusion of these EVs, adorned with the effigy of programmed death-ligand (PD-L1) upon their lipid bilayer. The advent of anti-PD1 antibodies, employed to obstruct the PD1/PD-L1 axis and forestall immune cell exhaustion, has indeed wrought a profound transformation in the therapeutic landscape for advanced malignancies [10, 11]. However, notwithstanding the substantial triumphs attributed to this therapeutic paradigm, a litany of both preclinical and clinical investigations has elucidated the emergence of resistance mechanisms, whether innate or acquired, to anti-PD1 therapy. PD-L1, recognized interchangeably as CD274 or B7-H1, manifests as a transmembrane protein of considerable complexity, spanning 290 amino acids and encoded by the CD274 gene. This multifaceted protein encompasses immunoglobulin V-like and C-like extracellular domains of intricate architecture [12]. Emerging evidence has suggested that tumor-derived EVs (TEVs) carry biologically active PD-L1 on their surface, leading to the suppression of immune responses [13]. It is worth noting, however, that the corpus of literature referenced herein exclusively scrutinizes tumor-derived extracellular vesicular PD-L1, and while the significance of PD-L1 expression in bone marrow cells in the context of immunosuppression is undeniable, no investigations pertaining to extracellular vesicular PD-L1 emanating from bone marrow cells have graced the scientific discourse thus far.
Non-coding RNAs (ncRNAs), a category of ribonucleic acids devoid of protein-coding potential [14]. Nonetheless, in recent times, a subset of these ncRNAs has been discovered to possess the ability to encode peptides and proteins [15]. This subset encompasses distinguished entities such as microRNA (miRNA), long-stranded non-coding RNA (lncRNA), and circular RNA (circRNA), all of which exhibit well-defined functional roles. Furthermore, there exist enigmatic RNAs whose functional significance remains elusive. It is worth noting that these entities are transcribed from the genome, yet they do not undergo translation into proteins [16,17,18]. Also, EVs shelters ncRNAs capable of traversing to neighboring or distant cells. There, they orchestrate gene expression by modulating the levels of transcription factors or miRNAs. In doing so, they intricately regulate a myriad of cellular processes, encompassing but not confined to cell proliferation, differentiation, and programmed cell death [19, 20].
This review aims to provide an extensive examination of the diverse effects of EVs from various sources on the distinct components of the TME, with particular emphasis on the role of PD-L1 and non-coding RNA from EVs in TME remodelling and their effects across different cancer types. Furthermore, we also discuss the remodeling of the lymph node microenvironment by EVs and the potential of engineering EVs to enhance tumour immunotherapy. Taken together, these findings highlight the promising potential of EVs-mediated remodeling of the TME as a powerful strategy for future cancer immunotherapy applications.
EVs and TME
EVs constitute a diverse array of membranous structures originating from various cellular sources, encompassing exosomes and microvesicles. They arise either from the endosomal system or shed from the plasma membrane, giving rise to this heterogeneous population [21, 22]. These vesicles serve as a crucial means of intercellular communication, facilitating the transfer of cellular components, such as lipids, proteins, and nucleic acids between cells [23,24,25]. They are characterized by high bioavailability, biostability, biocompatibility, and cargo loading capacity, which render them attractive candidates for therapeutic interventions. The unique features and functions of these vesicles are highly influenced by the cell type of origin, cellular state, and the surrounding microenvironment. These factors collectively contribute to the heterogeneous nature of EVs and emphasize the need for a comprehensive understanding of their biology to exploit their full potential in cancer therapy [26, 27]. EVs may be involved in various physiological and pathological processes, including cell cycle, apoptosis, angiogenesis, thrombosis, immune inflammation, fibrosis, and tumor development [28,29,30,31,32,33,34,35]. EVs deliver information to recipient cells through three different pathways: (1) direct contact of proteins on the EVs membrane with proteins on the recipient cell membrane, which then triggers an intracellular signaling cascade [36]; (2) fusion of the EVs membrane with the recipient cell membrane and release of its contents into the recipient cell [37]; (3) direct phagocytosis of the EVs by the target cell and internalization of the EVs into its components [38]. Based on the physical, chemical, and biological properties of EVs, different methods have been developed to isolate and purify EVs, including differential centrifugation, density gradient centrifugation, magnetic bead sorting, multimer-dependent, microfluidic, and EVs extraction kits with immunological methods [39] (Table 1). The most widely accepted method is differential centrifugation.
EVs play a key role in cellular communication between tumor cells and stromal cells in local and distant microenvironments. The TME constitutes a complex and heterogeneous milieu comprising various cellular and non-cellular components, such as blood vessels, immune cells, fibroblasts, extracellular matrix, metabolic waste products(e.g., lactic acid), and signaling molecules(e.g., chemokines, cytokines, growth factors, etc.) [50]. Low oxygen levels, high lactate levels, extracellular acidosis, and poor nutrient composition are prominent features of TME [51,52,53]. In vivo and ex vivo studies of the interaction of EVs with TME varied (Table 2). TME plays an integral role in cancer biology and is involved in tumorigenesis, progression, and response to therapy. TME harbors a plethora of immunosuppressive molecules and immunosuppressive cells, including regulatory T cells (Tregs), M2-type macrophages, TIM-3, IL-2, which collectively promote immune evasion and cancer progression. The intricate interplay between malignant, endothelial, stromal, and immune cells within the TME regulates its homeostasis and evolution. On the contrary, EVs represent pivotal factors that establish communication channels among these distinct cellular compartments.
Modulating different cells in TME with EVs to influence immunotherapy
Tumor tissues are like alien life forms that do whatever they can to survive in the organism. Tumor cells recruit normal tissue cells and induce conditions that produce favorable tumor growth. The effective amplification and metastasis of cancer cannot be separated from the immune escape ability of cancer cell or treatment-mediated immune surveillance [56,57,58,59]. EVs derived from tumor cells are crucial targets in the intricate network of tumor immunity [60]. These EVs have the ability to dampen immune function, foster the differentiation of regulatory T cells and tumor-associated macrophages, and even substitute tumor cells with immune cell assaults to bolster tumor cell immune tolerance and evade immune surveillance [61,62,63]. Cancer cell derived EVs help cancer cells grow, metastasize and become resistant to drug therapy [64, 65]. Herein, we elucidate pivotal factors in the current landscape of EVs reshaping the TME and their impact on immunotherapy (Table S1).
CD8 T cell
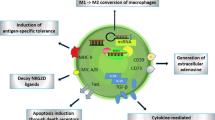
EVs assume a pivotal role in sculpting the intricate tapestry of the TME. These minute vesicles serve as discreet couriers, ferrying an array of immunosuppressive molecules that wield profound influence over the delicate balance of tumor immunity [66]. PD-L1, a linchpin molecule in the domain of immune regulation, serves as the harbinger of T cell exhaustion [67]. The indispensable role of CD8 T cells in the realm of tumor immunity should not be underestimated. Nevertheless, tumor tissues frequently pursue their own agenda by instigating the depletion of these CD8 T cells. It has been empirically validated that the interplay between PD-L1 adorning the tumor's surface and PD-1 residing on CD8 T cells, through their extracellular structural domain, culminates in the quelling of T-cell functionality [68,69,70]. Tumorous cells possess the capacity to liberate EVs, which enact direct influence upon CD8 T cells enmeshed within the intricate TME. Incipient investigations augur that extracellular vesicular PD-L1 may serve as a stratagem to counteract immune pressures during the effector stage, with a selective affinity for PD-1 + CD8 + T cells [55]. The PD-L1 adorning the surface of cancer cell EVs engages with PD-1 receptors on CD8 + T cells, resulting in the impediment of CD8 + T cell proliferation and activation [71, 72] (Fig. 1A). Notably, impeding the exodus of these EVs was accompanied by a marked deceleration in tumor progression [72]. EVs, derived from tumor cells, have emerged as pivotal arbiters in orchestrating the TME through the dissemination of their cargo. Upon internalization of TEVs, both the quantity and functionality of CD8 + T cells exhibit diminishment [73]. Furthermore, these EVs wield the capacity to modulate the expression of cell surface moieties within the TME. The bestowal of EVs upon CD8 + T cells culminates in their enervation and the concomitant suppression of tumor immunity, as evinced by a substantial reduction in the expression of TNF-α, IFN-γ, granzyme-B, and perforin [74]. It is noteworthy that the ICAM-1 expressed on cloaking PD-L1 engages with LFA-1 on the surface of CD8 + T cells, a pivotal interaction for the binding of TEXs with T cells. Simultaneously, the secretion of IFN-γ by CD8 + T cells upregulates the expression of ICAM-1 [75].

EVs regulating different cells in TME. A TEV induces CD8 T-cell depletion. B TEV activates Treg and thus suppresses CD8 T cell function. C CD4-derived EVs activate CD8 T cells and inhibit the cell cycle of tumour cells. D TEV induces a shift from M2 to M1 type macrophages. E Endocytosis of TEV-containing fatty acids leads to impaired DC cell function and reduced ability of antigen presentation to activate T cells. F TEV activates MDSCs. G TEV induces NK cell depletion. H TEV activates CAF, thereby recruiting MDSCs and Treg in TME
The impact of ncRNAs within EVs on immunotherapies in the TME is profound. These RNAs wield their influence by modulating T-cell activation and functionality, ultimately suppressing immune responses [30]. For instance, the transfer of EVs- derived circUSP7 from non-small cell lung cancer tissue to CD8 + T cells results in the suppression of miR-934 expression. This, in turn, upregulates downstream molecules of SHP2, impairing the function of CD8 + T cells. The consequence is a decrease in TNF-α and IFN-γ expression, leading to reduced CD8 + T cell counts and tumor immune evasion. Notably, in HuNSG mice endowed with a humanized immune system, high circUSP7 expression in NSCLC cells confers resistance to PD1 therapy [74]. Similarly, extracellular vesicular hsa-miR-498 originating from melanoma acts on the 3'UTR of TNF-α in T cells, resulting in downregulation of its expression. Likewise, hsa-miR-3187-3p targets the 3'UTR end of PTRRC, leading to transcriptional downregulation of its encoded CD45. These actions contribute to tumor immune evasion [73] (Fig. 2A). Given melanoma's pronounced metastatic propensity, researchers have delved into this domain and identified melanoma derived EVs that promote lymphatic vessel remodeling and expansion. These EVs also carry tumor antigens and induce CD8 + T cell apoptosis, thereby enhancing melanoma metastasis [76]. EVs derived from pancreatic cancer (PC) cells have been shown to activate the P38 MAPK signaling pathway, ultimately culminating in CD8 + T cell apoptosis. Unfortunately, the specific extracellular vesicular molecules responsible for this effect remain unclear [77]. These findings underscore the potential of extracellular vesicular ncRNAs as attractive therapeutic targets to enhance the efficacy of immunotherapies, albeit with the consequence of diminished T cell function and anti-tumor effects.

Mechanisms of remodelling TME by TEVs. A Mechanisms by which TEV affects T cells. TEV interferes with CD8 T cells and induces their depletion, while activating Treg, thereby affecting immunotherapy. B Mechanisms by which TEV affects macrophages and NK cells. TEV affects the expression of CD39 in macrophages and induces conversion to M2. TEV also induces depletion of NK cells, depriving them of their tumour-killing capacity
CD4 T cell
PD-L1 induces the generation of Tregs in a PD-1-dependent manner [78]. In head and neck squamous cell carcinoma (HNSCC), there is a significant positive correlation between the expression of FOXP3 mRNA and CD163 mRNA and TEV-PD-L1 features. Furthermore, it has been demonstrated that TEVs derived from PD-L1-rich HNSCC play a pivotal role in inducing Treg activation and differentiation [79]. Like other immune cells, the content of EVs can also upregulate PD-L1 expression in Tregs. Administration of EVs derived from oral squamous cell carcinoma (OSCC) results in an upregulation of PD-L1 expression in Tregs. Furthermore, upon uptake, these EVs carrying has_circ_0069313 facilitate Treg activation by impeding miR-325-3p-mediated Foxp3 degradation, thus promoting immune evasion [80] (Fig. 1B).
EVs derived from CD4 T cells, specifically carrying miR-25-3p, miR-155-5p, miR-215-5p, and miR-375, have been identified as key instigators of CD8 T cell-mediated anti-tumor responses. In a melanoma mouse model, these EVs effectively suppress tumor growth by activating CD8 T cells [81] ( Fig. 1C). Additionally, CD4 + T cells secrete IL-2, a crucial factor for initiating cytotoxic T lymphocyte (CTL) responses through CD8 + T cells [82]. It's worth noting that EVs derived from IL-2-stimulated CD4 T cells induce a more robust anti-tumor response in CD8 T cells when compared to those from unstimulated CD4 T cells [81]. This phenomenon represents a mechanism by which the immune system combats the immunosuppressive microenvironment in cancer.
Vδ2 T cells
Vδ2 T cells, due to their MHC-independent direct anti-tumor activity and their potential in adoptive cell therapy, hold promise as candidates for cancer immunotherapy [83,84,85,86]. Vδ2-T-Exos contain death-inducing ligands (FasL and TRAIL) as well as immune-stimulatory molecules (CD80, CD86, I and II class MHC). In immunodeficient and humanized mouse models, Vδ2-T-Exos effectively target EBV-related tumor cells and induce efficient killing of these cells through the FasL/Fas and TRAIL/DR5 pathways. This approach effectively controls EBV-related tumors, targets and efficiently eliminates EBV-related tumor cells through the FasL and TRAIL pathways and promotes the expansion of EBV antigen-specific CD4 and CD8 T cells [87].
Tumor-associated macrophage
In the cellular communication between tumor and CD8 + T cells, tumor-associated macrophage (TAM) acts as an "intermediate transduction station" to receive signals contained in EVs and deliver them to CD8 + T cells. The PD-L1 molecules exhibited on TAMs entangle with PD-1 receptors displayed on CD4 + and CD8 + effector T cells, instigating the curtailment of T-cell activity and instigating apoptosis through the inhibition of T Cell Receptor (TCR) signaling [88]. Nevertheless, the eradication of PD-L1-expressing TAM infiltration and the mitigation of CD8 + T cell suppression have been demonstrated to assume a pivotal role in enhancing the efficacy of anti-PD-L1 immunotherapy. GOLM1, through the upregulation of CSN5 expression, attenuates the ubiquitination of PD-L1, thereby enhancing the stability of PD-L1. Furthermore, GOLM1 facilitates the sorting of PD-L1 into EVs by restraining Rab27b in the trans-Golgi network region. TAMs internalize these EVs carrying PD-L1, resulting in an elevated expression of PD-L1 on TAMs compared to hepatocellular carcinoma (HCC) cells. This, in turn, induces the suppression of CD8 T cells [89]. EVs also translocate PD-L1 to other cell types in the TME, leading to immunosuppressive effects, such as monocytes、macrophages and CD4 T cells [55, 71, 72, 90]. Moreover, the utilization of HNSCC tumor cell supernatant containing TEVs precisely induce the differentiation of M0 macrophages into the M2 phenotype. Furthermore, the expression of TEV-PD-L1 characteristics is highly positively correlated with the M2 phenotype in the TME. Further investigation revealed that TEV-PD-L1 induces macrophages to differentiate into the M2 type [79]. Interestingly, extracellular vesicular components other than PD-L1 have been found to elevate PD-L1 expression in TAMs. Evidence indicates that HCC cells, under endoplasmic reticulum (ER) stress, release extracellular vesicular miR-23a-3p. This miRNA regulates PD-L1 via the PTEN-PI3K-AKT pathway. In vitro experiments confirmed that miR-23a-3p inhibits PTEN, leading to increased phosphorylated AKT and PD-L1 expression in macrophages [91].
As previously underscored, macrophages play a pivotal role in the interaction with tumor cells and CD8 + T cells. EVs originating from tumor tissues are internalized by macrophages and subsequently transmit signals to CD8 + T cells [92]. Recent investigations have unveiled that high expression of Tim-4 on lung-resident macrophages in lung cancer patients correlates with decreased levels of CD8 + T cells bearing characteristics associated with the tumor response, such as PD-1 and CD39 [93]. Adenosine suppresses immune cells, rendering them exhausted [94]. Recent studies have shown that TAMs receive EVs containing circTMEM181, leading to increased expression of MiR-488-3p, subsequently enhancing CD39 transcription. The combination of CD39 + TAMs and CD73 + tumor cells result in the degradation of extracellular ATP into adenosine monophosphate (APO), inducing CD8 + T cell exhaustion [95] (Fig. 2B). TAMs promote cancer cell proliferation and migration through extracellular vesicular signaling [96, 97]. Additionally, EVs foster an anti-inflammatory phenotype in TAMs, reshaping the immunosuppressive TME [98]. TAMs are recruited from normal macrophages by tumor tissues [99] and exhibit two distinct phenotypes: pro-inflammatory M1 and anti-inflammatory M2 [100]. EV-associated miR-145 induces polarization of macrophages towards an M2 phenotype [101] (Fig. 1D). Recent findings have demonstrated that extracellular vesicular circZNF451 derived from lung adenocarcinoma (LUAD) triggers an anti-inflammatory phenotype in macrophages, resulting in reduced cytotoxic CD8 + T cells. Transgenic mouse studies have shown that extracellular vesicular circZNF451 targeting macrophages elevates the levels of TRIM56, deubiquitinates the FXR1 protein, thereby enhancing the ELF4-IRF4 pathway, leading to macrophage differentiation into an M2 phenotype. Deletion of ELF4 in macrophages rescues the efficacy of immunotherapy [98] (Fig. 2B). EVs originating from PC cells stimulate macrophage polarization into the M2 phenotype [102,103,104,105]. This potent immunosuppressive milieu, coupled with the proliferation of immunosuppressive cells, may account for the limited success of immunotherapy in managing pancreatic cancer. However, recent research indicates that the M1/M2 classification of macrophages is not reliable, and CXCL9 and Spp1 are more persuasive markers for macrophage polarization [106]. This knowledge should be updated in future relevant studies.
Furthermore, TAMs also transmit EVs to tumor cells. In hypoxic glioblastoma, M2 macrophages transport EVs enriched with miR-501-3p to tumor cells, resulting in the downregulation of TGFBR3 expression in tumor cells. Ultimately, this process promotes tumor cell invasion and migration through the TGF-β signaling pathway, accelerating tumor progression [107]. EVs derived from TAMs are rich in miR-95 and are taken up by prostate cancer cells. In vitro and in vivo loss-of-function experiments suggest that miR-95 acts as a tumor initiator by promoting prostate cancer cell proliferation, invasion, and epithelial-mesenchymal transition by directly binding to its downstream target gene, JunB [108].
Dendritic cells
As the principal antigen-presenting cells, dendritic cells (DCs) wield undeniable influence in the realm of tumor immunotherapy. Transporting fatty acids, TEVs orchestrate a symphony of immune dysfunction within DCs, facilitating a ballet of immune evasion. Specifically, these fatty acids, in a manner contingent upon the peroxisome proliferator-activated receptor alpha (PPARα), incite an excess of lipid droplet biogenesis and augmented fatty acid oxidation (FAO), thereby shifting the metabolic course of DCs from glycolysis towards mitochondrial oxidative phosphorylation, ultimately propelling immune functional impediments in DCs [54] (Fig. 1E).
DCs derived EVs (DEX) are enriched in MHC-I molecules and are potent inducers of T-cell responses [109, 110]. Specifically, DEX generated from tumor peptide-pulsed DCs induce tumor-specific CTL response in vivo, which activates the TME and enhances anti-tumor effects. In animal models, treatment with EVs derived from DCs expressing the tumor-associated antigen afetoprotein (DEXAFP) improved the tumor immune microenvironment of primary tumors [111].
Myeloid-derived suppressor cells
While tumor progression, there is a notable increase in myeloid-derived suppressor cells (MDSCs), and TEVs play a pivotal inductive role in this process. In murine tumor models, miR-21a found within TDEs has the remarkable capacity to significantly promote the expansion and suppressive function of MDSCs by targeting Programmed Cell Death Protein 4 (PDCD4). This occurs through the activation of interleukin-6 (IL-6) autocrine signaling and the phosphorylation of the STAT3 signaling pathway, consequently expediting tumor growth [112] (Fig. 1F). Hypoxia promotes the secretion and upregulation of miR-10a and miR-21 in glioblastoma-derived extracellular vesicles (GDEs). Furthermore, hypoxia-induced glioblastoma-derived extracellular vesicles (H-GDEs) stimulate the activation and differentiation of MDSCs via miR-10a and miR-21, acting through the targeting of the Rora/IκBα/NF-κB and Pten/PI3K/AKT pathways [113].
Natural Killer cells
Natural Killer (NK) cells, owing to their potent antitumor responses, have emerged as promising candidates in cancer therapy [114, 115]. They discern tumor cells in an antigen-independent manner, endowing them with the ability to circumvent immune evasion mechanisms involving the downregulation of MHC-I expression [116, 117]. Within this context, TEVs containing circUHRF1 lead to the degradation of miR-449c-5p (Fig. 1G), subsequently diminishing the transcription of genes encoding downstream target TIM-3. This, in turn, suppresses the production of IFN-γ and TNF-α by NK cells. Furthermore, augmented intercellular communication between NK cells and HCC cells might bypass the upregulation of TIM-3, resulting in impaired NK cell functionality and a phenotypic state of exhaustion [118] (Fig. 2B).
In a select cohort of Non-Small Cell Lung Cancer (NSCLC) patients, NK cells and their extracellular vesicles, as well as circulating tumor cells (CTCs), were isolated. Interestingly, NSCLC patients exhibited a substantial abundance of NK cells and NK-derived extracellular vesicles in comparison to healthy donors. Furthermore, a negative correlation was observed between CTC numbers and NK cells, while a positive correlation existed between CTC numbers and NK-derived extracellular vesicles. This intricate relationship may stem from increased CTC quantities subjecting more circulating NK cells to stress, compounded by the immunosuppressive microenvironment, potentially inducing active release of extracellular vesicles by NK cells [119].
Cancer-associated fibroblasts
As a predominant component of TME matrix, EVs originating from cancer-associated fibroblasts (CAFs) intricately modulate cancer progression through mechanisms such as facilitating cancer cell invasion and metastasis, as well as promoting immune evasion [120,121,122,123]. Research on myofibroblasts in hepatic stellate cells (HSC) suggests that CAFs could serve as pivotal sources of PD-L1 in various cancer types. Moreover, HSC PD-L1 modulate tumor growth independently of PD-L1/PD-1-mediated immune suppression by regulating the release of paracrine factors [124]. Both CAF-EVs and normal fibroblasts-EVs contain PD-L1 protein and mRNA. These PD-L1-containing EVs can be internalized by tumor cells. Concurrently, it has been observed that CAFs-Exos upregulate PD-L1 on the surface of lung cancer cells, thereby inhibiting the peripheral blood mononuclear cells' (PBMC) ability to induce the killing of lung cancer cells [125]. Similarly, in breast cancer, CAF-Exos exhibit analogous functions. These EVs originating from CAFs significantly impede immune cell function in vivo and promote the expression of PD-L1 in breast cancer cells [124].
Tumor cells also employ EVs to activate CAFs and influence T cell functionality. Research has elucidated that within TEVs, the presence of miR-155 serves as an inducer, orchestrating the genesis of CAFs [126]. Exposed to TEVs, the invasive capacity of fibroblasts has markedly intensified, surpassing its previous magnitude by approximately 2.5-fold. Additional experimental evidence suggests that normal stromal fibroblasts undergo phenotypic and functional alterations upon contact with EVs, transforming into CAFs [127]. Moreover, in triple-negative breast cancer, miR-9 orchestrates a CAF-like phenotype in tumor cells via EV-mediated pathways [128] (Fig. 1H). CAFs foster tumor growth by establishing an immunosuppressive microenvironment that attracts MDSCs and Tregs, further dampening T cell functionality [129,130,131]. Nevertheless, it remains unclear whether this newly discovered mechanism impacted immunotherapy through extracellular vesicular signaling.
Mesenchymal stem cells
Mesenchymal stem cells (MSCs) have garnered considerable interest in the realm of cancer therapy due to their low immunogenicity and ease of isolation and cultivation from adult tissues [132]. Extracellular vesicles derived from CD90 low adipose-derived mesenchymal stem cells (ADSC-EVs) exhibit a remarkable capacity to impede tumor growth in murine models. This phenomenon is intricately linked to the reduction in tumor cell proliferation and migration mediated by ADSC-EVs, accompanied by enhanced tumor cell apoptosis. In vivo experiments underscore the significant deceleration of tumor growth when employing miR-16-5p-mimicking loaded CD90 low ADSC-EVs [133]. This implies the potential applicability of extracellular vesicles produced by mesenchymal stem cells in immunotherapy. However, harvesting a sufficient yield of endogenous EVs from MSCs for clinical and research purposes has proven to be a formidable challenge. Fortunately, recent research has unveiled that treatment with cell-relaxant B can augment vesicular membrane production [134]. Employing cell-relaxant B treatment on a specific line of human adipose tissue-derived mesenchymal stem cells (hADSC), which overexpress TRAIL, PTEN, and IFN-β1, led to the isolation of artificial vesicles. These artificial vesicles not only activate human immune cells, such as CD8 T cells, ex vivo, but also induce apoptosis in cancer cells [135].
Interestingly, EVs derived from MSCs also possess immunosuppressive properties, as exemplified by miR-222 sourced from MSC-EVs. Upon transcriptional regulation of ATF3, miR-222 activates the AKT pathway, thereby promoting the development of colorectal cancer (CRC), enhancing tumor growth, and facilitating in vivo immune evasion [136]. This peculiarity may be associated with the heterogeneous nature of MSC subpopulations.
Neurons
In tumors, those endowed with greater neural dominance exhibit heightened invasiveness [137]. As cancer progresses, neural fibers sprout and infiltrate the tumor microenvironment, and the density of these nerves within solid tumors correlates with poorer prognosis [138]. In vitro experiments have elucidated that EVs released by HNSCCs can indeed promote neural outgrowth, a phenomenon highly associated with HPV infection [139]. Given the pivotal link between HPV infection and cervical cancer, researchers have extended their observations to the realm of cervical malignancies. During cervical cancer progression, a discernible shift in neural innervation within the cervix is noted. This is exemplified by a marked reduction in the expression of β-III tubulin coinciding with a pronounced increase in TRPV1 expression, all mediated by tumor-dependent EVs [140]. The origin and mechanisms underlying the emergence of these newly formed adrenergic nerve fibers that bolster tumor growth have long been shrouded in mystery. Recent investigations have shed light on this mystery in the context of TP53-deficient oral cavity squamous cell carcinoma (OCSCC), which are notably enriched with adrenergic nerve fibers. In TP53-deficient tumors, the content of miR-34a within secreted EVs undergoes a significant reduction. Contrasting this against a control group, these EVs promote neo-adrenergic cancer-associated neurogenesis. MiR-34a, derived from OCSCC, is shuttled via EVs to cancer-associated neurons, whereby it exerts a negative modulation on EV-derived axon growth signals, thereby fostering resistance. Conversely, TP53-deficient tumors release EVs with diminished miR-34a content, neutralizing the negative axon growth signals and bolstering the number of adrenergic nerve fibers, ultimately facilitating tumor progression [141]. Emerging research has also unveiled that Schwann cell-derived EVs possess the capacity to instigate macrophage polarization towards the M2 phenotype, thereby promoting remyelination and axonal regeneration following peripheral nerve injury [142]. Regrettably, whether such a scenario unfolds within the tumor microenvironment remains elusive.
In a word, recent investigations into the mechanisms underlying EV-mediated remodeling of the tumor microenvironment have largely centered around extracellular vesicular PD-L1 and ncRNAs. Notably, the role of the adenosine pathway in immunotherapy represents a fascinating and emerging area of research that expands our understanding of this field. However, the specific genes and signaling pathways that mediate these effects remain unclear and will require further investigation in future studies. Overall, tumor cells exploit EVs to suppress immune function and evade immune surveillance, thus hampering the efficacy of immunotherapeutic approaches. Conversely, normal cells, particularly immune cells, also release EVs to counteract this phenomenon [143, 144]. The constant battle between normal and tumor cells relies heavily on the interplay of extracellular vesicular signaling. Should the immune system triumph, it eradicates the tumor and re-establish the body's homeostasis. In contrast, if the tumor cells gain the upper hand, they hijack immune cells via microenvironmental cues.
Hormonal effects in EVs remodelling TME
The interaction between tumor cells and the immune microenvironment relies upon intricate mechanisms, encompassing endocrine factors, pro-inflammatory states, and reciprocal influences among immune cells to regulate the TME [145]. Hormones are indispensable for the immune regulation within the body, and hormonal imbalances are, in part, factors contributing to tumor progression.
Estrogens are crucial drivers of estrogen receptor-positive (ER +) tumor progression. Estrogens increase the quantity of EVs in ER + breast cancer cells. ER + cell lines demonstrated a significant enrichment of Evs carrying either let-7a-5p, let-7c-5p, or let-7d-5p after treatment with 17β-estradiol, an effect not observed in ER-negative cells [146]. Interestingly, these 17β-estradiol-stimulated Evs subsequently attenuated the estrogen effect. Metastasis-associated protein 1 (MTA1) has been reported to prevent estrogen receptor-mediated transcription by interacting with histone deacetylases and nucleosome remodeling complexes [147]. MTA1 has also been identified in breast cancer-derived Evs, and it was confirmed to dampen estrogen signaling, as observed after stimulating breast cancer cells with 17β-estradiol [148]. In uterine corpus endometrial cancer, another estrogen-driven malignancy, low levels of miR-765 induced by estrogen triggered high proliferation, Epithelial–mesenchymal transition (EMT) processes, invasion, and poor prognosis via the activation of the PLP2-Notch signaling pathway. Interestingly, this effect could be partially suppressed by EVs carrying miR-765 released by CD8 T cells [149]. Estrogens offer a new perspective on the antitumor mechanisms of EVs in gynecological tumors, offering potential therapeutic avenues. Similar to gynecological tumors, prostate cancer presents issues related to androgen resistance. Unfortunately, the mechanisms of crosstalk between EVs and the immune microenvironment in this field remain unclear.
In non-hormone-driven tumors, hormones continue to play a significant role. Melatonin (MLT), known for its role in regulating circadian rhythms, has demonstrated antitumor activity in recent studies [150]. MLT modulates macrophage immune function and reduce cancer cell activity by regulating TEVs. EVs from gastric cancer(GC) cells treated with MLT promote the expression of TNF-α and CXCL10 in macrophages while suppressing IL-6 and MCP-1 expression, concurrently downregulating PD-L1 protein levels [151].
Surprisingly, hormones can also be contained within EVs. EVs from gallbladder cancer (GBC) harbor leptin and possess the capacity to upregulate leptin levels in TAMs. Elevated leptin levels activate the STAT3 signaling pathway, driving macrophage polarization towards the M2 phenotype, thereby promoting GBC cell invasion and migration [152].
In summary, hormones can modulate the reshaping of EVs in the tumor microenvironment, thereby influencing the immune system based on this principle. These mechanisms provide insights for future tumor immunotherapies.
EVs inducing pre-metastatic niche formation
The pre-metastatic niche (PMN) is an intricately orchestrated microenvironment pre-established in distant tumor-free regions or organs, primed by the primary tumor, in anticipation of widespread metastasis [153]. Its hallmarks encompass immune suppression, inflammation, angiogenesis, vascular permeability, lymphangiogenesis, extracellular matrix (ECM) remodeling, and extracellular matrix (ECM) deposition [154, 155]. As an integral facet of cellular communication, extracellular vesicles assume a paramount role in shaping the pre-metastatic niche, a function of undeniable significance.
The formation of Chronic inflammation and immunosuppressive microenvironment
The tumor microenvironment typically resides in a state of hypoxia, which stimulates the release of TEVs. Latent membrane protein 1 (LMP1) encapsulated by EVs promotes tumour growth in EBV-associated nasopharyngeal carcinomas [156] gastric cancer. In a liver metastasis model of colorectal cancer, these extracellular vesicles are absorbed by the liver. On one hand, they construct a pre-metastatic niche through the LATS2-YAP-MMP7 axis to facilitate CRC implantation in the liver, controlling the adhesion axis in hepatic tissue. On the other hand, they activate immune-suppressive signals mediated by the CD30-TRAF2-NF-κB pathway to remodel the metastatic microenvironment [157]. Within the PMN, macrophages often adopt the tumor-promoting M2 phenotype, characterized by increased Arg-1 expression and dependence on mitochondrial oxidative metabolism [158]. Macrophages treated with TEVs exhibit activation of the NF-κB pathway. This pathway utilizes HIF-1α/GLUT-1 to transport more glucose into macrophages and employs NOS2/NO to inhibit mitochondrial oxidative phosphorylation, consequently upregulating PD-L1 expression. Meanwhile, suppressed mitochondrial oxidative phosphorylation leads to the production of abundant lactate by macrophages, which, in a feedback loop, activates the NF-κB pathway to drive PD-L1 expression [159].
As previously mentioned, it is paramount not to overlook the pivotal role of let-7 s within EVs concerning the reshaping of the TME. In breast cancer, the content of Let-7 s in secreted EVs can be inhibited by Link28B, a type of RNA-binding protein. This induction prompts neutrophil infiltration into lung tissue and N2 transformation, and as the tumor progresses, this trend becomes increasingly pronounced. Furthermore, N2-transformed neutrophils suppress T cell proliferation, activation, and Th1 differentiation. Neutrophils undergoing N2 transformation exhibit high PD-L1 expression, limiting the tumor-killing capabilities of CD8 T cells. Additionally, this downregulation results in the upregulation of CXCL, IL-10, and IL-28 levels in lung fibroblasts, neutrophils, and macrophages, respectively [160]. These alterations in distant organ microenvironments create a conducive soil—an immunosuppressive PMN—that facilitates tumor metastasis.
Altered vascular permeability and angiogenesis
In both in vivo and in vitro experiments, research has demonstrated that EVs play a pivotal role in promoting angiogenesis and altering vascular permeability [161]. Among these mechanisms, sprouting stands out as a vital form of neoangiogenesis [162]. EVs rich in clathrin light chain A (CLTA) have been shown to stimulate endothelial cells, further enhancing their tube-forming and sprouting capabilities [163]. Tip cells, located at the leading edge of sprouting, respond to signals in the microenvironment, thus influencing the growth trajectory of nascent blood vessels [164]. Evidence suggests that EVs derived from colorectal cancer cells, specifically those carrying circTUBGCP4, promote tip cell formation, angiogenesis, and consequently, tumor metastasis [165].
Adjacent vascular endothelial cells are linked and solidify endothelial integrity through vascular endothelial cadherin (VE-Cad) at their extracellular domains [166]. EVs from mesenchymal CRC cells transport miR-27b-3p into vascular endothelial cells. Further investigation has revealed that miR-27b-3p exerts its effects by directly binding to the 3′UTR of VE-Cad and p120, leading to post-transcriptional suppression of VE-Cad/P120 protein. This disruption compromises endothelial junction integrity and increases vascular permeability [167].
Stromal component remodelling
Ovarian cancer derived EVs exhibit a proclivity for reprogramming stromal fibroblasts to shape the pre-metastatic niche (PMN) by secreting cytokines that promote metastasis via alterations in Hippo/YAP1 signaling cascade [168]. Following treatment with EVs rich in Cav-1 derived from breast cancer (BC), the expression of extracellular matrix (ECM) component proteins such as emilin1, nidogen, TnC, and FN increases in lung fibroblasts. Additionally, Cav-1-containing EVs mitigate the consequences of the silenced TnC gene in lung fibroblasts. This underscores the potential of Cav-1 in BC-derived EVs to transport TnC into lung stromal cells, facilitating the deposition of extracellular matrix proteins and acting as signaling molecules to promote the formation of the PMN during BC lung metastasis [169]. Fibroblasts, as vital constituents of the stroma, respond to signals conveyed by TEVs by upregulating CCL1 expression, thereby inducing the polarization of local microenvironmental Tregs [170]. In specific metastatic microenvironments, TEVs also play pivotal roles. In a bone metastasis model, the contents of TEVs, including miR-21, directly bind to PDCD4, promoting the activation and differentiation of osteoclasts without affecting osteoblasts. Consequently, this enhances local bone tissue osteoclast activity, leading to increased bone loss [171].
Remodeling lymph node microenvironment
Tumor-draining lymph nodes hold a pivotal role in the progression of cancer, serving as the initial destination of tumor metastasis and providing a significant prognostic indicator in various cancer types [172, 173]. These lymph nodes also have a crucial function in eliciting tumor-specific immunity and driving immunotherapeutic responses [174]. Tumor-secreted EVs play a regulatory role in the cytokine milieu within the lymph nodes, modulating the activity of immune cells. Specifically, they induce immunosuppression by upregulating pro-inflammatory cytokines such as IL-6 and TNF-α in lymph nodes, inhibiting tumor immune responses [175]. These EVs were taken up by macrophages and lymphocytes and decreased the cross-presentation of tumor antigens from DC cells, leading to a significant decrease in CD8 + T cell function in animal models [76] (Fig. 3a). Previous studies showed that lymphoepithelial cells in lymph nodes expressing PD-L1 triggered apoptosis of CD8 + T cells with tumor antigen specificity, thereby suppressing tumor immunity [176]. Another study found that TEVs, carrying PD-L1 reduced the number of CD8 + T cells infiltrating lymph nodes [55] (Fig. 3b).

Extracellular vesicular remodelling of the lymphatic vascular microenvironment. A Impaired DC antigen presentation due to poor internalization of TEVs carrying tumor antigens. B TEV impairs and depletes TAM function. C TEV induces apoptosis of CD8T cells. D TEV activates B-cell anti-tumour IgG responses. E TEV induces lymphatic vessel endothelial cell proliferation, lymphatic vessel dilation and gap enlargement. F Metastasis of tumour cells from the dilated lymphatic vessel gap
In the early stages of tumorigenesis, macrophages residing within the lymph nodes act as a physical barrier to hinder the dissemination of TEVs. However, as the tumor advances, this macrophage barricade is dismantled, enabling the entry of TEVs carrying tumor-specific antigens deep into the cortical regions (Fig. 3c). Herein, TEVs stimulate B cells to generate pro-tumorigenic IgG, thereby promoting tumor progression [177] (Fig. 3d). Recently, VCAM-1 was found to play a crucial role in the transport of tumor-derived EVs to lymphatic vessels in a mouse model of B16F10 melanoma, leading to lymphatic vessel endothelial cell proliferation and LN remodeling, which ultimately resulted in lymphatic vessel expansion [76] (Fig. 3e). Furthermore, EVs derived from CRC were found to upregulate VEGFC expression in macrophages located in lymphatic vessels [178], which promoted tumor-associated lymphangiogenesis [179] (Fig. 3f).
EVs as biomarkers in immunotherapy
Biomarkers in the realm of biology serve as indicators that reflect one or more physiological processes, pathogenic mechanisms, or responses to therapeutic interventions [180]. EVs play a pivotal role across a spectrum of malignancies (Fig. 4)). Tumor PD-L1 has already been employed as a predictive biomarker for clinical responses to anti-PD-1 therapies [181]. Intriguingly, PD-L1 encapsulated within EVs has garnered significant attention as an enhanced biomarker and has been validated across various cancer models [182, 183]. A groundbreaking development in this area involves the utilization of an immunogold biochip, enabling the quantification of RNA and proteins within individual EVs. This breakthrough substantially enhances the detection rate of EVs containing PD-1/PD-L1 mRNA [184].

The effect of TEV on TME in different cancer types. TEV remodels TME in different cancer types, thus contributing to the shaping of a tumor-favorable microenvironment
EVs, at their core, constitute carriers of substances that facilitate intercellular communication signals. Certain cargo within EVs themselves can signify the body's immune state and its anti-tumor potency. Reports have linked high expression of LINC02096 (RIME) in plasma-derived EVs from cancer patients to reduced sensitivity to PD-1 monoclonal antibody treatment and adverse prognoses. Elevated RIME expression also correlates with diminished tumor-killing capabilities of CTLs [185].
A risk score reflecting the prognosis of cancer patients has been established based on EV proteins TNFRSF10B and ILF3 secreted by CAFs. Interestingly, no significant positive correlation has been discovered between immune cells and this risk score. However, when the TIDE score was employed to predict the outcomes and immune therapy responses of cancer patients [186], it was found that a high-risk score was significantly associated with poorer immune therapy (anti-PD1) efficacy. Survival analysis results further indicate that the high-risk group exhibits a less favorable prognosis after receiving immune therapy [187]. While certain EVs may not exhibit a clear association with immune therapy outcomes, they are closely linked to immune-related adverse events during such therapy. For instance, EV-ICOS and EV-IDO1 have proven to be robust predictors of immune-related adverse events in GC patients undergoing ICI therapy [188].
Although the available body of research is quite limited, some studies have begun to explore whether the contents of novel EVs can serve as biomarkers for predicting immune therapy responses. Recent research has unveiled that EVs harboring the urokinase-type plasminogen activator receptor (uPAR) may serve as novel biomarkers for intrinsic resistance to ICIs [189]. uPAR is intricately linked to tumor progression and metastasis [190]. Among patients treated with Pembrolizumab and nivolumab for immune therapy, non-responders exhibited baseline levels of uPAR + EVs similar to responders, albeit with significant differences in the source of these EVs. In contrast to responders, non-responder uPAR EVs, originating from melanoma cells, CD8 T cells, and DC cells, displayed significantly lower baseline levels. Significantly, uPAR EVs are correlated with patients' progression-free survival (PFS) and overall survival (OS), inversely associated with treatment outcomes [189]. Similarly, a comprehensive follow-up investigation discovered that the baseline high expression of TGF-β in EVs is associated with non-responsiveness to ICI therapy, as well as shorter PFS and OS. As a predictive biomarker, its efficacy surpasses that of circulating TGF-β levels and tissue PD-L1 content [183].
Engineering EVs to reshape the TME to impact immunotherapy
Due to the pivotal role EVs play in tumor metastasis and therapy, researchers have embarked on the isolation and engineering of EVs to incorporate specific genes or proteins for employment in cancer treatment [191]. These bioengineered EVs are characterized by exceptional controllability, stability, and biocompatibility. They efficiently traverse vascular barriers and cellular membranes, enabling effective delivery of therapeutic agents into the intracellular milieu [192,193,194]. In recent years, engineered EVs have emerged prominently across various domains. They are manipulated via biotechnological approaches to harbor distinct cargoes, including therapeutic molecules or diagnostic markers, and are tailored for targeting specific cells or tissues. The versatility, stability, and biocompatibility of engineered EVs render them an appealing choice in biomedical applications, particularly in the realm of cancer research and treatment. Their potential to surmount biological barriers such as cell membranes and the blood–brain barrier holds exciting prospects for the development of novel therapies for a range of conditions, including oncology [195], cardiovascular diseases [196], tissue regeneration and repair [197], and neurological disorders [198], among others (Fig. 5).

Schematic representation of engineered EVs implementing immunotherapy in a mouse model. After intravenous injection, engineered EVs carrying therapeutic molecules are targeted to the tumour site, followed by the release of immunotherapy-related drugs into the TME
Engineered EVs offer promising therapeutic opportunities for targeting and inhibiting tumor growth. The antitumor effects of EVs derived from M1 cells have been demonstrated, as they induced apoptosis in tumor cells. However, the underlying mechanism remains unclear [199]. Recently, CAR-T-derived EVs were found to inhibit the growth of triple-negative breast cancer (TNBC) cells expressing MSLn, possibly through the actions of perforin and granzyme B [200]. Researchers loaded target SiRNA into bone marrow MSC-derived EVs via electroporation and exploited the tumor homing ability of these EVs to block signaling communication between tumor cells and macrophages, which in turn inhibits TAM polarization and enhances immunotherapy in pancreatic ductal carcinoma [201].
Engineered EVs demonstrated their potential in improving immune cell function. A novel hGLV EV overexpressing CD47 has been found to enhance macrophage-mediated tumor cell phagocytosis by blocking CD47 signaling in tumor cells. Furthermore, the fusion of immune adjuvant with hGLV facilitated the maturation of DC cells, which in turn increased the infiltration of CD8 + and CD4 + T cells in tumors, resulting in improved anti-tumor immunity [202]. The recent rise of chimeric antigen receptor T (CAR T) cell therapy as a personalized, sequential cell therapy has gained much attention in the "war" against tumors [203, 204]. Taking cues from the personalized and sequential CAR T cell therapy, researchers edited DEXs to contain the CAR component of the MHC antigen peptide complex and the CD86 co-stimulatory molecule to activate T cells. Introduction of aCD3 and aEGFR further enhanced the binding of T cells to tumor tissue, leading to effective anti-tumor effects, particularly in solid tumors, and inhibition of tumor metastasis [205]. CAFs are also another important target for research. Previous studies showed that CAFs play a crucial role in the TME, and it is also an important mesenchymal target for immunotherapy of solid tumors [206]. To target CAFs, researchers developed eNV-FAP-containing EVs that trigger a strong specific cytotoxic effect on CTL immune responses leading to CAFs depletion. Furthermore, EVs-induced antitumor immunity also promoted iron death of tumor cells, which may be related to the release of IFN-γ and FAP + CAFs depletion by CTL [207].
Of course, engineered EVs also are used to make tumor vaccines. DCs, being essential for innate and adaptive immunity regulation in the TME, are the target of several DC-targeted vaccines in previous clinical trials to improve cancer immunotherapy. In previous clinical trials, several DC-targeted vaccines developed to improve cancer immunotherapy [208, 209]. The most recently developed EV vaccine against DC cells achieved good tumor suppression in a mouse model of breast cancer [210]. In addition, engineered EVs are loaded with drugs from other therapies, providing a combination therapy effect. Incorporation of PH20 with FA into EVs and injection into a mouse model revealed a reduction in infiltration of immunosuppressive cells such as Treg. It also reduced hyaluronidase-induced tumor cell metastasis and allowed enhanced delivery of chemotherapy through FA-modified tumor targeting [211]. Additionally, genetically engineered EVs fused with drug-loaded thermosensitive liposomes, named hGLV, have enabled photothermal therapy (PTT) under laser irradiation to kill tumors and tumor cell lysis, producing tumor-associated antigens, and promoting the maturation of dendritic cells to trigger a robust immune response with the help of co-encapsulated immune adjuvants [202]. Surface modification of LGALS-9-containing EVs with oxaliplatin (OXA) prodrugs also showed significant therapeutic benefits in cancer treatment when used in combination with chemotherapy [201].
It is worth mentioning that studies have reported the use of engineered EVPD1 to target PD-L1 in tumor cells to block the immunosuppressive effects of PD-L1 [212]. However, no engineered EVs are available for this pathway for the time being.
Conclusion
In the intricate interplay between tumour and normal cells, EVs have emerged as key mediators of immune suppression and evasion. TEVs have been shown to subvert immune function, impairing the efficacy of immunotherapy. In contrast, normal cells, especially immune cells, also release EVs to counteract these effects, highlighting the crucial role of extracellular vesicular signals in regulating the immune response. The outcome of this struggle determines the fate of the tumour and the host. In the preclinical experimental phase, encompassing EVs including exosomes, their potential to impact immunotherapy in various cancers such as melanoma, lung cancer, liver cancer, colorectal cancer, breast cancer, and ovarian cancer has been observed (Table S1). Typically, TEVs harbor a plethora of immunosuppressive factors, such as PD-L1 and ncRNAs. However, it is noteworthy that interrupting EV release or taking intervention measures to impede the interaction between EV contents and receptor molecules can partially reverse this microenvironmental remodeling. Consequently, an augmentation in the efficacy of immunotherapy is evident. This immunotherapeutic sensitization is often reliant on the specific or signature molecules expressed by tumor cells or recipient cells. Deviating from this specificity may entail severe therapeutic side effects. As aforementioned, the body's immune system has persistently engaged in a relentless battle against tumors. Leveraging EVs generated by the immune system to combat cancer is another viable approach. Furthermore, the utilization of engineered EVs holds immense potential within the realm of tumor immunotherapy. These EVs possess distinctive targeting properties, allowing for the encapsulation of specific substances tailored to enhance therapeutic effects. Selecting appropriate EVs and cellular targets, standardizing engineered EVs, and developing reliable methods for EV extraction and quantification remain significant challenges. Regrettably, despite encouraging results in animal models, clinical translation remains in its infancy. The optimal therapeutic modalities, cargo selection, methods of EV cargo loading, and appropriate dosages for tumor treatment remain elusive. Perhaps drawing from the successful experiences of monoclonal antibody-based targeted therapies, proteins encapsulated within EVs may emerge as a predominant form of treatment in the future.
Nonetheless, the potential of EVs-mediated remodelling of the TME for enhancing immunotherapeutic sensitivity cannot be ignored. While more extensive research and validation are necessary, EVs offer unparalleled opportunities for revolutionizing tumour immunotherapy. As our understanding of EV biology deepens, we anticipate that these tiny vesicles will play an increasingly important role in the development of novel cancer treatments.
Availability of data and materials
Not applicable.
References
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70:86–104.
Frankel T, Lanfranca MP, Zou W. The role of tumor microenvironment in cancer immunotherapy. Adv Exp Med Biol. 2017;1036:51–64.
Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18:498–513.
Yang M, Yang F, Chen W, Liu S, Qiu L, Chen J. Bacteria-mediated cancer therapies: opportunities and challenges. Biomater Sci. 2021;9:5732–44.
Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17–35.
Sundararajan V, Sarkar FH, Ramasamy TS. The multifaceted role of exosomes in cancer progression: diagnostic and therapeutic implications [corrected]. Cell Oncol (Dordr). 2018;41:223–52.
Fontana S, Saieva L, Taverna S, Alessandro R. Contribution of proteomics to understanding the role of tumor-derived exosomes in cancer progression: state of the art and new perspectives. Proteomics. 2013;13:1581–94.
Wendler F, Favicchio R, Simon T, Alifrangis C, Stebbing J, Giamas G. Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene. 2017;36:877–84.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28.
Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. 2013;6:74.
Seo N, Akiyoshi K, Shiku H. Exosome-mediated regulation of tumor immunology. Cancer Sci. 2018;109:2998–3004.
Hombach S, Kretz M. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3–17.
Zhang Q, Wu E, Tang Y, Cai T, Zhang L, Wang J, Hao Y, Zhang B, Zhou Y, Guo X, et al. Deeply mining a universe of peptides encoded by long noncoding RNAs. Mol Cell Proteomics. 2021;20: 100109.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Guil S, Esteller M. RNA-RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci. 2015;40:248–56.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18.
Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–9.
Tang XH, Guo T, Gao XY, Wu XL, Xing XF, Ji JF, Li ZY. Exosome-derived noncoding RNAs in gastric cancer: functions and clinical applications. Mol Cancer. 2021;20:99.
Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). 2013;91:431–7.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Chevillet JR, Kang Q, Ruf IK, Briggs HA, Vojtech LN, Hughes SM, Cheng HH, Arroyo JD, Meredith EK, Gallichotte EN, et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A. 2014;111:14888–93.
Subra C, Laulagnier K, Perret B, Record M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie. 2007;89:205–12.
Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, Cao S, Mukhopadhyay D, Huebert RC, Shah VH. Exosome adherence and internalization by hepatic stellate cells triggers Sphingosine 1-Phosphate-dependent migration. J Biol Chem. 2015;290:30684–96.
Bell BM, Kirk ID, Hiltbrunner S, Gabrielsson S, Bultema JJ. Designer exosomes as next-generation cancer immunotherapy. Nanomedicine. 2016;12:163–9.
Syn NL, Wang L, Chow EK, Lim CT, Goh BC. Exosomes in cancer nanomedicine and immunotherapy: prospects and challenges. Trends Biotechnol. 2017;35:665–76.
Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinform. 2015;13:17–24.
Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. 2019;18:75.
Zhang L, Yu D. Exosomes in cancer development, metastasis, and immunity. Biochim Biophys Acta Rev Cancer. 2019;1871:455–68.
Ocansey DKW, Zhang L, Wang Y, Yan Y, Qian H, Zhang X, Xu W, Mao F. Exosome-mediated effects and applications in inflammatory bowel disease. Biol Rev Camb Philos Soc. 2020;95:1287–307.
Shen M, Shen Y, Fan X, Men R, Ye T, Yang L. Roles of macrophages and exosomes in liver diseases. Front Med (Lausanne). 2020;7: 583691.
Zheng D, Huo M, Li B, Wang W, Piao H, Wang Y, Zhu Z, Li D, Wang T, Liu K. The role of exosomes and exosomal MicroRNA in cardiovascular disease. Front Cell Dev Biol. 2020;8: 616161.
Zifkos K, Dubois C, Schäfer K. Extracellular vesicles and thrombosis: update on the clinical and experimental evidence. Int J Mol Sci. 2021;22:9317.
Xu Z, Chen Y, Ma L, Chen Y, Liu J, Guo Y, Yu T, Zhang L, Zhu L, Shu Y. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol Ther. 2022;30:3133–54.
Zhang B, Wang M, Gong A, Zhang X, Wu X, Zhu Y, Shi H, Wu L, Zhu W, Qian H, Xu W. HucMSC-Exosome mediated-Wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2015;33:2158–68.
Pironti G, Strachan RT, Abraham D, Mon-Wei YuS, Chen M, Chen W, Hanada K, Mao L, Watson LJ, Rockman HA. Circulating exosomes induced by cardiac pressure overload contain functional Angiotensin II type 1 receptors. Circulation. 2015;131:2120–30.
Mulcahy LA, Pink RC, Carter DR: Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles 2014;3.
Mateescu B, Kowal EJ, van Balkom BW, Bartel S, Bhattacharyya SN, Buzás EI, Buck AH, de Candia P, Chow FW, Das S, et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J Extracell Vesicles. 2017;6:1286095.
Gardiner C, Di Vizio D, Sahoo S, Théry C, Witwer KW, Wauben M, Hill AF. Techniques used for the isolation and characterization of extracellular vesicles: results of a worldwide survey. J Extracell Vesicles. 2016;5:32945.
Cvjetkovic A, Lötvall J, Lässer C: The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J Extracell Vesicles 2014;3.
Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015;65:783–97.
Cheng S, Li Y, Yan H, Wen Y, Zhou X, Friedman L, Zeng Y. Advances in microfluidic extracellular vesicle analysis for cancer diagnostics. Lab Chip. 2021;21:3219–43.
Li P, Kaslan M, Lee SH, Yao J, Gao Z. Progress in exosome isolation techniques. Theranostics. 2017;7:789–804.
Lin S, Yu Z, Chen D, Wang Z, Miao J, Li Q, Zhang D, Song J, Cui D. Progress in microfluidics-based exosome separation and detection technologies for diagnostic applications. Small. 2020;16: e1903916.
Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ, et al. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine. 2011;7:780–8.
Soo CY, Song Y, Zheng Y, Campbell EC, Riches AC, Gunn-Moore F, Powis SJ. Nanoparticle tracking analysis monitors microvesicle and exosome secretion from immune cells. Immunology. 2012;136:192–7.
Szatanek R, Baj-Krzyworzeka M, Zimoch J, Lekka M, Siedlar M, Baran J. The methods of choice for Extracellular Vesicles (EVs) characterization. Int J Mol Sci. 2017;18:1153.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
Goliwas KF, Deshane JS, Elmets CA, Athar M. Moving immune therapy forward targeting TME. Physiol Rev. 2021;101:417–25.
Paskeh MDA, Entezari M, Mirzaei S, Zabolian A, Saleki H, Naghdi MJ, Sabet S, Khoshbakht MA, Hashemi M, Hushmandi K, et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J Hematol Oncol. 2022;15:83.
Shanmugam MK, Warrier S, Kumar AP, Sethi G, Arfuso F. Potential role of natural compounds as anti-angiogenic agents in cancer. Curr Vasc Pharmacol. 2017;15:503–19.
Ma Z, Wang LZ, Cheng JT, Lam WST, Ma X, Xiang X, Wong AL, Goh BC, Gong Q, Sethi G, Wang L. Targeting Hypoxia-inducible factor-1-Mediated metastasis for cancer therapy. Antioxid Redox Signal. 2021;34:1484–97.
Yin X, Zeng W, Wu B, Wang L, Wang Z, Tian H, Wang L, Jiang Y, Clay R, Wei X, et al. PPARα inhibition overcomes tumor-derived Exosomal lipid-induced dendritic cell dysfunction. Cell Rep. 2020;33: 108278.
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6.
Parcesepe P, Giordano G, Laudanna C, Febbraro A, Pancione M. Cancer-associated immune resistance and evasion of immune surveillance in colorectal cancer. Gastroenterol Res Pract. 2016;2016:6261721.
Stewart TJ, Smyth MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev. 2011;30:125–40.
Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer. Cancer Lett. 2015;356:192–6.
Wu Q, Jiang L, Li SC, He QJ, Yang B, Cao J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol Sin. 2021;42:1–9.
Gao L, Wang L, Dai T, Jin K, Zhang Z, Wang S, Xie F, Fang P, Yang B, Huang H, et al. Tumor-derived exosomes antagonize innate antiviral immunity. Nat Immunol. 2018;19:233–45.
Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93.
Greening DW, Gopal SK, Xu R, Simpson RJ, Chen W. Exosomes and their roles in immune regulation and cancer. Semin Cell Dev Biol. 2015;40:72–81.
Webber J, Yeung V, Clayton A. Extracellular vesicles as modulators of the cancer microenvironment. Semin Cell Dev Biol. 2015;40:27–34.
Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32:623–42.
Milman N, Ginini L, Gil Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist Updat. 2019;45:1–12.
Czernek L, Düchler M. Functions of cancer-derived extracellular vesicles in immunosuppression. Arch Immunol Ther Exp (Warsz). 2017;65:311–23.
Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol. 2020;21:442–54.
Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–201.
Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, Cha JH, Hou J, Hsu JL, Sun L, Hung MC. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018;28:862–4.
Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, Blelloch R. Suppression of Exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177:414-427.e413.
Vignard V, Labbé M, Marec N, André-Grégoire G, Jouand N, Fonteneau JF, Labarrière N, Fradin D. MicroRNAs in tumor exosomes drive immune escape in melanoma. Cancer Immunol Res. 2020;8:255–67.
Chen SW, Zhu SQ, Pei X, Qiu BQ, Xiong D, Long X, Lin K, Lu F, Xu JJ, Wu YB. Cancer cell-derived exosomal circUSP7 induces CD8(+) T cell dysfunction and anti-PD1 resistance by regulating the miR-934/SHP2 axis in NSCLC. Mol Cancer. 2021;20:144.
Zhang W, Zhong W, Wang B, Yang J, Yang J, Yu Z, Qin Z, Shi A, Xu W, Zheng C, et al. ICAM-1-mediated adhesion is a prerequisite for exosome-induced T cell suppression. Dev Cell. 2022;57(329–343): e327.
Leary N, Walser S, He Y, Cousin N, Pereira P, Gallo A, Collado-Diaz V, Halin C, Garcia-Silva S, Peinado H, Dieterich LC. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J Extracell Vesicles. 2022;11: e12197.
Shen T, Huang Z, Shi C, Pu X, Xu X, Wu Z, Ding G, Cao L. Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the p38 MAPK-mediated endoplasmic reticulum stress. Faseb j. 2020;34:8442–58.
Zhao Y, Zhang Z, Lei W, Wei Y, Ma R, Wen Y, Wei F, Fan J, Xu Y, Chen L, et al. IL-21 Is an Accomplice of PD-L1 in the induction of PD-1-Dependent Treg generation in head and neck cancer. Front Oncol. 2021;11: 648293.
Wei F, Fang R, Lyu K, Liao J, Long Y, Yang J, Wen W, Sun W. Exosomal PD-L1 derived from head and neck squamous cell carcinoma promotes immune evasion by activating the positive feedback loop of activated regulatory T cell-M2 macrophage. Oral Oncol. 2023;145: 106532.
Chen Y, Li Z, Liang J, Liu J, Hao J, Wan Q, Liu J, Luo C, Lu Z. CircRNA has_circ_0069313 induced OSCC immunity escape by miR-325-3p-Foxp3 axes in both OSCC cells and Treg cells. Aging (Albany NY). 2022;14:4376–89.
Shin S, Jung I, Jung D, Kim CS, Kang SM, Ryu S, Choi SJ, Noh S, Jeong J, Lee BY, et al. Novel antitumor therapeutic strategy using CD4(+) T cell-derived extracellular vesicles. Biomaterials. 2022;289: 121765.
Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–47.
Sicard H, Ingoure S, Luciani B, Serraz C, Fournié JJ, Bonneville M, Tiollier J, Romagné F. In vivo immunomanipulation of V gamma 9V delta 2 T cells with a synthetic phosphoantigen in a preclinical nonhuman primate model. J Immunol. 2005;175:5471–80.
Hayday AC. γδ T cell update: adaptate orchestrators of immune surveillance. J Immunol. 2019;203:311–20.
Silva-Santos B, Mensurado S, Coffelt SB. γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat Rev Cancer. 2019;19:392–404.
Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, Lorenc A, Vantourout P, Gazinska P, Grigoriadis A, et al. An innate-like Vδ1(+) γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med. 2019;11:eaax9364.
Wang X, Xiang Z, Liu Y, Huang C, Pei Y, Wang X, Zhi H, Wong WH, Wei H, Ng IO, et al. Exosomes derived from Vδ2-T cells control Epstein-Barr virus-associated tumors and induce T cell antitumor immunity. Sci Transl Med. 2020;12:3426.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9.
Chen J, Lin Z, Liu L, Zhang R, Geng Y, Fan M, Zhu W, Lu M, Lu L, Jia H, et al. GOLM1 exacerbates CD8(+) T cell suppression in hepatocellular carcinoma by promoting exosomal PD-L1 transport into tumor-associated macrophages. Signal Transduct Target Ther. 2021;6:397.
Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL. Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res. 2018;24:896–905.
Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, Wang F, Li X, Liu Q, Li Y, et al. Endoplasmic reticulum stress causes liver cancer cells to release Exosomal miR-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology. 2019;70:241–58.
Cianciaruso C, Beltraminelli T, Duval F, Nassiri S, Hamelin R, Mozes A, Gallart-Ayala H, Ceada Torres G, Torchia B, Ries CH, et al. Molecular profiling and functional analysis of macrophage-derived tumor extracellular vesicles. Cell Rep. 2019;27:3062-3080.e3011.
Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N, Zago G, Shah NS, Sharma SK, Mattar M, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell. 2021;39:973-988.e979.
Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90:1600–10.
Lu JC, Zhang PF, Huang XY, Guo XJ, Gao C, Zeng HY, Zheng YM, Wang SW, Cai JB, Sun QM, et al. Amplification of spatially isolated adenosine pathway by tumor-macrophage interaction induces anti-PD1 resistance in hepatocellular carcinoma. J Hematol Oncol. 2021;14:200.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62.
Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6:18.
Gao J, Ao YQ, Zhang LX, Deng J, Wang S, Wang HK, Jiang JH, Ding JY. Exosomal circZNF451 restrains anti-PD1 treatment in lung adenocarcinoma via polarizing macrophages by complexing with TRIM56 and FXR1. J Exp Clin Cancer Res. 2022;41:295.
Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–91.
Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front Immunol. 2019;10:1084.
Shinohara H, Kuranaga Y, Kumazaki M, Sugito N, Yoshikawa Y, Takai T, Taniguchi K, Ito Y, Akao Y. Regulated polarization of tumor-associated macrophages by miR-145 via colorectal cancer-derived extracellular vesicles. J Immunol. 2017;199:1505–15.
Linton SS, Abraham T, Liao J, Clawson GA, Butler PJ, Fox T, Kester M, Matters GL. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS One. 2018;13: e0206759.
Chang YT, Peng HY, Hu CM, Huang SC, Tien SC, Jeng YM. Pancreatic cancer-derived small extracellular vesical Ezrin regulates macrophage polarization and promotes metastasis. Am J Cancer Res. 2020;10:12–37.
Wang S, Gao Y. Pancreatic cancer cell-derived microRNA-155-5p-containing extracellular vesicles promote immune evasion by triggering EHF-dependent activation of Akt/NF-κB signaling pathway. Int Immunopharmacol. 2021;100: 107990.
Xavier CPR, Castro I, Caires HR, Ferreira D, Cavadas B, Pereira L, Santos LL, Oliveira MJ, Vasconcelos MH. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett. 2021;501:210–23.
Bill R, Wirapati P, Messemaker M, Roh W, Zitti B, Duval F, Kiss M, Park JC, Saal TM, Hoelzl J, et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science. 2023;381:515–24.
Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, Zou Y, Chen S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-β signaling pathway. J Exp Clin Cancer Res. 2019;38:310.
Guan H, Peng R, Fang F, Mao L, Chen Z, Yang S, Dai C, Wu H, Wang C, Feng N, et al. Tumor-associated macrophages promote prostate cancer progression via exosome-mediated miR-95 transfer. J Cell Physiol. 2020;235:9729–42.
Théry C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147:599–610.
Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208.
Lu Z, Zuo B, Jing R, Gao X, Rao Q, Liu Z, Qi H, Guo H, Yin H. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol. 2017;67:739–48.
Zhang X, Li F, Tang Y, Ren Q, Xiao B, Wan Y, Jiang S. miR-21a in exosomes from Lewis lung carcinoma cells accelerates tumor growth through targeting PDCD4 to enhance expansion of myeloid-derived suppressor cells. Oncogene. 2020;39:6354–69.
Guo X, Qiu W, Liu Q, Qian M, Wang S, Zhang Z, Gao X, Chen Z, Xue H, Li G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten pathways. Oncogene. 2018;37:4239–59.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–86.
Gang M, Wong P, Berrien-Elliott MM, Fehniger TA. Memory-like natural killer cells for cancer immunotherapy. Semin Hematol. 2020;57:185–93.
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23.
Liu D, Jenkins RW, Sullivan RJ. Mechanisms of resistance to immune checkpoint blockade. Am J Clin Dermatol. 2019;20:41–54.
Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM, Cai JB, Ke AW. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110.
Kang YT, Niu Z, Hadlock T, Purcell E, Lo TW, Zeinali M, Owen S, Keshamouni VG, Reddy R, Ramnath N, Nagrath S. On-Chip biogenesis of circulating NK cell-derived exosomes in non-small cell lung cancer exhibits antitumoral activity. Adv Sci (Weinh). 2021;8:2003747.
Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, Tan P, Ishimoto T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38:4887–901.
Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16:282–95.
Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: cancer-associated fibroblasts and their markers. Int J Cancer. 2020;146:895–905.
Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev. 2021;101:147–76.
Sun L, Wang Y, Wang X, Navarro-Corcuera A, Ilyas S, Jalan-Sakrikar N, Gan C, Tu X, Shi Y, Tu K, et al. PD-L1 promotes myofibroblastic activation of hepatic stellate cells by distinct mechanisms selective for TGF-β receptor I versus II. Cell Rep. 2022;38: 110349.
Jiang Y, Wang K, Lu X, Wang Y, Chen J. Cancer-associated fibroblasts-derived exosomes promote lung cancer progression by OIP5-AS1/ miR-142-5p/ PD-L1 axis. Mol Immunol. 2021;140:47–58.
Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, Chen X, Yao W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015;106:1362–9.
Giusti I, Di Francesco M, D’Ascenzo S, Palmerini MG, Macchiarelli G, Carta G, Dolo V. Ovarian cancer-derived extracellular vesicles affect normal human fibroblast behavior. Cancer Biol Ther. 2018;19:722–34.
Baroni S, Romero-Cordoba S, Plantamura I, Dugo M, D’Ippolito E, Cataldo A, Cosentino G, Angeloni V, Rossini A, Daidone MG, Iorio MV. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016;7: e2312.
Lin Y, Li B, Yang X, Cai Q, Liu W, Tian M, Luo H, Yin W, Song Y, Shi Y, He R. Fibroblastic FAP promotes intrahepatic cholangiocarcinoma growth via MDSCs recruitment. Neoplasia. 2019;21:1133–42.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463-479.e410.
Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell. 2019;177:556-571.e516.
Chulpanova DS, Kitaeva KV, Tazetdinova LG, James V, Rizvanov AA, Solovyeva VV. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front Pharmacol. 2018;9:259.
Li T, Zhou X, Wang J, Liu Z, Han S, Wan L, Sun X, Chen H. Adipose-derived mesenchymal stem cells and extracellular vesicles confer antitumor activity in preclinical treatment of breast cancer. Pharmacol Res. 2020;157: 104843.
Oshchepkova A, Neumestova A, Matveeva V, Artemyeva L, Morozova K, Kiseleva E, Zenkova M, Vlassov V. Cytochalasin-B-inducible nanovesicle mimics of natural extracellular vesicles that are capable of nucleic acid transfer. Micromachines (Basel). 2019;10:750.
Chulpanova DS, Gilazieva ZE, Akhmetzyanova ER, Kletukhina SK, Rizvanov AA, Solovyeva VV. Cytochalasin B-induced membrane vesicles from human mesenchymal stem cells overexpressing TRAIL, PTEN and IFN-β1 can kill carcinoma cancer cells. Tissue Cell. 2021;73: 101664.
Li S, Yan G, Yue M, Wang L. Extracellular vesicles-derived microRNA-222 promotes immune escape via interacting with ATF3 to regulate AKT1 transcription in colorectal cancer. BMC Cancer. 2021;21:349.
Seifert P, Spitznas M. Axons in human choroidal melanoma suggest the participation of nerves in the control of these tumors. Am J Ophthalmol. 2002;133:711–3.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341:1236361.
Madeo M, Colbert PL, Vermeer DW, Lucido CT, Cain JT, Vichaya EG, Grossberg AJ, Muirhead D, Rickel AP, Hong Z, et al. Cancer exosomes induce tumor innervation. Nat Commun. 2018;9:4284.
Lucido CT, Wynja E, Madeo M, Williamson CS, Schwartz LE, Imblum BA, Drapkin R, Vermeer PD. Innervation of cervical carcinoma is mediated by cancer-derived exosomes. Gynecol Oncol. 2019;154:228–35.
Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, Anfossi S, Osman AA, Cai Y, Wang R, et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578:449–54.
Sun J, Liao Z, Li Z, Li H, Wu Z, Chen C, Wang H. Down-regulation miR-146a-5p in Schwann cell-derived exosomes induced macrophage M1 polarization by impairing the inhibition on TRAF6/NF-κB pathway after peripheral nerve injury. Exp Neurol. 2023;362: 114295.
Fabbri M. Natural killer cell-derived vesicular miRNAs: a new anticancer approach? Cancer Res. 2020;80:17–22.
Zhou Q, Wei S, Wang H, Li Y, Fan S, Cao Y, Wang C. T cell-derived exosomes in tumor immune modulation and immunotherapy. Front Immunol. 2023;14:1130033.
Dieci MV, Griguolo G, Miglietta F, Guarneri V. The immune system and hormone-receptor positive breast cancer: Is it really a dead end? Cancer Treat Rev. 2016;46:9–19.
Drula R, Pardini B, Fu X, De Los Santos MC, Jurj A, Pang L, El-Daly SM, Fabris L, Knutsen E, Dragomir MP, et al. 17β-estradiol promotes extracellular vesicle release and selective miRNA loading in ERα-positive breast cancer. Proc Natl Acad Sci U S A. 2023;120: e2122053120.
Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, Vadlamudi RK, Kumar R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol. 2001;3:30–7.
Hannafon BN, Gin AL, Xu YF, Bruns M, Calloway CL, Ding WQ. Metastasis-associated protein 1 (MTA1) is transferred by exosomes and contributes to the regulation of hypoxia and estrogen signaling in breast cancer cells. Cell Commun Signal. 2019;17:13.
Zhou WJ, Zhang J, Xie F, Wu JN, Ye JF, Wang J, Wu K, Li MQ. CD45RO(-)CD8(+) T cell-derived exosomes restrict estrogen-driven endometrial cancer development via the ERβ/miR-765/PLP2/Notch axis. Theranostics. 2021;11:5330–45.
Mu Q, Najafi M. Modulation of the tumor microenvironment (TME) by melatonin. Eur J Pharmacol. 2021;907: 174365.
Wang K, Cai R, Fei S, Chen X, Feng S, Zhang L, Liu H, Zhang Z, Song J, Zhou R. Melatonin enhances anti-tumor immunity by targeting macrophages PD-L1 via exosomes derived from gastric cancer cells. Mol Cell Endocrinol. 2023;568–569: 111917.
Zhao S, Liu Y, He L, Li Y, Lin K, Kang Q, Liu L, Zou H. Gallbladder cancer cell-derived exosome-mediated transfer of leptin promotes cell invasion and migration by modulating STAT3-mediated M2 macrophage polarization. Anal Cell Pathol (Amst). 2022;2022:9994906.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7.
Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007;109:1010–7.
Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–93.
Nkosi D, Sun L, Duke LC, Patel N, Surapaneni SK, Singh M, Meckes DG Jr. Epstein-Barr virus LMP1 promotes Syntenin-1- and Hrs-induced extracellular vesicle formation for its own secretion to increase cell proliferation and migration. mBio. 2020;11:e00589.
Sun H, Meng Q, Shi C, Yang H, Li X, Wu S, Familiari G, Relucenti M, Aschner M, Wang X, Chen R. Hypoxia-inducible exosomes facilitate liver-tropic Premetastatic Niche in colorectal cancer. Hepatology. 2021;74:2633–51.
Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, Lee B, Lee T, Mak SY, Ho YS, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5:1191731.
Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, Wang Z, Yuan F, Fox M, Zhang HG, et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021;33:2040-2058.e2010.
Qi M, Xia Y, Wu Y, Zhang Z, Wang X, Lu L, Dai C, Song Y, Xu K, Ji W, Zhan L. Lin28B-high breast cancer cells promote immune suppression in the lung pre-metastatic niche via exosomes and support cancer progression. Nat Commun. 2022;13:897.
Ma Z, Wei K, Yang F, Guo Z, Pan C, He Y, Wang J, Li Z, Chen L, Chen Y, Xia Y. Tumor-derived exosomal miR-3157-3p promotes angiogenesis, vascular permeability and metastasis by targeting TIMP/KLF2 in non-small cell lung cancer. Cell Death Dis. 2021;12:840.
Naito H, Iba T, Takakura N. Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int Immunol. 2020;32:295–305.
Xu Y, Yao Y, Yu L, Zhang X, Mao X, Tey SK, Wong SWK, Yeung CLS, Ng TH, Wong MY, et al. Clathrin light chain A-enriched small extracellular vesicles remodel microvascular niche to induce hepatocellular carcinoma metastasis. J Extracell Vesicles. 2023;12: e12359.
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–77.
Chen C, Liu Y, Liu L, Si C, Xu Y, Wu X, Wang C, Sun Z, Kang Q. Exosomal circTUBGCP4 promotes vascular endothelial cell tipping and colorectal cancer metastasis by activating Akt signaling pathway. J Exp Clin Cancer Res. 2023;42:46.
Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–54.
Dou R, Liu K, Yang C, Zheng J, Shi D, Lin X, Wei C, Zhang C, Fang Y, Huang S, et al. EMT-cancer cells-derived exosomal miR-27b-3p promotes circulating tumour cells-mediated metastasis by modulating vascular permeability in colorectal cancer. Clin Transl Med. 2021;11: e595.
Mo Y, Leung LL, Mak CSL, Wang X, Chan WS, Hui LMN, Tang HWM, Siu MKY, Sharma R, Xu D, et al. Tumor-secreted exosomal miR-141 activates tumor-stroma interactions and controls premetastatic niche formation in ovarian cancer metastasis. Mol Cancer. 2023;22:4.
Wang Y, Li Y, Zhong J, Li M, Zhou Y, Lin Q, Zong S, Luo W, Wang J, Wang K, et al. Tumor-derived Cav-1 promotes pre-metastatic niche formation and lung metastasis in breast cancer. Theranostics. 2023;13:1684–97.
Wang M, Qin Z, Wan J, Yan Y, Duan X, Yao X, Jiang Z, Li W, Qin Z. Tumor-derived exosomes drive pre-metastatic niche formation in lung via modulating CCL1(+) fibroblast and CCR8(+) Treg cell interactions. Cancer Immunol Immunother. 2022;71:2717–30.
Yuan X, Qian N, Ling S, Li Y, Sun W, Li J, Du R, Zhong G, Liu C, Yu G, et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics. 2021;11:1429–45.
Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922–8.
Nathanson SD, Shah R, Rosso K. Sentinel lymph node metastases in cancer: causes, detection and their role in disease progression. Semin Cell Dev Biol. 2015;38:106–16.
Francis DM, Manspeaker MP, Schudel A, Sestito LF, O’Melia MJ, Kissick HT, Pollack BP, Waller EK, Thomas SN. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci Transl Med. 2020;12:eaay3575.
Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, Manini I, Beltrami AP, Ius T, Skrap M, Di Loreto C, Gri G. Systemic T cells immunosuppression of Glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS One. 2017;12: e0169932.
Cousin N, Cap S, Dihr M, Tacconi C, Detmar M, Dieterich LC. Lymphatic PD-L1 expression restricts tumor-specific CD8(+) T-cell responses. Cancer Res. 2021;81:4133–44.
Pucci F, Garris C, Lai CP, Newton A, Pfirschke C, Engblom C, Alvarez D, Sprachman M, Evavold C, Magnuson A, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science. 2016;352:242–6.
Sun B, Zhou Y, Fang Y, Li Z, Gu X, Xiang J. Colorectal cancer exosomes induce lymphatic network remodeling in lymph nodes. Int J Cancer. 2019;145:1648–59.
Li S, Li Q. Cancer stem cells, lymphangiogenesis, and lymphatic metastasis. Cancer Lett. 2015;357:438–47.
Biomarkers and surrogate endpoints. preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95.
Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56.
Ayala-Mar S, Donoso-Quezada J, González-Valdez J. Clinical implications of Exosomal PD-L1 in cancer immunotherapy. J Immunol Res. 2021;2021:8839978.
de Miguel-Perez D, Russo A, Gunasekaran M, Buemi F, Hester L, Fan X, Carter-Cooper BA, Lapidus RG, Peleg A, Arroyo-Hernández M, et al. Baseline extracellular vesicle TGF-β is a predictive biomarker for response to immune checkpoint inhibitors and survival in non-small cell lung cancer. Cancer. 2023;129:521–30.
Nguyen LTH, Zhang J, Rima XY, Wang X, Kwak KJ, Okimoto T, Amann J, Yoon MJ, Shukuya T, Chiang CL, et al. An immunogold single extracellular vesicular RNA and protein ((Au) SERP) biochip to predict responses to immunotherapy in non-small cell lung cancer patients. J Extracell Vesicles. 2022;11: e12258.
Liu J, Zhou WY, Luo XJ, Chen YX, Wong CW, Liu ZX, Bo Zheng J, Yu Mo H, Chen JQ, Li JJ, et al. Long noncoding RNA regulating ImMune escape regulates mixed lineage leukaemia protein-1-H3K4me3-mediated immune escape in oesophageal squamous cell carcinoma. Clin Transl Med. 2023;13: e1410.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Wang Z, Zhang M, Liu L, Yang Y, Qiu J, Yu Y, Li J. Prognostic and immunological role of cancer-associated fibroblasts-derived exosomal protein in esophageal squamous cell carcinoma. Int Immunopharmacol. 2023;124: 110837.
Jiang F, Zhang Z, Chong X, Shen L, Fan M, Liu X, An J, Peng Z, Zhang C. Extracellular vesicle-derived protein file from peripheral blood predicts immune-related adverse events in gastric cancer patients receiving immunotherapy. Cancers (Basel). 2022;14:4167.
Porcelli L, Guida M, De Summa S, Di Fonte R, De Risi I, Garofoli M, Caputo M, Negri A, Strippoli S, Serratì S, Azzariti A. uPAR(+) extracellular vesicles: a robust biomarker of resistance to checkpoint inhibitor immunotherapy in metastatic melanoma patients. J Immunother Cancer. 2021;9:e002372.
Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted role of the Urokinase-type Plasminogen Activator (uPA) and its receptor (uPAR): diagnostic, prognostic, and therapeutic applications. Front Oncol. 2018;8:24.
Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846:75–87.
Kooijmans SAA, Fliervoet LAL, van der Meel R, Fens M, Heijnen HFG. van Bergen En Henegouwen PMP, Vader P, Schiffelers RM: PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Contr Rel. 2016;224:77–85.
Li S, Wu Y, Ding F, Yang J, Li J, Gao X, Zhang C, Feng J. Engineering macrophage-derived exosomes for targeted chemotherapy of triple-negative breast cancer. Nanoscale. 2020;12:10854–62.
Milbank E, Dragano NRV, González-García I, Garcia MR, Rivas-Limeres V, Perdomo L, Hilairet G, Ruiz-Pino F, Mallegol P, Morgan DA, et al. Small extracellular vesicle-mediated targeting of hypothalamic AMPKα1 corrects obesity through BAT activation. Nat Metab. 2021;3:1415–31.
Zhang X, Zhang H, Gu J, Zhang J, Shi H, Qian H, Wang D, Xu W, Pan J, Santos HA. Engineered extracellular vesicles for cancer therapy. Adv Mater. 2021;33: e2005709.
de Abreu RC, Fernandes H, da Costa Martins PA, Sahoo S, Emanueli C, Ferreira L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat Rev Cardiol. 2020;17:685–97.
Huang J, Xiong J, Yang L, Zhang J, Sun S, Liang Y. Cell-free exosome-laden scaffolds for tissue repair. Nanoscale. 2021;13:8740–50.
Xu M, Feng T, Liu B, Qiu F, Xu Y, Zhao Y, Zheng Y. Engineered exosomes: desirable target-tracking characteristics for cerebrovascular and neurodegenerative disease therapies. Theranostics. 2021;11:8926–44.
Fan Z, Xiao K, Lin J, Liao Y, Huang X. Functionalized DNA enables programming exosomes/Vesicles for tumor imaging and therapy. Small. 2019;15: e1903761.
Yang P, Cao X, Cai H, Feng P, Chen X, Zhu Y, Yang Y, An W, Yang Y, Jie J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021;360: 104262.
Zhou W, Zhou Y, Chen X, Ning T, Chen H, Guo Q, Zhang Y, Liu P, Zhang Y, Li C, et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials. 2021;268: 120546.
Cheng L, Zhang X, Tang J, Lv Q, Liu J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials. 2021;275: 120964.
Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625–35.
Topalian SL, Wolchok JD, Chan TA, Mellman I, Palucka K, Banchereau J, Rosenberg SA, Dane Wittrup K. Immunotherapy: The path to win the war on cancer? Cell. 2015;161:185–6.
Fan M, Liu H, Yan H, Che R, Jin Y, Yang X, Zhou X, Yang H, Ge K, Liang XJ, et al. A CAR T-inspiring platform based on antibody-engineered exosomes from antigen-feeding dendritic cells for precise solid tumor therapy. Biomaterials. 2022;282: 121424.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98.
Hu S, Ma J, Su C, Chen Y, Shu Y, Qi Z, Zhang B, Shi G, Zhang Y, Zhang Y, et al. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021;135:567–81.
Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–8.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22.
Huang L, Rong Y, Tang X, Yi K, Qi P, Hou J, Liu W, He Y, Gao X, Yuan C, Wang F. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol Cancer. 2022;21:45.
Feng C, Xiong Z, Wang C, Xiao W, Xiao H, Xie K, Chen K, Liang H, Zhang X, Yang H. Folic acid-modified Exosome-PH20 enhances the efficiency of therapy via modulation of the tumor microenvironment and directly inhibits tumor cell metastasis. Bioact Mater. 2021;6:963–74.
Han Y, Pan H, Li W, Chen Z, Ma A, Yin T, Liang R, Chen F, Ma Y, Jin Y, et al. T cell membrane mimicking nanoparticles with bioorthogonal targeting and immune recognition for enhanced photothermal therapy. Adv Sci (Weinh). 2019;6:1900251.
Acknowledgements
All illustrations were created with BioRender.com.
Funding
This study was supported by The National Natural Science Foundation of China (81972663, 82173055), Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (ZYCXTD2023018), The Excellent Youth Science Project of Henan Natural Science Foundation (212300410074), The Youth Talent Innovation Team Support Program of Zhengzhou University (32320290), The Provincial and Ministry Co-constructed Key Projects of Henan Medical Science and Technology (SBGJ202102134), Henan Medical Technology Popularization Project (SYJS2022109), Henan Provincial Health and Health Commission Joint Construction Project (LHGJ20200158), and Henan Province Young and Middle-aged Health Science and Technology Innovation Leading Talent Project (YXKC2022016).
Author information
Authors and Affiliations
Contributions
MY, SH, HS, and BT provided direction and guidance throughout the preparation of this manuscript. MY and HS wrote and edited the manuscript. CC and WW reviewed and made significant revisions to the manuscript. JL collected and prepared the related papers. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
EVs reshaping the TME and their impact on immunotherapy.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yue, M., Hu, S., Sun, H. et al. Extracellular vesicles remodel tumor environment for cancer immunotherapy. Mol Cancer 22, 203 (2023). https://doi.org/10.1186/s12943-023-01898-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01898-5