
Abstract
Background
The emergence and spread of malaria drug resistance have resulted in the need to understand disease mechanisms and importantly identify essential targets and potential drug candidates. Malaria infection involves the complex interaction between the host and pathogen, thus, functional interactions between human and Plasmodium falciparum is essential to obtain a holistic view of the genetic architecture of malaria. Several functional interaction studies have extended the understanding of malaria disease and integrating such datasets would provide further insights towards understanding drug resistance and/or genetic resistance/susceptibility, disease pathogenesis, and drug discovery.
Methods
This study curated and analysed data including pathogen and host selective genes, host and pathogen protein sequence data, protein–protein interaction datasets, and drug data from literature and databases to perform human-host and P. falciparum network-based analysis. An integrative computational framework is presented that was developed and found to be reasonably accurate based on various evaluations, applications, and experimental evidence of outputs produced, from data-driven analysis.
Results
This approach revealed 8 hub protein targets essential for parasite and human host-directed malaria drug therapy. In a semantic similarity approach, 26 potential repurposable drugs involved in regulating host immune response to inflammatory-driven disorders and/or inhibiting residual malaria infection that can be appropriated for malaria treatment. Further analysis of host–pathogen network shortest paths enabled the prediction of immune-related biological processes and pathways subverted by P. falciparum to increase its within-host survival.
Conclusions
Host–pathogen network analysis reveals potential drug targets and biological processes and pathways subverted by P. falciparum to enhance its within malaria host survival. The results presented have implications for drug discovery and will inform experimental studies.
Similar content being viewed by others

Background
Plasmodium falciparum malaria is a common infectious disease in Africa, and arguably the most important parasitic disease in the world, posing a significant public health burden as compared to other World Health Organization (WHO) disease-endemic regions. For instance, Africa contributed to about 93% (213 million of 228 million) and 94% (380,000 of 405,000) of global cases and deaths, respectively in 2018 [1].
The use of anti-malarial drugs has been the optimal avenue for controlling the disease. Currently, artemisinin-based combination therapy (ACT) is used as the first-line option for malaria treatment globally [2]. ACT was adopted in Africa after the decline in efficacy of previous widely used anti-malarial drugs, including chloroquine and sulfadoxine-pyrimethamine (SP) [2]. This was to ensure that, each component of the combinatorial drug acts through different mechanisms within the parasite, aiming to significantly reduce the likelihood of the emergence of multi-drug resistant parasites. Unfortunately, the parasite has shown tremendous ability to develop resistance and tolerance to these artemisinin derivatives and the long half-life partner drugs in some countries of the Greater Mekong Sub-region [2,3,4]. With several reports supporting parasite recrudescence and a significant decrease in their sensitivity to ACT, there has been continuous surveillance to monitor the emergence and spread of artemisinin-resistant parasite strains in Africa and elucidate whether it will follow a similar pattern observed for chloroquine and SP resistance where resistant strains originated from Southeast Asia [2, 4,5,6,7]. Interestingly, a study conducted by Uwimana et al. [7] has demonstrated the independent emergence and local spread of artemisinin partial resistance in Rwanda driven by R561H mutation in kelch gene. Another study conducted in Northern Uganda has also reported independent emergence and local spread of artemisinin-resistant parasite driven by mutations in the A675V or C469Y allele in the kelch13 gene [8]. These pieces of evidence suggest that artemisinin resistance has emerged independently in Eastern Africa.
Researchers have proposed that the emergence of artemisinin parasite-resistant strains in Africa would result in about 78 million additional cases [4] and over 100,000 deaths annually [9]. Evidence abounds to the fact that a major challenge to controlling, eliminating, and eradicating malaria is drug resistance. It is the principal reason for the expansion of this life-threatening disease.
The architectural framework of the parasite’s genome constitutes a major framework influencing variations in the levels of the drug susceptibility, particularly having elucidated that P. falciparum anti-malarial drug resistance involves a single major gene effect. Spontaneous alterations in the form of single nucleotide variation and multiple mutations in different genes within the parasite genome capacitate the pathogen’s ability to develop tolerance mechanisms or resist the drug action over time thus, yielding the unexpected result. Genetic polymorphisms of known drug-resistance genes, such as pfcrt, pfmdr1, pfk13, pfmrp1, pfdhfr, and pfdhps generally express effects that counteract drugs controlling the disease [7, 10,11,12]. Compared to the clinical phenotype of resistance to quinolones and SP which usually takes the form of reduced accumulation of drugs within the parasite, particularly targets, artemisinin resistance, manifests as slow parasite clearance in patients and is characterized by the parasite’s ability to alter intraerythrocytic cell cycle with an increased ring stage and a shortened trophozoite stage [8, 13].
Falciparum malaria is a multifactorial disease that involves the complex interplay between the host, vector, and the pathogen [14, 15]. The host–pathogen interactions have been a driving selective force influencing the genetic architecture of both species, particularly, on how their genes are involved in drug and/or genetic resistance, disease susceptibility, and the infection processes [14, 16, 17].
Understanding these interactions requires an in-depth analysis of the organism’s proteome which is regarded to execute the genetic programme. Proteins execute functions mostly through extended networks with each other thereby forming a framework of the sensitive and complex regulatory system underlying a wide degree of post-translational modifications and processes [18]. The complex physicochemical dynamic connections formed within the system facilitate the structural and functional organization of the organism. These connections make up the protein–protein interaction network (PPIN).
Recent advances in host and parasite genomics in terms of high-throughput proteomics studies, host and parasite genome sequencing have led to a corresponding increase in biological datasets that describe the transition of species over time, particularly, the metabolic and developmental stages of pathogens. As such, the application of computational approaches to efficiently mine the inter and intra-species functional interactions to address the challenges presented by the disease is critical [19]. A systematic and comprehensive study of these complex interactions is essential in elucidating relevant pathways, signalling, drug resistance patterns, genes-gene products inter-relationships, and drug targets as well as developing novel hypotheses and models to predict disease causality [20].
In this study, a network-based integrative computational framework was leveraged to predict protein targets that may be used to guide the rational design of pathogen- and host-directed therapies for malaria treatment. Following the target prediction, a semantic similarity approach was implemented to prioritize informed potentially repurposable drugs that can be engineered for malaria treatment. Further analysis of host–pathogen network shortest paths enabled the prediction of immune-related biological processes and pathways potentially subverted by P. falciparum to increase its within-host survival.
Methods
Study design and procedures
Various open access heterogeneous genomic and functional datasets retrieved from databases and literature using text mining techniques were used as inputs for analysis. The approach for this study (Fig. 1) consisted of five main steps: (1) data curation and pre-processing, (2) scoring and integrating functional datasets; (3) biological network assembling and structural analysis; (4) gene mapping and enrichment analysis (5) implicit semantic similarity approaches to predict malaria-similar diseases and repurposable drugs. Briefly, the framework uses integrative, scoring, and clustering algorithms coupled with statistical methods and biological knowledge to analyse and validate results.

An overview of the approach implemented in this study
Data pre-processing
The various datasets utilized for this study are described in Additional file 4: Table S1. To achieve uniform identifiers (IDs) and convenient data manipulation, all genes and protein IDs were mapped to only reviewed proteins from Swiss-Prot under the non-redundant UniProt identifier system for harmonization. Human and P. falciparum genes were mapped to UniProt proteins with taxon identifier 9609 and 36,329 (Plasmodium falciparum 3D7 strain), respectively. Genes with no corresponding UniProt protein ID as at the time of this study were discarded.
Human malaria susceptibility-associated single nucleotide polymorphisms (SNPs) were retrieved from GWAS summary statistics datasets obtained from MalariaGEN [21]. The summary statistics dataset comprised of 20,273,529 spanning across chromosome one (1) to twenty-two (22). In this study, approximately 690,000 significant SNPs (p-value < 0.05) were filtered for further analysis. These SNPs were then mapped onto 44 genes (herein referred to as host candidate genes, Additional file 5: Table S2) using the dbSNP annotated data [22, 23].
Scoring and integrating functional datasets
The study performed pathogen-pathogen, pathogen-host, and host-host protein sequence BLAST using their respective protein sequences retrieved from the UniProt database [24]. This was followed by implementing an information-theoretic based functional scoring scheme outlined by Mazandu and Mulder [25] and summarized in the Additional file 10: (Eqs. 1–8) to score the functional associations obtained from sequence BLAST and the conserved domains interaction datasets from the InterPro database [26].
Scoring high-throughput experimental datasets and interologs
To incorporate curated functional interaction datasets in the analysis, the following criteria were defined to prioritize and score pair-wise interactions from experimental and interolog datasets retrieved from Reactome [27], IntAct [28], MINT [29], BIOGRID [30], and literature [31,32,33,34,35,36]. The criteria for scoring were based on; (1) the number of experimental methods that have confirmed such pair-wise functional interaction, (2) the number of databases that have reported such pair-wise functional interaction, and (3) the number of times the pair-wise functional interaction has been reported in the literature. For every pair-wise functional interaction supported by one evidence, a reliability score of 0.3 was assigned, else, a reliability score of 0.7 if it is supported by two or more pieces of evidence.
Biological network assembling and structural analysis
Table 1 describes the number of proteins retrieved from each dataset, the number of reviewed proteins/genes considered from each input dataset and the pair-wise functional interaction implemented for further downstream analysis. From the pre-processed scored datasets, the functional interactions obtained were categorized, as low (scores less than 0.3), medium (scores ranging between 0.3 and 0.7), and high confidence levels (scores greater than 0.7). Biases may exist in the PPI network generated due to relatively high noise related to high-throughput data or experiments from which interactions are derived. In the absence of gold standard PPIs, integrating data from different sources and applying strict interaction reliability or confidence score cut-off are used to reduce the impact of these biases, leading to a PPI network of high confidence interactions with an increased coverage [37]. Further analyses only used medium and high confidence interactions or interactions predicted by two different sources. To evaluate the structural features of nodes (proteins) and edges (interactions), network centrality metrics including node degree, betweenness, and closeness (Additional file 10: Eqs. 9–11) were computed. High degree nodes with low betweenness describe degree-based or ‘local’ subnetwork interconnectivity mostly between functionally related proteins. High degree nodes with high betweenness contribute to structural-based or ‘global’ subnetwork interconnectivity and signal transmission thus, promoting system-level functional integration. Node closeness describes the average shortest length between neighbouring nodes determining the proximity to information sharing and biological process execution between functionally related nodes [38].
Community structure and hub classification
The study aimed to identify hub genes/proteins that establish links with multiple functional clusters (communities), thus, characterized by both ‘local’ and ‘global’ network interconnectivity, structural, and functional features. To predict the hubs, clustering analysis was performed to identify network communities of densely connected nodes using a variant of an integrative computational algorithm that implements the Blondel et al. [39] heuristic method based on modularity optimization. This clustering model is a scalable hierarchical agglomerative method based on modularity optimization and has been shown to outperform all other known community detection methods [40], including Smart Local Moving [41], Infomap [42], and Label Propagation [43], in terms of computation time or complexity and the quality of the communities detected (modularity). The parasite candidate genes (herein referring to known antimalarial resistant genes and reported genes expressing signature of selection towards drug resistance) retrieved from literature [2, 6, 10] and host candidate gene-encoded proteins (Additional file 5: Table S2) were mapped onto the assembled parasite and host networks to cluster the networks. The subnetworks were explored to identify global hubs, herein defined as candidate gene/proteins characterized by a high degree and high betweenness score.
Functional annotation analysis
Gene annotation and enrichment analysis were performed to elucidate statistically significant biological processes and pathways to which the hub genes are involved. Biological processes were inferred from the gene ontology database [44], whereas pathway information was obtained from PlasmoDB v46 [45] and the KEGG database [46]. By applying the hypergeometric test [47], p-values of processes and pathways were estimated, leveraging on their frequency of occurrence. The Bonferroni multiple correction test [47] was then implemented to estimate the adjusted p-values.
Semantic similarity
The development of human disease ontology terms [48] has provided an enriched platform of human disease data to evaluate similarities between various diseases of different disorder classes based on gene-related molecular functions. The analysis is based on the hypothesis that varying combinations of disease-associated genes can influence the pathogenicity of similar diseases [49]. To predict repurposable drugs for malaria treatment, an in-house python-based semantic model was implemented for disease and drug similarity. The model uses host candidate key proteins, disease-target datasets, and gene ontology datasets as input data to make predictions based on functional similarities inferred from associated gene ontology terms. The semantic similarity approach was further implemented to identify diseases that are biologically similar to malaria. In the analysis, the semantic similarity score between the pair of diseases was leveraged to identify and prioritize diseases similar to malaria. The similarity score was estimated by computing the Kappa statistic, Jaccard, and the Best Match Average (BMA) measures (Additional file 10). The score is a quantitative measure of the underlying shared biological processes among the disease targets. A higher score between disease enriched processes suggests that the disease-pair and their associated candidate proteins are functionally similar thus, the likelihood for similar treatment options. A similarity score threshold was defined based on the upper quartile and interquartile range of the distribution given by \(tr = Q3+\varepsilon *IQR\), where \(\varepsilon \), \(tr, Q3\) and \(IQR\) represent the tuning parameter \((0\le \varepsilon \le 1.5)\) threshold, upper quartile, and interquartile range, respectively.
Results
Network clustering and functional annotation analysis
The generated parasite network consists of 662 unique interactions among 140 characterized proteins (Fig. 2A). The unified host network assembled comprised of 4,133,136 unique functional interactions between 20,329 nodes. The host-parasite network consisted of 31,512 unique functional interactions between 8023 proteins. The topology properties of the generated networks were explored to investigate the relationships between the degree, betweenness, and closeness centrality measures. As shown in Additional file 1: Fig. S1, subnetworks were classified as either degree-based (subnetworks formed from nodes with a high degree but low betweenness) or structural-based (subnetworks formed from nodes with high degree, high betweenness, and high closeness). The nodes forming the degree-based and structural-based subnetworks are herein referred to as key proteins.

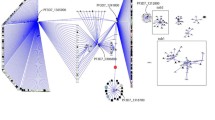
A Assembled parasite network and B Functional interactions between C6KTD2 and C6KTB7 subnetwork within the parasite network. The nodes common to the subnetworks are coloured in yellow
Network clustering analysis reveals disease candidate key proteins/genes as hubs
The purpose of clustering is to partition the complex network into subnetworks and identify essential communities and critical functional nodes. It is a way of grouping nodes in the network into modules sharing functional connectivity. The parasite network (Fig. 2A) consists of 8 clusters of which 5 contained key proteins whereas the dense human network consisted of 32 clusters of which 7 contained key proteins. From the network clustering (Additional file 2: Fig. S2A, Additional file 3: Fig. S2B), two parasite candidate key proteins were identified as hubs, C6KTD2 (SET1) and C6KTB7 (PFF1365c) both on chromosome 6. These parasite candidate key proteins are involved in the merozoite developmental stage where they invade red blood cells (RBCs), cause disease severity, and contribute to the exponential growth of the parasite population [50]. Analysis of the host network revealed 6 candidate key proteins as hubs; P22301 (IL10 [MIM: 124092]), P05362 (ICAM1 [MIM: 147840]), P01375 (TNF [MIM: 191160]), P30480 (HLA-B [MIM: 142830]), P16284 (PECAM1 [MIM: 173445]) and O00206 (TLR4 [MIM: 603030]). These proteins are cognate host receptors that respond to inflammation by releasing pro-inflammatory cytokines, enhancing adhesion of parasitized red blood cells (RBCs), parasite sequestration in organs rupture, and removal of infected RBCs [50, 51]. Most importantly, the identified host candidate key proteins are targets for drugs in DrugBank [52] and have been reported to offer higher opportunities for drug repurposing, although a smaller proportion of the human genome is druggable [53,54,55]. Additional file 6: Table S3 and Additional file 7: Table S4 describe the identified candidate key proteins prioritized by the degree, betweenness, and closeness scores.
Biological processes and pathway enrichment of hub genes
The identified hub genes within the subnetworks were used for the functional annotation process. The results revealed 4 statistically significant essential processes and an enriched pathway (Table 2) specific to the parasite key hub genes. A total of 23 significant biological processes and 21 enriched pathways (Table 3) were identified to underly host hub gene's contribution towards malaria infection. From the host perspective, the hub genes are mainly involved in immune regulatory biological processes within immune-related pathways (47.6%), parasitic disease-related pathways (23.8%), bacteria disease-related pathways (14.2%), endocrine and metabolic disease-related pathways (4.7%), viral disease-related pathway (4.7%) and transport and catabolism related pathway (4.7%)[44, 46]. Most importantly, the malaria pathway ranked the most significant pathway with both p-value and adjusted p-value of 0. This supports the association of these hub genes to malaria. The enriched pathways presented the likelihood of similarity between malaria and other diseases.
Shortest path analysis between hub genes reveals functional insights towards disease progression
The study investigated functional interactions between the host and pathogen targets in the context of parasite survival, host immune tolerance, and how it can inform drug discovery research. The immune tolerance machinery remains to be the natural driving force influencing the parasite's survival when host–pathogen recognition receptors sense infection. To contribute to this effort, the shortest paths between the parasite and host hub proteins within the host-parasite network were explored to gain insight into the most likely routes for innate immune response interference by the parasite.
Studies have shown that the shortest path analysis of a functional network yields high coverage compared to direct neighbours within the network [56]. The shortest path between host–pathogen disease-associated candidate key genes herein refer to the minimum number of edges required to connect these genes. Longer paths consist of more nodes (proteins) involved in a cascade of signalling processes to trigger innate immune responses by inducing the production of chemokines and cytokines upon parasite infection. It is, therefore, a measure of information relay between the hub genes thus, the shorter the path, the quicker the transmission and the relevance of the interaction in investigating immune adaptiveness and parasite pathogenesis [56]. It is noteworthy that, shortest path lengths between the pathogen disease-associated genes and human disease-associated genes conferring immunity in the functional network are the most feasible routes of parasite invasion of host immunity and escaping the contribution of host genetics towards drug action [56, 57]. Most importantly, shortest paths would trigger excessive activation which may be deleterious as it can cause systemic inflammation and disease [50]. This, therefore, suggests that developing immune-modulatory drugs that target the host targets can induce an immune response to avoid the state of been overwhelmed by the parasite.
The results showed that the shortest path between parasite hub proteins and any of the host hub proteins were between O00206—C6KTB7, and O00206-C6KTD2 as shown in Table 4. Such paths were characterized by mediators. These mediators are mostly signal receptors involved in cell regulatory activities, production of cytokines, transcription processes, and regulating cell survival and apoptosis. The shortest paths identified (Table 4) suggest that inhibition or alteration to the proper functioning of each path might help the parasite to survive immune responses, thus, the aggregation of small effects. The development of adaptive immunity is expected to happen when the parasite undergoes diversity throughout time such that they evade the host system when they become tolerant and establish different mechanisms to interfere with the host’s response [58]. These interferences can also be in the form of the production of effector mechanisms that can down-regulate innate immunity [59]. The results have shown that the dynamic patterns to parasite survival and immune adaptiveness are mediated by other human-specific genes or proteins conferring immunity.
Importantly, pfk13 is known to be associated with artemisinin resistance, but little is known of its interaction with host genes/proteins and how that influences drug resistance or parasite survival within the host. Further network analysis was performed to explore interactions between pfk13 and the host candidate key proteins. The results revealed no functional interactions between pfk13 and the host hub genes. However, the analysis showed interactions between pfk13 and highly expressed host kelch-like proteins and regulatory genes involved in essential processes such as transcription regulation, cell-surface, cell–cell signalling, and regulation of phosphorylation. Among the regulatory genes include the transcriptional regulator Kaiso (ZBTB33), Zinc finger and BTB domain-containing protein 17 (ZBTB17 [MIM: 604084]), BTB/POZ domain-containing protein 10 (KCTD10 [MIM: 613421]), Zinc finger and BTB domain-containing protein 10 (ZBTB10 [MIM: 618576]), Myoneurin (MYNN [MIM: 606042]), Nucleoprotein TPR (TPR [MIM: 189940]) and Gigaxonin (GAN [MIM: 605379]).
Predicting repurposable drugs for malaria treatment based on Implicit Semantic Similarity
After defining a semantic similarity score threshold (as illustrated in Fig. 3A), 1944 (8.04%) out of 24,166 diseases in the DisGeNet platform version 6 were identified to be semantically like malaria. The disease hits were filtered by maintaining those whose targets are involved in the same pathways of host Malaria hub genes. The disease hits were further filtered by maintaining diseases supported by biological evidence from the literature. The final filtered disease hits consisted of 113 diseases (Additional file 8: Table S5). These identified diseases fall in the category of infectious, inflammatory, and genetic neurological diseases which trigger the human immune machinery to overproduce cytokines; confirming the fact that malaria is an inflammatory response-driven disease. Among the top disease hits includes sickle cell anaemia [MIM: 603903], liver dysfunction [MIM: 613759], fever ([MIM: 142680], [MIM: 614371]), hepatitis ([MIM: 606518], [MIM: 609532]) and respiratory distress syndrome [MIM: 267450]. It is interesting to note that the disease hits described have been reported to be governed by the same pathologic principles as malaria infection [60, 61].Finally, to predict repurposable drugs, 1426 approved drugs and their corresponding targets were retrieved from the DrugBank database. Next, non-human drugs were excluded and were remained with 1282 drugs and their targets for further downstream analysis. The drugs were further filtered to retain those with target processes associated with malaria and the predicted malaria similar diseases. Then after, the semantic approach was implemented to predict putative repurposable drugs. From the identified drugs sharing some similarities in terms of processes, those that are over 1.5 of the interquartile range were extracted and ordered. With a defined similarity score threshold of 0.31099875 (Fig. 3B) based on similarity in terms of processes the drugs are involved in, the results revealed 26 potential repurposable drugs (Additional file 9: Table S6).The repurposable drugs categorized as known anti-malarial, monoclonal antibodies, immunomodulators, herbs, natural products, Janus kinase inhibitors, and thrombolytic agents act as either antagonist, agonists, inhibitors, or precursors targeting genes over-represented in immune response and cytokine-mediated signalling processes. Janus kinase inhibitors including ruxolitinib, are known for their ability to effectively inhibit the production of cytokines and cause eryptosis contributing to the clearance of erythrocytes infected with malaria, decreased parasitaemia, and protection against severe malaria [62]. The results showed that drugs involved in regulating host immune response to inflammatory-driven disorders target the Tumour necrosis factor and inhibit its activity to regulate downstream processes such as pro-inflammatory cascade signalling. Several of the potentially repurposable drugs are used for treating some diseases like malaria including rheumatoid arthritis, ischemic stroke, psoriatic arthritis, and idiopathic arthritis.

A Different distributions of disease similarity scores obtained in terms of frequencies (proportions) of disease matches vs similarity scores between disease-associated processes. The bigger rectangular bar indicates the threshold for the similarity between disease pairs of which the enriched similarity score (ESS) were used for further analysis. B Distributions of drug similarity scores obtained in terms of the relative frequency of drug matches against functional similarity scores between candidate gene and drug. The bigger rectangular bar indicates the threshold for the similarity between drug pairs of which the enriched similarity score (ESS) were used for further analysis
The drug hits include chloroquine, infliximab, hydroxychloroquine, glucosamine, ginseng, minocycline, ruxolitinib, and natalizumab which can be appropriated for malaria treatment. These drug hits have been reported to control malaria infection by inhibiting residual malaria infection, knocking parasite gene expression, and activating eryptosis. Furthermore, some of the hits such as adalimumab, Natalizumab, etanercept, thalidomide, ustekinumab, and canakinumab are anti-TNF monoclonal antibodies and anti-inflammatory agents that could modulate the immune response to severe and cerebral malaria. The analysis also predicted thrombolytic agents such as anistreplase, reteplase, alteplase, and tenecteplase which can play an essential role in the treatment of coagulopathy in malaria, particularly among severe and cerebral malaria infections [63]. Considering malaria as an inflammatory-response driven disease presenting with multiple manifestations, these putative drug hits can undergo both computational and experimental repositioning for adjunctive malaria therapy, particularly severe and cerebral malaria.
Discussion
In this study, an integrative network-based framework was implemented on the various heterogeneous experimental and in silico datasets retrieved from databases and literature to assemble Plasmodium falciparum, human, and human-Plasmodium falciparum functional protein–protein interaction network. Using host-malaria GWAS summary statistics datasets, host-disease-associated genes were identified by mapping nominally significant SNPs to their associated genes. The identified genes, malaria parasite selective variants, and parasite variants under strong signature of selection were mapped onto the host and pathogen functional network respectively to identify key subnetworks. The subnetworks of each assembled network were evaluated to investigate nodes (candidate key proteins) that contribute significantly to the stability and integrity of the network. Gene annotation and enrichment analysis of the identified hub genes were performed to elucidate underlying statistically significant biological processes and pathways. Also, shortest paths analysis was performed to elucidate pathways that could account for parasite adaptiveness to host response and potential drug resistance development. From the parasite assembled functional network, the analysis performed predicted C6KTD2 (SET1) and C6KTB7 (PFF1365c) as key targets. These targets are essential at specific developmental stages of the parasite and have been reported as candidates for drug and vaccine development. The results confirm the importance of these targets. Also, the analysis (Figs. 2B and 4A) showed that these targets could be critical for combinatorial drug design. There is an accumulation of evidence that C6KTB7 is a potential multi-stage target for a malaria vaccine and drug development [64,65,66,67,68]. C6KTB7 is mainly involved in ubiquitin-protein transferase activity (GO:0004842, GO:0019787) through the protein ubiquitination and modification pathway (UPA00143). Studies have shown that many biological processes and substrates are targeted by the ubiquitin pathway such that instability or modification in ubiquitination and deubiquitination reactions influences the pathogenesis of many eukaryotic system-related diseases [65]. For instance, the dysregulation of ubiquitin ligase is associated with neurodegenerative disorders, such as Parkinson’s disease and infectious diseases including tuberculosis [66]. This is usually associated with interference with immune response. C6KTB7 significantly influences the parasite’s development and malaria pathogenesis by regulating various cellular processes and pathways critical for the pathogen’s survival in the human host [69]. This phenomenon usually happens as a result of post-translational modifications within the biological system through processes such as transcriptional regulation and cell cycle progression [66]. For example, the protein is responsible for the positive regulation of DNA-templated transcription and epigenetic factors such as histone H3-K4 methylation, essential for transcription regulation [65]. Interestingly, studies have shown that inhibition of the activities of C6KTB7 and the ubiquitin–proteasome system has the potential for many disease treatments including P. falciparum malaria [65, 68, 69]. Of note, the parasite candidate proteins are essential during specific developmental stages. For instance, Aminake et al. [68] explored the role of the proteasome of P. falciparum for malaria drug research and revealed C6KTB7 as a component of the ubiquitin–proteasome which could serve as a promising multi-stage (liver, blood, and transmission stages of the pathogen) target, thus a supporting results presented by Chung et al. [70]. Additionally, Ponts et al. [65] showed that proteins involved in the ubiquitylation pathway including the ubiquitin ligases (E3) such as C6KTB7 (PFF1365c) influence parasite virulence, thus targeting such a pathway may represent new therapeutic targets for apicomplexan parasites, such as P. falciparum. This suggests that inhibiting parasite adaptation to the ubiquitylation pathway and the proteins involved (including putative E3 ubiquitin-protein ligase protein PFF1365c (C6KTB7)) is important for malaria drug research [65, 68]. C6KTD2 is a possible candidate for effective malaria vaccine development [67]. The protein plays an essential role in chromatin structure, protein domain-specific binding. and gene expression in the parasite [35, 71]. Also, it is mainly involved in the histone lysine methylation post-translational modification process (GO: 0051568) which usually involves the synergistic effect of histone-lysine methyltransferases and histone lysine demethylases [71, 72]. A gene knock-out study conducted by Jian et al. [73] revealed that C6KTD2 is essential particularly during the blood stage of the parasite, thus targeting it in drug research is important. Interactome analysis on the host functional network revealed (P22301 (IL10), P05362 (ICAM1), P01375 (TNF), P30480 (HLA-B), P16284 (PECAM1), O00206 (TLR4)) as key targets. These host candidate key proteins are involved in immune response and resistance against malaria infection including severe and cerebral malaria, thus, critical targets for adjunctive and antibody-based host-directed therapy for malaria control [74,75,76]. Importantly, studies have shown the need to complement artemisinin derivatives with host-directed therapy involved in immune modulation to help effectively control and treat severe malaria and cerebral malaria [77]. This may contribute significantly to improve treatment efficacy, reduce disease-associated complexity, reduce malaria-associated mortality and morbidity as well as slow artemisinin resistance development. In both the parasite and host-parasite functional network, the functional interactions between hubs formed by C6KTD2 and C6KTB7 were identified (Fig. 2B). This finding suggests the functional relatedness of these proteins and their modularity within the parasite to jointly regulate post-translational modification processes. Having established that nodes within a cluster might be involved in the same biological process, it is, therefore, possible that these key proteins within the clusters contribute significantly to similar processes [78].


A Functional interactions between C6KTD2 and C6KTB7 subnetwork in the unified host–pathogen functional network. The shared host proteins (yellow nodes) are involved in protein ubiquitination, positive regulation of cell apoptotic process, signal transduction, regulatory processes, and histone methylation. B Predicted shortest path network that could influence resistance and parasite adaptiveness between C6KTB7 (green node) and O00206 (bottom sky blue node) via co–targets (central sky blue nodes) in the host–pathogen network. C Predicted shortest path network that could influence resistance and parasite adaptiveness between C6KTD2 (green node) and O00206 (bottom sky blue node) via mediators (central sky blue nodes) in the host–pathogen network
23 significantly enriched malaria-related biological processes described in (Table 3) were identified. These gene ontology groups comprised of those involved in cell immune and inflammatory responses, regulation and production of transcription factors, biosynthetic processes, cell–cell adhesion, cell signalling, and cell apoptotic processes. Positive regulation of NIK/NF-kappaB signalling (GO:0042346) process responsible for the regulation of NF-kappaB importation has been studied to be involved in immune and inflammatory responses, particularly in eukaryotic cells. Down or negative regulation of NF-kappaB has been reported to be associated with P. falciparum-modulated endothelium transcriptome contributing to cerebral malaria [79]. Positive regulation of the MHC class II biosynthetic process (GO:0045348) process has been shown to regulate immune response to malaria [80]. Pre-erythrocytic immunity to malaria (cerebral malaria) is linked to MHC antigens such that variations in class I and class II in these antigens contribute significantly to malaria susceptibility thus, reduced, or increased host immune response [80]. Also, other processes such as negative regulation of interferon-gamma production (GO:0032689), negative regulation of interleukin-6 production (GO:0032715), negative regulation of cytokine secretion involved in immune response (GO:0002740), and positive regulation of interferon-gamma production (GO:0032729) serves as immunological mediating processes that influence disease susceptibility by either conferring protection or influencing disease progress. Activation and regulation of NLRP3 inflammasomes, immune system receptors, controls the activation of caspase-1 and induce inflammation in response to infectious pathogens [81]. Due to their influence on a wide range of diseases, their dysfunction results in the initiation or progression of diseases. Endothelial cell apoptosis has been studied to contribute to malaria severity. For instance, haem-induced microvasculature endothelial cell apoptosis mediated by proinflammatory and proapoptotic pathways contributes significantly to severe malaria.
In addition, the pathways of immune tolerance and potential resistance development among the host and pathogen key targets were investigated by analysing the shortest paths between these genes within the host–P. falciparum functional network. The results showed that these shortest paths between the candidate genes or proteins are mediated by host genes involved in cell regulatory activities and general cell integrity.
Shortest path analysis further revealed human immune-related genes and pathways that could be overwhelmed by the pathogen, knowing that the pathology of malaria is immune-mediated and inflammatory response-driven. Such inhibition could result in reduced anti-inflammatory responses thus limiting the production and possible cytopathic effects of cytokines [82]. The analysis revealed potential pathways between host malaria-associated candidate key protein O00206 (Toll-like receptor 4, TLR4) and pathogen proteins C6KTB7 (Putative E3 ubiquitin-protein ligase protein PFF1365c) and C6KTD2 (Putative histone-lysine N-methyltransferase 1, SET1) that could account for unrestrained parasite growth and severe complications. Experimental findings have revealed that activation of TLRs induces the production of nitric oxide and synthesis of pro-inflammatory cytokines, such as TNF and IL‑1β [50, 83]. Of note, activation of TLR4 induces macrophage release of pro-inflammatory mediators, such as TNF and nitric oxide [50, 83]. It also induces the expression of adhesion molecules on endothelial cells [50]. This may suggest that PECAM1, ICAM1, and TNF are from the downstream signalling cascade generated by TLR4 [83].
Severe malaria is associated with an increased level of pro-inflammatory cytokines (T helper 1 (Th1) cytokines) such as interleukin (IL)-12, IL-8, and interferon (IFN)-\(\upgamma \) in the affected person which helps to modulate defence against the infection and limit disease progression [59, 82]. This is attributed to the fact that the severity of malaria is proportional to the flawlessness in the host inflammatory response.
TLR4, a pathogen-recognition receptor, detects pathogen-associated molecular mechanisms in the body and initiates immune response through activation of signalling cascades such as nuclear factorkB, mitogen-activated protein kinase (MAPK), and Plasmodium antigens [59]. TLR4 and its immune-related signalling pathways have been reported to contribute significantly to P. falciparum growth and malaria pathogenesis, such that dysregulation and dysfunction of the gene increase malaria severity, symptomatic malaria, severe malaria anaemia, and resistance in Africa [84]. This suggests that deleterious activation of TLR4 by C6KTB7 and C6KTD2 will significantly contribute to parasite survival and disease susceptibility thereby causing severe pathological conditions.
Finally, a semantic similarity approach was implemented to identify 113 diseases like malaria (Additional file 8: Table S5) that facilitated the prediction of 26 potential repurposable drug hits, spanning across anti-malarials, monoclonal antibodies, immunomodulators, herbs, natural products, Janus kinase inhibitors, and thrombolytic agents, that can be computationally and experimentally modified for parasite or host-directed malaria treatment. Drug hits for each category were ranked based on the enriched similarity score. The results revealed certolizumab pegol and golimumab as hits for the monoclonal antibody category, pomalidomide for the immunomodulator category, ginseng for the herbs and natural product category, ruxolitinib for the Janus kinase inhibitors, anistreplase for the thrombolytic agent category, and chloroquine for the anti-malarial category. Additional file 9: Table S6 describes the known activity and the original therapeutic purpose of the potentially repurposable drugs identified.
Conclusions
With the gradual emergence and spread of malaria drug resistance, considering other potential drug targets and drug candidates are essential to increase the longevity of existing drugs as well as develop alternative treatment options. In this research, integrative computational methods were leveraged to (1) predict potential drug targets for both human host and pathogen-directed drug discovery, (2) predict drug candidates that could be re-engineered for malaria treatment and, (3) identify biological processes and pathways that could be overwhelmed by the pathogen to increase within-host survival.
The analysis revealed that repurposable drugs involved in regulating host immune response to inflammatory-driven disorders and/or inhibiting residual malaria infection may enable appropriate malaria treatment. Of note, the potential to treat malaria using inhibitors or drugs that target the proteasome component and/or proteins involved in the parasite’s post-translational modification such as C6KTB7 and C6KTD2 have been established. However, exploring these targets for drug and vaccine development is yet to be fully achieved. Both C6KTD2 and C6KTB7 proteins have no crystallized structure yet, but the availability of other homologs could be explored using homology modelling approach to model the proteins. The generated homology models could be the starting point for novel drug discovery and structure-based studies to identify potential inhibitors. Additionally, the host protein targets predicted have solved structures that can be harnessed for structure-based drug discovery to identify potential inhibitors for malaria research.
In summary, the uniqueness of the integrative network framework lies in the input datasets, scoring metrics/schemes, clustering algorithm, and the criteria defined for the various analysis which translates into the findings from this study. The integrative network-based approach incorporates interologs, sequence blast interactions, and protein–protein interaction data from the literature, as well as the STRING, IntAct, MINT, and BIOGRID databases. In addition, the network approach implements a scalable hierarchical agglomerative clustering model, based on modularity optimization, to cluster the network into communities by leveraging candidate genes. This is then followed by network topology analysis to evaluate the topological features (degree, betweenness, and closeness) of the malaria candidate genes to identify hubs genes/proteins. The semantic similarity measures implemented coupled with literature evidence helped to identify diseases similar to malaria and potential repurposable drug candidates.
Like other computational approaches which need validation through further functional study, our findings presented can inform functional study for potential experimental and clinical validation. Extended computational analysis of this work would consider incorporating non-reviewed protein data, other omics level datasets, and drug-drug interaction information.
Availability of data and materials
All the scripts and data used in this manuscript are available at https://github.com/francis-agamah/Network_Analysis_Malaria. Online Mendelian Inheritance in Man (http://www.omim.org). Supplementary data and figures are available online.
Abbreviations
- WHO:
-
World Health Organization
- SNP:
-
Single Nucleotide Polymorphism
- GWAS:
-
Genome-wide Association Study
- ESS:
-
Enriched Similarity Score
- PPIN:
-
Protein–Protein Interaction Network
- ACT:
-
Artemisinin-based Combination Therapy
References
WHO. World malaria report 2019. Geneva, World Health Organization, 2019.
Takala-Harrison S, Laufer MK. Antimalarial drug resistance in Africa: key lessons for the future. Ann N Y Acad Sci. 2015;1342:62–7.
Amor A, Toro C, Fernandez-Martinez A, Baquero M, Benito A, Berzosa P. Molecular markers in Plasmodium falciparum linked to resistance to anti-malarial drugs in samples imported from Africa over an eight-year period (2002–2010): impact of the introduction of artemisinin combination therapy. Malar J. 2012;11:100.
Ouji M, Augereau J-M, Paloque L, Benoit-Vical F. Plasmodium falciparum resistance to artemisinin-based combination therapies: a sword of Damocles in the path toward malaria elimination. Parasite. 2018;25:24.
Miraclin TA, Matthew A, Rupali P. Decreased response to artemisinin combination therapy in falciparum malaria: a preliminary report from South India. Trop Parasitol. 2016;6:85–6.
Antony HA, Parija SC. Antimalarial drug resistance: an overview. Trop Parasitol. 2016;6:30–41.
Uwimana A, Umulisa N, Venkatesan M, Svigel SS, Zhou Z, Munyaneza T, et al. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: an open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect Dis. 2021;21:1120–8.
Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana SI, Yamauchi M, et al. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med. 2021;385:1163–71.
Lubell Y, Dondorp A, Guérin P, Drake T, Meek S, Ashley E, et al. Artemisinin resistance–modelling the potential human and economic costs. Malar J. 2014;13:452.
Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. 2019;19:e338–51.
White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–92.
Gatton ML, Martin LB, Cheng Q. Evolution of resistance to sulfadoxine-pyrimethamine in Plasmodium falciparum. Antimicrob Agent Chemother. 2004;48:2116–23.
Fairhurst RM, Dondorp AM. Artemisinin-resistant Plasmodium falciparum malaria. Microbiol Spectr. 2016;4(10):1128.
Acharya P, Garg M, Kumar P, Munjal A, Raja KD. Host–parasite interactions in human malaria: clinical implications of basic research. Front Microbiol. 2017;8:889.
Clayton AM, Dong Y, Dimopoulos G. The Anopheles innate immune system in the defense against malaria infection. J Innate Immun. 2014;6:169–81.
Luckhart S, Pakpour N, Giulivi C. Host–pathogen interactions in malaria: cross-kingdom signaling and mitochondrial regulation. Curr Opin Immunol. 2015;36:73–9.
Su XZ, Zhang C, Joy DA. Host-malaria parasite interactions and impacts on mutual evolution. Front Cell Infect Microbiol. 2020;10:587933.
Ramaprasad A, Pain A, Ravasi T. Defining the protein interaction network of human malaria parasite Plasmodium falciparum. Genomics. 2012;99:69–75.
Agamah FE, Mazandu GK, Hassan R, Bope CD, Thomford NE, Ghansah A, et al. Computational/in silico methods in drug target and lead prediction. Brief Bioinform. 2020;21:1663–75.
Zuck M, Austin LS, Danziger SA, Aitchison JD, Kaushansky A. The promise of systems biology approaches for revealing host pathogen interactions in malaria. Front Microbiol. 2017;8:2183.
Network MGE. Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa. Asia and Oceania Nat Commun. 2019;10:5732.
Smigielski EM, Sirokin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28:352–5.
Sherry ST, Ward M, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38(Database issue):D142–8.
Mazandu GK, Mulder NJ. Scoring protein relationships in functional interaction networks predicted from sequence data. PLoS One. 2011;6:e18607.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37(Database issue):D211–5.
Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39(Database issue):D691–7.
Kerrien S, Aranda B, Breuza L, Bridge A, Broaches-Carter F, Chen C, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40(Database issue):D841-6.
Licata L, Briganti L, Peluso D, Perfetto L, Iannucelli M, Galeota E, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40(Database issue):D857–61.
Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45(Database issue):D369–79.
Wuchty S, Ipsaro JJ. A draft of protein interactions in the malaria parasite P. falciparum. J Proteome Res. 2007;6:1461–70.
Wuchty S. Topology and weights in a protein domain interaction network–a novel way to predict protein interactions. BMC Genomics. 2006;7:122.
Wuchty S. Rich-club phenomenon in the interactome of P. falciparum--artifact or signature of a parasitic life style? PLoS One. 2007;2:e335.
Wuchty S, Adams JH, Ferdig MT. A comprehensive Plasmodium falciparum protein interaction map reveals a distinct architecture of a core interactome. Proteomics. 2009;9:1841–9.
LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–7.
Bossi A, Lehner B. Tissue specificity and the human protein interaction network. Mol Syst Biol. 2009;5:260.
Mazandu GK, Mulder NJ. Generation and analysis of large-scale data-driven Mycobacterium tuberculosis functional networks for drug target identification. Adv Bioinformatics. 2011;2011:801478.
Mulder NJ, Akinola RO, Mazandu GK, Rapanoel H. Using biological networks to improve our understanding of infectious diseases. Comput Struct Biotechnol J. 2014;11:1–10.
Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech. 2008;10:P10008.
Emmons S, Kobourov S, Gallant M, Börner K. Analysis of network clustering algorithms and cluster quality metrics at scale. PLoS One. 2016;11:e0159161.
Waltman L, Van Eck NJ. A smart local moving algorithm for large-scale modularity-based community detection. Eur Phys J B. 2013;86:471.
Rosvall M, Bergstrom CT. Maps of random walks on complex networks reveal community structure. Proc Natl Acad Sci USA. 2008;105:1118–23.
Raghavan UN, Albert R, Kumara S. Near linear time algorithm to detect community structures in large-scale networks. Phys Rev E Stat. 2007;76:036106.
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, et al. The Gene Ontology (GO) database and informatics resource. Nucleaic Acids Res. 2004;32(Database issue):D258–61.
Aurrecoechea C, Brestelli J, Brunck BP, Dommer J, Fischer S, Garija B, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37(Database issue):D539–43.
Aoki KF, Kanehisa M. Using the KEGG database resource. Curr Protoc Bioinformatics. 2005;Chapt 1:Unit 1.12.
McDonald JH. Handbook of biological statistics. Vol. 2. 2009: Sparky House Publishing, Baltimore, MD.
Kibbe, W.A., et al., Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015;43((Database issue):D1071–8.
Mathur S, Dinakarpandian D. Finding disease similarity based on implicit semantic similarity. J Biomed Inform. 2012;45:363–71.
Gazzinelli RT, Kalantari P, Fitzgerald KS, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. 2014;14:744–57.
Bengtsson A, Joergensen L, Rask TS, Olsen RW, Andersen MA, Turner L, et al. A novel domain cassette identifies Plasmodium falciparum PfEMP1 proteins binding ICAM-1 and is a target of cross-reactive, adhesion-inhibitory antibodies. J Immunol. 2013;190:240–9.
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–72.
Fox CS. Using human genetics to drive drug discovery: a perspective. Am J Kidney Dis. 2019;74:111–9.
Chen Y, Xu R. Network-based gene prediction for Plasmodium falciparum malaria towards genetics-based drug discovery. BMC Genomics. 2015;16(Suppl 7):S9.
Hua S. Targeting sites of inflammation: intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Front Pharmacol. 2013;4:127.
Rives AW, Galitski T. Modular organization of cellular networks. Proc Natl Acad Sci USA. 2003;100:1128–33.
Chen LC, Yeh HY, Yeh CY, Arias CR, Soo VW. Identifying co-targets to fight drug resistance based on a random walk model. BMC Syst Biol. 2012;6:5.
Belachew EB. Immune response and evasion mechanisms of Plasmodium falciparum parasites. J Immunol Res. 2018;2018:6529681.
Gowda D, Wu X. Parasite recognition and signaling mechanisms in innate immune responses to malaria. Front Immunol. 2018;9:3006.
Clark IA, Alleva LM, Mills AC, Cowden WB. Pathogenesis of malaria and clinically similar conditions. Clin Microbiol Rev. 2004;17:509–39.
Murphy SC, Breman JG. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg. 2001;64(1_suppl):57–67.
Briglia M, Fazio A, Faggio C, Laufer S, Alzoubi K, Lang F. Triggering of suicidal erythrocyte death by ruxolitinib. Cell Physiol Biochem. 2015;37:768–78.
Francischetti IM, Seydel KB, Monteiro RQ. Blood coagulation, inflammation, and malaria. Microcirculation. 2008;15:81–107.
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, et al., UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32(Database issue):D115–9.
Ponts N, Yang J, Chung DK, Prudhomme J, Girke T, Horrocks P, et al. Deciphering the ubiquitin-mediated pathway in apicomplexan parasites: a potential strategy to interfere with parasite virulence. PLoS One. 2008;3:e2386.
Hamilton MJ, Lee M, Le Roch KG. The ubiquitin system: an essential component to unlocking the secrets of malaria parasite biology. Mol Biosyst. 2014;10:715–23.
Villard V, Agak GW, Frank G, Jafarshad A, Servis C, Nébié I, et al. Rapid identification of malaria vaccine candidates based on alpha-helical coiled coil protein motif. PLoS One. 2007;2:e645.
Aminake MN, Arndt HD, Pradel G. The proteasome of malaria parasites: a multi-stage drug target for chemotherapeutic intervention? Int J Parasitol Drugs Drug Resist. 2012;2:1–10.
Sharma M, Dhiman C, Dangi P, Singh S. Designing synthetic drugs against Plasmodium falciparum: a computational study of histone-lysine N-methyltransferase (PfHKMT). Syst Synth Biol. 2014;8:155–60.
Doug Chung D-W, Le Roch KG. Targeting the Plasmodium ubiquitin/proteasome system with anti-malarial compounds: promises for the future. Infect Disord Drug Targets. 2010;10:158–64.
Cui L, Fan Q, Cui L, Miao J. Histone lysine methyltransferases and demethylases in Plasmodium falciparum. Int J Parasitol. 2008;38:1083–97.
Kaur I, Zeeshan M, Saini E, Kaushik A, Mohmmed A, Gupta D, et al. Widespread occurrence of lysine methylation in Plasmodium falciparum proteins at asexual blood stages. Sci Rep. 2016;6:35432.
Jiang L, Mu J, Zhang Q, Ni T, Srinivasan P, Ryavara K, et al. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nature. 2013;499:223–7.
Dunst J, Kamena F, Matuschewski K. Cytokines and chemokines in cerebral malaria pathogenesis. Front Cell Microbiol. 2017;7:324.
Kumar R, Ng S, Engwerda C. The role of IL-10 in malaria: a double edged sword. Front Immunol. 2019;10:229.
Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, et al. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci USA. 2011;108:3689–94.
Varo R, Crowley VM, Sitoe A, Madrid L, Serguides L, Kain KC, et al. Adjunctive therapy for severe malaria: a review and critical appraisal. Malar J. 2018;17:47.
Mazandu GK, Chimusa ER, Rutherford K, Zekeng EG, Gebremariam ZZ, Onifade MY, et al. Large-scale data-driven integrative framework for extracting essential targets and processes from disease-associated gene data sets. Brief Bioinform. 2018;19:1141–52.
Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ. Plasmodium falciparum-infected erythrocytes induce NF-kappa B regulated inflammatory pathways in human cerebral endothelium. Blood. 2009;114:4243–52.
Lyke KE, Fernández-Vina MS, Cao K, Hollenbach J, Coulibaly D, Kone AK, et al. Association of HLA alleles with Plasmodium falciparum severity in Malian children. Tissue Antigens. 2011;77:562–71.
Guo HT, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Oyegue-Liabagui SL, Bouopda-Tuedom AG, Kouna LC, Maghendji-Nzondo S, Nzoughe H, Tchitoula-Makaya N, et al. Pro- and anti-inflammatory cytokines in children with malaria in Franceville. Gabon Am J Clin Exp Immunol. 2017;6:9–20.
Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280:8606–16.
Greene JA, Moormann AM, Vulule J, Bocharie MJ, Zimmerman PA, Kazura JW. Toll-like receptor polymorphisms in malaria-endemic populations. Malar J. 2009;8:50.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–61.
Acknowledgements
We acknowledge the staff, colleagues from the Division of Human Genetics and H3Africa Coordinating Center, University of Cape Town. We acknowledge members of the Trusted World of Corona (TWOC) Consortium. We also acknowledge the staff and colleagues from the Center for Molecular and Biomolecular Informatics (CMBI), Radboud University Medical Center, Nijmegen. Computations were performed using facilities provided by the Centre for High-Performance Computing (https://users.chpc.ac.za/, South Africa).
Funding
This work was supported through the DELTAS Africa Initiative [DELGEME grant 107740/Z/15/Z]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [DELGEME grant 107740/Z/15/Z] and the UK government. Also, this work was supported through the University of Cape Town, internal funding, and the National Research Foundation of South Africa for funding (NRF) [grant # RA171111285157/119056]. This work was partially funded by an LSH HealthHolland grant to the TWOC consortium, a large-scale infrastructure grant from the Dutch Organization of Scientific Research (NWO) to the Netherlands X-omics initiative (184.034.019), and a Horizon2020 research grant from the European Union to the EATRIS-Plus infrastructure project (grant agreement: No 871096). Some of the authors are supported in part by the National Institutes of Health (NIH) Common Fund under grant numbers 1U2RTW012131-01 (COBIP), U24HG006941 (H3ABioNet) and 1U01HG007459-01 (SADaCC). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Author information
Authors and Affiliations
Contributions
EC and GM designed the study, FA performed the data analysis and drafted the manuscript. EC, DD, MS, AG, GM contributed to the data analysis and revision of the manuscript and supervised the work. All authors read and approved the final manuscript.
Authors’ information
Francis E. Agamah, PhD student in Human Genetics at the division of Human Genetics, Department of Pathology University of Cape Town. Email: agmfra001@myuct.ac.za/francisagamahh@gmail.com.
Delesa Damena, PhD in Human Genetics at the division of Human Genetics, Department of Pathology University of Cape Town. Email: delesa_damenaa@yahoo.com /damenadelesa@gmail.com.
Michelle Skelton, PhD in Human Genetics at, Computational Biology Division, Department of Integrative Biomedical Sciences, University of Cape Town. Email: michelle.skelton@uct.ac.za.
Anita Ghansah, PhD in Genetic Epidemiology at the London School of Hygiene and Tropical Medicine. Senior Researcher at Noguchi Memorial Institute for Medical Research, University of Ghana. Email: aghansah@noguchi.ug.edu.gh.
Gaston K. Mazandu, PhD in Bioinformatics, Senior Lecturer at the Division of Human Genetics, Department of Pathology, University of Cape Town. Email: gmazandu@gmail.com.
Emile R. Chimusa, PhD in Bioinformatics. Associate Professor at the Division of Human Genetics, Department of Pathology, University of Cape Town. Email: emile.chimusa@uct.ac.za.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Therefore, the authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Additional file 1: Figure S1
. Relationship between the degree, betweenness, and closeness centrality measures in the host-parasite assembled functional network. Figures A, B and C show the relationship observed in the parasite network whereas Figures D, E, and F represent the host network. Figures A and D show that the majority of nodes are characterized by a relatively high betweenness and degree score. This depicts the small-world property of the network whereby non-neighboring nodes within the network can interact through influential nodes. Figures B and C show that lower degree nodes are usually in close interaction thus, suggesting that such nodes are involved in similar processes or pathways, thus execute the function within a smaller compartment (low-level modularity) of the system, and the effect is transmitted by central nodes with relatively higher degree and betweenness. Figures C and F suggest that signalling (flow of information) within the biological system is highly influenced by nodes with relatively high betweenness. Such nodes are characterized by relatively high degree and closeness and are known to transmit signals generated as a result of low-level modularity between nodes.

Additional file 2: Figure S2A
. Summary results for parasite network clustering.

Additional file 3: Figure S2B
. Summary results for host network clustering.
Additional file 4: Table S1
. Description of various datasets and databases used for the study.
Additional file 5: Table S2
. Malaria-associated genes were retrieved by mapping significant SNPs to the gene level. The table entails the gene’s functional network centrality scores, including betweenness, degree, and closeness.
Additional file 6: Table S3
. Degree, closeness, and betweenness centrality score of C6KTD2 and C6KTB7 within the parasite unified functional network.
Additional file 7: Table S4
. Degree, closeness, and betweenness centrality score for host candidate key proteins within the human functional network.
Additional file 8: Table S5
. Predicted malaria–similar diseases identified using semantic similarity approach. ESS represents the estimated enriched similarity scores.
Additional file 9: Table S6
. Predicted repurposable drug hits identified using semantic similarity approach.
Additional file 10
. Supplementary method.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Agamah, F.E., Damena, D., Skelton, M. et al. Network-driven analysis of human–Plasmodium falciparum interactome: processes for malaria drug discovery and extracting in silico targets. Malar J 20, 421 (2021). https://doi.org/10.1186/s12936-021-03955-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-021-03955-0