
Abstract
Transforming growth factor β (TGF-β) has long been identified with its intensive involvement in early embryonic development and organogenesis, immune supervision, tissue repair, and adult homeostasis. The role of TGF-β in fibrosis and cancer is complex and sometimes even contradictory, exhibiting either inhibitory or promoting effects depending on the stage of the disease. Under pathological conditions, overexpressed TGF-β causes epithelial-mesenchymal transition (EMT), extracellular matrix (ECM) deposition, cancer-associated fibroblast (CAF) formation, which leads to fibrotic disease, and cancer. Given the critical role of TGF-β and its downstream molecules in the progression of fibrosis and cancers, therapeutics targeting TGF-β signaling appears to be a promising strategy. However, due to potential systemic cytotoxicity, the development of TGF-β therapeutics has lagged. In this review, we summarized the biological process of TGF-β, with its dual role in fibrosis and tumorigenesis, and the clinical application of TGF-β-targeting therapies.
Similar content being viewed by others

Background
Transforming growth factor β (TGF-β) is a prototype of the TGF-β family, which is composed of TGF-β, Activin, Nodal, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), and other factors [1, 2]. As a multifunctional polypeptide cytokine, TGF-β plays a critical role in early embryonic development and adult homeostasis [3]. Three subtypes of TGF-β (TGF-βI-III) are only expressed in mammals with unique multifunctional growth factors. In the following paragraphs, TGF-β refers to TGF-βI if not otherwise specified. TGF-β is mainly secreted and stored in the extracellular matrix (ECM) as a latent complex [4], while only activated TGF-β binds to the TGF-β receptor (TβR) complex to lead to its biological functions. Therefore, TGF-β activation is critical for its operation.
In recent years, scientists found that overexpressed TGF-β causes a plethora of metabolic disorders and dysfunction, and promotes epithelial-mesenchymal transition (EMT) and excessive deposition of ECM [5, 6], which causes immune dysfunction, fibrosis, and cancers [7]. Because of the vital function of TGF-β in human fibrosis and cancers, anti-TGF-β approaches have been introduced to treat these diseases [8]. In recent years, many clinical trials have verified the therapeutic effect of TGF-β-targeted drugs on a variety of tumor and fibrotic diseases. By combining TGF-β-targeting drugs (anti-TGF-β antibody, TβR inhibitor, and recombinant proteins) with other antigens (programmed cell death one ligand 1 (PD-L1), M7824, SHR-1701, JS201, TST005, and COX-2 (STP705)) is the most popular treatment strategy currently. This review focuses on the biological process of TGF-β, its dual role in fibrosis and tumorigenesis, and the clinical application of TGF-β-targeting therapeutics.
The procession of TGF-β
Pro-TGF-β is synthesized as a latent complex in the ECM and is associated with a signal peptide in the large N-terminal portion called the latency-associated peptide (LAP) and a mature cytokine in the C-terminal fragment [9,10,11]. The large latent complex (LLC) comprises LAP, TGF-β, latent TGF-β binding proteins (LTBP) 1/3, and LTBP4. Latent TGF-β is activated by proteins and enzymes (thrombospondin 1 (TSP-1), glycoprotein A repetitions predominant protein (GARP), integrins, and other TGF-β-binding proteins) and transformed into disulfide-linked dimers and homodimeric ligands. The activated TGF-β interacts with the TβR complex or other cytokines to regulate biological responses through drosophila mothers against decapentaplegic (SMAD) and/or non-SMAD pathways [12].
TGF-β secretion
LAP binds to LTBPs covalently via two disulfide bonds with two cysteine residues [10]. LTBPs are the promoter of the folding of TGF-β precursor protein. In addition, LTBPs are crucial to latent TGF-β location and activation [13, 14]. LAP, in turn, is cleaved by furin (an indispensable proprotein convertase) from the mature TGF-β precursor in the trans-Golgi network (Fig. 1) [12], in which LTBPs are considered as the primary activator [13]. Although LAP is cleaved from the C-terminal portion, it remains associated with the mature cytokine TGF-β noncovalently [2].

A schematic representation of TGF-β activation The pro-TGF-β synthesized in the rough endoplasmic reticulum becomes latent TGF-β when cleaved by the convertase furin in the Golgi complex. Then the LAP dimer binds to mature TGF-β noncovalently to form a small latent complex (SLC). Then, SLC generally binds to LTBP, forming LLC, while binds to GARP in Treg cells. SLC is anchored to ECM proteins, including fibronectin and fibrillin, via LTBP. Both LTBP and GARP play a direct role in anchoring TGF-β for traction-driven activation by integrins. With the help of αβ integrins and mechanical force, latent TGF-β becomes active and connects to the TβR complex to regulate transcription
Location and activation of TGF-β
In general, LLC is secreted to the ECM and is located there via the unique biological properties of LTBP, which interacts with extracellular matrix fibers to keep TGF-β in an inactive form (Fig. 1) [13, 15]. A recent study showed that the crystals of pro-TGF-βI are a ring-shaped complex. When LAP-surrounded TGF-β monomers are freed under cytoskeletal force, the active cytokine interacts with TβR to regulate cellular responses (Fig. 1). This force-dependent activation requires the unfastening of a pro-domain named the ‘straitjacket’ element [16].
Enzymatic activation
In addition to the nonproteolytic mechanism for the activation of latent TGF-β, proteases are also involved in TGF-β activation. In addition, these proteases are divided into containing glycosidases (N-glycanase and neuraminidase) and serine proteases (plasmin, cathepsin D, and matrix metalloproteases) [10, 17, 18]. Summarily, in the cytoplasmic matrix, TGF-β can be activated by several proteases, integrins, and other TGF-β-binding proteins in different cell types, tissues, and disease milieus [19]. Notably, the proteolytic cleavage sites of latent TGF-β implicate the mechanism of how proteases cleave the LAP latency lasso. For instance, plasma kallikrein (PLK) cleaves residues between R58 and L59 of latent TGF-β [20]. Studies on the activation process of TGF-β at the molecular level helps to targeting-TGF-β therapy.
Regulation by fibrillin
LTBPs are structurally related to and physically bound to another family of proteins called fibrillins. Fibrillin 1 can function as an inhibitor of TGF-β signaling, but whether it works more directly in controlling the fibrillin-LTBP interaction or stability suppress latent LLC proteolytic activation still needs to be explored [21, 22]. As integral components of microfibrils, fibrillins play different roles in microfibril biology [23]. The microfibrils cover the elastin core of elastin-containing fibers and promote the temporal and spatial regulation of TGF-β activation [24]. Scientists previously suggested that fibrillin-1 can be presented to the surface-exposed loop when binding to the arginine-glycine-aspartic acid (RGD) integrin-binding motif [25,26,27]. While the remaining fibrillins showed little inhibitory effect on TGF-β activation. A number of studies indicated that fibrillin 2 expression is mainly restricted to developing fetal tissues, while fibrillin-1 expression endures throughout adult life [28]. Moreover, fibrillin-1, together with associated molecules, masks fibrillin-2 epitopes to block its bioactivity. Therefore, fibrillin 1 shows stronger anti-TGF-β activity. Notably, a recent study showed that when local fibrillin-1 was downregulated, fibrillin 2 molecules were exposed in the tumor endothelium with a lower capacity to block TGF-β [29]. Moreover, Heena Kumra et al. suggested that fibrillin-4 might regulate LTBP-4 matrix assembly to impact TGF-β signaling [30].
Regulation of TGF-β activation by GARP
Recent evidence demonstrated that regulatory T cells (Tregs) could promote latent TGF-β presented by GARP to integrin αVβ8 integrin (Fig. 1) [31]. Unlike LTBPs are abundantly presented in the ECM, GARP is retained only on the surface of Foxp3-expressing Tregs [32]. It is generally accepted that αVβ8 integrin is involved in GARP/TGF-β complex activation, but the exact mechanism is controversial. Some scientists indicated that cytoskeletal force was unnecessary for αVβ8-mediated TGF-β activation. Others believed that the regulation of TGF-β activation by GARP required the release and diffusion of mature TGF-β [33]. In addition, they discovered that mature TGF-β signals were involved in latent TGF-β, which indicated that αVβ8-mediated TGF-β activation may form a large multi-component cell–cell protein complex to induce the SMAD-dependent pathway [34, 35]. Regardless of the mechanism of GARP-induced TGF-β activation, targeting GARP is one of the approaches to avoid TGF-β activation, targeting GARP is one of the approaches to avoid TGF-β activation. Notably, a study showed that monoclonal antibodies against GARP in GARP/TGF-βI complexes could not recognize amino acids GARP137-139 within GARP/TGF-βI complexes could not inhibit Treg-associated TGF-β activation [36].
Activation of TGF-β by integrins
Integrin family members are implicated in the recognition and activation of TGF-β [37,38,39,40,41,42,43,44]. In addition, integrin-mediated TGF-β activation is essential in the immune system (integrins αvβ6 and αvβ8), tumorigenesis, and fibroblasts. Both Integrins αvβ6 and αvβ8 regulate TGF-β signaling by binding to a linear tripeptide RGD depending on actin cytoskeleton-generated tensile force [45]. In addition to integrins αvβ6 and αvβ8, integrins α8β1, α5β1, and αIIβ3 can also recognize the RGD site in the LAP region of TGF-β. This RGD recognition mechanism regulates the growth and differentiation factors of the TGF-β family to maintain morphogenesis and homeostasis [46].
However, the presence of integrin alone is insufficient for TGF-β activation. Considerable studies have suggested that actin-myosin contraction and mechanical deformation are of great importance for TGF-β activation. In addition, scientists widely believed that the contraction of the actin cytoskeleton previously generated integrin-mediated TGF-β activation by physical force. Furthermore, a study by Melody G. Campbell recently indicated that integrin αvβ6, along with its entire ectodomain, activates GARP to locate latent TGF-β without the release and diffusion of mature TGF-β [47]. In general, identifying a complete regulatory pathway would facilitate the development of more effective therapeutic strategies.
TGF-β signaling pathways
The low-affinity heteromeric receptor complex (tβR I with tβR II) conducted by activated TGF-β stimulates different downstream signaling pathways (SMAD pathways and no-SMAD pathways) to regulate context-dependent transcription (Fig. 2). Under different physiological and pathological conditions, different kinases or signaling pathways adjust the SMAD pathway to regulate protein expression [48].

SMAD and non-SMAD pathways Schematic of the TGF-β-induced canonical SMAD and noncanonical non-SMAD signaling pathways Mature TGF-β phosphorylates TβR II, which recruits TβR I to phosphorylate receptor-SMAD proteins. Then, co-SMAD with the R-SMAD complex translates into the nucleus to regulate CAGA gene transcription. TGF-β actives non-SMAD pathways when connected to other downstream factors, such as SHC/GRB2/SODS, TRAF4/6, PAR6, and PI3K
The canonical pathway—SMAD pathway
SMAD is a canonical pathway in which TGF-β is identified by TβR II equipped with an intracellular kinase domain, which recruits and phosphorylates TβR I with a conversed Gly/Ser-rich "GS sequence" from serine/threonine kinases. TβR II and TβR I then become a heteromeric complex [49]. Activated TβR I phosphorylates receptor-SMAD (R-SMAD) protein and promotes R-SMAD complex binding to Co-SMAD/SMAD4, forming a trimeric complex. The trimeric complex is then translated into and aggregates in the nucleus as a transcription factor to regulate target gene expression from embryonic development to adult organisms [48, 50].
In addition to being regulated by other signaling pathways or cytokines, TGF-β signaling is also automated. Downstream factors of SMAD signaling, especially Smad2/Smad3, are considered crucial mediators of TGF-β signaling in tissue fibrosis and tumorigenesis. At the same time, Smad6 and Smad7 are regarded as negative regulators to improve TGF-β-mediated fibrosis and tumorigenesis. For example, SMAD3-induced the upregulation of TSP-4, which stimulates tumor growth by mediating TGF-β-induced angiogenesis [51].
Noncanonical pathway—non-SMAD pathway
All the pathways and downstream cascades activated by TGF-β through phosphorylation, acetylation, sumoylation, ubiquitination, and protein–protein interactions are collectively referred to as non-SMAD signaling pathways [53, 54]. These interactions mediate the intracellular responses of TGF-β and/or its related factors are collectively referred as non-SMAD signaling pathways [52, 53]. Mature TGF-β activates the mitogen-activated protein kinase (MAPK) pathway [54], extracellular signal-regulated kinases 1/2 (Erk1/2) pathways, Rho-like signaling pathways, phosphatidylinositol-3-kinase (PI3K)/AKT pathways, c-Jun amino-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38/MAPK) signaling pathways [55]. The Erk signaling pathway (Fig. 3) is essential for embryonic development in adult organisms. For instance, it affects the development of embryos, especially nerves, and EMT to promote fibrosis and cancer metastasis in geriatric diseases [56,57,58,59]. Accumulating evidence has shown that diverse TGF-β signaling responses are related to the combinatorial usage of core pathway components, including ligands, receptors, SMADs, and transcription factors by cross interacting with other pathways to regulate target gene transcription [52].

TGF-β activated the Erk MAPK pathway. Activated TβR I recruits and phosphorylates the Shc adaptor protein ShcA. Actived TGF-β promotes the formation of the ShcA/Grb2/SOS complex, Ras connects, and degrades SPSB1 via mono- and deubiquitination. TGF-β-induced GTP loading on Ras helps recruit Raf to the plasma membrane, resulting in the activation of Erk1/2 through MEKs. The activated Erk MAPK signaling pathway further influences the SMAD signaling pathway
TGF-β in fibrosis
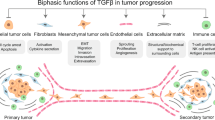
Fibrosis is a pathological process in which organ parenchyma cell necrosis and ECM deposit excessively, causing connective tissue hyperplasia, fibrosis, or even significantly producing organ sclerosis. In addition, fibrosis is usually accompanied by the transformation of fibroblasts into myofibroblasts, even CAFs. Normal fibroblasts are components of the paraneoplastic stroma, which are critical in supporting the homeostasis of tissue-resident cells and define the architecture of organs. Several cytokines and chemokines (miR-214 [60], IL-1 [61], α-SMA, integrin β-1), and signaling pathways (EGFR, Wnt/β-catenin, Hippo, TGF-β, and JAK/STAT cascades) reprogram normal fibroblasts into CAFs [62, 63]. However, the mechanisms underlying the transformation of CAFs are rarely known.
TGF-β I- III all have fibrogenic effects and share 70–82% homology at the amino acid level [64]. TGF-β I is considered as the primary factor in liver, kidney, and lung fibrosis through canonical and noncanonical signaling pathways. Usually, the cytokine TGF-β is up-regulated in tissue injury, inflammation, and wound healing [65]. The longer-term contractile state of the wound helps accelerate the expression of ECM proteins. Dysregulated TGF-β signaling promotes pathological fibrosis and tumorigenesis by excessive ECM deposition (Fig. 4). The abnormal accumulation of ECM triggers the process of fibrosis and immunosuppression by linking SMAD4, BRAF, and TP53 mutations and MYC amplification [6] and contributes to the cancer-associated fibroblast (CAF) phenotype. Scientists found that inhibiting TGF-β signaling and its downstream signaling pathways could significantly reduce fibrosis [66,67,68].

Essential functions of TGF-β in fibrosis Under pathological conditions, many different cell types, including macrophages, epithelial cells, lymphocytes, fibroblasts, and endothelial cells, can produce and secrete more TGF-β to mediate fibroblasts through SMAD and non-SMAD pathways. Although TGF-β plays a vital role in promoting fibrosis, few antifibrosis therapies target it in clinical practice
TGF-β in hepatic fibrosis
Acute and chronic liver injuries promote excessive expression of TGF-β from various cell types and activation of TGF-β in the ECM. Then, activated TGF-β promotes collagenase deposition and EMT to induce fibroblast mesenchymal transformation and the activation of HSCs. In addition, increased TGF-β can be directly generated in liver injury [69]. Hepatic stellate cells (HSCs) are turned into hepatocellular carcinoma (HCC) cells [70, 71]. The activated HSCs express α-SMA but do not have lipid droplets. In addition, they give rise to myofibroblasts (MFBs), which represent the primary producer of collagen and other ECM proteins [71, 72].
The composition of the hepatic ECM changes during liver fibrosis and interacts with factors in TGF-β signaling to regulate hepatic fibrosis. For instance, the disruption of SMAD2 and the composition of SMAD3 promote the transcription of type II collagen toward type I and III collagen [72,73,74]. Meanwhile, the phosphorylation of Smad2/3 also encourages the acceleration of MMP1, α-SMA, and collagen type I, which results in the overexpression of lysyl oxidase-like 1 (LOXL1) to promote liver fibrosis [75]. Despite SMAD pathways, TGF-β also promotes HSC activation through non-SMAD pathways (MAPK, ERK, p38, and JNK pathways). For instance, activated TGF-β increases the expression of kindlin-2 via p38 and MAPK signaling, and overexpressed kindlin-2 positively feedbacks the TGF-β pathway by up-regulating Smad2 and Smad3 phosphorylation [76, 77].
Given the vital role of TGF-β in liver fibrosis, baseline TGF-β is always regarded as a biomarker of prognostic indicators. Nevertheless, clinical trials targeting TGF-β for HCC have been rare in recent years. It may be because dysregulated TGF-β cascades are not the dominate factors for HCC occurrence [78]. Galunisertib, a small-molecule selective inhibitor of TβR I, has been shown to prolong overall survival when administered with sorafenib [79]. However, it is worth noting that not all combination drug therapies help to improve HCC (NCT00557856).
TGF-β in kidney fibrosis
Robust evidence suggests that TGF-β is a well-established central mediator of renal fibrosis. TGF-β can promote the accumulation of ECM proteins in progressive chronic kidney disease (CKD) [8, 80]. Similar to hepatic fibrosis, the development of renal fibrosis is also completed with phenotypic plasticity processes and migration, as well as invasion of epithelial cells [81], in which TGF-β has a central role. TGF-β causes progressive forms of human kidney disease by regulating apoptosis, activating ECM synthesis, and inhibiting ECM degradation through metalloproteinase inhibition [80]. TGF-β can also activate fibroblasts and translate other cell types into fibroblast-type cells directly or indirectly by SMAD or non-SMAD pathways [82, 83]. In addition, TGF-β can directly act on mesangial cells and fibroblasts to regulate cell proliferation, migration, and activation. TGF-β also mediates the accumulation of profibrotic molecules in ECM. Profibrotic molecules contain collagens, fibronectin, and plasminogen activator inhibitor-1 (PAI-1) [40, 84]. In contrast, overexpressed TGF-β indirectly prevents fibrosis. A recent study by Su J showed that TGF-β-stimulated human tubular epithelial cells and fibrotic kidneys express TGF-β/Smad3-interacting long noncoding RNA (lnc-TSI) to antagonize renal fibrosis [5].
Multiple drugs, including monoclonal antibodies (FG-3019, FG-4019), siRNAs (RXI-109, OLX-101, OLX-201), peptides (BLR-100/BLR-200), and antisense oligonucleotides, are under clinical trials, and other preclinical studies are trying to investigate more effective targets and therapies [85,86,87]. Furthermore, hepatocyte growth factor (HGF), BMP-7, SMAD7, and lnc-TSI can also be treated as antifibrotic targets. To date, scientists have identified multiple therapeutic targets for TGF-β-induced renal fibrosis, including microRNAs, proteins, genes, and transcription factors. For example, disrupting the recombination signal binding protein-Jκ (RBP-Jκ) could block Notch signaling, which regulates bone marrow-derived macrophages (BMDMs) to attenuate TGF-β-induced renal fibrosis [88]. MicroRNAs (miRNAs) containing 21–24 nucleotides (miR-34a, miR-30c, miR21, miR29, miR-101a, miR-34a, etc.) have been proved to play essential roles in the regulation of renal fibrosis through TGF-β signaling [89, 90]. Zhao et al. found that miR-30c inhibited the Snail 1-TGF-β axis in tubular epithelial cells to attenuate EMT, which was similar to paricalcitol [89].
TGF-β in lung fibrosis
Idiopathic pulmonary fibrosis (IPF) is a chronic and fibrotic lung disease with a periphery to center progression, characteristic imaging, irreversible structural alterations, and tissue stiffening [91]. The observation that alveolar epithelial cells (AECs) and fibroblasts in IPF produce aberrant ECM is implicated in the TGF-β signaling pathway [92]. TGF-β is mainly derived from alveolar macrophages and metaplastic type II AECs and driven by sustained elevated mechanical tension in IPF [93]. Scientists identified the up-regulated mature TGF-β and SMAD3, SMAD4, CTGF, together with the deregulated SMAD7 in IPF [92]. Through a study of fibrotic development and glutamate metabolism, scientists found that the connection between epigenetic and transcriptional processes was almost in a TGF-β-dependent manner [94]. Despite α-SMA, TGF-β-induced integrins, MMPs, protease inhibitors, tumor necrosis factor-α (TNF-α), and regulators of small GTPases are also participated in cell-ECM interactions [95, 96]. Meanwhile, TGF-β can not only inhibit the production of antifibrotic molecules [97] but also induce serum KL6/mucin 1 (MUC1) activation [98].
TGF-β is a key profibrotic factor in IPF, but inhibiting TGF-β causes multiple side effects due to its pleiotropic effects. Though not reported in clinical trials, some TβRI kinase inhibitors showed cardiac toxicity and skin toxicity when administrated at high dose [99]. Thus, searching downstream effectors of TGF-β signaling appears to be a new research direction. Long noncoding RNAs such as RNA H19X, dynamin three opposite strand (DNM3OS), and miRNAs including 199a-5p, miR-199-3p, and miR-214-3p are all crucial to TGF-β-mediated lung fibrosis [100,101,102,103]. DNM3OS is a fibroblast-specific critical downstream effector of TGF-β-induced lung fibrosis, and interfering with it may present new effective therapeutic targets [101]. In addition, TGF-β interacts with periostin to promote lung fibrosis through the αVβ3/β5-Smad3 pathway, which can be attenuated by the integrin low-molecular-weight inhibitor CP4715 [104].
TGF-β in cancer
TGF-β has been shown to play a crucial role in developing cancer by TGF-β pathway knockout in mice. Several experiments have demonstrated that TGF-β plays a dual role (a tumor suppressor in premalignant cells and a tumor promoter in carcinoma cells) in the process of cancer by modulating the cellular context and other effects of the cytokine [2]. TGF-β acts as a tumor suppressor by inhibiting proliferation and inducing apoptosis during the early stages of tumorigenesis [105]. Generally, TGF-β inhibits proliferation and promotes apoptosis through overexpressed cyclin-dependent kinase (CDK) inhibitors [106] and downregulated MYC expression [107]. Under this condition, premalignant cells become disseminated cancer cells, can self-impose a slow-cycling state to remain latent for extended periods [108]. The specific mechanism of how TGF-β promotes the immune escape of carcinoma cells will be described below.
Tumor cells escape antitumor surveillance of TGF-β by accumulating mutations in the TGF-β signaling cascades [109]. Examples of such escape include the mutation of SMAD4 in pancreatic ductal adenocarcinoma (PDAC) and gastric cancer (GC) [110, 111], the TβR I mutation in colon cancer [112], and even mutations in genes that encode TGF-β ligands (BMP5), receptors (TβR II, AVCR2A, BMPR2), and SMADs (SMAD2 and SMAD4) [113, 114]. Mutations in the TGF-β pathway in the head and neck, bladder, and endometrial adenocarcinomas occur in 10% to 20% of cases, compared to 25% to 50% of subjects in gastrointestinal cancer (esophageal, CRC and PDA) [111, 112, 115, 116]. Although a loss of TGF-β function mutation components is insufficient for tumor initiation, it promotes the transition of premalignant cells to a more overly malignant phenotype [2, 117].
In addition to the accumulated mutations of TGF-β signaling cascades, TGF-β-regulated immunosuppressive microenvironment also promotes tumor escape indirectly [118]. Adaptive immunity is one of three critical immune pathways implicated in disease, which is also regulated by TGF-β signaling [105, 114]. TGF-β signaling can not only control adaptive immunity by promoting the expansion of Treg cells directly, regulating the regulatory CD4 + T cell response, but also by controlling the function of effector T cells. In addition, TGF-β similarly controls the development and functions of the innate immune system by inhibiting natural killer (NK) cells [119] and regulating the proliferation of macrophages, antigen-presenting dendritic cells (DCs), and granulocytes [120]. Mutations of SMAD4 promote dysregulation of NK cell homeostasis and augment tumor cell metastases [121]. Actions on both adaptive immunity and innate immunity form a network of negative immune regulatory inputs. Luckily, scientists have indicated that TGF-β-induced immune tolerance and inflammatory responses can be flexibly treated by ionizing radiation combined with hyperthermia and checkpoint inhibitor therapies [122].
TGF-β in melanoma
Melanoma is the most aggressive type of skin cancer, accounting for 7% of all diagnosed cancers in men and 4% in women, with approximately 7,230 fatalities in 2019 [123]. Like other cancers, as a tumor suppressor, TGF-β exerts an anti-proliferative powerful impact in normal melanocytes. As a tumor promoter, TGF-β promotes EMT, proliferation, metastasis, and immune tolerance [124, 125]. The opposite effects of TGF-β in melanoma is associated with the deregulation of cytokines (TNF-α, VEPH1, SMAD4, INF-γ, SKI) and signaling pathways (Notch1, IL-6, and Erk/MAPK pathway), which in return regulate TGF-β signaling [121, 126,127,128,129,130,131,132,133].
Adipocyte-created IL-6 and TNF-αmiR-211 promote the miR-211-repressed translation of TβR I mRNA to enhance the cellular responsiveness and metastasis of melanoma [129]. The poorly expressed ventricular zone expressed PH domain-containing 1 (VEPH1) and up-regulated upstream transcription factor 1 (USF1) in melanoma tissues promoted EMT [127, 130]. TGF-β-induced transcription sustains actomyosin force is independent of EMT [134]. TGF-β-associated VEPH1 induces proliferation, migration, and invasion of conditioned medium (CM) cells by up-regulating the expression of E-cadherin and down-regulating the expression of N-cadherin, Vimentin, and SMAD4 [130, 135]. Notably, SMAD4 suppresses tumor metastasis and promotes antitumor immunity through up-regulated IFN-γ and granzyme B (GZMB) by non-SMAD in NK cells at early stages [119, 121]. Immune cells, such as TGF-β-sustained effector T cells, secrete CD73 to facilitate tumor resistance of anti-CD137 therapy [136]. In BRAF (V600E)-mutant melanoma, the sex-determining region Y-box 10 (SOX10) is suppressed, and BRAF signaling-activated TFEB S142 phosphorylation is promoted. Both of them help increase melanoma metastatic potential and drug resistance [137, 138].
Therapies targeting these deregulated cytokines and signaling pathways combined with radiation, chemotherapy, and other targeted therapies become revolutionary therapeutic strategies. In addition to MECOM and BMP5 in BRAF-mutated melanoma, GNAQ or CNA11 mutations in uveal melanoma are also associated with TGF-β signaling [139]. Furthermore, GNAQ or CNA11 mutations demonstrate low sensitivity or resistance to specific treatments [140, 141]. They indicate a suite of rationally designed clinical trials and potentially clinical targets. Scientists indicated that hydrophobic TGF-β inhibitor (SB-505124) and an adenoviral vector expressing IL-12 increase animal survival [142]. PD-1/PD-L1 antigen-specific checkpoints block siRNA entry into antigen-presenting cells. In addition, PD-1/PD-L1 antigen-specific checkpoints are associated with lipid-coated calcium phosphate (LCP) mRNA vaccine, which indicates a more robust immune response to melanoma growth and metastasis [143]. Overall, the rational development of multiple anticancer therapies, such as the combination of TGF-β inhibitors with checkpoint inhibitors and/or other biological treatments, holds excellent prospects.
TGF-β in pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most aggressive type of gastrointestinal tumor due to its rapid progression and resistance to traditional chemoradiotherapy [144]. Studies have shown that whether TGF-β acts as a tumor suppressor or a tumor promoter depends on the tumor microenvironment [145]. In the early stage of pancreatic cancer, TGF-β promotes apoptosis via ID1 [146], regulates cell cycle progression through G1 arrest [147], and inhibits the growth of epithelial cells. In addition, a decrease in VEGF and an increase in TSP-1 caused by TGF-β help inhibit pancreatic cancer [148]. However, during the advanced stage of PDAC, genetically inactivated TGF-β signaling has a potent growth promotor effect [149, 150]. Of note, TGF-β does not only promote evasion and metastasis in all advanced pancreatic cancer. Overexpressed TGF-β drives tumor suppression in SMAD4-positive PDA cells by repressing KLF5 [151].
More evidence is emerging that at least one mutation in the TGF-β signaling genes (TGFβRI, TGFβRII, Smad2, and Smad4 genes) occurs in all PDAC [152, 153]. SMAD mutation occurs in 60% of pancreatic cancer patients. An increased KRAS mutation and SMAD mutation in PDAC patients drive early tumorigenesis and metastasis. [154,155,156]. The mutated TGF-β signaling pathway has a much stronger ability to inhibit proliferation, promote angiogenesis and immune escape than simply shutting down the TGF-β signaling pathways [157, 158]. SMAD4 deletion leads to up-regulation of the oncogene (PGK1) [159]and down-regulation of the anticancer gene (SMAD4/DPC4). Such regulation promotes cell metastasis [148]. Meanwhile, deregulated TGF-β signaling leads to ECM deposition and immunosuppressive cell infiltration [160,161,162]. This kind of deposition and infiltration accelerates the metastasis of pancreatic cancer cells and rationalizes early PDAC dissemination [163, 164].
TGF-β plays a crucial role in the process and metastasis of PDAC, and therapies targeting TGF-β signaling hold great promise. Several strategies relevant to TGF-β signaling have been investigated in preclinical and clinical researches and have shown efficacy partially [165,166,167]. Therapeutic approaches are always associated with three levels of ligand, ligand-receptor binding, and intracellular transduction to disrupt TGF-β signaling. These approaches contain TβR II antagonists, sequence-targeted antifibrosis nanoparticles, anti-TGF-β recombinant protein, and DC vaccines [168,169,170]. Lipoxin A4 (LXA4), a metabolite derived from arachidonic acid, could significantly inhibit TGF-β signaling in PDAC [171]. Strategies targeting ligand-receptor binding levels, such as TGF-β inhibitors and monoclonal blocking antibodies, also show robust performance against PDAC [172]. TGF-β inhibitors are primarily TβR-targeted and SMADs-associated kinases at signal cell level. The most effective treatment is the combination of TGF-β inhibitors with chemotherapy and other biological agents. For example, vactosertib (activin receptor-like kinase 5 inhibitor) [173] in combination with nal-IRI plus 5-Fluorouracil/Leucovorin improved overall survival rates compared with vactosertib alone [174, 175]. Nanotargeted relaxin, an endogenous hormone, has also been shown to enhance the efficacy of gemcitabine in vivo [176]. Furthermore, the selection of correct dosage form and the establishment of a demonstration drug delivery system are critical for the treatment of desmoplastic tumors. Compared with traditional Chinese medicine dosage forms (decoction and powder), the targeted administration of nano-preparations (α-mangostein and triptolide) can overcome the permeation obstacles in PDAC and improve therapeutic effects. [176].
TGF-β in colorectal cancer
Colorectal cancer (CRC) is the leading cause of death among cancers of the digestive system (101,420 estimated new cases and 51,020 estimated deaths in 2019) [123], the poor prognosis of which is mainly associated with colorectal cancer metastasis and immune evasion. Many studies have indicated that malignant CRC is characterized by high stromal infiltration with innate immune cells, fibroblasts, and TGF-β activation [177]. TGF-β is involved in regulating CRC metastasis, tumor stroma, microenvironment, and immune system resistance.
Colorectal cancer is driven by the accumulation of mutations in APC, KRAS, TβR II, Trp53 [178,179,180,181,182], carcinoembryonic antigen-associated cell adhesion molecules (CEACAM) [183] and R-spondins (RSPOs) [184]. The four primary [185]mutations in intestinal tumors promote CRC metastasis, indicating a negative prognostic effect for recurrence of CRC [186,187,188] and regulating the tumor microenvironment [112, 189]. Despite these mutations demonstrate worse clinical outcomes, they also predict neoantigen-specific immunotherapeutic anti-TGF-β strategies [187].
It has been confirmed that inhibiting TGF-β signaling pathways in the preclinical and clinical treatment of CRC are effective [190]. However, anti-TGF-β therapy alone is insufficient to mediate antitumor immunity in CRC. In contrast, the combination of other biological agents or irradiated tumor vaccine with anti-TGF-β treatment can reduce CRC metastasis. Chemotherapies ginsenoside Rb2 [191] and tanshinone II A [192] showed therapeutic effects on CRC by inhibiting TGF-β-induced EMT and angiogenesis, respectively [193]. Nevertheless, the effect was mild. Monotherapy with galunisertib (LY2157299), an oral small-molecule inhibitor of the TβR I kinase, was also not significant [194]. Coadministration of TGF-β blocking agents and anti-PD-L1 antibodies indicated a dramatic response by promoting CD8 + T cells penetration into the tumor [189].
TGF-β in breast cancer
Along with lung and colon cancer, breast cancer is one of the most common cancers worldwide and is more malignant in females than in males. Although the mortality rates of breast cancer are decreasing in some developed countries, there are approximately 500,000 deaths because of breast cancer every year [195, 196]. Further understandment of the development, progression, and treatment of breast cancer is emergency.
The TGF-β signaling pathway is well known to play a vital role in cancer regulation, and breast cancer is no exception [115, 157]. TGF-β regulates the survival of cancer cells to influence breast cancer progression. On the one hand, TGF-β can induce the anti-apoptotic effects of mouse mammary carcinoma cells through up-regulated chondrocytes 1. Chondrocytes 1 is a basic helix-loop-helix (bHLH) transcription factor, which is tightly related to breast carcinomas [197]. On the other hand, the TGF-β signaling pathway can also disturb the immune system to induce immune evasion. In breast cancer, the lack of TβR III and its shed extracellular domain (sTβR III) will enhance TGF-β signaling within DCs. It finally results in Tregs infiltration and immune suppression [198]. In addition, TGF-β can also transactivate EGFR through the Smad3 and ERK/Sp1 signaling pathways to promote the migration and proliferation of breast cancer cells [199].
Moreover, we should highlight the contribution of TGF-β to breast cancer metastasis because breast cancer can quickly metastasize to the lung, brain, bone, and liver, which is lethal [200]. In addition to breast cancer, TGF-β is also critical in the metastasis of other cancers including bone, and gastric cancer [201,202,203]. TGF-β participates in breast cancer metastasis by up-regulating CXCR4 in monocytes. These attracted and differentiated tumor-associated macrophages (TAMs) assist tumor cell extravasation [204]. Additionally, miR-190 and OTU domain-containing protein 1 are two inhibitors of TGF-β signaling that target SMAD2 and SMAD7, respectively. The expression of SMAD2 and SMAD7 is associated with outcomes in breast cancer patients, for downregulated SMAD2 and SMAD7 promote breast cancer metastasis [205, 206].
The mechanism of TGF-β promoting breast cancer is manifold. Therefore, targeting TGF-β signaling is probably an effective way to treat breast cancer. Artemisinin derivatives, like artesunate (ARS) and dihydroartemisinin (DHA), are effective in suppressing TGF-β signaling and CAF activation. Breast cancer will be in remission because of the reduced interaction between the tumor and tumor microenvironment [207]. In addition, a bispecific receptor decoy containing TGF-β neutralizing the TβR II extracellular domain was designed. This decoy and ibalizumab were intended recently to inhibit TGF-β signaling in TH cells and decrease tumor burden in a breast cancer mouse model [208]. Due to the deficiency of SIRT7 in breast cancer metastasis mice, TGF-β signaling is activated to promote metastasis. It is already clear that resveratrol can activate SIRT7, regulate SMAD4 deacetylation, and most importantly inhibit metastasis [209].
TGF-β in glioma cancer
Glioma is a malignant primary brain tumor divided into four categories, including circumscribed gliomas (WHO grade I) and diffusely infiltrating gliomas (WHO grade II-IV). Diffusely infiltrating gliomas are more malignant than circumscribed gliomas, in which glioblastoma is the most lethal glioma, with a median overall survival of 14–17 months [210, 211].
Among the numerous signaling pathways that play a role in glioma, TGF-β signaling is being noted. The related mechanism and therapeutic strategies have been gradually clarified. It has already been found that the high proliferation and invasion of gliomas and the poor prognosis in glioma patients are usually accompanied by SMAD signaling in early studies, and Sox9 becomes an important regulatory target when TGF-β works in glioma progression [212, 213]. TGF-β plays an essential role in glioma progression by inducing the proliferation, invasion, EMT, and migration of glioma cells and depressing immune effector cells [214,215,216]. Furthermore, three kinds of TGF-β are all related to glioma. In a study of the relationship between TAMs and the progression of tumors, Z. Liu et al. found M2 phenotype TAMs to promote the stemness and migration of glioma cells by secreting TGF-β [217]. In addition, TGF-βII affects autophagy, a vital process connected with tumor growth, promoting glioma cells' invasion through the SMAD and non-SMAD pathways [218, 219]. Among those three isoforms, the expression of TGF-βIII was lower than that of the other two isoforms. However, it has an essential effect on SMAD phosphorylation and tumor invasiveness [220].
Previous studies have shown that overexpressed TGF-β in the glioma is involved in angiogenesis, tissue invasion, and cancer progression. Therapies targeting TGF-β are divided into three levels: TGF-β mRNA translation inhibitors, TGF-β neutralizing antibodies and receptor inhibitors, and regulators of TGF-β signaling pathway downstream factors. In a phase II clinical study (NCT00431561), intratumorally administered AP12009 alone exhibits one-third of the efficacy population [221]. AP12009 is a phosphorothioate antisense oligodeoxynucleotide specific for the mRNA of human TGF-βII [221]. RGFP966, along with an HDAC3 inhibitor, regulated SMAD7 acetylation rather than ubiquitination to promote gastric stump carcinoma (GSC) differentiation [222]. There appeared to be no difference in efficacy between monotherapy of TGF-β antibodies (GC1008, NCT01472731) or small-molecule TβR I inhibitors (LY2157299, NCT01220271), and their combination with chemotherapy (Table 1) [223]. The exploration of appropriate combination therapy is still the mainstream direction.
Clinical applications of TGF-β-targeting therapies
Extensive evidence suggests that targeting TGF-β cascades has the potential to treat patients with fibrosis and cancers. Numerous anti-cancer and anti-fibrosis pharmacological interventions targeting TGF-β have undergone pre-clinical and clinical stages. TGF-β-targeted drugs are mainly divided into neutralizing antibodies, small-molecule TGF-β inhibitors, ligand traps, antisense oligonucleotides, and vaccines (Table 1) [224]. Among all the TGF-β targeting drugs, Fresolimumab (GC1008), Galunisertib (LY2157299), Trabedersen (AP12009), and Vactosertib are the most striking drugs [224,225,226]. Moreover, Trabedersen, a TGF-βII specific phosphonothioate antisense oligodeoxynucleotide, also demonstrated a therapeutic effect on COVID-19 (NCT04801017).
Despite the encouraging potential displayed by TGF-β-targeted drugs in a part of pre-clinical animal studies, the results from subsequent clinical trials of those drugs seem to be disappointing. The application of TGF-β inhibition strategies in patients with fibrosis is challenging due to the systemic effects of TGF-β and the complexity of cancer and fibrosis formation [227]. Firstly, although TGF-β cascades are commonly activated to contribute to pathological processes, the physiological function of TGF-β cannot be ignored. Therefore, the wide defection of TGF-β may lead to the disturbance of normal physiological processes, which should be treated with caution [228]. Secondly, TGF-β modulates a wide range of signaling cascades to promote fibrosis and cancers, which increases the difficulty and complexity of the treatment. Exploring precise downstream TGF-β-activated factors for each disease is necessary. Thirdly, despite the key role of TGF-β in fibrosis and tumorigenesis, the onset and development of the disease is multifactorial. The combinational therapeutic strategies of TGF-β-targeted therapy with other traditional ones should be studied to achieve an ideal effect.
Conclusion
TGF-β plays a vital role from early embryonic development to adult homeostasis. However, dysregulation of TGF-β signaling is significantly associated with tumorigenesis and fibrosis. The exact mechanism is complex and mainly involves TGF-β as a tumor suppressor in premalignant cells and a tumor promoter in carcinoma cells by regulating EMT, ECM accumulation, immune invasion, and CAFs activation. TGF-β overexpression under pathological conditions directly promotes tissue lesions. In addition, TGF-β signaling cascade group mutation accumulation is also closely related to fibrosis and tumorigenesis.
The twenty-first century has witnessed a significant upgrade of precision medicine, among all, targeted therapy as the most promising one. Lots of preclinical researches have demonstrated the efficacy of TGF-β related pharmacological agents. In recent years, there have been various clinical experiments evaluating TGF-β-targeted antibody, small molecular receptor inhibitors, ligand traps, antisense oligonucleotides, and vaccines. Unfortunately, anti-TGF-β approaches achieved subtle efficacy due to the systemic biological effects of TGF-β and the complexity of fibrosis and tumorigenesis. It is known that most cancer patients die of metastasis after chemotherapy or radiotherapy, where the immunosuppressive TGF-β in the TME might be one of the factors. Therefore, the combination therapy of chemotherapy/ radiotherapy/targeted therapy with TGF-β-targeted therapies might be developed to achieve an enhanced antitumor efficacy by regulating tumor microenvironment. In addition, in future researches, researchers should further focus on the optimization of dosing and drug delivery systems in TGF-β-related therapies. Above all, the exploration of comprehensive mechanisms of TGF-β in diseases and the development of TGF-β based combination therapies might be very crucial for combatting fibrosis and cancer in future.
Availability of data and materials
Not applicable.
Abbreviations
- TGF-β:
-
Transforming growth factor β
- BMPs:
-
Bone morphogenetic proteins
- GDFs:
-
Growth and differentiation factors
- ECM:
-
Extracellular matrix
- TβR:
-
TGF-β receptor
- EMT:
-
Epithelial mesenchymal transition
- PD-L1:
-
Programmed cell death 1 ligand 1
- LAP:
-
Latency associated peptide
- LLC:
-
Large latent complex
- LTBP:
-
Latent TGF-β binding proteins
- TSP-1:
-
Thrombospondin 1
- GARP:
-
Glycoprotein a repetitions predominant protein
- SMAD:
-
Drosophila mothers against decapentaplegic
- SLC:
-
Small latent complex
- LTBP:
-
Latent TGF-β binding proteins
- MMP-2:
-
Matrix metalloproteinase-2
- RGD:
-
Arginine-glycine-aspartic acid
- Tregs:
-
Regulatory T cells
- ASCs:
-
Adipose-derived mesenchymal stem cell
- R-SMAD:
-
Receptor complex phosphorylates receptor-SMAD
- LMO7:
-
LIM domain only 7
- IL-2:
-
Interleukin-2
- CDK:
-
Cyclin-dependent kinase
- HK2:
-
Hexokinase2
- MAPK:
-
Mitogen-activated protein kinase
- Erk:
-
Extracellular signal regulated kinases
- PI3K:
-
Phosphatidylinositol-3-kinase
- JNK:
-
C-Jun amino terminal kinase
- CAFs:
-
Cancer-associated fibroblasts
- TIMP:
-
Tissue inhibitor of metalloproteinases
- α-SMA:
-
α-Smooth muscle actin
- LOXL1:
-
Lysyl oxidase-like 1
- HSCs:
-
Hepatic stellate cells
- MFBs:
-
Myofibroblasts
- HCC:
-
Hepatocellular carcinoma
- MFBs:
-
Myofibroblasts
- CTGF:
-
Connective tissue growth factor
- KCs:
-
Kupffer cells
- CKD:
-
Chronic kidney diseases
- PAI-1:
-
Plasminogen activator inhibitor-1
- UUO:
-
Unilateral ureteral obstruction
- HGF:
-
Hepatocyte growth factor
- RBP-Jκ:
-
Recombination signal binding protein-Jκ
- miRNAs:
-
MicroRNAs
- PTHrP:
-
Parathyroid hormone-related protein
- EGF:
-
Epidermal growth factor
- VEGF:
-
Vascular endothelial growth factor
- IPF:
-
Idiopathic pulmonary fibrosis
- AECs:
-
Alveolar epithelial cells
- TNF-α:
-
Tumor necrosis factor-α
- MUC1:
-
Mucin 1
- DNM3OS:
-
Dynamin 3 opposite strand
- PDAC:
-
Pancreatic ductal adenocarcinoma
- GC:
-
Gastric cancer
- DCs:
-
Dendritic cells
- VEPH1:
-
Ventricular zone expressed PH domain-containing 1
- USF1:
-
Upstream transcription factor 1
- CM:
-
Conditioned medium
- GZMB:
-
Granzyme B
- SOX10:
-
Sex determining region Y-box 10
- LCP:
-
Lipid-coated calcium phosphate
- PDAC:
-
Pancreatic ductal adenocarcinoma
- LXA4:
-
Lipoxin A4
- CRC:
-
Colorectal cancer
- CEACAM:
-
Carcinoembryonic antigen-related cell adhesion molecule
- RSPOs:
-
R-spondins
- bHLH:
-
Basic helix-loop-helix
- sTβR III:
-
Shed extracellular domain
- TAMs:
-
Tumor-associated macrophages
- ARS:
-
Artesunate
- DHA:
-
Dihydroartemisinin
- GSC:
-
Gastric stump carcinoma
References
Saito A, Horie M, Nagase T: TGF-beta Signaling in Lung Health and Disease. Int J Mol Sci 2018, 19.
David CJ, Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018;19:419–35.
Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, Cao X. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res. 2018;6:2.
Minton K. Extracellular matrix: Preconditioning the ECM for fibrosis. Nat Rev Mol Cell Biol. 2014;15:766–7.
Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, Basnet H, Zou Y, Shu W, Soni RK, et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. 2020;577:566–71.
Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD: TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nature Communications 2018, 9.
Liu S, Ren J, Ten Dijke P. Targeting TGFbeta signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6:8.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–38.
Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, Rifkin DB. TGF-beta latency: biological significance and mechanisms of activation. Stem Cells. 1997;15:190–7.
Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32:51–82.
Munger JS, Harpel JG, Gleizes P-E, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-β: Structural features and mechanisms of activation. Kidney Int. 1997;51:1376–82.
Cheifetz S, Weatherbee JA, Tsang ML, Anderson JK, Mole JE, Lucas R, Massagué J. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell. 1987;48:409–15.
Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol. 2015;47:44–53.
Rifkin D, Sachan N, Singh K, Sauber E, Tellides G, Ramirez F: The role of LTBPs in TGF beta signaling. Dev Dyn 2021.
Zigrino P, Sengle G. Fibrillin microfibrils and proteases, key integrators of fibrotic pathways. Adv Drug Deliv Rev. 2019;146:3–16.
Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-beta structure and activation. Nature. 2011;474:343–9.
Farhat YM, Al-Maliki AA, Easa A, O’Keefe RJ, Schwarz EM, Awad HA. TGF-beta1 Suppresses Plasmin and MMP Activity in Flexor Tendon Cells via PAI-1: Implications for Scarless Flexor Tendon Repair. J Cell Physiol. 2015;230:318–26.
Miyazono K, Heldin CH. Role for carbohydrate structures in TGF-beta 1 latency. Nature. 1989;338:158–60.
Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–50.
Li M, Qin XY, Furutani Y, Inoue I, Sekihara S, Kagechika H, Kojima S. Prevention of acute liver injury by suppressing plasma kallikrein-dependent activation of latent TGF-beta. Biochem Biophys Res Commun. 2018;504:857–64.
Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–11.
Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet. 2003;33:331–2.
Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Couroussé T, Sakai LY, Rifkin DB. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. 2012;227:3828–36.
Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113:410–8.
Jovanović J, Iqbal S, Jensen S, Mardon H, Handford P. Fibrillin-integrin interactions in health and disease. Biochem Soc Trans. 2008;36:257–62.
Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, Dietz HC. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503:126–30.
Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, Chillakuri CR, Macaya D, Coucke PJ, De Paepe A, et al: Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med 2010, 2:23ra20.
Ramirez F, Sakai LY. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339:71–82.
van Loon K, Yemelyanenko-Lyalenko J, Margadant C, Griffioen AW, Huijbers EJM: Role of fibrillin-2 in the control of TGF-beta activation in tumor angiogenesis and connective tissue disorders. Biochim Biophys Acta Rev Cancer 2020, 1873:188354.
Kumra H, Nelea V, Hakami H, Pagliuzza A, Djokic J, Xu J, Yanagisawa H, Reinhardt DP. Fibulin-4 exerts a dual role in LTBP-4L-mediated matrix assembly and function. Proc Natl Acad Sci U S A. 2019;116:20428–37.
Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM. Regulation of the expression of GARP/latent TGF-beta1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J Immunol. 2013;190:5506–15.
Stockis J, Dedobbeleer O, Lucas S. Role of GARP in the activation of latent TGF-beta1. Mol Biosyst. 2017;13:1925–35.
Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24.
Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, Marks JD, Baron JL, Cheng Y, Nishimura SL. Cryo-EM Reveals Integrin-Mediated TGF-beta Activation without Release from Latent TGF-beta. Cell. 2020;180:490-501 e416.
Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007;117:3551–62.
Cuende J, Liénart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, Huygens C, Colau D, Somja J, Delvenne P, et al: Monoclonal antibodies against GARP/TGF-β1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med 2015;7:284ra256.
Del Cid JS, Reed NI, Molnar K, Liu S, Dang B, Jensen SA, DeGrado W, Handford PA, Sheppard D, Sundaram AB. A disease-associated mutation in fibrillin-1 differentially regulates integrin-mediated cell adhesion. J Biol Chem. 2019;294:18232–43.
Nieberler M, Reuning U, Reichart F, Notni J, Wester HJ, Schwaiger M, Weinmüller M, Räder A, Steiger K, Kessler H: Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers (Basel) 2017, 9.
Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-β Pathway. Cell Rep. 2017;18:2480–93.
Olof Olsson P, Gustafsson R, Salnikov AV, Göthe M, Zeller KS, Friman T, Baldetorp B, Koopman LA, Weinreb PH, Violette SM, et al. Inhibition of integrin α(V)β(6) changes fibril thickness of stromal collagen in experimental carcinomas. Cell Commun Signal. 2018;16:36.
Hirota S, Clements TP, Tang LK, Morales JE, Lee HS, Oh SP, Rivera GM, Wagner DS, McCarty JH. Neuropilin 1 balances β8 integrin-activated TGFβ signaling to control sprouting angiogenesis in the brain. Development. 2015;142:4363–73.
Franco-Barraza J, Francescone R, Luong T, Shah N, Madhani R, Cukierman G, Dulaimi E, Devarajan K, Egleston BL, Nicolas E, et al: Matrix-regulated integrin α(v)β(5) maintains α(5)β(1)-dependent desmoplastic traits prognostic of neoplastic recurrence. Elife 2017, 6.
Breuss JM, Gallo J, DeLisser HM, Klimanskaya IV, Folkesson HG, Pittet JF, Nishimura SL, Aldape K, Landers DV, Carpenter W, et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci. 1995;108(Pt 6):2241–51.
Xia W, Springer TA. Metal ion and ligand binding of integrin α5β1. Proc Natl Acad Sci U S A. 2014;111:17863–8.
Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA. Force interacts with macromolecular structure in activation of TGF-beta. Nature. 2017;542:55–9.
Fan W, Liu T, Chen W, Hammad S, Longerich T, Hausser I, Fu Y, Li N, He Y, Liu C, et al: ECM1 Prevents Activation of Transforming Growth Factor beta, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology 2019;157:1352–1367 e1313.
Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, Marks JD, Baron JL, Cheng Y, Nishimura SL. Cryo-EM Reveals Integrin-Mediated TGF-β Activation without Release from Latent TGF-β. Cell. 2020;180:490-501.e416.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF- family signalling. Nature. 2003;425:577–84.
Xu P, Liu J, Derynck R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012;586:1871–84.
Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30.
Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, Habib N, Raman P, Plow E, Stenina-Adognravi O. Thrombospondin-4 mediates TGF-beta-induced angiogenesis. Oncogene. 2017;36:5189–98.
Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–84.
Derynck R, Zhang Y. Intracellular signalling: The Mad way to do it. Curr Biol. 1996;6:1226–9.
Bachegowda L, Gligich O, Mantzaris I, Schinke C, Wyville D, Carrillo T, Braunschweig I, Steidl U, Verma A. Signal transduction inhibitors in treatment of myelodysplastic syndromes. J Hematol Oncol. 2013;6:50.
Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–39.
Galvão F Jr, Grokoski KC, da Silva BB, Lamers ML, Siqueira IR. The amyloid precursor protein (APP) processing as a biological link between Alzheimer’s disease and cancer. Ageing Res Rev. 2019;49:83–91.
Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, Rosen B, Rodriguez BA, Crawford B, Swaminathan R, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2018;23:1051–65.
Muthusamy BP, Budi EH, Katsuno Y, Lee MK, Smith SM, Mirza AM, Akhurst RJ, Derynck R. ShcA Protects against Epithelial-Mesenchymal Transition through Compartmentalized Inhibition of TGF-beta-Induced Smad Activation. PLoS Biol. 2015;13:e1002325.
Johnson HE, Toettcher JE. Signaling Dynamics Control Cell Fate in the Early Drosophila Embryo. Dev Cell. 2019;48:361-370 e363.
Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, Lengyel E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012;2:1100–8.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9:282–301.
Yoshida GJ. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res. 2020;39:112.
Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593:575–9.
Yu L, Border WA, Huang Y, Noble NA. TGF-beta isoforms in renal fibrogenesis. Kidney Int. 2003;64:844–56.
Kolliopoulos C, Raja E, Razmara M, Heldin P, Heldin CH, Moustakas A, van der Heide LP. Transforming growth factor beta (TGFbeta) induces NUAK kinase expression to fine-tune its signaling output. J Biol Chem. 2019;294:4119–36.
Modi SJ, Kulkarni VM. Discovery of VEGFR-2 inhibitors exerting significant anticancer activity against CD44+ and CD133+ cancer stem cells (CSCs): Reversal of TGF-beta induced epithelial-mesenchymal transition (EMT) in hepatocellular carcinoma. Eur J Med Chem. 2020;207:112851.
Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, et al. Blockade of TGF-β inhibits mammary tumor cell viability, migration, and metastases. J Clin Investig. 2002;109:1551–9.
Schuliga M, Grainge C, Westall G, Knight D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int J Biochem Cell Biol. 2018;97:108–17.
Wanninger J, Neumeier M, Hellerbrand C, Schacherer D, Bauer S, Weiss TS, Huber H, Schaffler A, Aslanidis C, Scholmerich J, Buechler C. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim Biophys Acta. 2011;1811:626–33.
Yamazaki K, Masugi Y, Sakamoto M. Molecular pathogenesis of hepatocellular carcinoma: altering transforming growth factor-beta signaling in hepatocarcinogenesis. Dig Dis. 2011;29:284–8.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411.
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72.
Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S. TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006;44:57–66.
Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–24.
Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN: TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8.
Zhang Z, Mu Y, Zhang J, Zhou Y, Cattaneo P, Veevers J, Peter AK, Manso AM, Knowlton KU, Zhou X, et al. Kindlin-2 Is Essential for Preserving Integrity of the Developing Heart and Preventing Ventricular Rupture. Circulation. 2019;139:1554–6.
Yu J, Hu Y, Gao Y, Li Q, Zeng Z, Li Y, Chen H. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell Death Discovery. 2018;4:93.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma Nat Rev Dis Primers. 2021;7:6.
Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, Hourmand IO, Cleverly A, Zhao Y, Gueorguieva I, et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. 2019;10:e00056.
Humphreys BD. Mechanisms of Renal Fibrosis. Annu Rev Physiol. 2018;80:309–26.
Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83.
Yanagita M. Inhibitors/antagonists of TGF-beta system in kidney fibrosis. Nephrol Dial Transplant. 2012;27:3686–91.
Isaka Y: Targeting TGF-beta Signaling in Kidney Fibrosis. Int J Mol Sci 2018, 19.
Border WA, Okuda S, Languino LR, Ruoslahti E. Transforming growth factor-beta regulates production of proteoglycans by mesangial cells. Kidney Int. 1990;37:689–95.
Decleves AE, Sharma K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol. 2014;10:257–67.
Kölling M, Kaucsar T, Schauerte C, Hübner A, Dettling A, Park JK, Busch M, Wulff X, Meier M, Scherf K, et al. Therapeutic miR-21 Silencing Ameliorates Diabetic Kidney Disease in Mice. Mol Ther. 2017;25:165–80.
Lee SY, Kim SI, Choi ME. Therapeutic targets for treating fibrotic kidney diseases. Transl Res. 2015;165:512–30.
Jiang Y, Wang Y, Ma P, An D, Zhao J, Liang S, Ye Y, Lu Y, Zhang P, Liu X, et al. Myeloid-specific targeting of Notch ameliorates murine renal fibrosis via reduced infiltration and activation of bone marrow-derived macrophage. Protein Cell. 2019;10:196–210.
Zhao Y, Yin Z, Li H, Fan J, Yang S, Chen C, Wang DW. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell. 2017;16:387–400.
Sun SF, Tang PMK, Feng M, Xiao J, Huang XR, Li P, Ma RCW, Lan HY. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes. 2018;67:731–44.
Lederer DJ, Longo DL, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378:1811–23.
REN Y, JIAN X, ZHANG Z, NING Q, KAN B, KONG L: Effects of tacrolimus on the TGF‐β1/SMAD signaling pathway in paraquat‐exposed rat alveolar type II epithelial cells. MOLECULAR MEDICINE REPORTS 2019.
Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52.
Choudhury M, Yin X, Schaefbauer KJ, Kang JH, Roy B, Kottom TJ, Limper AH, Leof EB. SIRT7-mediated modulation of glutaminase 1 regulates TGF-β-induced pulmonary fibrosis. Faseb j. 2020;34:8920–40.
Kimura M, Hashimoto N, Kusunose M, Aoyama D, Sakamoto K, Miyazaki S, Ando A, Omote N, Imaizumi K, Kawabe T, Hasegawa Y. Exogenous induction of unphosphorylated PTEN reduces TGFbeta-induced extracellular matrix expressions in lung fibroblasts. Wound Repair Regen. 2017;25:86–97.
Saito A, Suzuki HI, Horie M, Ohshima M, Morishita Y, Abiko Y, Nagase T. An integrated expression profiling reveals target genes of TGF-β and TNF-α possibly mediated by microRNAs in lung cancer cells. PLoS One. 2013;8:e56587.
Wettlaufer SH, Scott JP, McEachin RC, Peters-Golden M, Huang SK. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am J Respir Cell Mol Biol. 2016;54:114–27.
Milara J, Ballester B, Montero P, Escriva J, Artigues E, Alós M, Pastor-Clerigues A, Morcillo E, Cortijo J. MUC1 intracellular bioactivation mediates lung fibrosis. Thorax. 2020;75:132–42.
Colak S, Ten Dijke P. Targeting TGF-beta Signaling in Cancer. Trends Cancer. 2017;3:56–71.
Coker RK, Laurent GJ, Jeffery PK, du Bois RM, Black CM, McAnulty RJ. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax. 2001;56:549–56.
Savary G, Dewaeles E, Diazzi S, Buscot M, Nottet N, Fassy J, Courcot E, Henaoui IS, Lemaire J, Martis N, et al. The Long Noncoding RNA DNM3OS Is a Reservoir of FibromiRs with Major Functions in Lung Fibroblast Response to TGF-β and Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;200:184–98.
Pachera E, Assassi S, Salazar GA, Stellato M, Renoux F, Wunderlin A, Blyszczuk P, Lafyatis R, Kurreeman F, de Vries-Bouwstra J, et al. Long noncoding RNA H19X is a key mediator of TGF-β-driven fibrosis. J Clin Invest. 2020;130:4888–905.
Jiang D, Liang J. A Long Noncoding RNA links TGF-β Signaling in Lung Fibrosis. Am J Respir Crit Care Med. 2019;200:123–5.
Zhang C, Hao Y, Wang Y, Xu J, Teng Y, Yang X. TGF-beta/SMAD4-Regulated LncRNA-LINP1 Inhibits Epithelial-Mesenchymal Transition in Lung Cancer. Int J Biol Sci. 2018;14:1715–23.
Moon H, Han KH, Ro SW. Pro-tumorigenic roles of TGF-β signaling during the early stages of liver tumorigenesis through upregulation of Snail. BMB Rep. 2017;50:599–600.
Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66.
Chen CR, Kang Y, Siegel PM, Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19–32.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massague J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell. 2016;165:45–60.
Korkut A, Zaidi S, Kanchi RS, Rao S, Gough NR, Schultz A, Li X, Lorenzi PL, Berger AC, Robertson G, et al. A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-β Superfamily. Cell Syst. 2018;7:422-437.e427.
Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93.
Pei Y, Chen L, Huang Y, Wang J, Feng J, Xu M, Chen Y, Song Q, Jiang G, Gu X, et al. Sequential Targeting TGF-beta Signaling and KRAS Mutation Increases Therapeutic Efficacy in Pancreatic Cancer. Small. 2019;15:e1900631.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–43.
Son HK, Kim D, Lim Y, Kim J, Park I. A novel TGF-β receptor II mutation (I227T/N236D) promotes aggressive phenotype of oral squamous cell carcinoma via enhanced EGFR signaling. BMC Cancer. 2020;20:1163.
Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, Piccolo S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell. 2005;121:87–99.
Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity. 2019;50:924–40.
Zhang Q, Xiao M, Gu S, Xu Y, Liu T, Li H, Yu Y, Qin L, Zhu Y, Chen F, et al. ALK phosphorylates SMAD4 on tyrosine to disable TGF-β tumour suppressor functions. Nat Cell Biol. 2019;21:179–89.
Seoane J, Gomis RR: TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol 2017, 9.
Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. 2014;32:51–82.
Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, White AJ, Gilfillan S, Cella M, Colonna M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol. 2017;18:995–1003.
Sanjabi S, Oh SA, Li MO: Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol 2017, 9.
Wang Y, Chu J, Yi P, Dong W, Saultz J, Wang Y, Wang H, Scoville S, Zhang J, Wu LC, et al. SMAD4 promotes TGF-β-independent NK cell homeostasis and maturation and antitumor immunity. J Clin Invest. 2018;128:5123–36.
Frey B, Rückert M, Deloch L, Rühle PF, Derer A, Fietkau R, Gaipl US. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol Rev. 2017;280:231–48.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21:123–32.
Liu EM, Martinez-Fundichely A, Diaz BJ, Aronson B, Cuykendall T, MacKay M, Dhingra P, Wong EWP, Chi P, Apostolou E, et al: Identification of Cancer Drivers at CTCF Insulators in 1,962 Whole Genomes. Cell Syst 2019, 8:446–455.e448.
Lauden L, Siewiera J, Boukouaci W, Ramgolam K, Mourah S, Lebbe C, Charron D, Aoudjit F, Jabrane-Ferrat N, Al-Daccak R. TGF-beta-induced (TGFBI) protein in melanoma: a signature of high metastatic potential. J Invest Dermatol. 2014;134:1675–85.
Ren YQ, Li QH, Liu LB. USF1 prompt melanoma through upregulating TGF-β signaling pathway. Eur Rev Med Pharmacol Sci. 2016;20:3592–8.
Javelaud D, van Kempen L, Alexaki VI, Le Scolan E, Luo K, Mauviel A. Efficient TGF-beta/SMAD signaling in human melanoma cells associated with high c-SKI/SnoN expression. Mol Cancer. 2011;10:2.
Golan T, Parikh R, Jacob E, Vaknine H, Zemser-Werner V, Hershkovitz D, Malcov H, Leibou S, Reichman H, Sheinboim D, et al: Adipocytes sensitize melanoma cells to environmental TGF-β cues by repressing the expression of miR-211. Sci Signal 2019, 12.
Feng H, Jia XM, Gao NN, Tang H, Huang W, Ning N. Overexpressed VEPH1 inhibits epithelial-mesenchymal transition, invasion, and migration of human cutaneous melanoma cells through inactivating the TGF-β signaling pathway. Cell Cycle. 2019;18:2860–75.
Yang Z, Qi Y, Lai N, Zhang J, Chen Z, Liu M, Zhang W, Luo R, Kang S. Notch1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-β1. J Exp Clin Cancer Res. 2018;37:1.
Eriksson E, Milenova I, Wenthe J, Moreno R, Alemany R, Loskog A. IL-6 Signaling Blockade during CD40-Mediated Immune Activation Favors Antitumor Factors by Reducing TGF-β, Collagen Type I, and PD-L1/PD-1. J Immunol. 2019;202:787–98.
Jenkins MH, Croteau W, Mullins DW, Brinckerhoff CE. The BRAF(V600E) inhibitor, PLX4032, increases type I collagen synthesis in melanoma cells. Matrix Biol. 2015;48:66–77.
Cantelli G, Orgaz JL, Rodriguez-Hernandez I, Karagiannis P, Maiques O, Matias-Guiu X, Nestle FO, Marti RM, Karagiannis SN, Sanz-Moreno V. TGF-β-Induced Transcription Sustains Amoeboid Melanoma Migration and Dissemination. Curr Biol. 2015;25:2899–914.
Shathasivam P, Kollara A, Ringuette MJ, Virtanen C, Wrana JL, Brown TJ. Human ortholog of Drosophila Melted impedes SMAD2 release from TGF-β receptor I to inhibit TGF-β signaling. Proc Natl Acad Sci U S A. 2015;112:E3000-3009.
Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, Wang G, Ma R, Li H, Zhang Y, et al. CD73 expression on effector T cells sustained by TGF-β facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat Commun. 2019;10:150.
Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118–22.
Li S, Song Y, Quach C, Guo H, Jang GB, Maazi H, Zhao S, Sands NA, Liu Q, In GK, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. 2019;10:1693.
Conway JR, Dietlein F, Taylor-Weiner A, AlDubayan S, Vokes N, Keenan T, Reardon B, He MX, Margolis CA, Weirather JL, et al. Integrated molecular drivers coordinate biological and clinical states in melanoma. Nat Genet. 2020;52:1373–83.
Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173:371-385 e318.
Guo X, Xu Y, Zhao Z. In-depth genomic data analyses revealed complex transcriptional and epigenetic dysregulations of BRAFV600E in melanoma. Mol Cancer. 2015;14:60.
Jiang J, Zhang Y, Peng K, Wang Q, Hong X, Li H, Fan G, Zhang Z, Gong T, Sun X. Combined delivery of a TGF-β inhibitor and an adenoviral vector expressing interleukin-12 potentiates cancer immunotherapy. Acta Biomater. 2017;61:114–23.
Wang Y, Zhang L, Xu Z, Miao L, Huang L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol Ther. 2018;26:420–34.
Parikh PY, Lillemoe KD. Surgical management of pancreatic cancer–distal pancreatectomy. Semin Oncol. 2015;42:110–22.
Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-β and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. 2017;15:19.
Huang YH, Hu J, Chen F, Lecomte N, Basnet H, David CJ, Witkin MD, Allen PJ, Leach SD, Hollmann TJ, et al. ID1 Mediates Escape from TGFβ Tumor Suppression in Pancreatic Cancer. Cancer Discov. 2020;10:142–57.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Schwarte-Waldhoff I, Volpert OV, Bouck NP, Sipos B, Hahn SA, Klein-Scory S, Lüttges J, Klöppel G, Graeven U, Eilert-Micus C, et al. Smad4/DPC4-mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:9624–9.
Lee JH, Mellado-Gil JM, Bahn YJ, Pathy SM, Zhang YE, Rane SG. Protection from β-cell apoptosis by inhibition of TGF-β/Smad3 signaling. Cell Death Dis. 2020;11:184.
Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grüssel S, Sipos B, Grützmann R, Pilarsky C, Ungefroren H, Saeger HD, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res. 2007;67:8344–50.
David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, Massague J. TGF-beta tumor suppression through a lethal EMT. Cell. 2016;164:1015–30.
Zhang Q, Xiao M, Gu S, Xu Y, Liu T, Li H, Yu Y, Qin L, Zhu Y, Chen F, et al. Author Crorrection: ALK phosphorylates SMAD4 on tyrosine to disable TGF-β tumour suppressor functions. Nat Cell Biol. 2019;23:179–89.
Riggins GJ, Kinzler KW, Vogelstein B, Thiagalingam S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997;57:2578–80.
Hahn SA, Hoque AT, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE: Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996, 56:490–494.
Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–52.
Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Öllinger R, Zwiebel M, Strong A, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554:62–8.
Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–24.
Gabitova-Cornell L, Surumbayeva A, Peri S, Franco-Barraza J, Restifo D, Weitz N, Ogier C, Goldman AR, Hartman TR, Francescone R, et al. Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell. 2020;38:567-583.e511.
Liang C, Shi S, Qin Y, Meng Q, Hua J, Hu Q, Ji S, Zhang B, Xu J, Yu XJ. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer. Gut. 2020;69:888–900.
Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, Halbrook CJ, Menjivar RE, Kemp SB, Sirihorachai VR, et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020;10:422–39.
Cave DD, Di Guida M, Costa V, Sevillano M, Ferrante L, Heeschen C, Corona M, Cucciardi A, Lonardo E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene. 2020;39:4271–85.
Hirai T, Yang Y, Zenke Y, Li H, Chaudhri VK, De La Cruz Diaz JS, Zhou PY, Nguyen BA, Bartholin L, Workman CJ, et al. Competition for Active TGFβ Cytokine Allows for Selective Retention of Antigen-Specific Tissue- Resident Memory T Cells in the Epidermal Niche. Immunity. 2021;54:84-98.e85.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23.
La Porta CAM, Zapperi S. Complexity in cancer stem cells and tumor evolution: Toward precision medicine. Semin Cancer Biol. 2017;44:3–9.
Hurtado de Mendoza T, Mose ES, Botta GP, Braun GB, Kotamraju VR, French RP, Suzuki K, Miyamura N, Teesalu T, Ruoslahti E, et al: Tumor-penetrating therapy for β5 integrin-rich pancreas cancer. Nat Commun 2021, 12:1541.
Han H, Hou Y, Chen X, Zhang P, Kang M, Jin Q, Ji J, Gao M. Metformin-Induced Stromal Depletion to Enhance the Penetration of Gemcitabine-Loaded Magnetic Nanoparticles for Pancreatic Cancer Targeted Therapy. J Am Chem Soc. 2020;142:4944–54.
Feng J, Xu M, Wang J, Zhou S, Liu Y, Liu S, Huang Y, Chen Y, Chen L, Song Q, et al. Sequential delivery of nanoformulated α-mangostin and triptolide overcomes permeation obstacles and improves therapeutic effects in pancreatic cancer. Biomaterials. 2020;241:119907.
Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, Lamping E, Marté JL, Donahue RN, Grenga I, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin Cancer Res. 2018;24:1287–95.
Pei Y, Chen L, Huang Y, Wang J, Feng J, Xu M, Chen Y, Song Q, Jiang G, Gu X, et al. Sequential Targeting TGF-β Signaling and KRAS Mutation Increases Therapeutic Efficacy in Pancreatic Cancer. Small. 2019;15:e1900631.
Ito Z, Kan S, Bito T, Horiuchi S, Akasu T, Yoshida S, Kajihara M, Hokari A, Saruta M, Yoshida N, et al. Predicted Markers of Overall Survival in Pancreatic Cancer Patients Receiving Dendritic Cell Vaccinations Targeting WT1. Oncology. 2019;97:135–48.
Zong L, Chen K, Jiang Z, Chen X, Sun L, Ma J, Zhou C, Xu Q, Duan W, Han L, et al. Lipoxin A4 reverses mesenchymal phenotypes to attenuate invasion and metastasis via the inhibition of autocrine TGF-β1 signaling in pancreatic cancer. J Exp Clin Cancer Res. 2017;36:181.
Murakami T, Hiroshima Y, Miyake K, Hwang HK, Kiyuna T, DeLong JC, Lwin TM, Matsuyama R, Mori R, Kumamoto T, et al. Color-coded intravital imaging demonstrates a transforming growth factor-β (TGF-β) antagonist selectively targets stromal cells in a human pancreatic-cancer orthotopic mouse model. Cell Cycle. 2017;16:1008–14.
Song KM, Chung DY, Choi MJ, Ghatak K, Minh NN, Limanjaya A, Kwon MH, Ock J, Yin GN, Kim DK, et al. Vactosertib, a Novel, Orally Bioavailable Activin Receptor-Like Kinase 5 Inhibitor, Promotes Regression of Fibrotic Plaques in a Rat Model of Peyronie’s Disease. World J Mens Health. 2020;38:552–63.
Bartscht T, Rosien B, Rades D, Kaufmann R, Biersack H, Lehnert H, Gieseler F, Ungefroren H. Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: implications for in vivo mode of action. Mol Cancer. 2015;14:199.
Hong E, Park S, Ooshima A, Hong CP, Park J, Heo JS, Lee S, An H, Kang JM, Park SH, et al. Inhibition of TGF-β signalling in combination with nal-IRI plus 5-Fluorouracil/Leucovorin suppresses invasion and prolongs survival in pancreatic tumour mouse models. Sci Rep. 2020;10:2935.
Mardhian DF, Storm G, Bansal R, Prakash J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J Control Release. 2018;290:1–10.
Ruland J. Colon Cancer: Epithelial Notch Signaling Recruits Neutrophils to Drive Metastasis. Cancer Cell. 2019;36:213–4.
Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004.
Levéen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjöstrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–8.
Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8.
Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–21.
Shen X, Hu X, Mao J, Wu Y, Liu H, Shen J, Yu J, Chen W. The long noncoding RNA TUG1 is required for TGF-β/TWIST1/EMT-mediated metastasis in colorectal cancer cells. Cell Death Dis. 2020;11:65.
Gu S, Zaidi S, Hassan MI, Mohammad T, Malta TM, Noushmehr H, Nguyen B, Crandall KA, Srivastav J, Obias V, et al. Mutated CEACAMs Disrupt Transforming Growth Factor Beta Signaling and Alter the Intestinal Microbiome to Promote Colorectal Carcinogenesis. Gastroenterology. 2020;158:238–52.
Lähde M, Heino S, Högström J, Kaijalainen S, Anisimov A, Flanagan D, Kallio P, Leppänen VM, Ristimäki A, Ritvos O, et al. Expression of R-Spondin 1 in Apc(Min/+) Mice Suppresses Growth of Intestinal Adenomas by Altering Wnt and Transforming Growth Factor Beta Signaling. Gastroenterology. 2021;160:245–59.
Sakai E, Nakayama M, Oshima H, Kouyama Y, Niida A, Fujii S, Ochiai A, Nakayama KI, Mimori K, Suzuki Y, et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2018;78:1334–46.
Nakayama M, Oshima M. Mutant p53 in colon cancer. J Mol Cell Biol. 2019;11:267–76.
van den Bulk J, Verdegaal EME, Ruano D, Ijsselsteijn ME, Visser M, van der Breggen R, Duhen T, van der Ploeg M, de Vries NL, Oosting J, et al. Neoantigen-specific immunity in low mutation burden colorectal cancers of the consensus molecular subtype 4. Genome Med. 2019;11:87.
Boutin AT, Liao WT, Wang M, Hwang SS, Karpinets TV, Cheung H, Chu GC, Jiang S, Hu J, Chang K, et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017;31:370–82.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Villalba M, Evans SR, Vidal-Vanaclocha F, Calvo A. Role of TGF-β in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370:29–39.
Dai G, Sun B, Gong T, Pan Z, Meng Q, Ju W. Ginsenoside Rb2 inhibits epithelial-mesenchymal transition of colorectal cancer cells by suppressing TGF-β/Smad signaling. Phytomedicine. 2019;56:126–35.
Sui H, Zhao J, Zhou L, Wen H, Deng W, Li C, Ji Q, Liu X, Feng Y, Chai N, et al. Tanshinone IIA inhibits β-catenin/VEGF-mediated angiogenesis by targeting TGF-β1 in normoxic and HIF-1α in hypoxic microenvironments in human colorectal cancer. Cancer Lett. 2017;403:86–97.
Xu J, Shao T, Song M, Xie Y, Zhou J, Yin J, Ding N, Zou H, Li Y, Zhang J. MIR22HG acts as a tumor suppressor via TGFbeta/SMAD signaling and facilitates immunotherapy in colorectal cancer. Mol Cancer. 2020;19:51.
Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba SC, Benhadji KA, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–99.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-386.
Harbeck N, Gnant M. Breast cancer. The Lancet. 2017;389:1134–50.
Ehata S, Hanyu A, Hayashi M, Aburatani H, Kato Y, Fujime M, Saitoh M, Miyazawa K, Imamura T, Miyazono K. Transforming growth factor-beta promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res. 2007;67:9694–703.
Hanks BA, Holtzhausen A, Evans KS, Jamieson R, Gimpel P, Campbell OM, Hector-Greene M, Sun L, Tewari A, George A, et al. Type III TGF-beta receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest. 2013;123:3925–40.
Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, Zhang F, Niu R. TGF-β transactivates EGFR and facilitates breast cancer migration and invasion through canonical Smad3 and ERK/Sp1 signaling pathways. Mol Oncol. 2018;12:305–21.
Medeiros B, Allan AL: Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int J Mol Sci 2019, 20.
Xia X, Zhang Z, Zhu C, Ni B, Wang S, Yang S, Yu F, Zhao E, Li Q, Zhao G. Neutrophil extracellular traps promote metastasis in gastric cancer patients with postoperative abdominal infectious complications. Nat Commun. 2022;13:1017.
Xi X, Hu Z, Wu Q, Hu K, Cao Z, Zhou J, Liao J, Zhang Z, Hu Y, Zhong X, Bao Y. High expression of small nucleolar RNA host gene 3 predicts poor prognosis and promotes bone metastasis in prostate cancer by activating transforming growth factor-beta signaling. Bioengineered. 2022;13:1895–907.
Li Q, Chen JX, Wu Y, Lv LL, Ying HF, Zhu WH, Xu JY, Ruan M, Guo Y, Zhu WR, Zheng L. The mechanism of FZXJJZ decoction suppresses colorectal liver metastasis via the VDR/TGF-β/Snail1 signaling pathways based on network pharmacology-TCGA data-transcriptomics analysis. J Ethnopharmacol. 2022;287:114904.
Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, Condeelis JS. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018;23:1239–48.
Yu Y, Luo W, Yang ZJ, Chi JR, Li YR, Ding Y, Ge J, Wang X, Cao XC. miR-190 suppresses breast cancer metastasis by regulation of TGF-β-induced epithelial-mesenchymal transition. Mol Cancer. 2018;17:70.
Zhang Z, Fan Y, Xie F, Zhou H, Jin K, Shao L, Shi W, Fang P, Yang B, van Dam H, et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat Commun. 2017;8:2116.
Yao Y, Guo Q, Cao Y, Qiu Y, Tan R, Yu Z, Zhou Y, Lu N. Artemisinin derivatives inactivate cancer-associated fibroblasts through suppressing TGF-β signaling in breast cancer. J Exp Clin Cancer Res. 2018;37:282.
Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, Shi W, Zhang X, Li P, Gao S, et al. Cancer immunotherapy via targeted TGF-beta signalling blockade in TH cells. Nature. 2020;587:121–5.
Tang X, Shi L, Xie N, Liu Z, Qian M, Meng F, Xu Q, Zhou M, Cao X, Zhu WG, Liu B. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat Commun. 2017;8:318.
Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. The Lancet. 2018;392:432–46.
Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019;15:405–17.
Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, Seoane J. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–60.
Chao M, Liu N, Sun Z, Jiang Y, Jiang T, Xv M, Jia L, Tu Y, Wang L. TGF-β Signaling Promotes Glioma Progression Through Stabilizing Sox9. Front Immunol. 2020;11:592080.
Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7–13.
Luo D, Xu X, Li J, Chen C, Chen W, Wang F, Xie Y, Li F. The PDK1/c-Jun pathway activated by TGF-β induces EMT and promotes proliferation and invasion in human glioblastoma. Int J Oncol. 2018;53:2067–80.
Wei L, Shao N, Peng Y, Zhou P. Inhibition of Cathepsin S Restores TGF-β-induced Epithelial-to-mesenchymal Transition and Tight Junction Turnover in Glioblastoma Cells. J Cancer. 2021;12:1592–603.
Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J Mol Med. 2018;42:3395–403.
Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417–20.
Zhang C, Zhang X, Xu R, Huang B, Chen AJ, Li C, Wang J, Li XG. TGF-β2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J Exp Clin Cancer Res. 2017;36:162.
Seystahl K, Papachristodoulou A, Burghardt I, Schneider H, Hasenbach K, Janicot M, Roth P, Weller M. Biological Role and Therapeutic Targeting of TGF-beta3 in Glioblastoma. Mol Cancer Ther. 2017;16:1177–86.
Uckun FM, Qazi S, Hwang L, Trieu VN: Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFβ2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers (Basel) 2019, 11.
Liang H, Wang Q, Wang D, Zheng H, Kalvakolanu DV, Lu H, Wen N, Chen X, Xu L, Ren J, et al. RGFP966, a histone deacetylase 3 inhibitor, promotes glioma stem cell differentiation by blocking TGF-β signaling via SMAD7. Biochem Pharmacol. 2020;180:114118.
Wick A, Desjardins A, Suarez C, Forsyth P, Gueorguieva I, Burkholder T, Cleverly AL, Estrem ST, Wang S, Lahn MM, et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest New Drugs. 2020;38:1570–9.
Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ. Novel therapies emerging in oncology to target the TGF-beta pathway. J Hematol Oncol. 2021;14:55.
Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One. 2014;9:e90353.
Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov. 2020;19:57–75.
Zhang M, Zhang YY, Chen Y, Wang J, Wang Q, Lu H. TGF-β Signaling and Resistance to Cancer Therapy. Front Cell Dev Biol. 2021;9:786728.
Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation. 2018;137:707–24.
Acknowledgements
Not applicable
Funding
This work was supported by the National Science Foundation for Excellent Young Scholars (32122052) and National Natural Science Foundation Regional Innovation and Development (No. U19A2003).
Author information
Authors and Affiliations
Contributions
Y.W. and X.W. designed this study. D.P. and M.F. drafted the manuscript. D.P. and M.F. and M.W. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No ethics approval was required for this review that did not involve patients or patient data.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peng, D., Fu, M., Wang, M. et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer 21, 104 (2022). https://doi.org/10.1186/s12943-022-01569-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01569-x