
Abstract
The gut microbiota have long been recognized to play a key role in human health and disease. Currently, several lines of evidence from preclinical to clinical research have gradually established that the gut microbiota can modulate antitumor immunity and affect the efficacy of cancer immunotherapies, especially immune checkpoint inhibitors (ICIs). Deciphering the underlying mechanisms reveals that the gut microbiota reprogram the immunity of the tumor microenvironment (TME) by engaging innate and/or adaptive immune cells. Notably, one of the primary modes by which the gut microbiota modulate antitumor immunity is by means of metabolites, which are small molecules that could spread from their initial location of the gut and impact local and systemic antitumor immune response to promote ICI efficiency. Mechanistic exploration provides novel insights for developing rational microbiota-based therapeutic strategies by manipulating gut microbiota, such as fecal microbiota transplantation (FMT), probiotics, engineered microbiomes, and specific microbial metabolites, to augment the efficacy of ICI and advance the age utilization of microbiota precision medicine.
Similar content being viewed by others

Background
The microbiota in the gastrointestinal tract, which produces a myriad of small molecules and metabolites, play an essential role in multiple human physiological processes, including metabolism, inflammation, immunity, and neurology [1,2,3,4,5]. The function of the microbiota and its metabolites in modulating local and systemic immune responses has led to the emergence of research on the effects of the cancer-immune system and immune checkpoint inhibitor (ICI) therapeutic response [6,7,8,9,10]. The widespread variability in the gut microbiota across adult individuals is another reason to consider the gut microbiome as a potential source of phenotypic variability in cancer progression and ICI treatment outcomes [11].
Mounting evidence supports the role of the gut microbiota in the ICI response in preclinical and clinical studies [12,13,14,15,16,17]. ICI immunotherapy has improved traditional cancer therapeutic medicine and has enabled breakthroughs in the treatment of solid metastatic malignancies [18,19,20,21]. ICIs unleash immune brake responses and effectively inhibit tumor immune escape by targeting programmed cell death 1 (PD-1) and its ligand (PD-L1), lymphocyte-activating gene-3 (LAG3), cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), and other targets [18, 22, 23]. However, responses to ICI therapy are heterogeneous and not robust, with patient objective response rates (ORRs) of only 10–30% [24,25,26,27]. Manipulation of the gut microbiota provides novel insight for improving the antitumor immune response and expanding ICI efficacy.
Nonetheless, the potential molecular mechanisms of the influence of the gut microbiome and its metabolites on ICI efficacy are still poorly understood in some cancer types. In this review, we have summarized studies of the influence of the gut microbiome on ICI efficacy and discussed the mechanisms of microbiota cross talk with innate and adaptive immune cells to ameliorate ICI responses. We will specifically highlight the mechanisms of microbiota-derived metabolites and molecule-mediated antitumor immune responses to ICI. Finally, we will review the therapeutic strategies and ongoing trials investigating manipulation of gut microbiota to improve ICI efficacy.
Gut microbiota and efficacy of immunotherapy: from discovery to applications
Early studies have shown that gut microbiota could stimulate antitumor immune responses by modulating CD8+ T cells [28], T helper 1 (Th1) [29], and tumor-associated myeloid cells [30]. Indeed, the effects of cancer therapy were attenuated in antibiotic-treated or germ-free mouse models and were affected by special gut microbiota species. Importantly, landmark publications in 2015 in mouse models first linked the gut microbiota to ICI responses [16, 17]. Gut microbiota composition influenced anti-PD-L1 therapy responses, and the difference in responses was eliminated upon FMT or cohousing [16]. Oral administration of Bifidobacterium restored the antitumor efficacy of PD-L1 blockade by enhancing dendritic cell (DC) maturation and increasing CD8+ T cell priming and accumulation in the tumor microenvironment (TME) [16]. Another contemporary parallel study on anti-CTLA-4 therapy suggested that antibiotics dampen the antitumor effect of ICI, and supplementation with Bacteroides fragilis in germ-free or antibiotic-treated melanoma mice could augment anti-CTLA-4 therapeutic efficacy [17]. The microbiota-dependent antitumor effect is dependent on eliciting Th1-cell activation in the tumor draining lymph node and inducing maturation of intratumoral DCs [17].
Human studies published in Science side by side in 2018 complemented these mouse studies. The three studies all demonstrated that gut microbiota composition and diversity were predictive of response to ICI immunotherapy [31,32,33]. A similar finding was that fecal microbiota transplantation (FMT) from ICI responding patients to germ-free or antibiotic-treated mice could improve tumor control and ameliorate responses to ICI, whereas FMT from non-responders failed to do so [31,32,33]. In non-small cell lung cancer (NSCLC) and renal cell carcinoma (RCC), patients with a higher diversity of bacteria were more sensitive to anti-PD-1 therapy [31]. Oral supplementation with Akkermansia muciniphila (A. muciniphila) after FMT from ICI non-responders restored anti-PD-1 therapy responses [31]. In melanoma patients, the diversity and composition of the gut microbiota were positively correlated with anti-PD-1 therapy responses [32]. Mostly, ICI responding patients with a higher abundance of Faecalibacterium and Ruminococcaceae in the gut displayed increased numbers of CD4+ T cells and CD8+ T cells in the periphery [32]. Another study involving patients with metastatic melanoma indicated that Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium were more abundant in the baseline feces of responders [33].
Prospective studies confirmed a significant association between gut microbiota and ICI outcomes in NSCLC, hepatocellular carcinoma (HCC), melanoma, and RCC patients from 2019 to 2020 [34,35,36,37,38]. At the same time, retrospective studies have implicated that antibiotics were associated with decreased survival and attenuated response to ICI in patients with advanced solid tumors [39,40,41,42,43,44], supporting a causal link between antibiotic-induced dysbiosis and poor therapeutic efficacy of ICI. Furthermore, two clinical trials unexpectedly found that FMT from ICI responders combined with anti-PD-1 therapy overcame resistance to PD-1 blockade in melanoma patients in 2021 [14, 15]. The timeline of gut microbiota and ICI efficacy is summarized in Fig. 1.

Timeline of gut microbiota and ICI efficacy: from discovery to application. From 2007 to 2013, mouse studies showed that the gut microbiota could stimulate antitumor immune responses. In 2015, two preclinical mouse studies first linked the gut microbiota to ICI responses. In 2018, mouse and human studies demonstrated that gut microbiota composition and diversity were predictive of the response to ICI immunotherapy. FMT from ICI responding patients to germ-free or antibiotic-treated mice could improve tumor control and ameliorate responses to ICI. From 2019 to 2020, prospective studies confirmed a significant association between gut microbiota and ICI outcomes in advanced solid tumors. Retrospective studies have implicated that antibiotics are associated with decreased survival and attenuated response to ICI. In 2021, two clinical trials found that FMT from ICI responders combined with anti-PD-1 therapy overcame resistance to PD-1 blockade in melanoma patients
In addition to modulating ICI immunotherapy, gut microbiota can influence adoptive T cell transfer (ACT) immunotherapy, CpG-oligodeoxynucleotide (CpG-ODN) immunotherapy, and cell-based immunotherapy. Antibiotic exposure decreased the efficacy of ACT therapy in mice [28, 45], while bacterial LPS supplementation restored the therapeutic effect via toll-like receptor (TLR) 4 signaling [28]. Another study demonstrated that the gut microbiota maintained ACT therapeutic efficacy by increasing the abundance of CD8α+ DCs, and upregulation of interleukin (IL)-1 [46]. In CpG-ODN immunotherapy, the gut microbiota activate TLR4, which directly or indirectly initiates the TLR9-dependent response of tumor-associated myeloid cells to CpG-ODNs [30]. The efficacy of CpG-ODN is abolished in GF and antibiotic-exposed mice by impairing the production of tumor necrosis factor (TNF) and IL-12 [30]. The gut microbiota also affect cell-based immunotherapy. Gut microbiota-mediated metabolism of bile acid increased the abundance of CXCR6+ natural killer T (NKT) cells in the liver and played an antitumor role in hepatocellular carcinomas [47].
The gut microbiota remodel the TME to improve ICI efficacy
Studies have shown that the gut microbiota modulate the ICI response, and detailed mechanistic exploring into the specific bacterial species and microbial metabolites on ICI is necessary. The gut microbiota can modulate innate and adaptive immunity and influence antitumor immune responses in the TME [2, 48]. Complex mechanisms by which specific bacterial species reprogram the TME to improve ICI efficacy in the context of immunity will be discussed herein (Table 1 and Fig. 2).

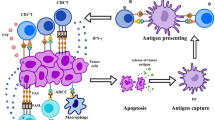
The gut microbiota modulate innate immunity, adaptive immunity, and tumor antigens to improve ICI responses. A Innate immunity. DCs: Bifidobacterium, eleven strains and their metabolites, and Bacteroides fragilis promote DC maturation or activation and subsequent activation of CD8+ T cells and Th1 cells. NK cells: Lactobacillus plantarum increases NK cell activation; a high-salt diet increases intestinal permeability and localization of intratumoral Bifidobacterium and enhances NK cell activation to induce antitumor immunity. Monocyte: Feeding a high-fiber diet, monocolonization with cdAMP-producing A. muciniphila or transferring fecal microbiota from ICI responders can trigger the monocyte-IFN-I-NK-cell-DC cascade; Bifidobacterium facilitates CD47-based immunotherapy in a STING signaling and IFN-I-dependent fashion; Bacteroides fragilis induces macrophage phenotype polarization to M1. B Adaptive immunity. CD8+ T cells: Bifidobacterium, Enterococcus, Faecalibacterium, Ruminococcus, and Clostridiales promote CD8+ T cell infiltrates in tumor tissues; Phyla Firmicutes and Actinobacteria improve the activation of CD56+CD8+ T cells in the peripheral blood of ICI responders; and eleven strains increase the proportion of effector IFNγ+CD8+ T cells in the systemic circulation. CD4+ T cells: B. pseudolongum and Bacteroides fragilis stimulate Th1 immune responses; A. muciniphila triggers CCR9+CXCR3+CD4+ T lymphocyte recruitment into tumor beds; and Faecalibacterium increases the CD4+ T cell proportion. C Tumor cross-antigen. The gut microbiota increase the immunogenicity of tumor cells by providing tumor cross-antigens to ameliorate the efficacy of ICIs, including the antigen epitope TMP1 and the antigen epitope SVY
Gut microbiota modulate innate immunity to ameliorate ICI responses
DCs
DCs are a group of special antigen-presenting cells that play an essential role in T cell activation and antitumor immunity [54,55,56,57]. Gut microbiota antigens or metabolites with immunomodulators were used to mobilize and activate DCs to reverse immature DC-induced immune tolerance [58] (Fig. 2A). For instance, oral administration of Bifidobacterium increased DC activation, which in turn supported improved tumor-specific CD8+ T cell responses and restored the therapeutic efficacy of anti-PD-L1 therapy in mice with an “unfavorable” gut microbiota [16]. Eleven strains combined with ICI robustly induced interferon (IFN) γ+ CD8+ T cells to inhibit tumor growth in a manner dependent on lamina propria cDC1 and major histocompatibility complex (MHC) class I [50]. Bacteroides fragilis enhanced the antitumor effect of CTLA-4 blockade by triggering DC maturation and stimulating IL-12-dependent Th1 cell immune responses [17]. Furthermore, vancomycin-mediated modulation of gut microbiome composition improved the activities of antitumor-specific effector T cells by increasing cDC1 and upregulating IL-12 [46].
Monocytes/macrophages
Skews in IFN-I and mononuclear phagocyte levels contribute to immune dysregulation and an immunosuppressive TME [59]. Microbiota-induced IFN-I signaling mediates the transition from innate immunity to adaptive immunity [7, 53, 60] (Fig. 2A). Microbiota-derived stimulator of interferon genes (STING) agonists (such as c-di-AMP) induce IFN-I signaling by intratumoral monocytes, which shift mononuclear phagocytes toward antitumor macrophages (Macs) and trigger monocyte-IFN-I-natural killer (NK) cell-DC cross talk [53]. Notably, feeding a high-fiber diet, monocolonization with cdAMP-producing A. muciniphila or transferring fecal microbiota from ICI responders improved the antitumor responses and ICI efficacy [53]. Similarly, it has been demonstrated that Bifidobacterium preferentially colonizes the tumor site and facilitates CD47-based immunotherapy in a STING signaling-and IFN-I-dependent fashion [52]. In addition, Bacteroides fragilis induced macrophage phenotype polarization to M1 and upregulated CD80 and CD86 expression on the cell, promoting innate immunity [61].
NK cells
It has been demonstrated that NK cells can regulate DC and CD8+ T cell abundance in the TME and influence responses to ICIs [54, 62,63,64]. Recently, an increasing number of studies have found an interplay between NK cells and gut microbiota (Fig. 2A). NSCLC patients with high microbial diversity had a higher abundance of unique memory CD8+ T cells and NK cell subsets in the periphery in response to PD-1 blockade [34]. Lactobacillus plantarum effectively increased expression of the natural cytotoxic receptor (NCR) protein and promoted NK cell activation to trigger innate immunity [65]. Interestingly, the suboptimal dose of PD-1 blockade combined with a high-salt diet significantly inhibited tumor growth in mice [66]. Mechanistic studies found that a high-salt diet increased intestinal permeability and the localization of intratumoral Bifidobacterium, which enhanced NK cell activation to induce antitumor immunity [66].
Gut microbiota modulate adaptive immunity to improve ICI responses
CD8+ T cells
Multiple publications have confirmed that specific gut microbiota induce CD8+ T cells in the systemic circulation or the TME (Fig. 2B). For example, melanoma patients with a high relative abundance of favorable microbiota, including Clostridiales, Ruminococcaceae, or Faecalibacterium, increased antigen presentation and improved effector CD4+ T cell and CD8+ T cell function in the peripheral blood and TME to ameliorate the antitumor efficacy of ICI [32]. Evidence from a clinical trial has implicated that the phyla Firmicutes and Actinobacteria were enriched in FMT combined with PD-1 blockade responders [14]. Combined FMT and PD-1 blockade stimulated mucosal-associated invariant T (MAIT) cells and CD56+CD8+ T cells in peripheral blood mononuclear cells (PBMCs) and upregulated expression of the human leucocyte antigen (HLA) class II genes CD74 and GZMK on CD8+ T cells at tumor sites [14]. In parallel, after using FMT combined with PD-1 blockade promoted a high relative abundance of Enterococcus in refractory metastatic melanoma and led to increased intratumoral CD8+ T cell infiltration and tumor cell necrosis [15]. Furthermore, Bifidobacterium and eleven strains could also increase the abundance of CD8+ T cells reliant on DCs to improve ICI therapy efficacy [16, 50].
CD4+ T cells
In mouse models, B. pseudolongum promoted Th1 transcriptional differentiation and antitumor immune responses to improve ICI efficacy mainly through the gut microbial metabolite inosine [51]. Bacteroides fragilis stimulated IL-12-dependent Th1 immune responses by facilitating the mobilization of lamina propria DCs, which restored the immune responses to ICI [17]. In addition, oral supplementation with A. muciniphila in FMT non-responsive mice recovered anti-PD-1 responses by triggering CCR9+CXCR3+CD4+ T lymphocyte recruitment into tumor beds [31]. In human patients, Faecalibacterium increased the CD4+ T cell proportion and serum CD25 production and reduced the Treg cell proportion in peripheral blood, which induced the long-term clinical benefit of ipilimumab [49] (Fig. 2B).
Gut microbiota modulate the immunogenicity of tumor cells to improve ICI responses
The decrease in tumor immunogenicity is an essential mechanism by which tumor cells resist T cell killing. On the one hand, the gut microbiota can directly enhance the innate immunogenicity of tumor cells by acting on UBA6 on the tumor cell surface to augment ICI responses [67]. On the other hand, the gut microbiota can indirectly increase the immunogenicity of tumor cells by providing tumor cross-antigens to promote the efficacy of ICI [50, 68, 69]. Some cross-reactivity between antigens expressed in gut microbes and tumor cells has been identified (Fig. 2C). The antigenic epitope tail length tape measure protein 1 (TMP1) in the genome of bacteriophage Enterococcus hirae had high similarity with the proteasome subunit beta type-4 (PSMB4) tumor antigen [70]. They activated CD8+ T cells simultaneously and improved the efficacy of PD-1 blockade therapy [70]. It has been demonstrated that the antigen epitope SVYRYYGL (SVY) expressed in the commensal bacterium Bifidobacterium breve was similar to the tumor-expressed antigen epitope SIYRYYGL (SIY), resulting in SVY-specific T cells recognizing SIY and inhibiting tumor growth [71].
Gut microbial metabolite-mediated antitumor immune responses to ICI
Notably, one of the primary modes by which the gut microbiota modulate antitumor immunity is by means of metabolites. The gut microbiota synthesize or transform a myriad of metabolites, which are small molecules that can spread from their original location in the gut and impact the local and systemic antitumor immune response to promote ICI efficacy [14, 72]. Accordingly, we further explored the mechanisms of the different gut microbial metabolites and other features of gut microbial signatures mediating antitumor immune responses.
Inosine
Inosine, the purine metabolite of Akkermansia muciniphila and Bifidobacterium pseudolongum (B. pseudolongum), plays a vital role in improving the efficacy of ICI [51]. Inosine is a normal metabolite of the human body and participates in nucleic acid metabolism, energy metabolism, and protein synthesis in a physiological state. It can activate immune cells and stimulate metabolism. In the past, it has been shown that inosine has immunosuppressive effects [73, 74]. However, in recent years, more studies have found that inosine could reprogram the TME and improve the response to ICI therapy [51, 75, 76]. Current studies indicate that inosine affects the efficacy of ICI mainly through the following mechanisms (Fig. 3).

Potential mechanisms by which the gut microbial metabolite inosine facilitates the efficacy of ICI. Inosine, purine metabolite of gut microbiota A. muciniphila and B. pseudolongum, combined with in ICI therapy exert synergistic antitumor effects. A Inosine increases the immunogenicity of tumor cells. Inosine can improve the ability of tumor cells to present tumor antigens so that cytotoxic immune cells can easily recognize and kill tumor cells. B Inosine promotes immune cell activation. Inosine could enhance ICI efficacy by acting on A2AR on T lymphocytes. It stimulates the phosphorylation of cAMP response element-binding protein (pCREB) through the inosine-A2AR-cAMP-PKA signaling pathway, which can upregulate IL12Rβ2 and IFNγ transcription and promote Th1-cell differentiation and accumulation in the TME. C Inosine provides an alternative carbon source for CD8+ T cells. Inosine can be used as an alternative carbon source for CD8+ T cells when glucose is limited and relieves the restrictions on CD8+ T cells energy metabolism in tumor cells
Inosine increases the immunogenicity of tumor cells. A research group found that inosine could significantly enhance the ability of tumor cells to present tumor antigens, so cytotoxic immune cells could easily recognize and kill tumor cells, achieving an antitumor effect [77]. Further mechanistic studies showed that activation of the IFNγ and TNFα signaling pathways was significantly increased in tumor cells treated with inosine. IFNγ can activate the cytotoxicity of tumor-specific T cells and NK cells through promoting their release of perforin and granzyme, and IFNγ can heighten antigen presentation to promote inosine-mediated antitumor effects [78]. Our recent work demonstrated that inosine sensitized tumor cells to T cell-mediated cytotoxicity by directly binding and inhibiting the ubiquitin-activating enzyme UBA6 to amplify tumor-intrinsic immunogenicity and enhance ICI efficacy [67] (Fig. 3A).
Inosine promotes immune cell activation. The gut microbial metabolite inosine could improve the efficacy of ICI by acting on adenosine 2A receptor (A2AR) on T lymphocytes in intestinal cancer, bladder cancer, and melanoma mouse models [51]. The inosine-A2AR-cAMP-PKA signaling cascade led to the phosphorylation of cAMP response element-binding protein (pCREB), which upregulated IL12Rβ2 and IFNγ transcription and promoted Th1-cell differentiation and accumulation in the TME [51]. Interestingly, the in vivo antitumor effects of ICI combined with inosine required a costimulus, such as CpG and IL-12 [51] (Fig. 3B). Inosine can also enhance immune responses mediated by phytohemagglutinin (PHA), increase tumor antigen levels, and strengthen T lymphocyte differentiation and proliferation [75, 79]. Furthermore, inosine stimulated B lymphocyte differentiation and antibody production by activating macrophages, exerting antiviral and antitumor actions [75, 79].
Inosine provides an alternative carbon source for CD8+ T cells. Oncogenic signaling pathway activation reprograms the metabolism of cancer cells [80]. The high metabolic demand of cancer cells can limit the capacity of effector T cells by reducing the levels of available nutrients and producing immunosuppressive metabolites [81, 82]. Inosine could be employed as an alternative carbon source for CD8+ T cells under energy metabolism limits to support the growth and capacity of CD8+ T cells [75] (Fig. 3C). T cells metabolize inosine into hypoxanthine and phosphorylated ribose by purine nucleoside phosphorylase (PNP) [75] (Fig. 3C). Importantly, the ribosomal subunit of inosine enters the central metabolic pathway, providing ATP and biosynthetic precursors for the glycolytic pathway and the pentose phosphate pathway (PPP) [75].
Short-chain fatty acids (SCFAs)
Colonic anaerobes produce SCFA from undigested and absorbed carbohydrates or glycoproteins secreted by gut epithelial cells. Recently, the association between gut microbiota-derived SCFAs and nivolumab or pembrolizumab treatment in solid tumors has been confirmed [83]. SCFAs were found to be significant physical and chemical barriers that stimulate Paneth cells and goblet cells to produce AMPs and mucus to support the integrity of the intestinal mucosal barrier [84, 85]. SCFAs play a key role in complex gut microbial immune and metabolic networks, affecting the activity of immune cells and tumor cells (Fig. 4).

Potential mechanisms by which the gut microbial metabolite SCFAs augment the efficacy of ICI. A SCFAs inhibit the proliferation and induce the apoptosis of cancer cells. The butyric acid of SCFAs, a metabolite of Faecalibaculum rodentium PB1 and H. biformis, inhibits the activity of HDAC and the calcineurin-mediated activation of NFATc3 transcription factor, thereby blocking the proliferation of tumor cells. Propionic acid produced by A. muciniphila activates the cell cycle inhibitor p21 through GPR43 and downregulates the IAP inhibitor, which inhibits cancer cell proliferation, induces apoptosis, and improves the antitumor effect of ICI. B SCFAs improve the antitumor immune response. Butyrate can directly enhance CD8+ T cell antitumor cytotoxicity by inducing ID2 expression in CD8+ T cells through IL-12 signaling. Valeric acid and butyric acid of SCFAs promote expression of effector molecules such as IFNγ and TNFα and enhance the antitumor effects of CTLs. C SCFAs provide energy for immune cells. SCFAs provide energy to B cells, memory T cells, and effector T cells by regulating metabolic pathways such as glycolysis, the TCA cycle, and β-oxidation to enhance ICI efficacy
SCFAs inhibit the proliferation and induce the apoptosis of tumor cells. The mechanism of the synergy of SCFAs and ICI is a research hotspot. Butyric acid, the metabolite of Faecalibaculum rodentium PB1 (F. PB1) and H. biformis, acts as a histone deacetylase (HDAC) inhibitor [86]. It increased acetylation and inhibited calcineurin-mediated nuclear factor of activated T cells C3 (NFATc3) activation, which blocked tumor cell proliferation [86] (Fig. 4A). In addition, the SCFA propionic acid produced by A. muciniphila activated the cell cycle inhibitor p21 through G protein-coupled receptor 43 (GPR43) and downregulated inhibitor of apoptosis protein (IAP), which restrained cancer cell proliferation, induced apoptosis, and improved the antitumor effect of ICI [87] (Fig. 4A).
SCFAs improve antitumor immune responses. SCFAs can directly promote the antitumor cytotoxicity of CD8 T cells in vivo and in vitro [6, 88] (Fig. 4B). Butyrate produced by gut microbiota metabolism could directly boost the antitumor cytotoxicity of CD8+ T cells by inhibiting DNA binding 2 (ID2)-dependent IL-12 signaling [6]. Another study confirmed that valeric acid and butyric acid enhanced the function of mTOR as central cellular metabolic sensors and inhibited the activity of class I HDACs through metabolic and epigenetic reprogramming [88]. Moreover, this led to an increase in effector molecules such as CD25, IFNγ, and TNFα, which significantly promoted the antitumor effects of antigen-specific cytotoxic T lymphocytes (CTLs) and chimeric antigen receptor (CAR) T cells in melanoma and pancreatic cancer mouse models [88]. Both studies identified valeric acid and butyric acid as two SCFAs with therapeutic effects in cancer immunotherapy.
SCFAs provide energy for immune cells. SCFAs derived from the gut microbiota can regulate glycolysis, the tricarboxylic acid (TCA) cycle, the PPP, and fatty acid oxidation (FAO) of antitumor effector cells to increase the efficiency of ICI (Fig. 4C). First, butyrate, an SCFA, is converted to butyryl-CoA, undergoes β-oxidation (β-OX), and participates in the TCA cycle and oxidative phosphorylation (OXPHOS). These processes provide intestinal mucosal epithelial cell energy, improve intestinal villus structure, inhibit autophagy, and preserve intestinal homeostasis [89, 90]. Second, in memory T cells, butyric acid activated intracellular β-OX, promoting the TCA cycle and OXPHOS [91]. At the same time, acetyl-CoA produced by acetic acid promoted glycolysis by acetylating glyceraldehyde-phosphate dehydrogenase (GAPDH) [92]. Importantly, in effector T cells, SCFAs increase the number of mitochondria in cells and stimulate glycolysis and OXPHOS [93, 94]. Finally, SCFAs provide energy for B-cell differentiation into plasma cells, antibody production, and overall changes in cell metabolism [95].
Anacardic acid
In addition to inosine and SCFAs (Fig. 5A, B), anacardic acid also modulates antitumor immune responses (Fig. 5C). In the prospective study, metabolomics analysis of fecal bacteria from melanoma patients treated with ICI showed that Bacteroides caccae was significantly enriched, and levels of anacardic acid were greatly increased in the ICI response group (62-fold, P < 0.01) [96] (Fig. 5C). Anacardic acid triggers the classic activation pathway in macrophages by phosphorylating mitogen-activated protein kinases (MAPKs), thereby activating innate immunity [97]. Anacardic acid can also induce production of a neutrophil extracellular trap (NET), which promotes creation of tumor-infiltrating immune cells with macrophages, NK cells, and T lymphocytes to modulate adaptive immunity and antitumor immunity [98, 99]. Indeed, anacardic acid has been confirmed to have antitumor effects in some preclinical models [100]. For example, in breast cancer models, anacardic acid increased levels of tumor-infiltrated NK cells and CTLs and induced apoptosis of tumor cells [101] (Fig. 5C).

Microbiota-derived metabolites and other gut microbial signature modulation of antitumor immune responses to improve ICI efficacy. A–E Microbiota-derived metabolites target immune cells and tumor cells to modulate antitumor immunity. Inosine, SCFA, and anacardic acid promote antitumor immunity and ICI efficacy; bile acid and tryptophan attenuate antitumor immune responses. F–H Other gut microbial signature modulation of antitumor immune responses. PG, PSA, and OMV promote anti-tumor immune responses by regulating immune cells, tumor cells, and cytokines
Bile acid and tryptophan
Gut microbiota-derived metabolites, such as secondary bile acid or tryptophan, have shown immunosuppressive effects in some studies (Fig. 5D, E). Gut microbiota that produce bile acid metabolite 3-oxolithocholic acid as well as an abundant gut metabolite isolithocholic acid inhibit Th17 cell differentiation [102]. Furthermore, the secondary bile acid 3β-hydroxydeoxycholic acid exhibits weakened immunostimulatory properties when acting on DCs, thus inducing the expression of Foxp3, upregulating the number of Tregs, and promoting immune escape [103]. Feeding secondary bile acids or bile acid-metabolizing bacteria Clostridium scindens attenuated NKT cell-mediated liver-selective tumor inhibition [47]. In addition, IL-2 induced CD8+ T cell exhaustion and strong expression of tryptophan hydroxylase 1 by activating the STAT5-5-hydroxytryptophan (5-HTP)-AhR axis [104]. Tryptophan metabolites effectively facilitated the motility and migration of tumor cells in breast cancer [105, 106]. The increase in the serum kynurenine/tryptophan ratio was associated with worse overall survival in melanoma and RCC patients receiving nivolumab therapy [107]. Low plasma tryptophan metabolite-3-hydroxyphthalate levels were significantly associated with prolonged median progression-free survival (PFS) in patients with NSCLC [108].
Other gut microbial signatures guide antitumor immune responses
Peptidoglycan (PG) and polysaccharide (PSA)
Enterococcus expressed and secreted orthologs NlpC/p60 PG hydrolase SagA and could promote expression of the innate immune sensor protein nucleotide-binding oligomerization domain containing 2 (NOD2) and augment ICI antitumor efficacy [109] (Fig. 5F). Recognition of microbiota-derived PG in a nucleotide-binding oligomerization domain containing 1 (NOD1)-dependent manner could facilitate systemic innate immunity [110]. Furthermore, PSA produced by Leuconostoc mesenteroides strain NTM048 or Bacteroides fragilis acts as an immunostimulant to enhance the mucosal barrier and influence systemic immune responses [111, 112] (Fig. 5G). PSA can be recognized by DCs in the small intestine and activated CD4+ T cells to secrete cytokines, thereby promoting T cell proliferation, improving Th1/Th2 cell imbalance, and promoting lymphoid tissue formation [113] (Fig. 5G). TLRs, such as TLR9 and its agonist CpG-ODN, play an essential role in pathogen recognition and initiation of immune responses [114,115,116,117,118]. Clostridium difficile toxin A-bound DNA activated TLR9 signaling and the innate immune response [119].
Outer membrane vesicle (OMV)
Microbiota-derived OMVs naturally secreted by bacteria can reprogram the TME and have been developed into tumor immunotherapeutic reagents, bacterial vaccines, adjuvants, and drug delivery carriers [120, 121]. OMVs express tumor antigens, inducing innate immune responses and antigen-specific T cell-mediated antitumor immunity; bioengineered bacterial OMVs expressing multiple tumor antigens can trigger a synergistic antitumor immune response [122] (Fig. 5H). OMVs with calcium phosphate (CaP) shells promoted cytotoxic T cell infiltration and M2 to M1 macrophage polarization, effectively improving the immunosuppressive TME [123] (Fig. 5H). Additionally, systematically administered bacterial OMVs specifically targeted and accumulated in the tumor bed and subsequently induced the production of the antitumor cytokines CXCL10 and IFN-γ to effectively augment antitumor immune responses [124] (Fig. 5H).
Gut microbiota and ICI therapy toxicity
ICI therapy disrupts the host immune balance while killing tumor cells, which may result in immune-related colitis, immune-related pneumonia, and even life-threatening immune-related myocarditis. Several lines of evidence suggest that the role of the gut microbiota in immune-related adverse events (irAEs) is a double-edged sword. Certain gut bacteria may be protective against immunotherapy-induced toxicity. Mouse models have verified that Bifidobacteria, Bacteroides fragilis, and Burkholderia cepacia can ameliorate intestinal immunopathology in the context of anti-CTLA-4 therapy [17, 125, 126]. A prospective study demonstrated that increased representation of the Bacteroidetes phylum and microbial genetic pathways involved in polyamine transport and vitamin B biosynthesis was associated with developed resistance to ICI-induced colitis in metastatic melanoma patients treated with ipilimumab [127]. In contrast, some gut microbiota are associated with a high risk of ICI-induced toxicity. The enrichment of two microbes, Lachnospiraceae spp. and Streptococcus spp., was associated with an increase in irAEs in melanoma patients treated with anti-PD-1 [128]. Interestingly, a distinct baseline of gut microbiota may also be associated with favorable anticancer response as well as ICI-induced toxicity. Among 26 patients with metastatic melanoma treated with ipilimumab, patients with Faecalibacterium and Firmicutes enrichment at baseline were prone to develop immunotherapy-induced colitis and enhanced ICI sensitivity simultaneously [49]. This is known as the efficacy–toxicity coupling effect in the context of ICI [126, 129, 130]. Gut microbes, such as Bacteroides, could also be a biomarker for predicting ICI therapeutic toxicity in advanced melanoma patients treated with combined CTLA-4 and PD-1 blockade [131]. The beneficial effect of FMT as a therapeutic modality for immunotherapy-induced toxicity in two patients has been demonstrated by reconstructing the gut microbiome and increasing the proportion of Tregs in the colonic mucosa [132]. These studies indicate that the gut microbiome has complex positive and negative effects on ICI-induced toxicity. More evidence is required before adequately filtering and manipulating the gut microbiota to augment the ICI response and attenuate irAEs.
Therapeutic strategies utilizing the gut microbiome combined with ICI
Understanding the biological mechanisms of the gut microbiome and its metabolites on antitumor immunity and immunotherapy responses is essential for rationally manipulating microbial activities to improve ICI efficacy [133, 134]. The therapeutic strategies utilizing the gut microbiome combined with ICI are delineated in Fig. 6.

Therapeutic strategies utilizing the gut microbiome combined with ICI. Therapeutic strategies for manipulating gut microbiota include FMT, probiotics, engineered microbiome, and other strategies to increase ICI responses
Fecal microbiota transplantation
FMT treatment refers to the whole transplantation of an individual's gut microbiome, usually from ICI responders. FMT preparations can be administered directly by oral lyophilized pills or by colonoscopy or gastroscopy. FMT was initially used to treat Clostridium difficile infection resistant to traditional therapy [135]. Recently, several studies have indicated that FMT could augment the antitumor effect of ICI and overcome resistance to immunotherapy [31,32,33]. In a phase I clinical trial (NCT03353402), researchers performed FMT and reinduction anti-PD-1 therapy for ten melanoma patients who were unresponsive to PD-1 blockade [15]. The results showed that three of the ten patients showed tumor volume decline, including two partial responses (PR) and one complete response (CR) [15]. In the same period, another clinical trial focused on 15 melanoma patients resistant to anti-PD-1 therapy (NCT03341143), and the results showed that three patients showed PR after using FMT combined with pembrolizumab, and three patients had stable disease (SD) for more than 12 months [14]. The application of FMT is a novel approach for reversing ICI immunotherapy resistance and decreasing irAEs. Some clinical trials to evaluate the safety and efficacy of the combination of FMT with ICI treatment are underway (Table 2).
Despite the promising results of FMT therapy in patients treated with ICI, there are still concerns about its long-term safety. In 2019, it was reported that in two independent clinical trials, two patients developed bacteremia with extended-spectrum β-lactamase (ESBL)-producing Escherichia coli after receiving FMT from the same donor, and one patient died [136]. This study led the US Food and Drug Administration to issue a safety bulletin warning of the infection risk with FMT therapy. Furthermore, a recent retrospective cohort study of collected donor feces tested for multidrug-resistant organisms showed that 6 of 66 tested individuals (9%) were positive for multidrug-resistant organisms at any timepoint [137]. Therefore, periodic screening of donor feces should be performed to strictly limit the spread of organisms that may lead to adverse infection events, which is especially relevant for immunodeficient patients. Further clinical investigations enabling a better understanding of the source, transplantation procedure, and recipient phenotype of donor FMT are critical for successful ICI-FMT combination therapy.
Probiotics
Probiotics are “live microorganisms which when administered in adequate amounts confer a health benefit on the host” [138]. Early clinical trials in patients with cancer mainly evaluated how probiotics modified microbiota composition or modulated antitumor immunity. A clinical trial in patients with breast cancer assessed the effects of the administration of probiotics (13 strains of beneficial bacteria) on CD8+ T cell infiltration in the TME (NCT03358511). Patients with CRC administered probiotics (Bifidobacterium lactis and Lactobacillus acidophilus) had an increased abundance of butyrate-producing bacteria in the tumor, mucosa, and feces (NCT03072641) [139]. Another trial demonstrated that patients treated with probiotics have lower IL-1b, IL-10, and IL-23A mRNA levels in the colonic mucosa (NCT01895530) [140]. Moreover, specific gut microbes, such as Bifidobacterium [16, 33, 51, 141], Akkermansia [31, 38], Enterococcus [15, 33, 141], Faecalibacterium [49, 142], and Ruminococcaceae [32], play the role of immune adjuvants in ICI immunotherapy. Several clinical trials to assess the safety and efficacy of probiotics combined with ICIs are underway (Table 3). For example, MRx0518 (Enterococcus gallinarum capsule) mainly relies on free flagellin to activate the TLR5 and NF‐κB signaling pathways and exert antitumor effects [143]. The phase I/II open-label clinical trial (NCT03637803) explored the synergistic effects of the oral probiotic MRx0518 in combination with pembrolizumab in NSCLC, RCC, bladder cancer, or melanoma, and the results have not yet been published. EDP1503 is a novel adjuvant for the immunotherapy of cancer-based Bifidobacterium. Clinical trials (NCT03775850) showed that EDP1503 in combination with pembrolizumab was safe and well tolerated, and biomarker studies found that EDP1503 works by upregulating the ratio of CD8+ T cells/Treg cells [144].
Engineered microbiomes
With the development of bacterial genetic engineering technology, it is possible to improve antitumor responses to ICI by modifying gut microbiota or metabolites. Genetically engineered drugs have specificity that FMT cannot achieve. To date, genetically attenuated, nutrient deficient, and inducible Escherichia coli [146], Bifidobacteria [147], Listeria [148], and Salmonella [148] have been transformed and have shown antitumor effects in preclinical models of intravenous, intratumoral, and oral administration routes [149]. Guo et al. [150] made use of the abilities of several strains, including Salmonella and Clostridium, to elicit specific targeting in solid tumors, making them ideal carriers for the delivery and induction of immune stimulants to delay tumor growth and metastasis. In addition, SYNB1891 is a dual innate immune agonist designed based on the biology of E. coli. SYNB1891 has been modified to express the STING agonist cyclic adenosine diphosphate ribose (CADPR), which can stimulate expression of IFNs and achieve antitumor effects [151]. Clinical trials exploring the safety and efficacy of SYNB1891 in combination with atezolizumab in advanced solid tumors are ongoing (Table 3). Furthermore, L-arginine enhances T cell survival and antitumor activity by modulating T cell metabolism [152, 153]. The engineered probiotic Escherichia coli Nissle 1917 strain colonizes tumor sites and continuously converts the metabolite ammonia to L-arginine in the tumor bed [154]. Intertumoral injection of this strain in mice increased the intracellular L-arginine concentration, triggered intratumoral CD4+ T cell and CD8+ T cell infiltration, and exerted synergistic antitumor effects when combined with anti-PD-L1 [154]. These results show that engineered microbial therapies enable metabolic modulation of the TME to enhance the efficacy of immunotherapies.
Other strategies
Therapeutic strategies involving the manipulation of gut microbiota to enhance ICI efficacy also include adjusting diet and lifestyle, taking prebiotics, and avoiding antibiotics [155,156,157,158]. Additionally, the discovery of tumor cross-antigens and gut microbiota-derived immune activators provides insights into the development of tumor-therapeutic vaccines such as Ty21a [159, 160], JNJ-64041809 [161], and VXM01 [162]. EO2401 is a microbial-derived polypeptide drug with a structure homologous to that of tumor-associated antigens. The clinical trial of EO2401 combined with nivolumab is ongoing in glioblastoma multiforme (GBM) and adrenocortical carcinoma patients (Table 3). It is noteworthy that oral administration of microbiome-derived compounds, such as bacteriophages and microbial metabolites, may be more practical and precise than administration of full or unique live bacteria transplants [70, 163]. Indeed, bacteriophages as therapeutic strategies could modulate the gut microbiota, immunity, and TME [70, 164,165,166]. Importantly, the SCFA valproic acid (VPA, 2-propylpetanoic acid) is a type of microbiota-derived metabolite. Clinical trials to assess the safety and efficacy of VPA in combination with ICI in patients with solid tumors are already underway (NCT02446431, NCT01106872, NCT02624128).
Further directions
We have gained insights into the potential mechanisms of gut microbiome influences on the antitumor immune response and ICI efficacy and into their use in therapeutic strategies from preclinical and clinical research. In the future, we still need to consider the following factors. First, the accuracy of the findings of the microbiome influence on ICI efficacy should be demonstrated. The effects of gut microbiota on tumor ICI therapy are multifaceted and even bidirectional. We should accurately classify favorable and unfavorable microbiome features that target immune cells or pathways and fully understand their effects in different TMEs. Second, this approach facilitates the improved precision of therapeutic strategies. Understanding the microbiota-metabolite-immune axis helps us directly manipulate specific immune-stimulating metabolites or compounds derived from gut microbiota, rather than whole or unique live bacteria transplants, to improve the ICI response. In addition, the effects of other microbiota-derived molecules that promote the antitumor immune response may become a future direction of preclinical or clinical research on ICIs. Third, gut microbiota may act as a predictive biomarker for ICI efficacy or safety. A patient's microbiota data could be combined with those of other known related predictive markers, such as PD-L1 expression and tumor mutation load, to predict immunotherapy's potential efficacy or adverse events. Finally, the effectiveness of clinical trials should be improved. In human clinical trials, it is necessary to comprehensively monitor the factors influencing the gut microbiome, such as diet, probiotics, antibiotics, drugs, mental health, host immune system, host genetics, geographical factors, tumor type, and stage. Notably, multilevel and multidimensional research designs integrating microbiology, genetics, immunology, metabolomics, molecular pathology, and epidemiology will become a part of personalized cancer therapy in the future.
Conclusion
In conclusion, the gut microbiome cross talk with innate and adaptive immune cells occurs, augmenting the intermediate effects of innate immune cells, enhancing the antitumor effect of adaptive immune cells, and increasing the immunogenicity of tumor cells, which reprograms the immunity of the TME and ameliorate ICI responses. Notably, microbiota-derived circulating metabolites modulate multiple human physiologies and spread from their original location in the gut to mediate local and systemic antitumor immune responses to promote ICI efficiency. Therapeutic strategies utilizing gut microbiota combined with ICI, such as appropriate antibiotic selection, probiotic intake, FMT, and bacterial genetic engineering, may provide novel possibilities for gut microbiota and their metabolites to become excellent adjuvants for ICI. Further understanding the mechanisms of synergy between ICI and the gut microbiome and accurate identification of immunostimulant and immunosuppressive strains or pathways is expected to enable the development of more effective combination therapy strategies for ICI and the advancement of precision medicine strategies.
Availability of data and materials
Not applicable.
Abbreviations
- 5-HTP:
-
5-Hydroxytryptophan
- ACT:
-
T cell transfer
- A. muciniphila :
-
Akkermansia muciniphila
- A2AR:
-
Adenosine 2A receptor
- B. pseudolongum :
-
Bifidobacterium pseudolongum
- CADPR:
-
Cyclic adenosine diphosphate ribose
- CaP:
-
Calcium phosphate
- CAR:
-
Chimeric antigen receptor
- CpG-ODN:
-
CpG-oligodeoxynucleotide
- CR:
-
Complete response
- CRC:
-
Colorectal cancer
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen-4
- CTLs:
-
Cytotoxic T lymphocytes
- DCs:
-
Dendritic cells
- dMMR:
-
Mismatch-repair deficiency
- DTR:
-
Diphtheria toxin receptor
- ESBL:
-
Extended-spectrum β-lactamase
- F. PB1:
-
Faecalibaculum rodentium PB1
- FA:
-
Fatty acid
- FAO:
-
Fatty acid oxidation
- FMT:
-
Fecal microbiota transplantation
- GAPDH:
-
Glyceraldehyde-phosphate dehydrogenase
- GBM:
-
Glioblastoma multiforme
- GPR43:
-
G protein-coupled receptor 43
- HCC:
-
Hepatocellular carcinoma
- HDAC:
-
Histone deacetylase
- HLA:
-
Human leucocyte antigen
- IAP:
-
Inhibitor of apoptosis protein
- ICI:
-
Immune checkpoint inhibitors
- ID2:
-
Inhibiting DNA binding 2
- IFN:
-
Interferon
- IL:
-
Interleukin
- IrAEs:
-
Immune-related adverse events
- LAG3:
-
Lymphocyte-activating gene-3
- Macs:
-
Macrophages
- MAIT:
-
Mucosal-associated invariant T
- MAPKs:
-
Mitogen-activated protein kinases
- MHC:
-
Major histocompatibility complex
- NA:
-
Not applicable
- NCR:
-
Natural cytotoxic receptor
- NET:
-
Neutrophil extracellular trap
- NFATc3:
-
Nuclear factor of activated T cells C3
- NK:
-
Natural killer
- NOD1:
-
Nucleotide-binding oligomerization domain containing 1
- NOD2:
-
Nucleotide-binding oligomerization domain containing 2
- NSCLC:
-
Non-small cell lung cancer
- OMV:
-
Outer membrane vesicle
- ORR:
-
Objective response rates
- OXPHOS:
-
Oxidative phosphorylation
- PBMCs:
-
Peripheral blood mononuclear cells
- pCREB:
-
Phosphorylation of cAMP responses element-binding protein
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand-1
- PFS:
-
Progression-free survival
- PG:
-
Peptidoglycan
- PHA:
-
Phytohemagglutinin
- PNP:
-
Purine nucleoside phosphorylase
- PPP:
-
Pentose phosphate pathway
- PR:
-
Partial responses
- PSA:
-
Polysaccharide
- PSMB4:
-
Proteasome subunit beta type-4
- RCC:
-
Renal cell carcinoma
- SCFAs:
-
Short-chain fatty acids
- SD:
-
Stable disease
- SIY:
-
SIYRYYGL
- STING:
-
Stimulator of interferon genes
- SVY:
-
SVYRYYGL
- TCA:
-
Tricarboxylic acid
- TCR:
-
T cell receptor
- Th1:
-
T helper 1
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TMP1:
-
Tape measure protein 1
- TNF:
-
Tumor necrosis factor
- β-OX:
-
β-Oxidation
References
Jugder BE, Kamareddine L, Watnick PI. Microbiota-derived acetate activates intestinal innate immunity via the Tip60 histone acetyltransferase complex. Immunity. 2021;54(8):1683-97.e3.
Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30(6):492–506.
Miyauchi E, Kim SW, Suda W, Kawasumi M, Onawa S, Taguchi-Atarashi N, et al. Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature. 2020;585(7823):102–6.
Erny D, Dokalis N, Mezö C, Castoldi A, Mossad O, Staszewski O, et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021;33(11):2260-76.e7.
Morais LH, Schreiber HL, Mazmanian SK. The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021;19(4):241–55.
He Y, Fu L, Li Y, Wang W, Gong M, Zhang J, et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8(+) T cell immunity. Cell Metab. 2021;33(5):988-1000.e7.
Microbiota-induced IFN-I signaling promotes an antitumor microenvironment. Cancer Discov. 2021. https://doi.org/10.1158/2159-8290.CD-RW2021-145.
Stower H. Microbiome transplant-induced response to immunotherapy. Nat Med. 2021;27(1):21.
McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019;20(2):e77–91.
Fehervari Z. Microbiota shape tumor immunity. Nat Immunol. 2021;22(12):1469.
Mallott EK, Amato KR. Host specificity of the gut microbiome. Nat Rev Microbiol. 2021;19(10):639–53.
Allen-Vercoe E, Coburn B. A microbiota-derived metabolite augments cancer immunotherapy responses in mice. Cancer Cell. 2020;38(4):452–3.
Xu X, Lv J, Guo F, Li J, Jia Y, Jiang D, et al. Gut microbiome influences the efficacy of PD-1 antibody immunotherapy on MSS-type colorectal cancer via metabolic pathway. Front Microbiol. 2020;11:814.
Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science (New York, NY). 2021;371(6529):595–602.
Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science (New York, NY). 2021;371(6529):602–9.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, NY). 2015;350(6264):1084–9.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (New York, NY). 2015;350(6264):1079–84.
He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–9.
Nathan P, Hassel JC, Rutkowski P, Baurain JF, Butler MO, Schlaak M, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196–206.
Goldberg SB, Schalper KA, Gettinger SN, Mahajan A, Herbst RS, Chiang AC, et al. Pembrolizumab for management of patients with NSCLC and brain metastases: long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(5):655–63.
Xin YuJ, Hubbard-Lucey VM, Tang J. Immuno-oncology drug development goes global. Nat Rev Drug Discov. 2019;18(12):899–900.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–86.
Wang M, Du Q, Jin J, Wei Y, Lu Y, Li Q. LAG3 and its emerging role in cancer immunotherapy. Clin Transl Med. 2021;11(3): e365.
Qin S, Ren Z, Meng Z, Chen Z, Chai X, Xiong J, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020;21(4):571–80.
Nayak L, Molinaro AM, Peters K, Clarke JL, Jordan JT, de Groot J, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27(4):1048–57.
Jiang X, Li L, Li Y, Li Q. Molecular mechanisms and countermeasures of immunotherapy resistance in malignant tumor. J Cancer. 2019;10(7):1764–71.
Wang M, Zhen H, Jiang X, Lu Y, Wei Y, Jin J, et al. Clinical observation of the efficacy of PD-1/PD-L1 inhibitors in the treatment of patients with advanced solid tumors. Immunity Inflamm Dis. 2021;9(4):1584–95.
Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Investig. 2007;117(8):2197–204.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (New York, NY). 2013;342(6161):971–6.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (New York, NY). 2013;342(6161):967–70.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (New York, NY). 2018;359(6371):91–7.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (New York, NY). 2018;359(6371):97–103.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (New York, NY). 2018;359(6371):104–8.
Jin Y, Dong H, Xia L, Yang Y, Zhu Y, Shen Y, et al. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in Chinese patients with NSCLC. J Thorac Oncol. 2019;14(8):1378–89.
Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):193.
Song P, Yang D, Wang H, Cui X, Si X, Zhang X, et al. Relationship between intestinal flora structure and metabolite analysis and immunotherapy efficacy in Chinese NSCLC patients. Thorac Cancer. 2020;11(6):1621–32.
Hakozaki T, Richard C, Elkrief A, Hosomi Y, Benlaïfaoui M, Mimpen I, et al. The gut microbiome associates with immune checkpoint inhibition outcomes in patients with advanced non-small cell lung cancer. Cancer Immunol Res. 2020;8(10):1243–50.
Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol. 2020;78(2):195–206.
Lalani AA, Xie W, Braun DA, Kaymakcalan M, Bossé D, Steinharter JA, et al. Effect of antibiotic use on outcomes with systemic therapies in metastatic renal cell carcinoma. Eur Urol Oncol. 2020;3(3):372–81.
Pinato DJ, Howlett S, Ottaviani D, Urus H, Patel A, Mineo T, et al. Association of prior antibiotic treatment with survival and response to immune checkpoint inhibitor therapy in patients with cancer. JAMA Oncol. 2019;5(12):1774–8.
Elkrief A, El Raichani L, Richard C, Messaoudene M, Belkaid W, Malo J, et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology. 2019;8(4): e1568812.
Zhao S, Gao G, Li W, Li X, Zhao C, Jiang T, et al. Antibiotics are associated with attenuated efficacy of anti-PD-1/PD-L1 therapies in Chinese patients with advanced non-small cell lung cancer. Lung Cancer. 2019;130:10–7.
Tinsley N, Zhou C, Tan G, Rack S, Lorigan P, Blackhall F, et al. Cumulative antibiotic use significantly decreases efficacy of checkpoint inhibitors in patients with advanced cancer. Oncologist. 2020;25(1):55–63.
Wilson BE, Routy B, Nagrial A, Chin VT. The effect of antibiotics on clinical outcomes in immune-checkpoint blockade: a systematic review and meta-analysis of observational studies. Cancer Immunol Immunother CII. 2020;69(3):343–54.
Kuczma MP, Ding ZC, Li T, Habtetsion T, Chen T, Hao Z, et al. The impact of antibiotic usage on the efficacy of chemoimmunotherapy is contingent on the source of tumor-reactive T cells. Oncotarget. 2017;8(67):111931–42.
Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI insight. 2018;3(4): e94952.
Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science (New York, NY). 2018;360(6391):eaan5931.
Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune profiling of human gut-associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region-specific immunity. Immunity. 2020;52(3):557-70.e6.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–79.
Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565(7741):600–5.
Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (New York, NY). 2020;369(6510):1481–9.
Shi Y, Zheng W, Yang K, Harris KG, Ni K, Xue L, et al. Intratumoral accumulation of gut microbiota facilitates CD47-based immunotherapy via STING signaling. J Exp Med. 2020;217(5): e20192282.
Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell. 2021;184(21):5338-56.e21.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022-37.e14.
Wculek SK, Amores-Iniesta J, Conde-Garrosa R, Khouili SC, Melero I, Sancho D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J Immunother Cancer. 2019;7(1):100.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24.
Overacre-Delgoffe AE, Bumgarner HJ, Cillo AR, Burr AHP, Tometich JT, Bhattacharjee A, et al. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity. 2021;54(12):2812-24.e4.
Schaupp L, Muth S, Rogell L, Kofoed-Branzk M, Melchior F, Lienenklaus S, et al. Microbiota-induced type I interferons instruct a poised basal state of dendritic cells. Cell. 2020;181(5):1080-96.e19.
Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell. 2020;183(3):771-85.e12.
Di Domizio J, Belkhodja C, Chenuet P, Fries A, Murray T, Mondéjar PM, et al. The commensal skin microbiota triggers type I IFN-dependent innate repair responses in injured skin. Nat Immunol. 2020;21(9):1034–45.
Deng H, Li Z, Tan Y, Guo Z, Liu Y, Wang Y, et al. A novel strain of Bacteroides fragilis enhances phagocytosis and polarises M1 macrophages. Sci Rep. 2016;6:29401.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100.
Nicolai CJ, Wolf N, Chang IC, Kirn G, Marcus A, Ndubaku CO, et al. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci Immunol. 2020;5(45):eaaz2738.
Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24(8):1178–91.
Qiu Y, Jiang Z, Hu S, Wang L, Ma X, Yang X. Lactobacillus plantarum enhanced IL-22 production in Natural Killer (NK) cells that protect the integrity of intestinal epithelial cell barrier damaged by enterotoxigenic Escherichia coli. Int J Mol Sci. 2017;18(11):2409.
Rizvi ZA, Dalal R, Sadhu S, Kumar Y, Kumar S, Gupta SK, et al. High-salt diet mediates interplay between NK cells and gut microbiota to induce potent tumor immunity. Sci Adv. 2021;7(37):eabg5016.
He B, Zhang L, Jiang L, Yu L, Li Q, Tian X, et al. Inhibiting tumor cell-intrinsic UBA6 by inosine augments tumor immunogenicity. Nature Portfolio. 2021. https://www.researchsquare.com/article/rs-620390/v1.
Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell. 2016;165(2):276–87.
Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–6.
Fluckiger A, Daillère R, Sassi M, Sixt BS, Liu P, Loos F, et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science (New York, NY). 2020;369(6506):936–42.
Bessell CA, Isser A, Havel JJ, Lee S, Bell DR, Hickey JW, et al. Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI insight. 2020;5(8): e135597.
Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol. 2021;19(2):77–94.
He B, Hoang TK, Wang T, Ferris M, Taylor CM, Tian X, et al. Resetting microbiota by Lactobacillus reuteri inhibits T reg deficiency-induced autoimmunity via adenosine A2A receptors. J Exp Med. 2017;214(1):107–23.
Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol. 2019;10:925.
Wang T, Gnanaprakasam JNR, Chen X, Kang S, Xu X, Sun H, et al. Inosine is an alternative carbon source for CD8(+)-T-cell function under glucose restriction. Nat Metab. 2020;2(7):635–47.
Lioux T, Mauny MA, Lamoureux A, Bascoul N, Hays M, Vernejoul F, et al. Design, synthesis, and biological evaluation of novel Cyclic Adenosine-Inosine Monophosphate (cAIMP) analogs that activate Stimulator of Interferon Genes (STING). J Med Chem. 2016;59(22):10253–67.
Bird L. Microbial metabolite boosts immunotherapy. Nat Rev Immunol. 2020;20(11):648–9.
Harjes U. Tumour-reactive T cells work remotely using IFNγ. Nat Rev Cancer. 2020;20(5):261.
Shinohara Y, Tsukimoto M. Guanine and inosine nucleotides/nucleosides suppress murine T cell activation. Biochem Biophys Res Commun. 2018;498(4):764–8.
DeNicola GM, Cantley LC. Cancer’s fuel choice: new flavors for a picky eater. Mol Cell. 2015;60(4):514–23.
Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 2019;29(1):135-50.e9.
Xu X, Gnanaprakasam JNR, Sherman J, Wang R. A metabolism toolbox for CAR T therapy. Front Oncol. 2019;9:322.
Nomura M, Nagatomo R, Doi K, Shimizu J, Baba K, Saito T, et al. Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid cancer tumors. JAMA Netw Open. 2020;3(4): e202895.
Tan B, Luo W, Shen Z, Xiao M, Wu S, Meng X, et al. Roseburia intestinalis inhibits oncostatin M and maintains tight junction integrity in a murine model of acute experimental colitis. Scand J Gastroenterol. 2019;54(4):432–40.
Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, et al. Nod-Like Receptor Pyrin-Containing Protein 6 (NLRP6) is up-regulated in Ileal Crohn’s disease and differentially expressed in goblet cells. Cell Mol Gastroenterol Hepatol. 2018;6(1):110-2.e8.
Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat Microbiol. 2020;5(3):511–24.
Santoni M, Piva F, Conti A, Santoni A, Cimadamore A, Scarpelli M, et al. Re: gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Eur Urol. 2018;74(4):521–2.
Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8(+) T cell responses and improve adoptive immunotherapy for cancer. Nat Commun. 2021;12(1):4077.
Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13(5):517–26.
Feng Y, Wang Y, Wang P, Huang Y, Wang F. Short-chain fatty acids manifest stimulative and protective effects on intestinal barrier function through the inhibition of NLRP3 inflammasome and autophagy. Cell Physiol Biochem. 2018;49(1):190–205.
Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8(+) T cells. Immunity. 2019;51(2):285-97.e5.
Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity. 2016;44(6):1312–24.
Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against flu by shaping Ly6c(-) patrolling monocyte hematopoiesis and CD8(+) T cell metabolism. Immunity. 2018;48(5):992-1005.e8.
Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci Rep. 2018;8(1):14430.
Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. 2016;20(2):202–14.
Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia (New York, NY). 2017;19(10):848–55.
Gnanaprakasam JNR, Estrada-Muñiz E, Vega L. The anacardic 6-pentadecyl salicylic acid induces macrophage activation via the phosphorylation of ERK1/2, JNK, P38 kinases and NF-κB. Int Immunopharmacol. 2015;29(2):808–17.
Hollands A, Corriden R, Gysler G, Dahesh S, Olson J, Raza Ali S, et al. Natural product anacardic acid from cashew nut shells stimulates neutrophil extracellular trap production and bactericidal activity. J Biol Chem. 2016;291(27):13964–73.
Erpenbeck L, Schön MP. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene. 2017;36(18):2483–90.
Hemshekhar M, Sebastin Santhosh M, Kemparaju K, Girish KS. Emerging roles of anacardic acid and its derivatives: a pharmacological overview. Basic Clin Pharmacol Toxicol. 2012;110(2):122–32.
Gnanaprakasam JNR, López-Bañuelos L, Vega L. Anacardic 6-pentadecyl salicylic acid induces apoptosis in breast cancer tumor cells, immunostimulation in the host and decreases blood toxic effects of taxol in an animal model. Toxicol Appl Pharmacol. 2021;410: 115359.
Paik D, Yao L, Zhang Y, Bae S, D’Agostino GD, Zhang M, et al. Human gut bacteria produce Τ(Η)17-modulating bile acid metabolites. Nature. 2022;603(7903):907–12.
Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. 2020;581(7809):475–9.
Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22(3):358–69.
D’Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Can Res. 2015;75(21):4651–64.
Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol Pharmacol. 2016;90(5):674–88.
Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bossé D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019;10(1):4346.
Karayama M, Masuda J, Mori K, Yasui H, Hozumi H, Suzuki Y, et al. Comprehensive assessment of multiple tryptophan metabolites as potential biomarkers for immune checkpoint inhibitors in patients with non-small cell lung cancer. Clin Transl Oncol. 2021;23(2):418–23.
Griffin ME, Espinosa J, Becker JL, Luo JD, Carroll TS, Jha JK, et al. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science (New York, NY). 2021;373(6558):1040–6.
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16(2):228–31.
Matsuzaki C, Hayakawa A, Matsumoto K, Katoh T, Yamamoto K, Hisa K. Exopolysaccharides produced by Leuconostoc mesenteroides strain NTM048 as an immunostimulant to enhance the mucosal barrier and influence the systemic immune response. J Agric Food Chem. 2015;63(31):7009–15.
Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe. 2012;12(4):509–20.
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–18.
Pelka K, Shibata T, Miyake K, Latz E. Nucleic acid-sensing TLRs and autoimmunity: novel insights from structural and cell biology. Immunol Rev. 2016;269(1):60–75.
Adamus T, Kortylewski M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp Oncol (Poznan, Poland). 2018;22(1a):56–60.
Moreira D, Adamus T, Zhao X, Su YL, Zhang Z, White SV, et al. STAT3 inhibition combined with CpG immunostimulation activates antitumor immunity to eradicate genetically distinct castration-resistant prostate cancers. Clin Cancer Res. 2018;24(23):5948–62.
Ribas A, Medina T, Kirkwood JM, Zakharia Y, Gonzalez R, Davar D, et al. Overcoming PD-1 blockade resistance with CpG-A toll-like receptor 9 agonist vidutolimod in patients with metastatic melanoma. Cancer Discov. 2021. https://doi.org/10.1158/2159-8290.CD-21-0425.
Huang Y, Ma SF, Espindola MS, Vij R, Oldham JM, Huffnagle GB, et al. Microbes are associated with host innate immune response in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(2):208–19.
Chen X, Yang X, de Anda J, Huang J, Li D, Xu H, et al. Clostridioides difficile toxin A remodels membranes and mediates DNA entry into cells to activate toll-like receptor 9 signaling. Gastroenterology. 2020;159(6):2181-92.e1.
Sartorio MG, Pardue EJ, Feldman MF, Haurat MF. Bacterial outer membrane vesicles: from discovery to applications. Annu Rev Microbiol. 2021;75:609–30.
Li M, Zhou H, Yang C, Wu Y, Zhou X, Liu H, et al. Bacterial outer membrane vesicles as a platform for biomedical applications: an update. J Control Release. 2020;323:253–68.
Cheng K, Zhao R, Li Y, Qi Y, Wang Y, Zhang Y, et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug-and-Display technology. Nat Commun. 2021;12(1):2041.
Qing S, Lyu C, Zhu L, Pan C, Wang S, Li F, et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv Mater (Deerfield Beach, Fla). 2020;32(47): e2002085.
Kim OY, Park HT, Dinh NTH, Choi SJ, Lee J, Kim JH, et al. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat Commun. 2017;8(1):626.
Wang F, Yin Q, Chen L, Davis MM. Bifidobacterium can mitigate intestinal immunopathology in the context of CTLA-4 blockade. Proc Natl Acad Sci USA. 2018;115(1):157–61.
Sun S, Luo L, Liang W, Yin Q, Guo J, Rush AM, et al. Bifidobacterium alters the gut microbiota and modulates the functional metabolism of T regulatory cells in the context of immune checkpoint blockade. Proc Natl Acad Sci USA. 2020;117(44):27509–15.
Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391.
McCulloch JA, Davar D, Rodrigues RR, Badger JH, Fang JR, Cole AM, et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat Med. 2022. https://doi.org/10.1038/s41591-022-01698-2.
Naqash AR, Ricciuti B, Owen DH, Florou V, Toi Y, Cherry C, et al. Outcomes associated with immune-related adverse events in metastatic non-small cell lung cancer treated with nivolumab: a pooled exploratory analysis from a global cohort. Cancer Immunol Immunother CII. 2020;69(7):1177–87.
Verzoni E, Cartenì G, Cortesi E, Giannarelli D, De Giglio A, Sabbatini R, et al. Real-world efficacy and safety of nivolumab in previously-treated metastatic renal cell carcinoma, and association between immune-related adverse events and survival: the Italian expanded access program. J Immunother Cancer. 2019;7(1):99.
Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021;27(8):1432–41.
Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24(12):1804–8.
Inamura K. Gut microbiota contributes towards immunomodulation against cancer: new frontiers in precision cancer therapeutics. Semin Cancer Biol. 2021;70:11–23.
Daillère R, Derosa L, Bonvalet M, Segata N, Routy B, Gariboldi M, et al. Trial watch : the gut microbiota as a tool to boost the clinical efficacy of anticancer immunotherapy. Oncoimmunology. 2020;9(1):1774298.
Littmann ER, Lee JJ, Denny JE, Alam Z, Maslanka JR, Zarin I, et al. Host immunity modulates the efficacy of microbiota transplantation for treatment of Clostridioides difficile infection. Nat Commun. 2021;12(1):755.
DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. 2019;381(21):2043–50.
Vendrik KEW, Terveer EM, Kuijper EJ, Nooij S, Boeije-Koppenol E, Sanders I, et al. Periodic screening of donor faeces with a quarantine period to prevent transmission of multidrug-resistant organisms during faecal microbiota transplantation: a retrospective cohort study. Lancet Infect Dis. 2021;21(5):711–21.
Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11(8):506–14.
Hibberd AA, Lyra A, Ouwehand AC, Rolny P, Lindegren H, Cedgård L, et al. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017;4(1): e000145.
Consoli ML, da Silva RS, Nicoli JR, Bruña-Romero O, da Silva RG, de Vasconcelos GS, et al. Randomized clinical trial: impact of oral administration of Saccharomyces boulardii on gene expression of intestinal cytokines in patients undergoing colon resection. JPEN J Parenter Enteral Nutr. 2016;40(8):1114–21.
Rong Y, Dong Z, Hong Z, Jin Y, Zhang W, Zhang B, et al. Reactivity toward Bifidobacterium longum and Enterococcus hirae demonstrate robust CD8(+) T cell response and better prognosis in HBV-related hepatocellular carcinoma. Exp Cell Res. 2017;358(2):352–9.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2019;30(12):2012.
Lauté-Caly DL, Raftis EJ, Cowie P, Hennessy E, Holt A, Panzica DA, et al. The flagellin of candidate live biotherapeutic Enterococcus gallinarum MRx0518 is a potent immunostimulant. Sci Rep. 2019;9(1):801.
McHale D, Francisco-Anderson L, Sandy P, Shariffudin S, Goldberg M, Gardner H, et al. Oral delivery of a single microbial strain, EDP1503, induces anti-tumor responses via gut-mediated activation of both innate and adaptive immunity. Ann Oncol. 2020;31:S195. https://doi.org/10.1016/j.annonc.2020.04.407.
Dizman N, Meza L, Bergerot P, Alcantara M, Dorff T, Lyou Y, et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med. 2022. https://doi.org/10.1038/s41591-022-01694-6.
Chang Y, Yuan L, Liu J, Muhammad I, Cao C, Shi C, et al. Dihydromyricetin attenuates Escherichia coli lipopolysaccharide-induced ileum injury in chickens by inhibiting NLRP3 inflammasome and TLR4/NF-κB signalling pathway. Vet Res. 2020;51(1):72.
Taniguchi S, Shimatani Y, Fujimori M. Tumor-Targeting therapy using gene-engineered anaerobic-nonpathogenic Bifidobacterium longum. Methods Mol Biol. 2016;1409:49–60.
Liang K, Liu Q, Li P, Luo H, Wang H, Kong Q. Genetically engineered Salmonella typhimurium: recent advances in cancer therapy. Cancer Lett. 2019;448:168–81.
Zhou S, Gravekamp C, Bermudes D, Liu K. Tumour-targeting bacteria engineered to fight cancer. Nat Rev Cancer. 2018;18(12):727–43.
Guo Y, Chen Y, Liu X, Min JJ, Tan W, Zheng JH. Targeted cancer immunotherapy with genetically engineered oncolytic Salmonella typhimurium. Cancer Lett. 2020;469:102–10.
Leventhal DS, Sokolovska A, Li N, Plescia C, Kolodziej SA, Gallant CW, et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat Commun. 2020;11(1):2739.
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829-42.e13.
Martí i Líndez AA, Dunand-Sauthier I, Conti M, Gobet F, Núñez N, Hannich JT, et al. Mitochondrial arginase-2 is a cell-autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI insight. 2019;4(24):32975.
Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature. 2021;598(7882):662–6.
Malczewski AB, Ketheesan N, Coward JIG, Navarro S. Enhancing checkpoint inhibitor therapy in solid tissue cancers: the role of diet, the microbiome & microbiome-derived metabolites. Front Immunol. 2021;12: 624434.
Ansaldo E, Belkaid Y. How microbiota improve immunotherapy. Science (New York, NY). 2021;373(6558):966–7.
Matson V, Chervin CS, Gajewski TF. Cancer and the microbiome-influence of the commensal microbiota on cancer, immune responses, and immunotherapy. Gastroenterology. 2021;160(2):600–13.
Wolter M, Grant ET, Boudaud M, Steimle A, Pereira GV, Martens EC, et al. Leveraging diet to engineer the gut microbiome. Nat Rev Gastroenterol Hepatol. 2021;18(12):885–902.
Pennington SH, Ferreira DM, Caamaño-Gutiérrez E, Reiné J, Hewitt C, Hyder-Wright AD, et al. Nonspecific effects of oral vaccination with live-attenuated Salmonella Typhi strain Ty21a. Sci Adv. 2019;5(2):eaau6849.
Domingos-Pereira S, Cesson V, Chevalier MF, Derré L, Jichlinski P, Nardelli-Haefliger D. Preclinical efficacy and safety of the Ty21a vaccine strain for intravesical immunotherapy of non-muscle-invasive bladder cancer. Oncoimmunology. 2017;6(1): e1265720.
Drake CG, Pachynski RK, Subudhi SK, McNeel DG, Antonarakis ES, Bauer TM, et al. Safety and preliminary immunogenicity of JNJ-64041809, a live-attenuated, double-deleted Listeria monocytogenes-based immunotherapy, in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2021. https://doi.org/10.1038/s41391-021-00402-8.
Schmitz-Winnenthal FH, Hohmann N, Schmidt T, Podola L, Friedrich T, Lubenau H, et al. A phase 1 trial extension to assess immunologic efficacy and safety of prime-boost vaccination with VXM01, an oral T cell vaccine against VEGFR2, in patients with advanced pancreatic cancer. Oncoimmunology. 2018;7(4): e1303584.
Skelly AN, Sato Y, Kearney S, Honda K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol. 2019;19(5):305–23.
Cieplak T, Soffer N, Sulakvelidze A, Nielsen DS. A bacteriophage cocktail targeting Escherichia coli reduces E. coli in simulated gut conditions, while preserving a non-targeted representative commensal normal microbiota. Gut Microbes. 2018;9(5):391–9.
Zuo T, Wong SH, Lam K, Lui R, Cheung K, Tang W, et al. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut. 2018;67(4):634–43.
Maciejewska B, Olszak T, Drulis-Kawa Z. Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: an ambitious and also a realistic application? Appl Microbiol Biotechnol. 2018;102(6):2563–81.
Acknowledgements
The authors would like to thank Y.W., X.J., and H.Z. for the critical reading of the manuscript and helpful discussions.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81301912); the Beijing Municipal Health and Scientific and Technological Achievements and Appropriate Technology Promotion Project (Grant No. BHTPP202008); the “High-value Patent Cultivation” project of the Beijing Friendship Hospital Affiliated to the Capital Medical University (Grant No. yyzscq202003); the Research Foundation of Beijing Friendship Hospital (Grant No. yyqdkt2019-40); the General program of National Natural Science Foundation of China (Grant No. 81973715 and 82174243); and the Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (Grant No. ZYYCXTD-C-202001).
Author information
Authors and Affiliations
Contributions
QL, JW, XL, HL, and YL conceived and designed the article. YL, QL, JW, and HL wrote and edited the original draft. ZH and MW collected the clinical data. YL, ZH, MW, and HL polished and modified the English. YL and XL visualized the article. All the authors contributed to the review and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Y., Yuan, X., Wang, M. et al. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J Hematol Oncol 15, 47 (2022). https://doi.org/10.1186/s13045-022-01273-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-022-01273-9