
Abstract
Background
Malaria is the most deadly parasitic infectious disease. Existing drug treatments have limited efficacy in malaria elimination, and the complex pathogenesis of the disease is not fully understood. Detecting novel malaria-associated genes not only contributes in revealing the disease pathogenesis, but also facilitates discovering new targets for anti-malaria drugs.
Methods
In this study, we developed a network-based approach to predict malaria-associated genes. We constructed a cross-species network to integrate human-human, parasite-parasite and human-parasite protein interactions. Then we extended the random walk algorithm on this network, and used known malaria genes as the seeds to find novel candidate genes for malaria.
Results
We validated our algorithms using 77 known malaria genes: 14 human genes and 63 parasite genes were ranked averagely within top 2% and top 4%, respectively among human and parasite genomes. We also evaluated our method for predicting novel malaria genes using a set of 27 genes with literature supporting evidence. Our approach ranked 12 genes within top 1% and 24 genes within top 5%. In addition, we demonstrated that top-ranked candied genes were enriched for drug targets, and identified commonalities underlying top-ranked malaria genes through pathway analysis. In summary, the candidate malaria-associated genes predicted by our data-driven approach have the potential to guide genetics-based anti-malaria drug discovery.
Similar content being viewed by others


Background
Malaria is the most deadly parasitic infectious disease, which killed 627,000 people and caused 482,000 childhood deaths worldwide in 2012 [1]. Existing drug treatments show limited efficacy in malaria elimination [2–6]. Detecting the novel genetic basis for malaria not only reveals the disease pathogenesis, but also facilitates discovering new targets for anti-malaria drugs [7–11].
The pathogen causing malaria is the Plasmodium species. After being injected by mosquitos into human skin, these parasites infect the liver and multiply using the host's cell resources. Then they invade the red blood cells and cause the disease symptoms [12–14]. In both the liver and blood stage, the parasites trigger the host's innate immune responses and remodel the host cells to survive from the immune responses [15–19]. The complex pathogenesis of malaria involves both human and parasite genomes [20–24], and is not fully understood yet [25–27].
Studies of the human-parasite protein interactions have provided insights into the molecular signatures for malaria-specific host immune responses [20, 28, 29]. For example, studies show that the parasite protein PfEMP1 binds the human protein CD36 [30–32] and ICAM1 [33–35], which play critical roles in the adhesion of the infected red blood cells to the endothelial cells, and eventually lead to the disruption of bloodbrain barrier in cerebral malaria patients [36, 37]. Another example shows that the PfRh family of proteins in the parasites directly interacting with the human protein CR1 during the invasion of red blood cells, and CR1 has the potential to become the target of blood-stage vaccines [38, 39].
Currently, large-scale data have been accumulated on the human genome, parasite genome and their interactions. Integration and systematic analysis of the cross-species genomic data may lead to novel discoveries in genetic basis of malaria. In this study, we designed a data-driven approach to infer novel malariaassociated genes. Recent computational disease gene discovery algorithms have shown great potential in predicting disease causes [40–48]. They exploited the protein interactome in human genome and assumed that genes related to a disease phenotype tend to be located in a neighborhood in the protein-protein interaction network [49]. However, traditional methods are not sufficient for predicting genes for malaria, which naturally involves human-parasite protein interactions. Our approach represented the interacting human and parasite genomes with a heterogeneous network. We prioritized genes that are functionally related to the known malaria genes in the heterogeneous network and investigated if the top-ranked genes have the potential to guide drug discovery for malaria. We made our results publicly accessible at http://nlp.case.edu/public/data/malaria
Methods
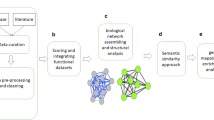
Our experiment work flow is depicted in Figure 1 and consists of two steps: (1) prioritize genes through network analysis and (2) analyze the result. We first constructed separate genetic networks for human genome and parasite genome, and then connected them with host-pathogen protein interactions. We used genes that are known to be associated with malaria as the seeds and applied a random-walk based algorithm to rank genes in the cross-species network. To validate our method in prioritizing malaria genes, we performed a "leave-one-out" cross validation analysis and examined the ranks of a set of malaria genes extracted from literature. Then we evaluated if the top-ranked genes are druggable. Finally, we analyzed the functions of the prioritized genes by extracting pathways on the basis of gene ranking.

Our methods contain two parts: gene prediction and result analysis. We predicted genes associated with malaria from the cross-species genetic networks. We also evaluated the method validity in predicting malaria genes, distribution of druggable genes among the rank, and the pathways associated with the top-ranked genes.
Construct cross-species gene network
We constructed the genetic network for human and Plasmodium falciparum (the species that causes the most dangerous form of malaria) from the STRING [50, 51] database. STRING includes gene relationships over a thousand species from four sources:protein-protein interactions (PPIs) databases, PPIs mined from literature abstracts, curated pathway databases and co-expressed genes.We used the four sources to build comprehensive networks for both human and Plasmodium falciparum.The human network contains 20,770 proteins and 4,850,628 interactions; and the Plasmodium falciparum network contains 4,913 proteins and 1,007,938 interactions. In addition, we used the scores from STRING to weight the edges in the two genetic networks.
We connected these two protein networks with 36 interactions from PathogenPortal [52] and literature [29, 30, 33]. These interactions are binary and cover physical associations, direct interactions and chemical reactions between the two species. The interaction pairs from literature were curated manually. We unified the gene identifiers with the genetic networks for human and parasites through HUGO Gene Nomenclature Committee [53] and PlasmoDB [54].
Predict candidate genes for malaria
We manually collected 77 known malaria genes and used them as the seeds in our algorithm to find additional malaria genes. Among the 77 seed genes, 14 human genes were extracted from Online Mendelian Inheritance in Man (OMIM). In addition, extensive literature evidence suggests that the Plasmodium falciparum proteins--PfEMP1 [55–57], PfRh4 [38, 58, 59] and PfRh5 [60–62]-- are essential for parasite growth and red blood cell invasion. We extracted 63 parasite genes encoding these three proteins and added them into the seed list.
We initiated a random walk on the cross-species genetic network from the seeds, and ranked all the genes by the probabilities of being reached from the seeds. We extended the algorithm by regulating the movements of the random walker between networks with the jumping probabilities λ. We represented the human and parasite genetic network with H and P , respectively. When the random walker stands on a node in H, which is connected with a node in P , it may jump to P with the probability λ or stay in H with the probability of 1 − λ.
We calculated the ranking scores for each node as follows. Assume p0 is a vector of initial scores for each node, p k is the score vector at step k and was iteratively updated by:
where γ is the probability that the random walker restarts from the seeds at each step, and M is the transition matrix of the cross-species genetic network:
The diagonal sub-matrices M H and M P consist of intra-network transition probabilities and were calculated as:
where i ∈ {H, P }, A i is the adjacency matrix of the network H or P , k is the index of row, l is the index of column, and x is an indicator variable, which equals to 1 if and 0 otherwise. The off-diagonal sub-matrices MHP and MHP T consist of inter-network transition probabilities and were calculated as:
where j ∈ {HP, HP T } and × is the same indicator variable. While the method could obtain a score for each human and parasite gene, we focus on ranking and analyzing the human genes in this study.
Evaluate the validity in predicting malaria genes
Before we used our method to predict genes for malaria, we performed the "leave-one-out" cross validation analysis to validate the method. Each time, we left out one malaria gene from the seed list, used the rest seeds as the input, and examined the rank of the excluded seed among the genes from the human or parasite genome. We repeated the same procedure for each of the 77 seeds, and assumed that the excluded seeds can be ranked highly if the method works well.
Then we used all the 77 seeds as the input, and evaluated if our gene ranking can prioritize novel malaria genes (other than the seeds). We manually constructed an independent set of 27 human genes involving malaria resistance and the host immune responses triggered by malaria parasites. These genes were extracted from literature references, which were mentioned in the textual descriptions of malaria in OMIM, and have zero overlap with the seed genes. We used this set as a proxy of novel malaria genes and evaluated the rank of this gene set among all human genes.
Evaluate the ranks of druggable genes
Currently, only a subset of the human genome is druggable [63]. In this study, we investigated if the topranked genes represent opportunities for drug discovery for malaria. We first extracted 1,935 human genes that were targets of all drugs from DrugBank [64]. All these drug target genes appear in our genetic network and have no overlap with the seeds. We used all 77 seeds as the input and ranked the human genes. Then we calculated the number of target genes among every 500 human genes in the rank from the top to the bottom, and plotted the variation of this number.
Extract and analyze malaria-specific pathways based on gene ranking
To better understand the functions of the prioritized genes, we linked the top 10% of human genes to their pathways. We downloaded 1320 canonical pathways from MSigDB [65] and ranked them based on the average of random walk scores for all the genes in each pathway. We manually examined if the top pathways are associated with the host response to the pathogen invasion.
In addition, we evaluated the impact of introducing the parasite genome into our gene prediction method. We removed the parasite genetic network and hostparasite interactions from our method, and calculated the random walk scores for human genes. Then we re-ranked the pathways containing the top 10% genes again. We compared the rank of pathways before and after using the parasite genetic network, and extracted the ones with largest rank difference.
Result
Network-based approach allowed the prioritization of known malaria genes from both human and parasite genomes
Among the 77 seed genes, 14 were human genes and 63 were parasite genes. We evaluated the performances of our algorithms in ranking human and parasite seed genes separately with a leave-one-out cross validation analysis. Our method required two parameters: the jumping probability λ between human and parasite genetic networks and the probability γ that the random walker restarts from the seeds. We chose λ=0.8 and γ=0.3 to achieve the best performance in the cross validation, but different parameter values only slightly affect the result. We used the same values for the two parameters through all the analyses.
Table 1 shows that the ranks of the excluded human seed genes were high. In nine cases, the excluded genes directly interact with another seed and were ranked within the top 1% amongst all the human genes. Of these, two genes (CD36, ICAM1) were ranked in the top five. In 13 out of 14 cases, the excluded genes were ranked within top 3%. The average rank for the excluded human seed genes is 336 (top 2% among all human genes).
We also evaluated the 63 parasite seed genes, and our approach ranked the excluded nodes within the top 5% in 56 out of 62 cases. Table 2 shows the top 10 parasite genes and their ranks in the cross validation. The average rank for the excluded parasite genes is 199 (top 4% among all parasite genes). Less comprehensive data in the parasite genome than in the human genome may contribute to the lower rank (in percentage) of the parasite seed genes. Overall, this analysis demonstrated the utility of the extended random walk to accurately prioritize known malaria genes.
Network-based approach prioritized novel malaria genes other than the seeds
Large amounts of literature have demonstrated strong associations between individual genes and malaria through transcriptional profiling, biological experimenting and genome-wide association studies. These genes include inflammatory responding genes, such as NFκB and CXCL1 [66], parasite protein receptors, such as BSG [67] and PROCR [57], and the genes involving protection against malaria, such as HLA-B [68] and HAVCR1 [69]. We then used all the seeds to generate our ranking for human genes, and examined the rank of 27 malaria genes, which have been validated in previous published studies. Table 3 shows that 12 out of 27 genes were ranked within the top 1%, and a total of 24 genes within the top 5%.
We also manually examined the top 50 human genes and found interesting predictions. Among them, TLR4 has been suggested to be protective against malaria in certain populations [70, 71]. In addition, a recent mouse model experiment [72] has demonstrated that P53 was critical in the liver-stage infection of malaria. Together, the result demonstrated that our gene ranking prioritized novel malaria-associated genes other than the seeds.
Prioritized genes are enriched by druggable genes
Figure 2 shows that the top-ranked genes are enriched for drug targets. The top 500 human genes in our ranking have 235 overlaps with the drug targets, which is a 4.3 fold enrichment compared with the average of 100 random rankings (p < 10−8). Among the 235 druggable genes, only 5 have been targeted by FDA-approved anti-malaria drugs, such as chloroquine, proguanil and mefloquine. This result indicated that the top-ranked candidate genes for malaria may provide unique opportunities for malaria drug discovery through novel disease genetics.

The count of drug target genes among every 500 genes in our rank from the top to the bottom.
Pathway analysis shows functions of prioritized genes
In order to gain insight into the commonalities underlying predicted malaria candidate genes, we analyzed the pathways associated with top-ranked genes. The top-ranked pathways are associated with different aspects of malaria. For example, malaria parasites actively alter the immune function of B cells and BIOCARTA BLYMPHOCYTE PATHWAY [73]. BIOCARTA LYM PATHWAY is a pathway of lymphocytes adhesion, and plays a central role in binding bacteria, parasites, viruses and tumor cells [74]. Also, BIOCARTA STEM PATHWAY regulates the hematopoiesis and induce hematopoietic activities in the presence of infection [75].
We compared the pathway ranking before and after introducing the parasite genetic network and found nine pathways increased the rank by over 50%. Table 4 lists these pathways and their plausible associations with malaria pathogenesis and protection. Several of these pathways are directly related with the parasite infection and inflammatory responses. REACTOME BASIGIN INTERACTIONS was prioritized through the interaction with the parasite protein PfRh5. Other pathways that were brought up by less than 50% also may have associations with malaria. For example, the rank of the REACTOME HDL MEDIATED LIPID TRANSPORT pathway were improved by 40%. A recent meta-analysis showed that host lipid profile alteration has a link with malaria pathogenesis, though the precise pathway has not been elucidated yet [76].
Discussion
Malaria is caused by the invasion of deadly parasites into human skin, liver and blood. The parasites trigger the human immune responses, but can manipulate human cells for nutrient uptake and cell growth. Recent studies have shown that host-pathogen protein interactions illuminate the malaria-specific pathways in the human host. With the accumulation of data in both human and parasite genome, systematically analyzing these two interacting genomes may potentially discover new malaria-associated genes, which will pave the way to identify novel anti-malaria drugs.
We developed a data-driven method to infer malaria genes based on random walking on the cross-species genetic networks. We demonstrated that the method can prioritize genes that are both drug targets and associated with malaria. Through comparing the result before and after adding the parasite genetic network into our method, we extracted specific pathways involving human-parasite interactions.
Our approach can be improved with a more comprehensive database of host-pathogen protein interactions. We currently manually curated 36 interactions, mostly from literature, to connect the human and parasite genetic network. Compared with the humanhuman protein interactions, the coverage of humanparasite interaction is much lower and might be biased. As more data are introduced into the method, the global structure of the cross-species genetic network may change, which will affect the result of gene ranking. In the future, we plan to automatically mine the human-parasite interaction from literature and construct a database with better coverage.
Since our approach prioritized a set of druggable genes, which are associated with malaria, one example of subsequent work is to perform drug repositioning through matching the targets of approved drugs to predicted genes. In this way, however, a part of the candidate drugs may target generic inflammatory responses and may not be specific enough to kill the parasites. In addition, malaria is associated with different pathways when human are infected by different parasite species (other than Plasmodium falciparum) or different strains [28]. To develop more effective agents against malaria, we need to dissect the genetic basis using more specific data.
Conclusions
The lack of effective anti-malaria drugs and the poorlyunderstood disease genetics has motivated our study of detecting novel malaria-associated genes from both human and parasite genomes, with the ultimate goal of discovering innovative anti-malaria drugs based on a new genetic understanding of the disease. We developed a data-driven approach to infer malariaassociated genes. Since malaria is caused by the interactions between parasites and human, we constructed a cross-species genetic network to model these interactions, and prioritized relative genes using network analysis. We demonstrated the validity of the method in predicting malaria genes, and showed the potential of the predicted genes in drug discovery. We also extracted pathways from the result of gene ranking, and found these pathways reflect different aspects of malaria pathogenesis.
References
World Health Organization. World Malaria Report 2013[http://www.who.int/malaria/publications/worldmalariareport2013/en/]
Kar S, Kar S: Control of malaria. Nature Reviews Drug Discovery. 2010, 16 (7): 511-512.
Biamonte MA, Wanner J, Le Roch KG: Recent advances in malaria drug discovery. Bioorg Med Chem Lett. 2013, 23 (10): 2829-2843. 10.1016/j.bmcl.2013.03.067.
Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, et al: Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet. 2012, 379 (9814): 413-431. 10.1016/S0140-6736(12)60034-8.
Kim Y, Schneider K: Evolution of drug resistance in malaria parasite populations. Nature Education Knowledge. 2013, 4 (8): 6-
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, et al: Artemisinin resistance in Plasmodium falciparum malaria. New England Journal of Medicine. 2009, 361 (5): 455-467. 10.1056/NEJMoa0808859.
Miller LH, Ackerman HC, Su Xz, Wellems TE: Malaria biology and disease pathogenesis: insights for new treatments. Nature Med. 2013, 19 (2): 156-167. 10.1038/nm.3073.
Horn D, Duraisingh MT: Antiparasitic chemotherapy: From genomes to mechanisms. Pharmacology and Toxicology. 2014, 54 (1): 71-94.
Flannery EL, Chatterjee AK, Winzeler EA: Antimalarial drug discovery [MDASH] approaches and progress towards new medicines. Nature Reviews Microbiology. 2013, 11 (12): 849-862. 10.1038/nrmicro3138.
Hanson KK, Ressurreicao AS, Buchholz K, Prudencio M, Herman-Ornelas JD, Rebelo M, et al: Torins are potent antimalarials that block replenishment of Plasmodium liver stage parasitophorous vacuole membrane proteins. Proceedings of the National Academy of Sciences. 2013, 110 (30): 2838-2847. 10.1073/pnas.1306097110.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al: A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014, 505 (7481): 50-55.
Kwiatkowski DP: How malaria has affected the human genome and what human genetics can teach us about malaria. The American Journal of Human Genetics. 2015, 77 (2): 171-192.
Menard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R: Looking under the skin: the first steps in malarial infection and immunity. Nature Reviews Microbiology. 2013, 11 (10): 701-712. 10.1038/nrmicro3111.
Crompton PD, Moebius J, Portugal S, Waisberg M, Hart G, Garver LS, et al: Malaria immunity in man and mosquito: Insights into unsolved mysteries of a deadly infectious disease*. Annu Rev Immunol. 2014, 32: 157-187. 10.1146/annurev-immunol-032713-120220.
Zheng H, Tan Z, Xu W: Immune evasion strategies of pre-erythrocytic malaria parasites. Mediators of Inflammation. 2014, 2014:
Engwerda CR, Kumar R: Mast cells fuel the fire of malaria immunopathology. Nature Med. 2013, 19 (6): 672-674. 10.1038/nm.3227.
Kaushansky A, Metzger P, Douglass A, Mikolajczak S, Lakshmanan V, Kain H, Kappe SH: Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis. 2013, 4 (8): e762-10.1038/cddis.2013.286.
Sicard A, Semblat JP, Doerig C, Hamelin R, Moniatte M, Dorin-Semblat D, et al: Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cell Microbiol. 2011, 13 (6): 836-845. 10.1111/j.1462-5822.2011.01582.x.
Austin LS, Kaushansky A, Kappe SH: Susceptibility to Plasmodium liver stage infection is altered by hepatocyte polyploidy. Cellular Microbiol. 2014, 16 (5): 784-795. 10.1111/cmi.12282.
Khor CC, Hibberd ML: Revealing the molecular signatures of host-pathogen interactions. Genome Biol. 2011, 12 (10): 229-10.1186/gb-2011-12-10-229.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419 (6906): 498-511. 10.1038/nature01097.
Elsworth B, Matthews K, Nie CQ, Kalanon M, Charnaud SC, Sanders PR, et al: PTEX is an essential nexus for protein export in malaria parasites. Nature. 2014, 511 (7511): 587-591. 10.1038/nature13555.
Beck JR, Muralidharan V, Oksman A, Goldberg DE: PTEX component HSP101 mediates export of diverse malaria effectors into host erythrocytes. Nature. 2014, 511 (7511): 592-595. 10.1038/nature13574.
Bongfen SE, Laroque A, Berghout J, Gros P: Genetic and genomic analyses of host-pathogen interactions in malaria. Trends Parasitol. 2009, 25 (9): 417-422. 10.1016/j.pt.2009.05.012.
Hedrick PW: Population genetics of malaria resistance in humans. Heredity. 2011, 107 (4): 283-304. 10.1038/hdy.2011.16.
Driss A, Hibbert JM, Wilson NO, Iqbal SA, Adamkiewicz TV, Stiles JK: Genetic polymorphisms linked to susceptibility to malaria. Malar J. 2011, 10: 271-10.1186/1475-2875-10-271.
Venkatesh S, Workman JL, Wahlgren M, Bejarano MT: Malaria: Molecular secrets of a parasite. Nature. 2013, 499 (7457): 156-157. 10.1038/nature12407.
Wu J, Tian L, Yu X, Pattaradilokrat S, Li J, Wang M, et al: Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proceedings of the National Academy of Sciences. 2014, 111 (4): E511-E520. 10.1073/pnas.1316467111.
Janes JH, Wang CP, Levin-Edens E, Vigan-Womas I, Guillotte M, Melcher M, et al: Investigating the host binding signature on the plasmodium falciparum PfEMP1 protein family. PLoS Pathog. 2011, 7 (5): e1002032-10.1371/journal.ppat.1002032.
Robinson BA, Welch TL, Smith JD: Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Molecular Microbiol. 2003, 47 (5): 1265-1278. 10.1046/j.1365-2958.2003.03378.x.
Baruch DI, Ma XC, Pasloske B, Howard RJ, Miller LH: CD36 peptides that block cytoadherence define the CD36 binding region for Plasmodium falciparum-infected erythrocytes. Blood. 1999, 94 (6): 2121-2127.
Baruch DI, Gormely JA, Ma C, Howard RJ, Pasloske BL: Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proceedings of the National Academy of Sciences. 1996, 93 (8): 3497-3502. 10.1073/pnas.93.8.3497.
Bengtsson A, Joergensen L, Rask TS, Olsen RW, Andersen MA, Turner L, et al: A novel domain cassette identifies plasmodium falciparum PFEMP1 proteins binding ICAM-1 and is a target of cross-reactive, adhesion-inhibitory antibodies. The Journal of Immunology. 2013, 190 (1): 240-249. 10.4049/jimmunol.1202578.
Smith JD, Craig AG, Kriek N, Hudson-Taylor D, Kyes S, Fagen T, et al: Identification of a Plasmodium falciparum intercellular adhesion molecule-1 binding domain: a parasite adhesion trait implicated in cerebral malaria. Proceedings of the National Academy of Sciences. 2000, 97 (4): 1766-1771. 10.1073/pnas.040545897.
Brown A, Turner L, Christoffersen S, Andrews KA, Szestak T, Zhao Y, et al: Molecular architecture of a complex between an adhesion protein from the malaria parasite and intracellular adhesion molecule 1. Journal of Biological Chemistry. 2013, 288 (8): 5992-6003. 10.1074/jbc.M112.416347.
Chakravorty S, Hughes K, Craig A: Host response to cytoadherence in Plasmodium falciparum. Biochemical Society Transactions. 2008, 36 (Pt 2): 221-228.
Medana IM, Turner GD: Human cerebral malaria and the blood-brain barrier. Int J Parasitol. 2006, 36 (5): 555-568. 10.1016/j.ijpara.2006.02.004.
Tham WH, Wilson DW, Lopaticki S, Schmidt CQ, Tetteh-Quarcoo PB, Barlow PN, et al: Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proceedings of the National Academy of Sciences. 2010, 107 (40): 17327-17332. 10.1073/pnas.1008151107.
Spadafora C, Awandare GA, Kopydlowski KM, Czege J, Moch JK, Finberg RW, et al: Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathogens. 2010, 6 (6): e1000968-10.1371/journal.ppat.1000968.
Barabasi AL, Gulbahce N, Loscalzo J: Network medicine: a network-based approach to human disease. Nature Reviews Genetics. 2011, 12 (1): 56-68. 10.1038/nrg2918.
Kohler S, Bauer S, Horn D, Robinson PN: Walking the interactome for prioritization of candidate disease genes. The American Journal of Human Genetics. 2008, 82 (4): 949-958. 10.1016/j.ajhg.2008.02.013.
Wang X, Gulbahce N, Yu H: Network-based methods for human disease gene prediction. Brief Funct Genomics. 2011, 10 (5): 280-293. 10.1093/bfgp/elr024.
Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, et al: Gene prioritization through genomic data fusion. Nature Biotechnol. 2006, 24 (5): 537-544. 10.1038/nbt1203.
Lage K, Karlberg EO, Størling ZM, Olason PI, Pedersen AG, Rigina O, et al: A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature Biotechnol. 2007, 25 (3): 309-316. 10.1038/nbt1295.
Wu X, Jiang R, Zhang MQ, Li S: Network-based global inference of human disease genes. Mol Syst Biol. 2008, 4 (1): 189-
Li Y, Patra JC: Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics. 2010, 26 (9): 1219-1224. 10.1093/bioinformatics/btq108.
Wu X, Liu Q, Jiang R: Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics. 2009, 25 (1): 98-104. 10.1093/bioinformatics/btn593.
Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R: Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010, 6 (1): e1000641-10.1371/journal.pcbi.1000641.
Gandhi T, Zhong J, Mathivanan S, Karthick L, Chandrika K, Mohan SS, et al: Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nature Genetics. 2006, 38 (3): 285-293. 10.1038/ng1747.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al: String v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41 (D1): 808-815. 10.1093/nar/gks1094.
Snel B, Lehmann G, Bork P, Huynen MA: String: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28 (18): 3442-3444. 10.1093/nar/28.18.3442.
Aurrecoechea C, Brestelli J, Brunk BP, Fischer S, Gajria B, Gao X, et al: EuPathDB: a portal to eukaryotic pathogen databases. Nucleic Acids Res. 2010, 38 (Database issue): D415-D419.
Gray KA, Daugherty LC, Gordon SM, Seal RL, Wright MW, Bruford EA: Genenames. org: the hgnc resources in 2013. Nucleic Acids Res. 2012, D545-D552. Database issue
Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, et al: PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37 (Database issue): D539-D543.
Pasternak ND, Dzikowski R: PfEMP1: An antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum. Int J Biochem Cell Biol. 2009, 41 (7): 1463-1466. 10.1016/j.biocel.2008.12.012.
Flick K, Chen Q: var genes, PfEMP1 and the human host. Mol Biochem Parasitol. 2004, 134 (1): 3-9. 10.1016/j.molbiopara.2003.09.010.
Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al: Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013, 498 (7455): 502-505. 10.1038/nature12216.
Park HJ, Guariento M, Maciejewski M, Hauhart R, Tham WH, Cowman AF, et al: Using mutagenesis and structural biology to map the binding site for the Plasmodium falciparum merozoite protein PfRh4 on the human immune adherence receptor. Journal of Biological Chemistry. 2014, 289 (1): 450-463. 10.1074/jbc.M113.520346.
Tham WH, Wilson DW, Reiling L, Chen L, Beeson JG, Cowman AF: Antibodies to reticulocyte binding protein-like homologue 4 inhibit invasion of Plasmodium falciparum into human erythrocytes. Infect Immun. 2009, 77 (6): 2427-2435. 10.1128/IAI.00048-09.
Williams AR, Douglas AD, Miura K, Illingworth JJ, Choudhary P, Murungi LM, et al: Enhancing blockade of Plasmodium falciparum erythrocyte invasion: assessing combinations of antibodies against PfRH5 and other merozoite antigens. PLoS Pathogens. 2012, 8 (11): e1002991-10.1371/journal.ppat.1002991.
Douglas AD, Williams AR, Illingworth JJ, Kamuyu G, Biswas S, Goodman AL, et al: The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nature Commun. 2011, 2: 601-
Douglas AD, Baldeviano GC, Miura K, Wright GJ, Draper SJ: PfRH5 vaccine efficacy against heterologous strain blood-stage Plasmodium falciparum. The Lancet. 2014, 383 (Special issue): S43-
Hopkins AL, Groom CR: The druggable genome. Nat Rev Drug Discov. 2002, 1 (9): 727-730. 10.1038/nrd892.
Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, et al: DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42 (D1): 1091-1097. 10.1093/nar/gkt1068.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005, 102 (43): 15545-15550. 10.1073/pnas.0506580102.
Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ: Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood. 2009, 114 (19): 4243-4252. 10.1182/blood-2009-06-226415.
Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, et al: Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011, 480 (7378): 534-537.
Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, et al: Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991, 352 (6336): 595-600. 10.1038/352595a0.
Nuchnoi P, Ohashi J, Kimura R, Hananantachai H, Naka I, Krudsood S, et al: Significant association between TIM1 promoter polymorphisms and protection against cerebral malaria in Thailand. Ann Hum Genet. 2008, 72 (3): 327-336. 10.1111/j.1469-1809.2007.00424.x.
Ferwerda B, McCall MB, Alonso S, Giamarellos-Bourboulis EJ, Mouktaroudi M, Izagirre N, et al: TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proceedings of the National Academy of Sciences. 2007, 104 (42): 16645-16650. 10.1073/pnas.0704828104.
Sawian CE, Lourembam SD, Banerjee A, Baruah S: Polymorphisms and expression of TLR4 and 9 in malaria in two ethnic groups of Assam, northeast India. Innate Immunity. 2013, 19 (2): 174-183. 10.1177/1753425912455675.
Kaushansky A, Ye AS, Austin LS, Mikolajczak SA, Vaughan AM, Camargo N, et al: Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Reports. 2013, 3 (3): 630-637. 10.1016/j.celrep.2013.02.010.
Scholzen A, Sauerwein RW: How malaria modulates memory: activation and dysregulation of B cells in Plasmodium infection. Trends Parasitol. 2013, 29 (5): 252-262. 10.1016/j.pt.2013.03.002.
Vestweber D, Blanks JE: Mechanisms that regulate the function of the selectins and their ligands. Physiological Reviews. 1999, 79 (1): 181-213.
Abdalla S: Hematopoiesis in human malaria. Blood Cells. 1989, 16 (2-3): 401-416.
Visser BJ, Wieten RW, Nagel IM, Grobusch MP: Serum lipids and lipoproteins in malaria-a systematic review and meta-analysis. Malaria Journal. 2013, 12 (1): 442-10.1186/1475-2875-12-442.
Ayi K, Min-Oo G, Serghides L, Crockett M, Kirby-Allen M, Quirt I, et al: Pyruvate kinase deficiency and malaria. New England Journal of Medicine. 2008, 358 (17): 1805-1810. 10.1056/NEJMoa072464.
Beiting DP, Park PW, Appleton JA: Synthesis of syndecan-1 by skeletal muscle cells is an early response to infection with Trichinella spiralis but is not essential for nurse cell development. Infection and Immunity. 2006, 74 (3): 1941-1943. 10.1128/IAI.74.3.1941-1943.2006.
Howells R, Maxwell L: Citric acid cycle activity and chloroquine resistance in rodent malaria parasites: the role of the reticulocyte. Ann Trop Med Parasitol. 1973, 67 (3): 285-300.
Campos FM, Santos ML, Kano FS, Fontes CJ, Lacerda MV, Brito CF, Carvalho LH: Genetic variability in platelet integrin α2β1 density: Possible contributor to Plasmodium vivax-induced severe thrombocytopenia. Am J Trop Med Hyg. 2013, 88 (2): 325-328. 10.4269/ajtmh.2012.12-0297.
Mota MM, Jarra W, Hirst E, Patnaik PK, Holder AA: Plasmodium chabaudi-infected erythrocytes adhere to CD36 and bind to microvascular endothelial cells in an organ-specific way. Infection and Immunity. 2000, 68 (7): 4135-4144. 10.1128/IAI.68.7.4135-4144.2000.
Nacer A, Movila A, Baer K, Mikolajczak SA, Kappe SH, Frevert U: Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathogens. 2012, 8 (10): e1002982-10.1371/journal.ppat.1002982.
Zeba AN, Sorgho H, Rouamba N, Zongo I, Rouamba J, Guiguemd´e RT, et al: Major reduction of malaria morbidity with combined vitamin a and zinc supplementation in young children in burkina faso: a randomized double blind trial. Nutr J. 2008, 7 (7): 7-
Chua CLL, Brown G, Hamilton JA, Rogerson S, Boeuf P: Monocytes and macrophages in malaria: protection or pathology?. Trends in Parasitology. 2013, 29 (1): 26-34. 10.1016/j.pt.2012.10.002.
Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, et al: Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. Journal of Infectious Diseases. 2003, 187 (3): 461-466. 10.1086/367960.
Pain A, Ferguson DJ, Kai O, Urban BC, Lowe B, Marsh K, Roberts DJ: Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proceedings of the National Academy of Sciences. 2001, 98 (4): 1805-1810. 10.1073/pnas.98.4.1805.
Wassmer SC, Lepolard C, Traore B, Pouvelle B, Gysin J, Grau GE: Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. Journal of Infectious Diseases. 2004, 189 (2): 180-189. 10.1086/380761.
Declarations
Publication charges for this article have been funded by the Training grant in Computational Genomic Epidemiology of Cancer (CoGE) (R25 CA094186-06).
This article has been published as part of BMC Genomics Volume 16 Supplement 7, 2015: Selected articles from The International Conference on Intelligent Biology and Medicine (ICIBM) 2014: Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S7.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RX conceived the study. YC designed the methods, performed the experiments and wrote the manuscript. Both authors have participated study discussion and manuscript preparation.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Chen, Y., Xu, R. Network-based gene prediction for Plasmodium falciparum malaria towards genetics-based drug discovery. BMC Genomics 16 (Suppl 7), S9 (2015). https://doi.org/10.1186/1471-2164-16-S7-S9
Published:
DOI: https://doi.org/10.1186/1471-2164-16-S7-S9