
Abstract
Background
Immunotherapy has recently emerged as a treatment strategy which stimulates the human immune system to kill tumor cells. Tumor immunotherapy is based on immune editing, which enhances the antigenicity of tumor cells and increases the tumoricidal effect of immune cells. It also suppresses immunosuppressive molecules, activates or restores immune system function, enhances anti-tumor immune responses, and inhibits the growth f tumor cell. This offers the possibility of reducing mortality in triple-negative breast cancer (TNBC).
Main body
Immunotherapy approaches for TNBC have been diversified in recent years, with breakthroughs in the treatment of this entity. Research on immune checkpoint inhibitors (ICIs) has made it possible to identify different molecular subtypes and formulate individualized immunotherapy schedules. This review highlights the unique tumor microenvironment of TNBC and integrates and analyzes the advances in ICI therapy. It also discusses strategies for the combination of ICIs with chemotherapy, radiation therapy, targeted therapy, and emerging treatment methods such as nanotechnology, ribonucleic acid vaccines, and gene therapy. Currently, numerous ongoing or completed clinical trials are exploring the utilization of immunotherapy in conjunction with existing treatment modalities for TNBC. The objective of these investigations is to assess the effectiveness of various combined immunotherapy approaches and determine the most effective treatment regimens for patients with TNBC.
Conclusion
This review provides insights into the approaches used to overcome drug resistance in immunotherapy, and explores the directions of immunotherapy development in the treatment of TNBC.
Similar content being viewed by others

Introduction
Breast cancer has become the commonest malignancy among women all over the world, and is related to the highest mortality rates from the disease. Triple-negative breast cancer (TNBC) is an entity that tests negative for the expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (Her-2), it is characterized by low differentiation, high invasiveness, a propensity for local and distant metastases, poor prognosis, and high recurrence rates [1]. Gene expression analysis shows that immune markers, mesenchymal phenotypes, androgen receptors, stem cell markers and basic markers are all associated with TNBC [2]. Based on findings from transcriptome research, TNBC may be divided into six subtypes, namely, basal-like 1, basal-like 2, mesenchymal stem cell, immunomodulatory, mesenchymal, and luminal androgen receptor [3]. In addition to the extracellular matrix (ECM), numerous immune cells in the tumor microenvironment (TME), including tumor-infiltrating lymphocytes (TILs), antigen-presenting cells, and fibroblasts contribute to the progression and metastasis in TNBC. At present, immunotherapy has become a new choice for many refractory solid tumors. Immune checkpoint inhibitors (ICIs) include inhibitors of programmed death receptor-1 (PD-1), programmed death receptor-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), which is the most well-known immunotherapeutic agents. Breast cancer has long been considered a “cold” tumor due to its limited T-cell infiltration and low tumor mutation burden. Nevertheless, TNBC exhibits a greater presence of infiltrating lymphocytes, thereby establishing a favorable immune microenvironment for the potential utilization of ICIs. TNBC also demonstrates a relatively substantial tumor mutation load, thereby offering an antigenic foundation for immune cell recognition. The expression of PD-L1 is notably elevated in TNBC, thereby presenting a promising target for the implementation of ICIs [4]. Immunotherapy holds significant clinical relevance for TNBC, as the combination of ICIs with chemotherapy has yielded promising therapeutic outcomes in TNBC patients. Subsequent investigations should be conducted to elucidate efficacious immunotherapy regimens, thereby offering novel approaches to enhance the therapeutic efficacy of TNBC.
TME and therapeutic resistance in TNBC
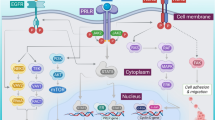
The tumor microenvironment (TME) encompasses tumor cells, cancer-associated fibroblasts (CAFs), immune cells including B and T lymphocytes, natural killer (NK) cells, tumor-associated macrophages (TAMs), the vasculature, and extracellular components including ECM, cytokines, chemokines, metabolites, and exosomes. The heterogeneity of TME within different subtypes of TNBC is notably pronounced [5]. The constituents of TME possess the ability to interact with one another, thereby altering the internal environment of the tumor and ultimately facilitating the development of resistance in TNBC. Research has indicated that non-cancerous cells within the TME play an important part in the development of cancer by facilitating tumor survival, progression, metastasis, and therapeutic resistance. The interaction between tumor cells and stromal cells in the TME, which are also implicated in tumor growth and drug resistance [2]. In addition, the proliferation, the metabolic remodeling, and the inhibition of cell apoptosis in tumor cells of TNBC can lead to hypoxia, acidosis, and oxidative stress within TME [6, 7]. Hypoxia, along with oxidative stress and acidosis, can induce the activation of lysyl oxidase (LOX) and subsequently reshape the ECM, and ultimately leading to the formation of chemotherapy resistance [8]. The activation of the collagen prolyl 4-hydroxylase P4H-α1/HIF-1 axis has been found to enhance the stemness of TNBC cells, resulting in a reduction in oxidative phosphorylation (OXPHOS) and reactive oxygen species (ROS) levels [9]. CAFs play a crucial role in promoting cancer progression and resistance to treatment through the secretion of cytokines, chemokines and ECM remodeling factors [10, 11]. Furthermore, CAF-induced lipid-associated macrophages (LAM) have been identified as mediators of immune suppression in breast cancer, particularly in TNBC [12]. The presence of ACSL3 in TNBC has been observed to result in the protection of TNBC cells from ferroptosis induced by mammary adipocytes [13]. Qiong et al. discovered that M2 macrophages secrete VEGF and activate PCAT6, thereby promoting the growth and metastasis of the cancer cells through the modulation of VEFGR2 in TNBC [14]. Zhang et al. have divided TNBC into macrophage-enriched subtype (MES) and neutrophil-enriched subtype (NES), revealing distinct mechanisms of immunotherapy resistance among different myeloid cell subtypes of TNBC [15]. The interactions between components of TME induce therapeutic resistance in TNBC are shown in Fig. 1.

Interactions between components of TME induce therapeutic resistance in TNBC. The hypoxia of TME induces the activation of LOX and reshapes the ECM, leading to the formation of chemotherapy resistance. The activation of the P4H-α1/HIF-1 axis has been found to enhance the stemness of TNBC cells, resulting in a reduction in OXPHOS and ROS. CAF-induced lipid-associated macrophages induce immunosuppression of TNBC. The adipocytes induce the production of oleic acid to result in the protection of TNBC cells from ferroptosis by ACSL3. The M2 macrophages secrete VEGF and activate PCAT6 to promote the proliferation and metastasis of tumor cells through the modulation of VEFGR2
Remodeling of the TME
Abnormalities in the TME act as barriers to drug delivery, and are the main reasons for poor efficacy of chemotherapy and immunotherapy [16]. TNBC tissue exhibits severe fibrosis and abundant ECM deposition, which induces tumor vascular compression and reduces perfusion, which leads to poor drug delivery [17]. The Cytokines secreted by cancer-associated fibroblasts (CAFs) cause immunosuppression, tumor cell proliferation, epigenetic changes and ECM [18]. As TILs in the TME are closely related to the prognosis of TNBC, TME remodeling (recruiting CD4 + T cells, CD8 + T cells, and NK cells) improves the effect of immunotherapy which is a crucial approach used for treating breast cancer. The targets for reshaping the TME are therefore important for the treatment and prognosis of TNBC, including ameliorating tumor hypoxia, modulating tumor blood vessels, regulating CAFs and ECM, as well as influencing TAMs and dendritic cells (DCs). By either weakening the matrix barrier or enhancing the immunosuppressive microenvironment, the remodeling of TME can be achieved through the utilization of phytochemicals, targeting gut microbiota and metabolites ,and manipulating immunocytes and cytokines. These approaches collectively contribute to the optimization of TNBC anti-tumor therapy. (Fig. 2)

The diagram of remodeling of the TME in TNBC immunotherapy. Rg3-PTX-LPs induced apoptosis by NF-κB. Rg3-LP-Doxetaxel reduces CAFs, TGF-β, and collagen fibers in the TME to enhance the anti-tumor effect of Doxetaxel. 6SA triggers macrophages by MAPK as well as enhancing the secretion of NO and pro-inflammatory cytokine and inducing the induction of Caspase-8-mediated apoptosis, which activate macrophages and inhibit Treg and TAM. TMAO induces tumor cell pyrosis mediated by GSDME through activation of PERK, and releases IL-1β and IL-18, thereby enhancing the infiltration and killing function of CD8 + T cells. MYC protein expression is slightly downregulated after treatment with IFN-γ, IFNs may increase MHC-I expression in tumor cells that demonstrate high MYC expression. CpG-ODN offers synergistic effects with PD-1 inhibitors by producing IFN α and β as well as increasing CD8 + T cell infiltration in the TME
The phytochemicals remodel TME
The chemotherapy drugs encapsulated in cholesterol liposomes generally have a limited effect on TNBC. This is mainly due to the formation of an impenetrable physical barrier by CAFs and their secreted products; these cells also secrete interleukin (IL)-10 and IL-6 and transforming growth factor-β (TGF-β), that collectively inhibit the expression of CD8 + T cells and have immunosuppressive effects within the TME [19, 20]. Notably, TGF-β signaling pathways inhibit CAFs, activate CD8 + T cells, enhance ECM degradation, increase blood perfusion and immune cell infiltration, and improve drug delivery, thereby improving the efficacy of immunochemotherapy [21, 22]. Ginsenoside Rg3, a component of ginseng, exhibits anti-tumor activity and inhibits growth and angiogenesis in human breast invasive ductal carcinoma xenografts in nude mice [23]. Rg3 has been used instead of cholesterol to develop a docetaxel-loaded Rg3 liposome, which can penetrate more deeply into the tumor, inhibit the CAFs ,TGF-β, and collagens to offer better chemosensitization in TNBC [24]. The combination of ginsenoside Rg3 with paclitaxel (PTX) can inhibit the nuclear factor-κB (NF-κB) pathway to enhance the cytotoxicity of chemotherapeutic agents in TNBC [25]. The liposomes based on Rg3 offer excellent encapsulation efficiency and drug-loading ability and can achieve tumor targeting and TME remodeling without any synthetic processes. They offer good clinical applicability and are expected to provide higher drug delivery efficiency for the treatment of TNBC.
Anacardic acid(6-Pentadecyl Salicylic Acid, 6SA) is found in the bark of dipteran plants and elicits the activation of macrophages by augmenting the MAPK signaling pathway, particularly ERK and NF-κB, as well as enhancing the release of nitric oxide (NO) and cytokines [26], which facilitates the activation of immune cells. 6SA reduces tumor volume without diminishing the number of TILs by inducing caspase-8 mediated cell apoptosis, and also concurrently augmenting the secretion of interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α), and improves the immune microenvironment of TNBC [26]. 6SA exhibits potent anti-tumor effects on TNBC in vivo with intact immune function, while also mitigating the myelosuppressive and leukopenic effects induced by paclitaxel [27]. Additionally, it enhances the tumor immune microenvironment in TNBC.
Gut microbiota activate anti-tumor immunity
The response to ICIs is enhanced by the modulation of innate immunity, adaptive immunity, and the immunogenicity of tumor cells by gut microbiota. Specifically, the gut microbiota can influence the activity of DCs, mononuclear macrophages, and NK cells in terms of innate immune regulation. Additionally, the role of CD8 + T cells and CD4 + T cells could be influenced by the gut microbiota in terms of adaptive immune regulation. The metabolites, which are small molecules capable of disseminating from the intestinal primary site, promote the efficacy of ICI by influencing local and systemic anti-tumor immune responses [28]. Certain studies have identified subtypes of immunomodulatory organisms that induce a relatively active immune microenvironment in TNBC. The immunomodulatory (IM) subtype constitutes approximately 24% of TNBC cases and exhibits a more favorable prognosis in comparison to other subtypes. This particular subtype is more inclined to derive benefits from immunotherapy interventions, which involve the enhancement of immune signaling pathways and the augmentation of TILs. It has been found that the enrichment of Clostridium species and related metabolite trimethylamine oxide (TMAO) increases in tumor tissues of the IM subtype. TMAO can activate anti-tumor immunity and higher plasma TMAO levels are associated with better immune response. TMAO can induce tumor cell pyrosis mediated by GSDME protein through activation of endoplasmic reticulum kinase PERK, and secret inflammatory factors (IL-18 and IL-1β) into the TME, thereby enhancing the infiltration and cytotoxicity of CD8 + T cells. TMAO levels correlate positively with the amount of CD8 + T cells in the TME. In this context, TNBC patients with high CD8 + T cell counts demonstrate favorable IFN responses and higher immune cell infiltration [29]. In a clinical trial, choline (a precursor of TMAO) was employed to improve the efficacy of immunotherapy; this provided a new strategy for precision immunotherapy in TNBC. More metabolites of the gut microbiota have been shown to reshape the TME by regulating immune cells and affecting the efficacy of cancer immunotherapy, including ICIs. Therapeutic strategies for intestinal microbiome combined with ICI include FMT, probiotics, prebiotics, engineering bacteria, bacteriophage, and other strategies to enhance ICI response [30]. However, little is known regarding the potential molecular mechanisms related to gut microbiota and the impact of their metabolites on the efficacy of ICI in TNBC. Future studies are therefore needed to further explore the efficacy of microbiota in the treatment of TNBC, as this may help improve patient prognosis.
IFNs activate anti-tumor immunity
The involvement of IFNs in the immunotherapy response against tumors presents a dual nature. Initially, IFNs can stimulate dendritic cells, thereby facilitating the cross-activation of CD8 + T cells specific to the tumor. However, prolonged exposure to IFNs can elicit negative feedback mechanisms, which faciliates the depletion of T cells and the induction of immunosuppressive effects. By modulating the immune response of IFNs, it is possible to augment the body’s immune response to tumors [31]. Studies have demonstrated that IFN signaling of tumor cells can restrict immune responses, whereas IFN signaling of adaptive and innate immune cells can enhance the immune response [32]. IFN-α and IFN-β, known as Type I IFN, have a wide range of impacts on different immune cells. such as augmenting the cytotoxic potential of NK cells and their ability to secrete IFN-γ, as well as facilitating the differentiation, maturation, and migration of antigen-presenting cells. On the other hand, IFN-γ, classified as a type II IFN, possesses antiviral, anti-tumor, and immunomodulatory properties [33], which are capable of activating other immune cells and fostering the immune response. Previous studies have found that MYC is overexpressed in some cases of TNBC and it promotes immunosuppression by inhibiting the IFN signaling pathway [34]. The MYC signaling pathway is related to low PD-L1 expression in cancer cells. Some studies suggest that patients with TNBC who demonstrate high MYC expression have lower major histocompatibility complex class I (MHC-I) expression [35]. MYC inactivation may therefore restore MHC-I expression, CD8 + T cell infiltration, and the anti-PD-L1 response. As MYC protein expression is slightly downregulated after treatment with IFN-γ, IFNs may increase MHC-I expression in tumor cells that demonstrate high MYC expression.
Unmethylated cytosine-phosphorothioate-guanine (CpG) oligodeoxynucleotides are synthetic toll-like receptor 9 (TLR9) agonists that stimulate plasmacytoid dendritic cells (DCs) to produce IFN α and β. They also activate T and B cells, recruit NK cells, induce humoral and cellular immunity, and upregulate IFN release; these promote the infiltration of antigen-specific CD8 + T cells [36]. Based on their chemical composition and in-vitro activity, CpG-oligodeoxynucleotides are mainly divided into three categories: A, B, and C. Studies have found that low doses of CpG-B can significantly inhibit tumor growth and offer synergistic effects with PD-1 inhibitors. Although higher doses of CpG-C may achieve the same anti-tumor effect as CpG-B, CpG-C offers more efficacy than CpG-B when combined with anti-PD-1 inhibitors [37]. The challenge of TNBC treatment interventions lies in effectively targeting IFNs to reshape the immune microenvironment, to guide rational combination therapy.
Targeting immunotherapy resistance in TNBC
Although ICI is effective in some TNBC patients who experience a prolonged response, certain patients either have no response or acquire drug resistance [38]. Current understanding of the mechanisms of acquired resistance to ICIs is considerably limited; this inhibits the effective development of next-generation immunotherapy. Relevant studies on immunotherapy resistance have been described in Fig. 3.

The diagram of overcoming TNBC Immunotherapy Resistance. Pos3Aa-p53 also significantly enhances the sensitivity of p53- deficient TNBC cells to the chemotherapy drug, 5-fluorouracil, and increases IFN. The combination of the CXCR2 antagonist, AZD5069, with doxorubicin, has been found to inhibit doxorubicin-mediated CXCR2 overexpression and reduce chemoresistance and TGF-β expression in TNBC. PmTriTNE@CDA releases STING agonist CDA to upregulate the expression of IFN-γ, promote the maturation of DCs, and recruit CD4 + T cells, CD8 + T cells, and NK cells. CSCs are suppressed by PmTriTNE@CDA to recruit CD8 + T cells
p53 delivery combined with PD-1 inhibitors
Numerous researches have shown that treatment with PD-1/PD-L1 inhibitors is more effective in cancers with more CD8 + T cells which may be used as predictive and therapeutic biomarkers for anti-PD-1 therapy [39, 40]. Notably, certain advanced TNBC cases lacking CD8 + infiltrating T cells demonstrate resistance to PD-1/PD-L1 inhibitors. The mutation frequency of tumor suppressor protein p53 may reach up to 80% in TNBC [41]; this may therefore be used as a potential biomarker. As the deletion or mutation of p53 can promote immune escape of cancer cells, restoring P53 activity in deficient tumors may potentially overcome resistance to PD-1/PD-L1 therapy. In this context, the naturally formed Pos3Aa protein crystal in Bacillus thuringiensis can be used as a carrier. Protein crystal-mediated p53 delivery can restore function in p53 deficient cancer cells, inducing significant apoptosis and cell cycle arrest. It is worth noting that Pos3Aa-p53 also significantly enhances the sensitivity of p53- deficient TNBC cells to the chemotherapy drug, 5-fluorouracil, and provides a new combined approach for the treatment of TNBC [42]. The strategy of Pos3Aa-p53 crystals combined with anti-PD-1 antibodies is safe, and significantly increases the number of IFNs, CD4+, CD44+, and CD8 + CD44 + memory T cells, thereby enhancing the curative effect to anti-PD-1 therapy [43]. In summary, these data confirm that intracellular p53 transmission may be an efficacious approach for improving the immunogenicity of p53 mutant tumors. The combination therapy of p53 restoration and ICIs in TNBC has considerable potential in overcoming resistance to ICIs. However, further studies are needed regarding the mechanisms involved; high-level evidence-based clinical trials are also needed for future clinical application.
CXCR2 inhibitor combined with ICIs
CXCR2 chemokine receptor 2 (CXCR2) is mainly expressed by neutrophils. Notably, TNBC demonstrates higher levels of CXCR2 expression than other types of breast cancer [44]. In this context, CXCR2 participates in the treatment of drug resistance by maintaining of stemness of cancer stem cells (CSCs). The combination of the CXCR2 antagonist, AZD5069, with doxorubicin, has been found to inhibit doxorubicin-mediated CXCR2 overexpression and reduce chemoresistance and TGF-β expression in TNBC. An in vitro study found the combination of 30 nM of AZD5069 and 200 nM of atezolizumab to induce a notable cytotoxic effect (p = 0.0065); this indicates that in addition to the reduction of chemoresistance, CXCR2 inhibition enhances the efficacy of PD-L1 inhibitor [45]. Clinical trials and in vivo experiments are needed to evaluate CXCR2 inhibitor-based combination therapy in specific patient cohorts. This may provide breakthroughs and improve survival rates in TNBC patients.
PmTriTNE@CDA nanotechnology
CD44 and PD-L1, which have overexpression in TNBC, play a important role in CSC stemness immune escape [1].In this context, a methoxy polyethylene glycol-covered CD44 × PD-L1/CD3 three specific T cell nano adapter (TriTNE) was loaded with c-di-AMP (CDA), the agonist of STING pathway, by using nanotechnology to develop PmTriTNE@CDA. Methoxy polyethylene glycol coverage blocks the antigen-binding of TriTNE; this dual targeting strategy (of CD44 and PD-L1) may improve selectivity and binding affinity to tumors. STING agonists can upregulate the expression of type I IFNs in TME, promote DCs maturation, activate T cells via tumor antigen presentation, and recruit CD8 + T cells and NK cells to the tumor lesion, thereby improving the immunogenicity of TNBC [46]. PmTriTNE@CDA targets CD3 and PD-L1 while inherently activating IFN signals; CSCs are suppressed and eliminated, and they interact synergistically with CDA to overcome the heterogeneity of TNBC and immunotherapeutic resistance.
ICI monotherapy
ICIs eliminate cancer cells by releasing the “braking” mechanism of the immune system against cancer-attacking molecules. The two most prominent ICIs block CTLA-4 (CD152) and target PD-1 (CD279) and PD-L1 (CD274 or B7 homologue 1) [47]. A large-scale multi-omics analysis demonstrated the unique microenvironment characteristics and immune escape mechanism of TNBC. The types of microenvironment included the following: (1) immune desert type: unable to attract immune cells (related to MYC gene amplification), (2) immunosuppression type: chemotaxis present, but innate immunity is inactivated and tumor antigen levels are low, possibly contributing to immune escape (possibly linked to mutations in the phosphoinositide-3-kinase (PI3K)-AKT pathway), and (3) immunoinflammatory type: demonstrating high expression of immune checkpoint molecules. The results of this study are helpful for individualizing immunotherapy in TNBC patients. Notably, ICIs may be effective for the immunoinflammatory type and may transform the TME from an “immune cold” to an “immune hot” state in the immune desert and immunesuppression types.
Anti-PD-1/PD-L1 therapy
PD-1 is an important immunosuppressive molecule that is mainly found on immune cells [48] and is typically expressed on the surface of T, B, NK, and myeloid cells. PD-L1 may be expressed by tumor and activated T cells, macrophages, and CAFs [49]. The binding of PD-1 to its ligand PD-L1 trigger SH2 protein tyrosine phosphatase 2 (SHP2), this downregulates the PI3K/AKT axis, inhibits the proliferation of T and B cells in peripheral tissue, induces programmed T cell death, and prevents autoimmune diseases, thereby maintaining immune homeostasis. However, this also provides the possibility of immune escape in tumor cells [50]. The Food and Drug Administration (FDA) of United States has approved the following PD-1/PD-L1 inhibitors: nivolumab, pembrolizumab, atezolizumab, cemiplimab, avelumab, and durvalumab [51]. Certain studies have reported that PD-L1 is present in around 20–30% of cases of TNBC, and is associated with lymphocyte infiltration and histological grades. In combination with extracellular signal-regulated kinase 1/2 inhibitors, PD-1/PD-L1 inhibitors show greater effects in TNBC cell lines than in non-TNBC cells [52]. Phase I clinical trial KEYNOTE-012 showed that pembrolizumab has good safety and object response rate (ORR) in TNBC patients [53]. Studies have confirmed that monotherapy of pembrolizumab offers sustained anti-tumor activity in patients with early and advanced PD-L1-positive TNBC (with a combined positive score [CPS] of ≥ 1) [54,55,56]. In phase I clinical trial (NCT02838823), PD-1 inhibitor JS001 shows good safety and effectiveness in metastatic TNBC patients who failed in multi-line treatment [57]. In the Phase I clinical trial (NCT01375842), the monotherapy of PD-L1 inhibitor atezolizumab provides long-lasting clinical benefits in mTNBC patients [58]. Phase I JAVELIN study showed that Avelumab, a PD-L1 inhibitor, had an ORR of 44.4% and 2.6% in 58 TNBC patients with PD-L1 ≥ 10% and < 10%, respectively [59].
However, the monotheraputic efficacy of PD-1/PD-L1 inhibitors is limited in TNBC and influenced by numerous factors. The resistance of breast cancer to ICIs also hinders their clinical application.
Anti-CTLA-4 therapy
The CD28 and CTLA-4 receptors on T cells bind to CD80/CD86 to provide positive and negative feedback, respectively, for T cell activation. CTLA-4 is considered to be a necessary suppressor of the adaptive immune response, and is capable of maintaining peripheral tolerance, binding to CD80/CD86 on activated T cells, competitively inhibiting CD28, and binding to B7 co-stimulatory molecules expressed on antigen-presenting or tumor cells; this reduces the binding of B7 to CD28 and downregulates the expression of T cells [60]. CTLA-4 hinders the growth of T cells, progression of the cell cycle, production of IL-2, and differentiation of T cells. It is therefore a specific regulator of the CD4 + T cell response, and thereby maintains T cell homeostasis [61]. Some studies have analyzed the CTLA-4 expression among different types of breast cancer in The Cancer Genome Atlas (TCGA), it is demonstrated that the expression of CTLA-4 is found to be the highest in TNBC. Notably, it may be controlled by hsa-mir-92a to form a competing endogenous ribonucleic acid (RNA) network, which affects the prognosis of TNBC patients via the leukocyte and T cell activation pathways [61]. In this context, CTLA-4 expression is negatively associated with the T-cell activation score and survival rate (P = 0.0061) [62]. Notably, a study found CTLA-4 levels to be significantly higher in tissue samples from lymph node metastases than in those from the primary breast tumor. Additionally, the patients with high expression of CTLA-4 demonstrated a significantly increasing in axillary lymph node metastases [63]. CTLA-4 blocking may therefore activate or restore T cell function. In addition to inhibiting axillary lymph node metastasis, it can also enhance immune defense mechanisms and tumoricidal effects in breast cancer. In a study, combined preoperative tumor cryoablation and ipilimumab therapy demonstrated potential intratumoral and systemic immune effects with better safety; this suggests the possibility of a synergistic anti-TNBC immunity effect [64]. None of the CTLA-4 inhibitors has been currently approved for sole use in TNBC. However, combined pembrolizumab and chemotherapy with subsequent pembrolizumab monotherapy has been used for the adjuvant treatment of early high-risk TNBC with a PD-L1 CPS of ≥ 20.
Combination of two ICIs
Both PD-1 and CTLA-4 are highly expressed in regulatory T cells (Treg). In this context, PD-1 can promote Treg cell proliferation in the presence of ligands. Numerous tumors demonstrate high infiltration by Treg cells (including CD8 + and NK cells), which inhibit the immune response. Blocking the PD-1 pathway may therefore enhance the immune response by reducing the inhibitory activity of Treg cells [65]. As CTLA-4 and PD-1 prohibit T cell function via different mechanisms, it may be reasonable to use CTLA-4 and PD-1 inhibitors to treat TNBC. Some studies have shown that the combination of two types of ICIs or ICIs with chemotherapy drugs offers a better response rate than monotherapy with either agent [66, 67]. However, 55% of grade 3 or 4 side effects occur in patients receiving ipilimumab and nivolumab; the corresponding incidence is 16.3% and 27.3% in those who receive nivolumab and ipilimumab, respectively. Notably, dual blockade with durvalumab (anti-PD-L1 monoclonal antibody) and tremelimumab (anti-CTLA-4 monoclonal antibody) activates and effectively expands T cell populations. PD-L1 and CTLA4 inhibitors may influence the crosstalk between B and T cells, inducing plasma cells to produce specific antibodies in the TME. These findings suggest that patients with TNBC are prone to benefit from combined treatment with durvalumab and tremelimumab [66]. Although this study had an exploratory design and a small sample size for verification of reproducibility, it confirmed the feasibility and effectiveness of combining two ICIs to a certain extent. The findings may be validated by future preclinical or clinical studies including larger sample sizes.
Combination of ICIs and other treatments
ICIs combined with chemotherapy
A meta-analysis that included early-stage TNBC patients showed that the use of ICIs in combination with anthracyclines and taxanes significantly increased the pathological complete response rate (pCR); in addition, ICI-chemotherapy drug combinations offered a lower incidence of adverse events compared with platinum chemotherapy [68]. Metastatic TNBC (mTNBC) is usually treated using chemotherapy; however, the objective response rate (ORR) with single-agent chemotherapy is only 10 − 30% and that of multidrug combination therapy can reach 63% [69, 70]. Clinical trials have found that cisplatin and doxorubicin significantly increase T-cell infiltration in patients with TNBC who subsequently receive nivolumab. Based on the high response rate to anti-PD-1 treatment and the upregulation of immune-related genes, cisplatin or doxorubicin induction may be considered to initiate tumor responses to PD-1 therapy. Clinical data on TNBC indicate that these chemotherapy agents can induce the formation of a more favorable TME in the short term, thereby improving the efficacy of ICIs [71]. Several clinical trials show that multiple neoadjuvant chemotherapy combined with Pembrolizumab regimens showed a favorable PCR rate in the treatment of early TNBC [72, 73]. In a study on early-stage TNBC, the combination of chemotherapy with pembrolizumab in the neoadjuvant setting provided a higher pCR (64.8%) than that of placebo (51.2%) [74]. The combination of anthracycline and taxane chemotherapy with durvalumab as a new adjuvant treatment can improve the prognosis of early TNBC patients [75, 76]. In the phase III clinical trial NeoTRIPaPDL1, the addition of PD-L1 inhibitor atezolizumab to neoadjuvant chemotherapy haven’t significantly improved the pCR rate of TNBC patients compared to neoadjuvant chemotherapy alone [77]. In the IMpassion031 trial, which compared atezolizumab combined with albumin-bound PTX and anthracycline with placebo and chemotherapy, the pCR was higher in the atezolizumab group than in the placebo group (58% vs. 41%, respectively) [78].
In IMpassion130 trial, which compared the efficacy of atezolizumab combined with albumin-bound PTX (experimental group) with that of albumin-bound PTX monotherapy (control group), the experimental group exhibited a longer median progression-free survival (PFS) and the median overall survival (OS) compared to the control group. In PD-L1 positive patients, the median PFS was longer in the experimental group than in the control group [79]. The results showed that atezolizumab combined with albumin-bound PTX offered a better prognosis than single-drug therapy in advanced TNBC patients, especially in PD-L-positive patients. However, in Phase III clinical study IMpassion131, the combination of atezolizumab and paclitaxel in the treatment of metastatic TNBC did not significantly improved PFS in the PD-L1-positive population. In another trial that evaluated pembrolizumab with chemotherapy against placebo with chemotherapy for treating advanced TNBC, the subgroup with CPS of > 10 demonstrated a median OS of 23.0 months in the pembrolizumab group and 16.1 months in the placebo group. Chemotherapy combined with ICIs was, therefore, more effective than chemotherapy in patients with PD-L1-expressing (CPS ≥ 10) advanced TNBC [80]. The therapeutic effect and prognosis of immunotherapy may therefore be predicted before treatment in advanced TNBC patients (based on the expression of PD-L1). KEYNOTE-355 showed that the first-line treatment of pembrolizumab combined with chemotherapy significantly improved the OS for advanced TNBC with PD-L1 (CPS ≥ 10) [81].
An open-label, non-randomized, single-arm phase I/II clinical trial demonstrated favorable tolerability and efficacy of the combination therapy comprising eribulin and pembrolizumab for managing metastatic TNBC [82]. Similar clinical studies have achieved different results, such as in the IMpassion031 study and NeoTRIPaPDL1 study, which may be related to factors such as the pathological characteristics of the enrolled population, PD-L1 detection methods, and compatibility protocols.
ICIs combined with radiotherapy
Despite significant advances in chemotherapy, endocrine therapy, and targeted therapy for breast cancer, radiotherapy continues to play an important role in TNBC. Radiotherapy recruits immune cells into the TME in various ways. It induces dying tumor cells to release danger signals. DCs combine with these danger signals to ingest antigens from cancer cells and transmit the antigens to lymph nodes. These are then presented to the initial T cells, thereby activating CD8 + and CD4 + T cells. This in turn triggers chemokine-induced recruitment of effector T cells to tumors [83]. Preclinical data indicate that radiotherapy combined with ICIs can not only enhance anti-tumor efficacy but also induce responses outside the radiation field. In a low immunogenicity mouse model of TNBC, radiotherapy upregulated the expression of genes containing immunogenic mutations. Vaccination with new epitopes encoded by these genes induced CD8 + and CD4 + T cells, thereby enhancing the immune response [84]. A study compared radiotherapy, anti-CTLA-4 antibody, and combined treatment with both in low- immunogenicity metastatic mouse breast cancer 4T1 cells; the findings suggested that combined treatment with ICIs and chemotherapy enhanced T cell infiltration, delayed growth of the primary irradiated tumor, improved survival rates, and inhibited lung metastasis [85].
In the TONIC trial, which had an adaptive design, mTNBC patients received nivolumab for 2 weeks and were then administered low-dose radiotherapy, the final ORR was 10%. The results indicate that CTLA-4 inhibitor therapy before low-dose radiotherapy offers better outcomes than other regimens, as the consumption of immunosuppressive Treg cells limits the function of CD8 effector cells and anti-tumor immunity. In this context, a multi-center phase 2 study evaluated the effectiveness and safety of combined radiotherapy and pembrolizumab in mTNBC patients. The study found that in the intention-to-treat cohort, the ORR of the unselected PD-L1 population was 17.6%; this was higher than that of mTNBC patients who had previously received ICI monotherapy [86]. However, the combination of immunotherapy and radiotherapy may be a double-edged sword. Owing to ICI-induced alterations in the balance between immune resistance and tolerance, increased tumor recognition is accompanied by higher immune response rates in normal tissues. On administration of radiotherapy, both tumor-specific and non-tumor-specific antigens may be released into the TME; this may activate autoreactive T cells, and possibly attack and damage normal tissues. Therefore, the combination of the two may increase the severity of adverse effects. Future large-scale clinical trials are therefore needed to further explore the best combination strategy for radiotherapy and immunotherapy.
ICIs combined with gene therapy
There has been some progress in the administration of gene therapy with ICIs in TNBC, and studies are evaluating its immunotherapeutic effect (whether similar or superior). In the early stages, gene therapy utilized the oncolytic effect of Ad.sT, a protein created by fusing an adenovirus and a TGF β receptor II IgG Fc fragment (sTGFβ RIIFc). The Ad.sT interfered with TGF β-1 binding and inhibited TGF-β-dependent transcription when it was infected in MDA-MB-231 cells. More than 85% of the tumors of MDA-MB-231 xenografts in nude mice showed a reduction by the administration of Ad.sT [87]. Ad.sT could increase the amounts of CD4 + and CD8 + T lymphocytes in peripheral blood, thereby improving the immunotherapeutic effect of ICIs. Notably, a combination of PD-1 inhibitors and CTLA-4 inhibitors increases the efficacy of Ad.sT in inhibiting the metastasis and proliferation of tumor [88]. In this context, the LyP-1 receptor is highly expressed in breast cancer and three oncolytic viruses, namely, Ad.sT, AdLyp.sT, and mHAdLyp.sT; the latter two produce higher levels of the sTGFβRIIFc protein compared to viruses without LyP-1 modification. Intravenous injection of AdLyp.sT and mHAdLyp.sT has been found to induce a strong anti-tumor response in a bone metastasis model of TNBC, the delivery of AdLyp.sT in a 4T1 mouse model with normal immune function also enhanced the efficacy of anti- CTLA-4 and anti-PD-1treatment [89]. These findings suggest that it may be possible to develop more potential targeted immunotherapeutic drugs for TNBC.
In some studies, adenovirus-mediated herpes simplex virus(HSV)thymidine kinase was injected into mTNBC tumors; the injection site was then irradiated using stereotactic body radiotherapy (SBRT) and the patients subsequently received pembrolizumab. The median duration of treatment and OS in this study were 9.6 and 14.7 months, respectively. Notably, the median OS had more than doubled in patients who experienced clinical benefits; this indicated that the use of SBRT in conjunction with HSV thymidine kinase and gene therapy prior to pembrolizumab can improve the therapeutic efficacy of the latter in patients with mTNBC. This combined treatment regimen has also shown improved anti-tumor efficacy in a study on patients with liver metastases who had a low or negative PD-L1 status [90]. However, the patients in this study were not randomized, and the response rate to oncolytic viruses (alone or in combination with ICIs) did not exceed 12%. In addition, the combination of ICIs and SBRT only improved the response in preconditioned patients without liver metastasis (and demonstrated a shorter duration of response) [86]. Larger future randomized studies are therefore needed to evaluate the effect of each modality and its impact on the tumor microenvironment.
ICIs combined with targeted therapy
ICIs combined with poly (adenosine diphosphate ribose) polymerase inhibitors
The enzyme, poly (adenosine diphosphate ribose) polymerase (PARP), plays a role in the repair of deoxyribonucleic acid (DNA) single-strand breaks. Notably, PARP inhibitors (PARPi) have been widely used in treating cancers with homologous recombination repair defects. In this context, TNBC is usually found in cases of familial breast cancer; approximately 50% of cases demonstrate BRCA1/2 mutations, which can disrupt DNA repair pathways [91]. Four PARPIs have been currently approved by the FDA of the United States, namely olaparib, rucaparib, niraparib, and talazoparib. Olaparib, and talazoparib monotherapy have been approved for advanced or metastatic human epidermal growth factor receptor 2-negative breast cancers with BRCA mutations [91,92,93]. Notably, PARPIs affect the TME by activating the IFN gene stimulating factor (STING) pathway in cancer cells. Certain studies have confirmed that in cases of BRCA1 deletion/mutation, PARPIs induce T cell recruitment, upregulate IFN responses through the cGAS-cGAMP-STING pathway, increase tumor neoantigens, upregulate PD-L1 expression, and increase tumor immunogenicity in TNBC cell lines.
In advanced or mTNBC, combined pembrolizumab and niraparib have demonstrated an ORR of 21% and a disease control rate of 49% [94]. In a cohort study where TNBC patients were treated with ICIs, combined avelumab and talazoparib offered an ORR of 18.2% [95]. The combination olaparib and durvalumab is safe in BRCA-mutated mTNBC [96]. It offered a high response rate in patients with homologous recombinant DNA repair pathway gene changes [97]. The mentioned studies have evaluated the potential synergistic effect of ICIs and PARPIs in the treatment of TNBC. The reports from other studies using combination therapy, including the NCT03101280 and NCT03544125, have not yet been published. Combined treatment needs to be based upon the identification of TNBC molecular subtypes (immune checkpoint positivity or negativity and high or low expression) and homologous recombinant defective gene types.
ICIs combined with anti-angiogenic drugs
Vascular endothelial growth factor (VEGF) is crucial in facilitating growth, survival, migration, invasion, angiogenesis, and increased vascular permeability in vascular endothelial cells. Studies have found that higher angiogenic enrichment scores are closely associated with higher age, lower TIL counts, and the immunomodulatory subtype of TNBC. Anti-VEGF therapy may therefore be used to promote immune checkpoint blockade. Notably, the immunostimulatory effects and toxicities of anti-VEGF drugs are highly dose-dependent. Low-dose anti-VEGF therapy can normalize blood vessels and improve anti-tumor immunity, while high doses can induce hypoxia and create an immunosuppressive TME [98]. Low doses of the anti-VEGF receptor (VEGFR)2 antibodies may induce more immune cell infiltration than regular doses; this provokes CD8 + T cells and macrophages to secrete osteopontin (OPN), a crucial bone matrix protein in cell movement, cytokine generation, and immune control [99]. OPN may promote the production of TGF-β, which upregulates the expression of PD-1 on immune cells. In patients with advanced TNBC, the combination of low-dose VEGFR inhibitors (apatinib and camrelizumab) has shown good tolerance and efficacy with a higher ORR than that of monotherapy. Notably, the approach of combining camrelizumab and VEGFR2 inhibitors has even shown good efficacy in patients with PD-L1 negative tumors or those receiving advanced lines of chemotherapy [100]. Certain studies have combined the VEGFR inhibitors famitinib and camrelizumab to treat patients with advanced TNBC. The combination provided an ORR of 81.3% and a median PFS of 13.6 months. All PD-L1 positive patients achieved an objective response, while only 69.2% of PD-L1 negative patients achieved an objective response (P = 0.030); this indicated that PD-L1 positive patients obtained more benefit from this protocol [101]. In addition, the PFS was higher in CD8 + patients than in CD8- patients; CD8- PD-L1- tumors showed the most unfavorable prognosis with an ORR of 60% and a median OS of 15.2 months. This suggests that immunohistochemistry for CD8 + and PD-L1 may be used for predicting the markers of clinical response. This study confirmed the effectiveness, safety, and feasibility of triple therapy in TNBC and demonstrated the potential of combined anti-VEGF, CD8+, and anti-PD-L1 therapy in the clinic. The prognosis of the combination may be evaluated in future studies.
ICIs combined with PI3K/AKT pathway inhibitors and chemotherapy
The PI3K/AKT/protein kinase B (PKB)/ mammalian target of rapamycin (mTOR) (referred to as the PAM pathway) regulates the proliferation, differentiation, apoptosis, and angiogenesis of tumor cells [102]. The activation of proto-oncogenes (PIK3CA, AKT, and mTOR) and inactivation of tumor suppressor genes (PTEN) are often observed in TNBC and are closely associated with proliferation and chemoresistance in these tumors [103]. An albumin nanoparticle containing PI3K-γ inhibitor eganelisib (IPI-549) and PTX enhanced the efficacy of α-PD1 with longer PFS and a better remission rate in TNBC [104]. The combination of Ipatasertib, an AKT inhibitor, with Atezolizumab + Paclitaxel/Nab-paclitaxel has demonstrated favorable efficacy in managing advanced and metastatic TNBC [105].
ICIs combined with 2-fluoro-L-focus (2 F-Fuc)
The B7 homologous 3 (B7H3) protein is a type I transmembrane protein. N-glycosylation of B7H3 induces immunosuppression in TNBC, indicating a poor prognosis in these patients; the glycosylated protein, therefore, represents a potential new target for immunotherapy. Compared with non-glycosylated B7H3, glycosylated B7H3 induces a reduction in total CD4 + T, CD8 + T, and NK cell counts in tumors and activates fewer cytotoxic CD8 + TILs; this suggests that glycosylated B7H3 interferes with the proliferation and activation of T cells. In this context, 2-fluoro-L-focus (2 F-Fuc) is an effective inhibitor of fucosylation, which enhances the activation of T cells by reducing B7H3 glycosylation and restoring the vulnerability of cancer cells to cytotoxic T-cell-mediated death. A study found combined treatment with 2 F-Fuc and anti-PD-L1 to significantly inhibit tumor growth in vivo [106]. Notably, 2 F-Fuc may enhance PD-L1 immunotherapy by blocking B7H3 core focusing, and combined treatment with PD-L1 may enhance the therapeutic effect in B7H3-positive TNBC. Targeting the FUT8-B7H3 axis may therefore represent an effective strategy for improving the therapeutic effect of ICIs in TNBC patients.
Furthermore, the efficacy of various pathway inhibitors, such as MEK1 inhibitor cobimetinib, HDAC inhibitor entinostat, ADC agents such as ladiratuzumab vedotin and trastuzumab deruxtecan in conjunction with ICIs, has been demonstrated to be promising in the treatment of TNB [107,108,109,110].
ICIs combined with nanotechnology
Nanoscale materials have small particle sizes, stable properties, and high bioavailability. As a new area of interest in the field of cancer research, nano delivery systems are applied for drug delivery and early detection. Certain studies have shown that the combination of nanotechnology with immunotherapy ensures targeted delivery and stability of loaded drugs (via the application of nanocarriers) and enhances drug uptake and biocompatibility in breast cancer.
ICIs combined with recombinant nanoparticles
Nanoparticles carrying ozone (O3) were designed and synthesized in a laboratory and polylactic co-glycolic acid was used to wrap an O3-rich perfluoronaphthalane carrier; this was modified with iRGD or isotactic polypropylene oxide (iPPO) [111]. O3 is prepared by ionizing oxygen and blowing into an iPP (polyacrylic acid polymer lactic acid and glycolic acid perfluoronaphthalane) solution to form iPPO (O3-loaded iPP particles), a microwave (MW)-controlled release nanosystem. Notably, MWs have good penetration ability as electromagnetic waves [112], and iPP particles can stably carry O3 and store it in the carrier perfluoronaphthalane, which accumulates in tumor tissue after intravenous injection. After internalization of the nanoparticles into tumor cells, low-energy MW irradiation promotes the release and decomposition of O3 from perfluoronalkanes to produce ROS (especially highly toxic hydroxyl radicals), which induce tumor cell apoptosis and cell lysis without damaging normal tissues and organs [113]. The findings showed the combined treatment of iPPO and MW resulted in a 3.3-fold increase in the total apoptosis rate of 4T1 cells compared to the control group. The total apoptosis rate even reached 98.8% in MDA-MB-468 cells treated with iPPO, this proportion was higher than that of the control group (6.36%). Systemic use of anti-PD-1 inhibitors can inhibit the growth of primary and peripheral tumors to a certain extent. In this context, combining anti-PD-1 therapy with iPPO and MW offers inhibitory rates of 77.65% and 68.17% in primary and peripheral tumors, respectively. Anti-PD-1 therapy combined with iPPO and MW has shown the most effective tumor growth inhibition effect in all groups. It significantly improved CD4 + and CD8 + T cell infiltration in primary tumors and increased immunogenic cell death (ICD) and new antigens release; this effectively promoted the anti-tumor efficacy of PD-1 inhibitors. Notably, hydroxyl free radicals induce ICD, thereby triggering an anti-tumor immune response. Their binding to ICIs can promote the maturation of DCs, reverse immunosuppression in the TME, and enhance the systemic anti-tumor immune response [114].
Another approach for improving anti-PD-L1 therapy via ICD is the construction of a self-expanding biomimetic nanosystem (mEHGZ), created by glucose oxidase, wrapping epirubicin (EPI), and heme in zeolitic imidazolate framework-8 nanoparticles and coating them with calreticulin-overexpressing tumor cell membranes. EPI can induce ICD, glucose oxidase, and heme; this enhances ICD via cascade generation of ROS and promoting DC maturation and CTL infiltration [115]. Compared with the controls (such as EPI monotherapy), mEHGZ nanoparticles demonstrated the highest efficacy (86.0%) in inducing 4T1 cell apoptosis in a study. In combination with anti-PD-L1 therapy, mEHGZ effectively induced IFN-γ positivity. The CD8 + T cell tumor infiltration rate was 31.2%; this was 11.5-fold more than that observed in the EPI-treatment group. Compared with EPI-treated group, the tumor growth inhibition rate (82.02%) was also higher (36.41%). These results confirm that mEHGZ combined with anti PD-L1 therapy can induce a strong adaptive immune response in vivo. These two nanotechnology approaches enhance the sensitivity and efficacy of ICIs against TNBC via the PD1/PDL1 pathway and offer a promising approach for improving the response rate to ICIs.
ICIs combined with Mucin 1 messenger RNA nanovaccine
Mucin 1 (MUC1), a highly glycosylated tumor-associated antigen expressed on the external surface of epithelial cells [116], is a target in breast cancer immunotherapy. It binds to oxidized mannan and targets the mannose receptor on DCs to induce specific antibody and CD8 + T cell responses in breast cancer. In this context, a type of lipid/calcium/phosphate (LCP) nanoparticle was developed and used for creating a vaccine, which can effectively release the messenger RNA encoding MUC1 into the cytoplasm to activate tumor-specific T cells. In a study, the mice in the LCP-messenger RNA treatment group showed the highest MUC1-specific CTL-induced cytotoxicity; notably, immunization with the target MUC1 antigen improved CD8 + cytotoxicity. In another study on a 4T1 mouse model, LCP vaccines (with or without mRNA) were injected subcutaneously and anti-CTLA-4 antibodies were injected intraperitoneally. After multiple measurements of the tumor size within 25 days, the group receiving the MUC1 vaccine in combination with the CTLA-4 antibody demonstrated the smallest tumor size and the strongest anti-tumor activity (p < 0.001) as compared with the vaccine (p < 0.001) and anti CTLA-4 antibody (p < 0.01) groups. The findings support the potential of nanoparticle-based messenger RNA vaccines in combination with CTLA-4 inhibitors in immunotherapy for TNBC. Although the mechanism responsible for enhancing the efficacy of the combination is unclear, the nanoparticle delivery system improves the stability, sustainability, and expression level of messenger RNA-based vaccines [117]. Further studies on the injection of CTLA-4 inhibitors prior to vaccination with the MUC1 messenger RNA nanovaccine have shown that treatment with the combination is associated with lower expression of cytokines including IL-6, TGF-β, and TNF-α, and low Treg cell counts and bone marrow-derived inhibitory cells, this may reshape the immunosuppressive TME. The combination of ICIs and other treatments enhanced the antitumor effect in TNBC as shown in Fig. 4.

The diagram of combination of ICIs and other treatment. Anthracyclines, taxanes, cisplatin, doxorubicin and Albumin-PTX, and eribulin are used to combine with ICIs to enhance the clinical response in early and metastatic TNBC. Low-dose RT with Nivolumab and RT with Pembrolizumab enhance anti-tumor efficacy in TNBC. The Ad.sT interfered with TGF β-1 binding and improved the immunotherapeutic effect of ICIs. The regimen of adenovirus-mediated HSV thymidine kinase, SBRT, and pembrolizumab improve the therapeutic efficacy in patients with mTNBC, PARPIs upregulate IFN responses through the cGAS-cGAMP-STING pathway to upregulate PD-L1 expression. Low doses of VEGFR2 antibodies provoke CD8 + T cells and macrophages to secrete OPN and promote the production of TGF-β which upregulates the expression of PD-1. PI3K-γ inhibitor eganelisib (IPI-549) and PTX enhanced the efficacy of α-PD1 in TNBC. 2 F-Fuc enhances the activation of T cells by reducing B7H3 glycosylation and restoring the susceptibility of tumor cells to enhance PD-L1 efficacy. Olaparib, Ipatasertib, cobimetinib, entinostat, ladiratuzumab vedotin, and trastuzumab deruxtecan combined with ICIs and chemo drugs has demonstrated favorable efficacy in managing TNBC. The combining anti-PD-1 therapy with iPPO and MW offers an effective tumor growth inhibition effect. in a study. In combination with anti-PD-L1 therapy, mEHGZ effectively induced IFN-γ positivity in TNBC. The combination treatment with the CTLA-4 and MUC1 messenger RNA nanomachines is associated with lower expression of IL-6 and TNF-α, which may reshape the immunosuppressive TME
Tumor vaccine
Tumor vaccines use cancer cells, antigens, or other related biological components to confer the host and produce a potent anti-tumor immune response. The vaccines may be used to treat and prevent the progression of cancer [118]. Compared with other treatment methods such as radiotherapy and chemotherapy, tumor vaccines offer the advantages of high specificity and a low incidence of adverse effects. However, owing to current technological limitations, responses to therapeutic tumor vaccines remain unsatisfactory.
Adagloxad simolenin
Adagloxad simolenin is an active anti-cancer immunogen, which targets the Globo-H antigen which is highly prevalent on the surface of various cancers including TNBC [119]. It can be used as a target to identify cancer cells (which may stimulate the human immune response) and eliminate malignant tumors. On combined with the saponin adjuvant, OBI-821, adagloxad simolenin can induce an immune response to challenge tumor cells. An ongoing international multicenter phase III study (NCT03562637) is evaluating the anti-Globo-H vaccine in high-risk early Globo H-positive TNBC [120].
α- lactalbumin engineered vaccine
The α-lactalbumin is being considered a potential target for developing a vaccine to treat and prevent TNBC. Although this protein does not exist in normal and aging tissues after lactation, it exists in most TNBCs. Activating the immune system against this redundant protein may provide pre-emptive immune protection against α- lactalbumin protein expression. In addition, the vaccine contains an adjuvant that can stimulate the innate immune response, thereby promoting the production of a specific immune response against tumors and preventing their further progression. In a study performed in a mouse model, α- lactalbumin engineered vaccine boosts antitumor immunity in breast cancer which could prevent breast cancer recurrence [121]. An open-label phase I clinical trial (NCT04674306)is ongoing to determine the safety as well as the optimal dose of α- lactalbumin vaccine to treat TNBC patients with a high risk of recurrence.
DR5 DNA vaccine
DR5, also referred to as TNF-related apoptosis-inducing ligand receptor 2, has the ability to attach to TNF-related apoptosis-inducing ligands and facilitate cellular apoptosis. In a study using the BALB/c mouse model, a DR5 DNA vaccine induced the expression of proapoptotic antibodies, thereby triggering tumor cell apoptosis. In addition, the vaccine-induced DR5-specific T cells to secrete IFN-γ. The investigators concluded that DR5 can be used as an immunogenic target for a TNBC vaccine [122].
DC fusion vaccine
A study evaluated a DC fusion vaccine in TNBC. Compared with the control group, the group that was administered a co-culture of DC, T cells, and TNBC cells (created by electrofusion technology) demonstrated significant stimulation of T cell expansion. The increased levels of IL-12 and IFN-γ demonstrated the specific cytotoxic effect of the DC fusion vaccine on tumor cells [123].
Personalized peptide vaccine
Research has indicated the efficacy of personalized peptide vaccination (PPV) in managing TNBC [124]. During a phase II study involving patients with metastatic and recurrent TNBC, 1/18 patients each developed a complete and partial immune response after PPV administration (NCT02427581). PPV appeared to be safe, as it did not lead to any serious adverse effects in these patients. These results reveal that PPV has a potential effect in treating patients with TNBC [125].
Cellular immunotherapy
Chimeric antigen receptor T cell therapy
Chimeric antigen receptor T cell (CAR-T) therapy has shown excellent therapeutic effects or potential in treating diverse forms of cancer. Although breakthroughs have been achieved in hematological tumors, their use in the field of solid tumor therapy remains in the explorative stage. Studies in patients with TNBC have identified an antigen target related to breast CSCs (disialoganglioside GD2), which is widely found in highly invasive breast cancer subtypes; GD2-CAR-T cells were therefore created. In a study on a mouse model, GD2-CAR-T cells demonstrated a significantly higher inhibitory effect on tumor cell growth compared to CD19-CAR-T cells. Notably, no metastatic cancer cells were observed in the lungs of mice treated with GD2-CAR-T cells, all mice treated with CD19-CAR-T cells exhibited metastatic cancer cells in the lung [126]. Investigators have used TAB004 (a highly specific monoclonal antibody against the tumor-associated form of MUC1) to engineer the MUC28z (a chimeric antigen receptor) fusion molecule and generate CAR T cells; they also performed phenotypic and functional analyses. The results showed that MUC28z CAR-T cells can efficiently suppress the proliferation of TNBC tumors, under both in-vivo and in-vitro conditions. The CAR-T cells exhibited significant therapeutic promise in combating MUC1-positive TNBC tumors [127]. In a study on a TNBC mouse model, a fibrin gel containing CAR-T cells was applied to the surgical wound after partial tumor resection. The CAR-T cells significantly eliminated residual tumor cells, allowing the mice to survive. The results indicate that CAR-T cells could potentially act as a supplementary approach to enhance the efficiency and success rate of surgery in TNBC [128].
NK cell therapy
The regulation of NK cell activities, such as degranulation, cytokine secretion, and cytotoxicity, is determined by the combination of signals received from activating and inhibitory receptors. NK cells possess the ability to identify and eliminate tumor cells that exhibit diminished or suppressed expression of MHC-I, a phenomenon referred to as the “missing self” recognition mechanism [129]. NK cells possess the ability to not only secrete cytotoxic particles, such as perforin and granase, via exocytosis but also directly induce the lysis of target cells and initiate cell apoptosis by activating the expressionof FASL/TRAIL in NK cells. Furthermore, other immune cells can be recruited to generate secondary immune responses by synthesizing and secreting IFN-γ, TNF-α, and chemokines [130, 131]. Additionally, another significant mechanism employed by NK cells in tumor eradication is antibody-dependent cell-mediated cytotoxicity (ADCC), which triggers the degranulation response of NK cells to eliminate target cells that are coated with antibodies. Presently, the repertoire of NK cell approaches employed in tumor mmunotherapy encompasses the following: in vitro administration of activated autologous or allogeneic NK cells; the induction of antibody-specific cytotoxicity through the amalgamation of NK cells and monoclonal agents (e.g., ICIs); and the implementation of CAR-NK cell immunotherapy. Notably, NK cells derived from cancer patients exhibit comparable expansion potential to those derived from healthy donors, exhibit cytotoxicity against TNBC cell lines and xenograft tumor models, and can be harmoniously integrated with existing.
cancer treatments [132]. To enhance the functionality of NK cells in patients with TNBC, it has been observed that the combined approach of radiotherapy (RT) and NK cell therapy can effectively facilitate the infiltration and migration of NK cells into the tumor lesion within the xenograft tumor model of TNBC. Consequently, this approach leads to the reduction of tumor burden and tumor growth. Notably, compared to the NK group, the RT + NK group demonstrated a significantly prolonged presence of NK cells within the primary tumor tissue. Furthermore, the combined therapy of RT + NK cells effectively suppressed the dissemination and distant metastasis of breast cancer cells to the lymph nodes, specifically in the lung and liver [133]. Avelumab, a checkpoint inhibitor targeting PD-L1, demonstrated the ability to elicit cytotoxic effects mediated by NK cells in TNBC patients [134]. Chen et al. developed a nano-system incorporating selenium, which effectively increased NKG2D expression on NK cells and NKG2DLs expression on tumor cells. This augmentation enhanced the recognition and cytotoxicity of NK cells towards tumor cells, thereby bolstering the immune response against tumors facilitated by NK cells [135]. Additionally, studies have shown that Ruthenium complexes and Aptamer-engineered approaches can enhance the sensitivity of TNBC to NK cell therapy and modulate the immune microenvironment [136, 137]. The presence of an immature NK cell population (CD11b CD27 Socs3high) in TNBC has been found to diminish granules-mediated cytolysis and enhance the expression of Wnt16 ligand, which has been implicated in tumor progression and metastasis [138]. The promotion of TNBC progression can be facilitated by the depletion of NK cells, inhibition of Wnt secretion, utilization of immunotherapy involving anti-PD-L1 antibodies, or a combination of chemotherapy and immunotherapy [139].
CAR-NK is an application of genetic engineering cellular therapy that incorporates a chimeric antibody into NK cells, which can significantly improve the specificity of NK cell’ s therapeutic effects. Notably, a study demonstrated that EGFR-CAR NK cells exhibited cytotoxicity and anti-tumor properties against TNBC cell lines with high EGFR expression.
Optimizing the cytotoxic potential of NK cells appears to heavily rely on the tailored design of CAR structures dedicated to NK cells [140]. In addition, it is imperative to optimize the procedures for amplifying and activating harvested NK cells to obtain a large number of memory-like, unexhausted, and homogeneous populations of NK cells in clinical practice. Moreover, the effectiveness of CAR-NK cells in TNBC treatment might necessitate additional modifications to enhance the trafficking capabilities and reduce the sensitivity to immunosuppression in the TME.
Cytokine-induced killer cell therapy
Cytokine-Induced Killer (CIK) cells are a diverse group of polyclonal T lymphocytes acquired by expanding lymphocytes in vitro, and exhibit functional and phenotype characteristics of both T cells and NK cells. These cells express two membrane protein molecules, CD3 and CD56, simultaneously, earning them the designation of NK cell-like T lymphocytes. Due to their potent tumor cell recognition abilities, CIK cells are often likened to “cell missiles” that can accurately target tumor cells without harming normal cells. In the case of patients with TNBC, repeated administration of CIK cells and cetuximab to patients with TNBC has been shown to significantly impede the growth, metastasis, and dissemination of TNBC cells and xenotransplanted tumors in lymph nodes and lungs [141]. The use of the treatment approach successfully inhibits the reoccurrence and extends the lifespan of patients with TNBC who have lymph node spread, an advanced TNM stage, and poor histological grade [142].
CIK cells have demonstrated potential benefits in patients with various tumor types, however, their effectiveness varies among individuals. The role of Focal adhesion kinase (FAK).
in controlling cell invasion and migration is vital, which in turn affects the vulnerability of tumor cells to CIK cells. Current evidence suggests a favorable connection between PD-L1 and FAK mRNA expression in PD-L1 positive TNBC, indicating a possible link.
between FAK and the function of immune checkpoints. Furthermore, studies have shown.
that the anti-PD-L1 antibody atezolizumab greatly augments the suppressive impact of FAK inhibitors on cancer cells [143]. The application of FAK inhibitors in the treatment of TNBC cells, along with co-culturing with CIK cells, resulted in a higher incidence of cell death, suggesting that FAK enhances the susceptibility of tumor cells to CIK cells [144]. A study conducted in vivo has substantiated that the combined administration of FAK inhibitors and CIK cells surpasses the efficacy of either treatment in inhibiting tumor growth [145]. Consequently, the integration of CIK cell therapy with PD-L1 inhibitors and FAK inhibitors holds promise as a potentially efficacious and innovative treatment approach for patients with TNBC. The immunotherapy of CAR-T cell, NK cell and CIK cell separately or with others anticancer approaches to treat TNBC is shown in Fig. 5.

The diagram CAR-T cell, NK cell and CIK cell separately or with others anticancer approaches to treat TNBC. GD2-CAR-T cells, MUC28z CAR-T cells and a fibrin gel containing CAR-T cells demonstrate a significantly higher inhibitory effect on tumor cell growth in TNBC. NK cells derived from cancer patients exhibit heightened cytotoxicity against TNBC cell lines and xenograft tumor models. The combination therapy of RTand NK cells, Avelumab, Selenocystine/TGF-β Inhibitor, Ruthenium complexes, Aptamer-engineered approach enhance the cytotoxic effects mediated by NK cells. EGFR-CAR NK cells exhibited cytotoxicity and anti-tumor properties against TNBC cell lines. The combination administration of CIK cells and cetuximab or CIK cells and FAK inhibitor has been shown to significantly impede the growth, metastasis, and dissemination of TNBC
Bispecific antibody approaches for TNBC therapy
Bispecific antibody (BsAb) refers to an artificially engineered antibody that can specifically bind two antigens or antigenic epitopes simultaneously, serving as a bridge connecting two antigens (epitopes). This antibody does not occur naturally but is synthesized through cell fusion or recombinant DNA techniques. The main mechanism of BsAb in anti-tumor therapy involves (1) The recruitment of T cells or NK cells and augmenting cytotoxicity effect on tumor cells (2) Simultaneously blocking two different signaling pathways to synergistically inhibit the growth and proliferation of tumor cells (3) Concurrently targeting different antigens or epitopes on the cell surface to enhance the specific binding with tumor cells. F7AK3 is a BsAb that has been developed and can specifically bind to the trophoblast cell surface antigen 2 (TROP2) and CD3 of TNBC cell lines and primary tumor cells, thereby recruiting T cells. A study found that F7AK3 inhibits the growth of a xenograft NCG immunodeficient mouse model in TNBC [146]. Consequently, the potential utilization of F7AK3 alone or in combination with ICIs for immunotherapy in advanced TNBC necessitates further investigation through clinical trials. MesobsFab, a Fab-like format, targeting mesothelin and CD16, which facilitates the recruitment and infiltration of NK cells into tumor spheres, leading to strong, dose-dependent cell-mediated cytotoxicity in mesothelin-positive TNBC [147]. BiTP, a kind of BsAb, was developed for TGF-β and human PD-L1 using the Check-BODY platform. The administration of BiTP in the humanized TNBC model resulted in the elevated infiltration of TILs, CD8 + T cells, and NK cells, thereby reshaping the TME [148]. The structure and effect of F7AK3, MesobsFab, and BiTP was illustrated in Fig. 6. Furthermore, numerous therapeutic targets for TNBC have been integrated into BsAb constructs, including CEACAM5, EphA10, P-cadherin, EpCAM, and EGFR [149,150,151,152,153,154]. Recently, there has been an evaluation of BsAb that specifically target receptors, such as EGFR, HER3, and Notch on TNBC cells [147, 155,156,157,158,159]. The utilization of BsAb for the treatment of TNBC is experiencing a persistent surge in momentum. Numerous targets specific to TNBC have emerged as potential candidates for the future advancement of BsAb [159].
Currently, diverse technologies for BsAbs production exist, which can be broadly categorized into two groups: BsAbs with the Fc domain and BsAbs without the Fc domain. The inclusion of the Fc domain in BsAbs serves to enhance stability and prolong their half-life. Nevertheless, the interaction between the Fc domain and its receptors or complements may trigger ADCC, resulting in non-specific immune responses. On the other hand, Bispecific T cell engagers (BiTEs) are a type of BsAb that lack the Fc domain, thereby circumventing cytotoxicity. However, BiTEs exhibit a short half-life in the bloodstream, necessitating continuous infusion. One arm of the BiTE molecule specifically binds to the CD3 antigen on the surface of T cells, while the other arm binds to the tumor-associated antigen (TAA). Upon binding to their respective targets, the formation of synapses between T cells and cancer cells occurs, leading to the release of perforin and granzyme [160], which ultimately results in the death of cancer cells. In comparison to conventional antibodies, BiTE exhibits higher tissue permeability, increased efficiency in killing tumor cells, requires lower dosages, and produces stronger therapeutic effects [161]. However, the full extent of the benefits of this combination therapy in TNBC remains unclear due to the inherent heterogeneity, necessitating further investigation.
The utilization of BsAb has emerged as a significant and promising constituent of the forthcoming therapeutic antibody generation, owing to its capacity to effectively target two epitopes within tumor cells or the TME. Presently, numerous preclinical and clinical trials, including NCT05403554, NCT03219268, and NCT04424641, among others, are either in progress or have been concluded. Nevertheless, the utilization of BsAb therapeutics in the context of tumor treatment encounters significant hurdles, encompassing tumor heterogeneity and mutation burden, the imperative for repeated administration, deleterious adverse reactions, and off-target effects.

The diagram of bsAb types applied to treat TNBC. F7AK3 with cell surface antigen TROP2 and CD3 facilitates the recruitment of T cells. MesobsFab elicits the recruitment and infiltration of NK cells and leads to the induction of ADCC effect in mesothelin-positive tumors. BiTP was developed for TGF-β and human PD-L1 by using the Check-BODY platform, which increase the collagen deposition, TILs infiltration, CD8 + T cells penetrating
In this summary, we have provided an overview of the fundamental and clinical investigations conducted on immunotherapy in TNBC (Tables 1 and 2). Moreover, the scope of immunotherapy application in TNBC is expected to expand, encompassing the entire domain of breast cancer, ultimately benefiting a substantial proportion of patients.
Discussion
The IMpassion130 study was the first to indicate the value of immunotherapy in the management of TNBC. Notably, TNBC is the most studied malignant tumor in the field of immunotherapy. Although immunotherapy offers better safety and tolerability and a longer median PFS and OS, many issues remain to be resolved. Firstly, effective immune markers for excluding TNBC immune-dominant populations are lacking. Studies categorizing high, medium, and low-risk populations based on marker threshold values (and thereby demonstrating the mechanism of immunotherapy) are also lacking. Secondly, immunotherapy remains at an early stage of development, and not all immune checkpoints that may be relevant to patients with TNBC have been identified. The ICIs mentioned in this review are unlikely to apply to all patients with TNBC. In order to identify the recipients of precise treatment, it is imperative to discover more precise predictive biomarkers. Additionally, the reports from the literature show that immunotherapy is associated with risks of developing irreversible endocrine disorders including hypoglycemia, hyperthyroidism, type I diabetes, adrenal insufficiency, and hypophysitis. it is crucial to prioritize the management of adverse reactions in clinical trials. Should OS and DFS be considered alternative endpoints for neoadjuvant immunotherapy in TNBC? In other types of solid tumors, the true advantages of immunotherapy are often observed through the extension of OS. Consequently, pCR may not be the most optimal alternative endpoint for the neoadjuvant treatment phase. Therefore, it is highly recommended to investigate appropriate research endpoints in future clinical studies on neoadjuvant immunotherapy in TNBC. In order to enhance the efficacy of immunotherapy for TNBC, it is imperative to explore a more potent combination therapy that can effectively eradicate tumors, augment tumor antigen exposure, facilitate antigen presentation, and prevent immune evasion.
Prospect
The combination with other treatment modalities currently offers an effective approach for immunotherapy in TNBC. The enhancement of immunotherapy response rates can be achieved through the integration of various treatment strategies, encompassing combination chemotherapy, targeted therapy, alternative immunotherapy approaches, cellular therapy, radiotherapy, etc. Future advancements in the field lie within the realm of immunosuppressive combined chemotherapy/targeting/novel immunotherapies, including inhibitors targeting BRCA/PI3K/AKT/mTOR/MEK/CDK4/6, androgen receptor inhibitors, ADC drugs, tumor vaccines, oncolytic viruses, and adoptive immunotherapies such as TIL and CAR-T. Future clinical trials need to consider the use of traditional therapies, ICIs, and combination therapies based on the specific molecular subtypes of TNBC, the number of TILs, and PD-L1 expression. It is also necessary to identify patients who may safely receive immunotherapy and even benefit from different combination therapies.
Conclusion
Although studies have explored the molecular characteristics of TNBC and their dynamic relationship with the TME and immune cells in detail, the considerable heterogeneity in this entity warrants further studies to understand its molecular characteristics and microenvironmental structure. Existing multi-dimensional omics data need to be integrated to ensure a more accurate classification of TNBC subtypes. New targets are being continually developed to achieve good efficacy and improve individualized precision treatment approaches. Their clinical application is expected to confer better therapeutic outcomes in TNBC patients.
Data Availability
Not applicable.
Change history
14 September 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12943-023-01858-z
Abbreviations
- 2 F-Fuc:
-
2-fluoro-L-focus
- ACSL3:
-
Acyl-CoA Synthetase Long Chain Family Member 3
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- ADV/HSV-tk:
-
Adenovirus mediated herpes simplex virus thymidine kinase
- APCs:
-
Antigen presenting cells
- B7H3:
-
B7 homologous 3
- BiTEs:
-
Bispecific T cell engagers
- BsAb:
-
Bispecific antibody
- CAFs:
-
Cancer-associated fibroblasts
- CAR-T:
-
Chimeric antigen receptor T cell
- CDA:
-
C-di-AMP
- CIK:
-
Cytokine-induced killer
- CpG-ODN:
-
CpG oligodeoxynucleotides
- CpG:
-
Cytosine-phosphorothioate-guanine
- CPS:
-
Comprehensive positive score
- CSCs:
-
Tumor stem cells
- CTLA-4:
-
Cytotoxic T-lymphocyte associated antigen 4
- CXCR2:
-
Chemokine receptor 2
- DCR:
-
Disease control rate
- DCs:
-
Dendritic cells
- ECM:
-
Extracellular matrix
- EPI:
-
Epirubicin
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- FAK:
-
Focal adhesion kinase
- HSV:
-
Herpes simplex virus
- ICD:
-
Immunogenic cell death
- ICIs:
-
Immune checkpoint inhibitors
- IFN-:
-
γ interferon γ
- IFNs:
-
Interferon
- IL-12:
-
Interleukin-12
- IM:
-
Immunomodulatory
- iPP:
-
Polyacrylic acid polymer lactic acid and glycolic acid perfluoronaphthalane
- iPPO:
-
O3 loaded iPP particles
- LAM:
-
Lipid-associated macrophage
- LCP:
-
Lipid calcium phosphate
- LOX:
-
Lysyl oxidase
- MAPK:
-
Mitogen activated protein kinases
- MES:
-
Macrophage-enriched subtype
- MHC-I:
-
Major histocompatibility complex class I
- mPEG:
-
Methoxy polyethylene glycol
- mTNBC:
-
Metastatic triple negative breast cancer
- MUC1:
-
Mucin 1
- MW:
-
Microwave
- NES:
-
Neutrophil-enriched subtype
- NK:
-
NK
- NO:
-
Nitric oxide
- OPN:
-
Osteopontin
- ORR:
-
Objective response rate
- OXPHOS:
-
Oxidative phosphorylation
- PARP:
-
Poly ADP-ribose polymerase
- PARPi:
-
PARP inhibitors
- PCR:
-
Pathological complete response rate
- PD-1:
-
Programmed death receptor-1
- PD-L1:
-
Programmed death receptor-ligand 1
- PFS:
-
Progression-free survival
- PI3K/AKT/mTOR:
-
Phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin
- PPV:
-
Personalized peptide vaccination
- PPV:
-
Personalized peptide vaccination
- PTX:
-
Paclitaxel
- Rg3-DTX-LPs:
-
Docetaxel loaded Rg3 liposome
- RNA:
-
Ribonucleic acid
- ROS:
-
Reactive oxygen species
- SBRT:
-
Stereotactic body radiotherapy
- SHP2:
-
SH2 protein tyrosine phosphatase 2
- STING:
-
Interferon gene stimulating factor
- TAA:
-
Tumor associated antigen
- TAMs:
-
Tumor-associated macrophages
- TGF:
-
β RIIFc TGF β Receptor II IgG Fc Fragment
- TIL:
-
Tumor infiltrating lymphocyte
- TLR9:
-
Toll-like receptor 9
- TMAO:
-
Trimethylamine oxide
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple negative breast cancer
- TNF-α:
-
Tumor necrosis factor-α
- Treg:
-
Regulatory T cells
- TriTNE:
-
Nanotechnology × PD-L1/CD3 three specific T cell nano adapter
- TROP2:
-
Trophoblast cell surface antigen 2
- VEGF:
-
Vascular endothelial growth factor
References
Park SY, Choi JH, Nam JS. Targeting Cancer Stem cells in Triple-Negative breast Cancer. Cancers (Basel) 2019, 11.
Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat Rev Clin Oncol. 2022;19:91–113.
Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, Xiao Y, Yu KD, Liu YR, Yu Y, et al. Genomic and Transcriptomic Landscape of Triple-Negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35:428–440e425.
Heeke AL, Tan AR. Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 2021;40:537–47.
Bareche Y, Buisseret L, Gruosso T, Girard E, Venet D, Dupont F, Desmedt C, Larsimont D, Park M, Rothe F, et al. Unraveling triple-negative breast Cancer Tumor Microenvironment Heterogeneity: towards an Optimized Treatment Approach. J Natl Cancer Inst. 2020;112:708–19.
Du Y, Wei N, Ma R, Jiang S, Song D. A mir-210-3p regulon that controls the Warburg effect by modulating HIF-1α and p53 activity in triple-negative breast cancer. Cell Death Dis. 2020;11:731.
Li H, Sun X, Li J, Liu W, Pan G, Mao A, Liu J, Zhang Q, Rao L, Xie X, Sheng X. Hypoxia induces docetaxel resistance in triple-negative breast cancer via the HIF-1α/miR-494/Survivin signaling pathway. Neoplasia. 2022;32:100821.
Saatci O, Kaymak A, Raza U, Ersan PG, Akbulut O, Banister CE, Sikirzhytski V, Tokat UM, Aykut G, Ansari SA, et al. Targeting lysyl oxidase (LOX) overcomes chemotherapy resistance in triple negative breast cancer. Nat Commun. 2020;11:2416.
Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, O’Connor K, Lane AN, Xu R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1alpha stabilization and TNBC chemoresistance. Nat Commun. 2018;9:4456.
Broad RV, Jones SJ, Teske MC, Wastall LM, Hanby AM, Thorne JL, Hughes TA. Inhibition of interferon-signalling halts cancer-associated fibroblast-dependent protection of breast cancer cells from chemotherapy. Br J Cancer. 2021;124:1110–20.
Lawal B, Wu AT, Chen CH, T AG, Wu SY. Identification of INFG/STAT1/NOTCH3 as gamma-mangostin’s potential targets for overcoming doxorubicin resistance and reducing cancer-associated fibroblasts in triple-negative breast cancer. Biomed Pharmacother. 2023;163:114800.
Timperi E, Gueguen P, Molgora M, Magagna I, Kieffer Y, Lopez-Lastra S, Sirven P, Baudrin LG, Baulande S, Nicolas A, et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated fibroblasts and mediate Immune suppression in breast Cancer. Cancer Res. 2022;82:3291–306.
Xie Y, Wang B, Zhao Y, Tao Z, Wang Y, Chen G, Hu X. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J Hematol Oncol. 2022;15:72.
Dong F, Ruan S, Wang J, Xia Y, Le K, Xiao X, Hu T, Wang Q. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis. 2020;11:728.
Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113–26.
Yamauchi M, Gibbons DL, Zong C, Fradette JJ, Bota-Rabassedas N, Kurie JM. Fibroblast heterogeneity and its impact on extracellular matrix and immune landscape remodeling in cancer. Matrix Biol. 2020;91–92:8–18.
Fan Y, He S. The characteristics of Tumor Microenvironment in Triple negative breast Cancer. Cancer Manag Res. 2022;14:1–17.
Sarkar M, Nguyen T, Gundre E, Ogunlusi O, El-Sobky M, Giri B, Sarkar TR. Cancer-associated fibroblasts: the chief architect in the tumor microenvironment. Front Cell Dev Biol. 2023;11:1089068.
Joensuu H, Kellokumpu-Lehtinen PL, Huovinen R, Jukkola-Vuorinen A, Tanner M, Kokko R, Ahlgren J, Auvinen P, Paija O, Helle L, et al. Adjuvant capecitabine, docetaxel, cyclophosphamide, and epirubicin for early breast cancer: final analysis of the randomized FinXX trial. J Clin Oncol. 2012;30:11–8.
Desmedt C, Fornili M, Clatot F, Demicheli R, De Bortoli D, Di Leo A, Viale G, de Azambuja E, Crown J, Francis PA, et al. Differential Benefit of Adjuvant Docetaxel-Based chemotherapy in patients with early breast Cancer according to Baseline Body Mass Index. J Clin Oncol. 2020;38:2883–91.
Zhang P, Qin C, Liu N, Zhou X, Chu X, Lv F, Gu Y, Yin L, Liu J, Zhou J, Huo M. The programmed site-specific delivery of LY3200882 and PD-L1 siRNA boosts immunotherapy for triple-negative breast cancer by remodeling tumor microenvironment. Biomaterials. 2022;284:121518.
Shi X, Young CD, Zhou H, Wang X. Transforming growth factor-beta signaling in Fibrotic Diseases and Cancer-Associated fibroblasts. Biomolecules 2020, 10.
Chen D, Zhao Y, Bai S, Shi Z, Zhang J. [Effect of ginsenoside Rg3 on the progression of orthotopically xenotransplanted human breast cancer in nude mice and its mechanism]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2003;34:546–8.
Xia J, Zhang S, Zhang R, Wang A, Zhu Y, Dong M, Ma S, Hong C, Liu S, Wang D, Wang J. Targeting therapy and tumor microenvironment remodeling of triple-negative breast cancer by ginsenoside Rg3 based liposomes. J Nanobiotechnol. 2022;20:414.
Yuan Z, Jiang H, Zhu X, Liu X, Li J. Ginsenoside Rg3 promotes cytotoxicity of Paclitaxel through inhibiting NF-kappaB signaling and regulating Bax/Bcl-2 expression on triple-negative breast cancer. Biomed Pharmacother. 2017;89:227–32.
Gnanaprakasam JNR, Estrada-Muniz E, Vega L. The anacardic 6-pentadecyl salicylic acid induces macrophage activation via the phosphorylation of ERK1/2, JNK, P38 kinases and NF-kappaB. Int Immunopharmacol. 2015;29:808–17.
Gnanaprakasam JNR, Lopez-Banuelos L, Vega L. Anacardic 6-pentadecyl salicylic acid induces apoptosis in breast cancer tumor cells, immunostimulation in the host and decreases blood toxic effects of taxol in an animal model. Toxicol Appl Pharmacol. 2021;410:115359.
Lu Y, Yuan X, Wang M, He Z, Li H, Wang J, Li Q. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J Hematol Oncol. 2022;15:47.
Oshi M, Asaoka M, Tokumaru Y, Yan L, Matsuyama R, Ishikawa T, Endo I, Takabe K. CD8 T cell score as a Prognostic Biomarker for Triple negative breast Cancer. Int J Mol Sci 2020, 21.
Zhou CB, Zhou YL, Fang JY. Gut microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer. 2021;7:647–60.
Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus infection and Cancer. Trends Immunol. 2017;38:542–57.
Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. Opposing functions of Interferon Coordinate Adaptive and Innate Immune responses to Cancer Immune Checkpoint Blockade. Cell. 2019;178:933–948e914.
Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the crossroads of Tumor Immune Surveillance or Evasion. Front Immunol. 2018;9:847.
Zimmerli D, Brambillasca CS, Talens F, Bhin J, Linstra R, Romanens L, Bhattacharya A, Joosten SEP, Da Silva AM, Padrao N, et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat Commun. 2022;13:6579.
Lee JV, Housley F, Yau C, Nakagawa R, Winkler J, Anttila JM, Munne PM, Savelius M, Houlahan KE, Van de Mark D, et al. Combinatorial immunotherapies overcome MYC-driven immune evasion in triple negative breast cancer. Nat Commun. 2022;13:3671.
Tseng JC, Yang JX, Liu YL, Su YW, Lee AY, Chen YW, Liu KJ, Luo Y, Hong YR, Chuang TH. Sharpening up tumor microenvironment to enhance the efficacy of immune checkpoint blockade on head and neck cancer using a CpG-oligodeoxynucleotide. Cancer Immunol Immunother. 2022;71:1115–28.
Li T, Hua C, Yue W, Wu J, Lv X, Wei Q, Zhu S, Zang G, Cui J, Liu YJ, Chen J. Discrepant antitumor efficacies of three CpG oligodeoxynucleotide classes in monotherapy and co-therapy with PD-1 blockade. Pharmacol Res. 2020;161:105293.
Schoenfeld AJ, Hellmann MD. Acquired Resistance to Immune Checkpoint inhibitors. Cancer Cell. 2020;37:443–55.
Kepp O, Zitvogel L, Kroemer G. Clinical evidence that immunogenic cell death sensitizes to PD-1/PD-L1 blockade. Oncoimmunology. 2019;8:e1637188.
Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, Nandre R, Gaur P, Lopez J, Sade-Feldman M, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol. 2019;20:1231–43.
Duffy MJ, Synnott NC, Crown J. Mutant p53 in breast cancer: potential as a therapeutic target and biomarker. Breast Cancer Res Treat. 2018;170:213–9.
Yang Z, Lee MMM, Chan MK. Efficient intracellular delivery of p53 protein by engineered protein crystals restores tumor suppressing function in vivo. Biomaterials. 2021;271:120759.
Yang Z, Sun JK, Lee MM, Chan MK. Restoration of p53 activity via intracellular protein delivery sensitizes triple negative breast cancer to anti-PD-1 immunotherapy. J Immunother Cancer 2022, 10.
Boissiere-Michot F, Jacot W, Fraisse J, Gourgou S, Timaxian C, Lazennec G. Prognostic value of CXCR2 in breast Cancer. Cancers (Basel) 2020, 12.
Ghallab AM, Eissa RA, El Tayebi HM. CXCR2 small-molecule antagonist combats Chemoresistance and enhances immunotherapy in Triple-Negative breast Cancer. Front Pharmacol. 2022;13:862125.
Shen M, Chen C, Guo Q, Wang Q, Liao J, Wang L, Yu J, Xue M, Duan Y, Zhang J. Systemic delivery of mPEG-Masked trispecific T-Cell nanoengagers in synergy with STING agonists overcomes Immunotherapy Resistance in TNBC and generates a vaccination effect. Adv Sci (Weinh). 2022;9:e2203523.
Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy - immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13:195–207.
Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, Wang X. The diverse function of PD-1/PD-L pathway Beyond Cancer. Front Immunol. 2019;10:2298.
Ahmed FS, Gaule P, McGuire J, Patel K, Blenman K, Pusztai L, Rimm DL. PD-L1 protein expression on both Tumor cells and macrophages are Associated with response to Neoadjuvant Durvalumab with Chemotherapy in Triple-negative breast Cancer. Clin Cancer Res. 2020;26:5456–61.
Twomey JD, Zhang B. Cancer Immunotherapy Update: FDA-Approved checkpoint inhibitors and Companion Diagnostics. AAPS J. 2021;23:39.
Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39.
Brautigam K, Kabore-Wolff E, Hussain AF, Polack S, Rody A, Hanker L, Koster F. Inhibitors of PD-1/PD-L1 and ERK1/2 impede the proliferation of receptor positive and triple-negative breast cancer cell lines. J Cancer Res Clin Oncol. 2021;147:2923–33.
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, et al. Pembrolizumab in patients with Advanced Triple-Negative breast Cancer: phase ib KEYNOTE-012 study. J Clin Oncol. 2016;34:2460–7.
Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort a of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:397–404.
Ganguly S, Gogia A. Pembrolizumab monotherapy in advanced triple-negative breast cancer. Lancet Oncol. 2021;22:e224.
Tarantino P, Corti C, Schmid P, Cortes J, Mittendorf EA, Rugo H, Tolaney SM, Bianchini G, Andre F, Curigliano G. Immunotherapy for early triple negative breast cancer: research agenda for the next decade. NPJ Breast Cancer. 2022;8:23.
Bian L, Zhang H, Wang T, Zhang S, Song H, Xu M, Yao S, Jiang Z. JS001, an anti-PD-1 mAb for advanced triple negative breast cancer patients after multi-line systemic therapy in a phase I trial. Ann Transl Med. 2019;7:435.
Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, Kuter I, Nanda R, Cassier PA, Delord JP, et al. Long-term clinical outcomes and biomarker analyses of Atezolizumab Therapy for patients with metastatic triple-negative breast Cancer: a phase 1 study. JAMA Oncol. 2019;5:74–82.
Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, Boccia R, Lippman ME, Somer R, Smakal M, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN solid tumor study. Breast Cancer Res Treat. 2018;167:671–86.
Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: from mechanism to autoimmune therapy. Int Immunopharmacol. 2020;80:106221.
Peng Z, Su P, Yang Y, Yao X, Zhang Y, Jin F, Yang B. Identification of CTLA-4 associated with tumor microenvironment and competing interactions in triple negative breast cancer by co-expression network analysis. J Cancer. 2020;11:6365–75.
Lu L, Bai Y, Wang Z. Elevated T cell activation score is associated with improved survival of breast cancer. Breast Cancer Res Treat. 2017;164:689–96.
Kaewkangsadan V, Verma C, Eremin JM, Cowley G, Ilyas M, Eremin O. Tumour-draining axillary lymph nodes in patients with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC): the crucial contribution of immune cells (effector, regulatory) and cytokines (Th1, Th2) to immune-mediated tumour cell death induced by NAC. BMC Cancer. 2018;18:123.
McArthur HL, Diab A, Page DB, Yuan J, Solomon SB, Sacchini V, Comstock C, Durack JC, Maybody M, Sung J, et al. A pilot study of preoperative single-dose Ipilimumab and/or cryoablation in women with early-stage breast Cancer with Comprehensive Immune Profiling. Clin Cancer Res. 2016;22:5729–37.
Abdeladhim M, Karnell JL, Rieder SA. In or out of control: modulating regulatory T cell homeostasis and function with immune checkpoint pathways. Front Immunol. 2022;13:1033705.
Santa-Maria CA, Kato T, Park JH, Kiyotani K, Rademaker A, Shah AN, Gross L, Blanco LZ, Jain S, Flaum L, et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget. 2018;9:18985–96.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative breast Cancer. N Engl J Med. 2018;379:2108–21.
He Q, Peng Y, Sun J, Liu J. Platinum-based chemotherapy and immunotherapy in early triple-negative breast Cancer: a Meta-analysis and Indirect Treatment comparison. Front Oncol. 2021;11:693542.
Zhang J, Wang Z, Hu X, Wang B, Wang L, Yang W, Liu Y, Liu G, Di G, Hu Z, et al. Cisplatin and gemcitabine as the first line therapy in metastatic triple negative breast cancer. Int J Cancer. 2015;136:204–11.
Ge J, Zuo W, Chen Y, Shao Z, Yu K. The advance of adjuvant treatment for triple-negative breast cancer. Cancer Biol Med. 2021;19:187–201.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25:920–8.
Schmid P, Salgado R, Park YH, Munoz-Couselo E, Kim SB, Sohn J, Im SA, Foukakis T, Kuemmel S, Dent R, et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann Oncol. 2020;31:569–81.
Schmid P, Dent R, O’Shaughnessy J. Pembrolizumab for early triple-negative breast Cancer reply. N Engl J Med 2020, 382.
Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. Pembrolizumab for early triple-negative breast Cancer. N Engl J Med. 2020;382:810–21.
Loibl S, Untch M, Burchardi N, Huober J, Sinn BV, Blohmer JU, Grischke EM, Furlanetto J, Tesch H, Hanusch C, et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol. 2019;30:1279–88.
Loibl S, Schneeweiss A, Huober JB, Braun M, Rey J, Blohmer JU, Furlanetto J, Zahm DM, Hanusch C, Thomalla J et al. Durvalumab improves long-term outcome in TNBC: results from the phase II randomized GeparNUEVO study investigating neodjuvant durvalumab in addition to an anthracycline/taxane based neoadjuvant chemotherapy in early triple-negative breast cancer (TNBC). J Clin Oncol 2021, 39.
Gianni L, Huang C-S, Egle D, Bermejo B, Zamagni C, Thill M, Anton A, Zambelli S, Bianchini G, Russo S et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple negative, early high-risk and locally advanced breast cancer. NeoTRIPaPDL1 Michelangelo randomized study. Cancer Res 2020, 80.
Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, Koehler A, Sohn J, Iwata H, Telli ML, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090–100.
Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Henschel V, Molinero L, Chui SY, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59.
Cortes J, Rugo HS, Cescon DW, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Perez-Garcia J, Iwata H, et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative breast Cancer. N Engl J Med. 2022;387:217–26.
Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396:1817–28.
Tolaney SM, Kalinsky K, Kaklamani VG, D’Adamo DR, Aktan G, Tsai ML, O’Regan RM, Kaufman PA, Wilks ST, Andreopoulou E, et al. Eribulin Plus Pembrolizumab in patients with metastatic triple-negative breast Cancer (ENHANCE 1): a phase Ib/II study. Clin Cancer Res. 2021;27:3061–8.
Charpentier M, Spada S, Van Nest SJ, Demaria S. Radiation therapy-induced remodeling of the tumor immune microenvironment. Semin Cancer Biol. 2022;86:737–47.
Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, Dephoure N, Clement CC, Santambrogio L, Zhou XK et al. Radiotherapy-exposed CD8 + and CD4 + neoantigens enhance tumor control. J Clin Invest 2021, 131.
Song HN, Jin H, Kim JH, Ha IB, Kang KM, Choi HS, Jeong HJ, Kim MY, Kim HJ, Jeong BK. Abscopal Effect of Radiotherapy enhanced with Immune Checkpoint inhibitors of Triple negative breast Cancer in 4T1 mammary carcinoma model. Int J Mol Sci 2021, 22.
Ho AY, Barker CA, Arnold BB, Powell SN, Hu ZI, Gucalp A, Lebron-Zapata L, Wen HY, Kallman C, D’Agnolo A, et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer. 2020;126:850–60.
Seth P, Wang ZG, Pister A, Zafar MB, Kim S, Guise T, Wakefield L. Development of oncolytic adenovirus armed with a fusion of soluble transforming growth factor-beta receptor II and human immunoglobulin fc for breast cancer therapy. Hum Gene Ther. 2006;17:1152–60.
Yang Y, Xu W, Peng D, Wang H, Zhang X, Wang H, Xiao F, Zhu Y, Ji Y, Gulukota K, et al. An oncolytic adenovirus targeting transforming growth factor beta inhibits protumorigenic signals and produces Immune activation: a Novel Approach to enhance Anti-PD-1 and Anti-CTLA-4 therapy. Hum Gene Ther. 2019;30:1117–32.
Xu W, Yang Y, Hu Z, Head M, Mangold KA, Sullivan M, Wang E, Saha P, Gulukota K, Helseth DL, et al. LyP-1-Modified oncolytic adenoviruses targeting transforming growth factor beta inhibit Tumor Growth and Metastases and augment Immune checkpoint inhibitor therapy in breast Cancer Mouse Models. Hum Gene Ther. 2020;31:863–80.
Sun K, Xu Y, Zhang L, Niravath P, Darcourt J, Patel T, Teh BS, Farach AM, Guerrero C, Mathur S, et al. A phase 2 trial of enhancing Immune Checkpoint Blockade by Stereotactic Radiation and in situ Virus Gene Therapy in Metastatic Triple-Negative breast Cancer. Clin Cancer Res. 2022;28:4392–401.
Clark CA, Yang ES. Harnessing DNA repair defects to Augment Immune-Based therapies in Triple-Negative breast Cancer. Front Oncol. 2021;11:703802.
Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, Gelber RD, de Azambuja E, Fielding A, Balmana J, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-Mutated breast Cancer. N Engl J Med. 2021;384:2394–405.
Batalini F, Xiong N, Tayob N, Polak M, Eismann J, Cantley LC, Shapiro GI, Adalsteinsson V, Winer EP, Konstantinopoulos PA, et al. Phase 1b clinical trial with Alpelisib plus Olaparib for patients with Advanced Triple-Negative breast Cancer. Clin Cancer Res. 2022;28:1493–9.
Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM, et al. Open-label clinical trial of Niraparib Combined with Pembrolizumab for treatment of Advanced or Metastatic Triple-Negative breast Cancer. JAMA Oncol. 2019;5:1132–40.
Yap TA, Bardia A, Dvorkin M, Galsky MD, Beck JT, Wise DR, Karyakin O, Rubovszky G, Kislov N, Rohrberg K, et al. Avelumab Plus Talazoparib in patients with Advanced Solid Tumors: the JAVELIN PARP Medley Nonrandomized Controlled Trial. JAMA Oncol. 2023;9:40–50.
Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, Alexandre J, You B, Bastian S, Krebs MG, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64.
Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via Intratumoral STING pathway activation in BRCA-Deficient models of triple-negative breast Cancer. Cancer Discov. 2019;9:722–37.
Li Q, Wang Y, Jia W, Deng H, Li G, Deng W, Chen J, Kim BYS, Jiang W, Liu Q, Liu J. Low-dose anti-angiogenic therapy sensitizes breast Cancer to PD-1 blockade. Clin Cancer Res. 2020;26:1712–24.
Zhao H, Chen Q, Alam A, Cui J, Suen KC, Soo AP, Eguchi S, Gu J, Ma D. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018;9:356.
Liu J, Liu Q, Li Y, Li Q, Su F, Yao H, Su S, Wang Q, Jin L, Wang Y et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label phase II trial. J Immunother Cancer 2020, 8.
Wu SY, Xu Y, Chen L, Fan L, Ma XY, Zhao S, Song XQ, Hu X, Yang WT, Chai WJ, et al. Combined angiogenesis and PD-1 inhibition for immunomodulatory TNBC: concept exploration and biomarker analysis in the FUTURE-C-Plus trial. Mol Cancer. 2022;21:84.
Miricescu D, Totan A, Stanescu S, Badoiu II, Stefani SC, Greabu C. M: PI3K/AKT/mTOR signaling pathway in breast Cancer: from Molecular Landscape to clinical aspects. Int J Mol Sci 2020, 22.
Kriegsmann M, Endris V, Wolf T, Pfarr N, Stenzinger A, Loibl S, Denkert C, Schneeweiss A, Budczies J, Sinn P, Weichert W. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget. 2014;5:9952–65.
Song Y, Bugada L, Li R, Hu H, Zhang L, Li C, Yuan H, Rajanayake KK, Truchan NA, Wen F, et al. Albumin nanoparticle containing a PI3Kgamma inhibitor and paclitaxel in combination with alpha-PD1 induces tumor remission of breast cancer in mice. Sci Transl Med. 2022;14:eabl3649.
Schmid P, Loirat D, Savas P, Espinosa E, Boni V, Italiano A, White S, Singel SM, Withana N, Mani A et al. Phase ib study evaluating a triplet combination of ipatasertib (IPAT), atezolizumab (atezo), and paclitaxel (PAC) or nab-PAC as first-line (1L) therapy for locally advanced/metastatic triple-negative breast cancer (TNBC). Cancer Res 2019, 79.
Huang Y, Zhang HL, Li ZL, Du T, Chen YH, Wang Y, Ni HH, Zhang KM, Mai J, Hu BX, et al. FUT8-mediated aberrant N-glycosylation of B7H3 suppresses the immune response in triple-negative breast cancer. Nat Commun. 2021;12:2672.
O’Shaughnessy J, Moroose RL, Babu S, Baramidze K, Chan D, Leitner SP, Nemsadze G, Ordentlich P, Quaranto C, Meyers ML et al. Results of ENCORE 602 (TRIO025), a phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab with or without entinostat in patients with advanced triple-negative breast cancer (aTNBC). J Clin Oncol 2020, 38.
Brufsky A, Kim SB, Zvirbule Z, Eniu A, Mebis J, Sohn JH, Wongchenko M, Chohan S, Amin R, Yan Y, et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): primary analysis. Ann Oncol. 2021;32:652–60.
Li Q, Wang J, Mu Y, Zhang T, Han Y, Wang J, Li Q, Luo Y, Ma F, Fan Y, et al. Dose-dense paclitaxel plus carboplatin vs. epirubicin and cyclophosphamide with paclitaxel as adjuvant chemotherapy for high-risk triple-negative breast cancer. Chin J Cancer Res. 2020;32:485–96.
Schmid P, Im S-A, Armstrong A, Park YH, Chung W-P, Nowecki Z, Lord S, Wysocki PJ, Lu Y-S, Dry H et al. BEGONIA: phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from arm 1, d + paclitaxel (P), and arm 6, d + trastuzumab deruxtecan (T-DXd). 2021, 39:1023–3.
Song L, Zheng D, Xu J, Xu T, Liu Z, Zhang H, Li Y, Peng Y, Shi H. Improvement of TNBC immune checkpoint blockade with a microwave-controlled ozone release nanosystem. J Control Release. 2022;351:954–69.
Zhou W, Yu M, Mao X, Pan H, Tang X, Wang J, Che N, Xie H, Ling L, Zhao Y, et al. Landscape of the Peripheral Immune Response Induced by local microwave ablation in patients with breast Cancer. Adv Sci (Weinh). 2022;9:e2200033.
Huo M, Wang L, Chen Y, Shi J. Tumor-selective catalytic nanomedicine by nanocatalyst delivery. Nat Commun. 2017;8:357.
Chen Q, Zhou S, Ding Y, Chen D, Dahiru NS, Tang H, Xu H, Ji M, Wang X, Li Z, et al. A bio-responsive, cargo-catchable gel for postsurgical tumor treatment via ICD-based immunotherapy. J Control Release. 2022;346:212–25.
Li Z, Cai H, Li Z, Ren L, Ma X, Zhu H, Gong Q, Zhang H, Gu Z, Luo K. A tumor cell membrane-coated self-amplified nanosystem as a nanovaccine to boost the therapeutic effect of anti-PD-L1 antibody. Bioact Mater. 2023;21:299–312.
Gebregiworgis T, Purohit V, Shukla SK, Tadros S, Chaika NV, Abrego J, Mulder SE, Gunda V, Singh PK, Powers R. Glucose limitation alters glutamine metabolism in MUC1-Overexpressing pancreatic Cancer cells. J Proteome Res. 2017;16:3536–46.
Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, Huang L. Combination immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade effectively inhibits growth of Triple negative breast Cancer. Mol Ther. 2018;26:45–55.
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, Guo C, Wu X, Li Y, Li X, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
Rugo HS, Cortes J, Barrios CH, Cabrera P, Xu B, Huang CS, Kim SB, Melisko M, Nanda R, Pienkowski T et al. GLORIA: phase III, open-label study of adagloxad simolenin/OBI-821 in patients with high-risk triple-negative breast cancer. Future Oncol 2022.
Hung JT, Chen IJ, Ueng SH, Huang CS, Chen SC, Chen MY, Lin YC, Lin CY, Campbell MJ, Rugo HS, Yu AL. The clinical relevance of humoral immune responses to Globo H-KLH vaccine adagloxad simolenin (OBI-822)/OBI-821 and expression of Globo H in metastatic breast cancer. J Immunother Cancer 2022, 10.
Jaini R, Kesaraju P, Johnson JM, Altuntas CZ, Jane-Wit D, Tuohy VK. An autoimmune-mediated strategy for prophylactic breast cancer vaccination. Nat Med. 2010;16:799–803.
Piechocki MP, Wu GS, Jones RF, Jacob JB, Gibson H, Ethier SP, Abrams J, Yagita H, Venuprasad K, Wei WZ. Induction of proapoptotic antibodies to triple-negative breast cancer by vaccination with TRAIL death receptor DR5 DNA. Int J Cancer. 2012;131:2562–72.
Huo W, Yang X, Wang B, Cao L, Fang Z, Li Z, Liu H, Liang XJ, Zhang J, Jin Y. Biomineralized hydrogel DC vaccine for cancer immunotherapy: a boosting strategy via improving immunogenicity and reversing immune-inhibitory microenvironment. Biomaterials. 2022;288:121722.
Takahashi R, Toh U, Iwakuma N, Takenaka M, Otsuka H, Furukawa M, Fujii T, Seki N, Kawahara A, Kage M, et al. Feasibility study of personalized peptide vaccination for metastatic recurrent triple-negative breast cancer patients. Breast Cancer Res. 2014;16:R70.
Zhu Y, Zhu X, Tang C, Guan X, Zhang W. Progress and challenges of immunotherapy in triple-negative breast cancer. Biochim Biophys Acta Rev Cancer. 2021;1876:188593.
Seitz CM, Schroeder S, Knopf P, Krahl AC, Hau J, Schleicher S, Martella M, Quintanilla-Martinez L, Kneilling M, Pichler B, et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology. 2020;9:1683345.
Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, Mukherjee P. CAR T cells targeting the Tumor MUC1 glycoprotein reduce triple-negative breast Cancer Growth. Front Immunol. 2019;10:1149.
Uslu U, Da T, Assenmacher CA, Scholler J, Young RM, Tchou J, June CH. Chimeric antigen receptor T cells as adjuvant therapy for unresectable adenocarcinoma. Sci Adv. 2023;9:eade2526.
Yokoyama WM, Kim S. How do natural killer cells find self to achieve tolerance? Immunity 2006, 24:249–57.
Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188:2375–80.
Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2:850–61.
Shenouda MM, Gillgrass A, Nham T, Hogg R, Lee AJ, Chew MV, Shafaei M, Aarts C, Lee DA, Hassell J, et al. Ex vivo expanded natural killer cells from breast cancer patients and healthy donors are highly cytotoxic against breast cancer cell lines and patient-derived tumours. Breast Cancer Res. 2017;19:76.
Kim KW, Jeong JU, Lee KH, Uong TNT, Rhee JH, Ahn SJ, Kim SK, Cho D, Quang Nguyen HP, Pham CT, Yoon MS. Combined NK Cell Therapy and Radiation Therapy exhibit Long-Term Therapeutic and Antimetastatic Effects in a human triple negative breast Cancer Model. Int J Radiat Oncol Biol Phys. 2020;108:115–25.
Julia EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 Immune checkpoint inhibitor, Triggers NK cell-mediated cytotoxicity and cytokine production against Triple negative breast Cancer cells. Front Immunol. 2018;9:2140.
Liu C, Lai H, Chen T. Boosting natural killer cell-based Cancer immunotherapy with Selenocystine/Transforming growth factor-Beta inhibitor-encapsulated nanoemulsion. ACS Nano. 2020;14:11067–82.
Chen Q, He L, Li X, Xu L, Chen T. Ruthenium complexes boost NK cell immunotherapy via sensitizing triple-negative breast cancer and shaping immuno-microenvironment. Biomaterials. 2022;281:121371.
Chen Z, Zeng Z, Wan Q, Liu X, Qi J, Zu Y. Targeted immunotherapy of triple-negative breast cancer by aptamer-engineered NK cells. Biomaterials. 2022;280:121259.
Thacker G, Henry S, Nandi A, Debnath R, Singh S, Nayak A, Susnik B, Boone MM, Zhang Q, Kesmodel SB, et al. Immature natural killer cells promote progression of triple-negative breast cancer. Sci Transl Med. 2023;15:eabl4414.
Arianfar E, Shahgordi S, Memarian A. Natural killer cell defects in breast Cancer: a key pathway for Tumor Evasion. Int Rev Immunol. 2021;40:197–216.
Nikoo M, Rudiansyah M, Bokov DO, Jainakbaev NT, Suksatan W, Ansari MJ, Thangavelu L, Chupradit S, Zamani A, Adili A, et al. Potential of chimeric antigen receptor (CAR)-redirected immune cells in breast cancer therapies: recent advances. J Cell Mol Med. 2022;26:4137–56.
Sommaggio R, Cappuzzello E, Dalla Pieta A, Tosi A, Palmerini P, Carpanese D, Nicole L, Rosato A. Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. Oncoimmunology. 2020;9:1777046.
Pan K, Guan XX, Li YQ, Zhao JJ, Li JJ, Qiu HJ, Weng DS, Wang QJ, Liu Q, Huang LX, et al. Clinical activity of adjuvant cytokine-induced killer cell immunotherapy in patients with post-mastectomy triple-negative breast cancer. Clin Cancer Res. 2014;20:3003–11.
Mohan N, Hosain S, Zhao J, Shen Y, Luo X, Jiang J, Endo Y, Wu WJ. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1(+) triple negative breast cancer cells. Oncoimmunology. 2019;8:e1624128.
Pan MR, Wu CC, Kan JY, Li QL, Chang SJ, Wu CC, Li CL, Ou-Yang F, Hou MF, Yip HK, Luo CW. Impact of FAK expression on the cytotoxic Effects of CIK Therapy in Triple-Negative breast Cancer. Cancers (Basel) 2019, 12.
Wu CC, Pan MR, Shih SL, Shiau JP, Wu CC, Chang SJ, Kao CN, Chen FM, Hou MF, Luo CW. Combination of FAK inhibitor and cytokine-induced killer cell therapy: an alternative therapeutic strategy for patients with triple-negative breast cancer. Biomed Pharmacother. 2023;163:114732.
Liu H, Bai L, Huang L, Ning N, Li L, Li Y, Dong X, Du Q, Xia M, Chen Y et al. Bispecific antibody targeting TROP2xCD3 suppresses tumor growth of triple negative breast cancer. J Immunother Cancer 2021, 9.
Del Bano J, Flores-Flores R, Josselin E, Goubard A, Ganier L, Castellano R, Chames P, Baty D, Kerfelec B. A bispecific antibody-based Approach for Targeting Mesothelin in Triple negative breast Cancer. Front Immunol. 2019;10:1593.
Yi M, Wu Y, Niu M, Zhu S, Zhang J, Yan Y, Zhou P, Dai Z, Wu K. Anti-TGF-beta/PD-L1 bispecific antibody promotes T cell infiltration and exhibits enhanced antitumor activity in triple-negative breast cancer. J Immunother Cancer 2022, 10.
Chang CH, Wang Y, Li R, Rossi DL, Liu D, Rossi EA, Cardillo TM, Goldenberg DM. Combination therapy with bispecific antibodies and PD-1 blockade enhances the Antitumor Potency of T cells. Cancer Res. 2017;77:5384–94.
Nagano K, Maeda Y, Kanasaki S, Watanabe T, Yamashita T, Inoue M, Higashisaka K, Yoshioka Y, Abe Y, Mukai Y, et al. Ephrin receptor A10 is a promising drug target potentially useful for breast cancers including triple negative breast cancers. J Control Release. 2014;189:72–9.
Kamada H, Taki S, Nagano K, Inoue M, Ando D, Mukai Y, Higashisaka K, Yoshioka Y, Tsutsumi Y, Tsunoda S. Generation and characterization of a bispecific diabody targeting both EPH receptor A10 and CD3. Biochem Biophys Res Commun. 2015;456:908–12.
Fisher TS, Hooper AT, Lucas J, Clark TH, Rohner AK, Peano B, Elliott MW, Tsaparikos K, Wang H, Golas J, et al. A CD3-bispecific molecule targeting P-cadherin demonstrates T cell-mediated regression of established solid tumors in mice. Cancer Immunol Immunother. 2018;67:247–59.
Kubo M, Umebayashi M, Kurata K, Mori H, Kai M, Onishi H, Katano M, Nakamura M, Morisaki T. Catumaxomab with activated T-cells efficiently lyses Chemoresistant EpCAM-positive triple-negative breast Cancer cell lines. Anticancer Res. 2018;38:4273–9.
Stamm H, Oliveira-Ferrer L, Grossjohann EM, Muschhammer J, Thaden V, Brauneck F, Kischel R, Muller V, Bokemeyer C, Fiedler W, Wellbrock J. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. Oncoimmunology. 2019;8:e1674605.
Cheng Q, Shi X, Han M, Smbatyan G, Lenz HJ, Zhang Y. Reprogramming exosomes as Nanoscale Controllers of Cellular Immunity. J Am Chem Soc. 2018;140:16413–7.
Rau A, Lieb WS, Seifert O, Honer J, Birnstock D, Richter F, Aschmoneit N, Olayioye MA, Kontermann RE. Inhibition of Tumor Cell Growth and Cancer Stem Cell expansion by a bispecific antibody targeting EGFR and HER3. Mol Cancer Ther. 2020;19:1474–85.
Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, Weitsman G, Barber P, Vojnovic B, Ellis H, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7:ra29.
Fu W, Lei C, Yu Y, Liu S, Li T, Lin F, Fan X, Shen Y, Ding M, Tang Y, et al. EGFR/Notch Antagonists enhance the response to inhibitors of the PI3K-Akt pathway by decreasing tumor-initiating cell frequency. Clin Cancer Res. 2019;25:2835–47.
Tang J, Li B, Howard CB, Mahler SM, Thurecht KJ, Wu Y, Huang L, Xu ZP. Multifunctional lipid-coated calcium phosphate nanoplatforms for complete inhibition of large triple negative breast cancer via targeted combined therapy. Biomaterials. 2019;216:119232.
Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17:418–34.
Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10:1237–44.
Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:405–11.
Winer EP, Lipatov O, Im SA, Goncalves A, Muñoz-Couselo E, Lee KS, Schmid P, Tamura K, Testa L, Witzel I, et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:499–511.
Han H, Diab S, Alemany C, Basho R, Brown-Glaberman U, Meisel J, Pluard T, Cortes J, Dillon P, Ettl J et al. Open label phase 1b/2 study of ladiratuzumab vedotin in combination with pembrolizumab for first-line treatment of patients with unresectable locally-advanced or metastatic triple-negative breast cancer. Cancer Res 2020, 80.
Schmid P, Im S-A, Armstrong A, Park YH, Chung W-P, Nowecki Z, Lord S, Wysocki PJ, Lu Y-S, Dry H et al. BEGONIA: phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)-Initial results from arm 1, d + paclitaxel (P), and arm 6, d + trastuzumab deruxtecan (T-DXd). J Clin Oncol 2021, 39.
Tolaney SM, Kalinsky K, Kaklamani VG, D’Adamo DR, Aktan G, Tsai ML, O’Regan R, Kaufman PA, Wilks S, Andreopoulou E et al. A phase Ib/II study of eribulin (ERI) plus pembrolizumab (PEMBRO) in metastatic triple-negative breast cancer (mTNBC) (ENHANCE 1). J Clin Oncol 2020, 38.
Sohn J, Park KH, Ahn HK, Lee KS, Kim JH, Kim S-B, Lee KE, Kim JH, Yang SH, Le NT et al. Preliminary safety and efficacy of GX-17, a long-acting interleukin-7, in combination with pembrolizumab in patients with refractory or recurrent metastatic triple negative breast cancer (mTNBC): dose escalation period of phase Ib/II study (KEYNOTE-899). J Clin Oncol 2020, 38.
Lwin Z, Gomez-Roca C, Saada-Bouzid E, Yanez E, Muñoz FL, Im SA, Castanon E, Senellart H, Graham D, Voss M, et al. LBA41 LEAP-005: phase II study of lenvatinib (len) plus pembrolizumab (pembro) in patients (pts) with previously treated advanced solid tumours. Ann Oncol. 2020;31:1170.
Rugo HS, Schmid P, Cescon DW, Nowecki Z, Im S-A, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Perez-Garcia J, et al. Abstract GS3-01: additional efficacy endpoints from the phase 3 KEYNOTE-355 study of pembrolizumab plus chemotherapy vs placebo plus chemotherapy as first-line therapy for locally recurrent inoperable or metastatic triple-negative breast cancer. Cancer Res. 2021;81:GS3–01.
Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, Chien AJ, Forero-Torres A, Ellis E, Han H, et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete response in women with early-stage breast Cancer an analysis of the Ongoing phase 2 adaptively randomized I-SPY2 trial. Jama Oncol. 2020;6:676–84.
Adams S, Diamond JR, Hamilton E, Pohlmann PR, Tolaney SM, Chang CW, Zhang W, Iizuka K, Foster PG, Molinero L, et al. Atezolizumab Plus nab-Paclitaxel in the treatment of metastatic triple-negative breast Cancer with 2-Year Survival Follow-up: a phase 1b clinical trial. JAMA Oncol. 2019;5:334–42.
Emens LA, Adams S, Barrios CH, Dieras V, Iwata H, Loi S, Rugo HS, Schneeweiss A, Winer EP, Patel S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol. 2021;32:983–93.
Miles D, Gligorov J, Andre F, Cameron D, Schneeweiss A, Barrios C, Xu B, Wardley A, Kaen D, Andrade L, et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol. 2021;32:994–1004.
Bachelot T, Filleron T, Bieche I, Arnedos M, Campone M, Dalenc F, Coussy F, Sablin MP, Debled M, Lefeuvre-Plesse C, et al. Durvalumab compared to maintenance chemotherapy in metastatic breast cancer: the randomized phase II SAFIR02-BREAST IMMUNO trial. Nat Med. 2021;27:250–5.
Yi M, Wu Y, Niu M, Zhu S, Zhang J, Yan Y, Zhou P, Dai Z, Wu K. Anti-TGF-β/PD-L1 bispecific antibody promotes T cell infiltration and exhibits enhanced antitumor activity in triple-negative breast cancer. J Immunother Cancer 2022, 10.
Sommaggio R, Cappuzzello E, Dalla Pietà A, Tosi A, Palmerini P, Carpanese D, Nicolè L, Rosato A. Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. Oncoimmunology. 2020;9:1777046.
Del Bano J, Florès-Florès R, Josselin E, Goubard A, Ganier L, Castellano R, Chames P, Baty D, Kerfelec B. A bispecific antibody-based Approach for Targeting Mesothelin in Triple negative breast Cancer. Front Immunol. 2019;10:1593.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science foundation of China (Grant No.82103468 to YL), Basic Research Project for Universities of Liaoning Provincial Department of Education (Grant No.LJKMZ20221155 to YL) and 345 Talent Project of Shengjing Hospital of China Medical University (to YL).
Author information
Authors and Affiliations
Contributions
XG, YL, ZZ and JL contribute to the conception and design. YH, JQ and YL draft the main manuscript text. YJ, JB, PQ and ZZ draw Figs. 1, 2, 3, 4, 5 and 6. XG eviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., Hu, Y., Xue, J. et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer 22, 145 (2023). https://doi.org/10.1186/s12943-023-01850-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01850-7