
Abstract
Background
Latic acid bacteria (LAB) are exploited for development of gene expression system owing to its health promoting properties and a high degree of safety status. Most of the expression systems were constructed in Lactobacillus lactis with inducible promoters. It is necessary to exploit novel promoters to develop LAB host platforms which are indispensable in dairy and health application to satisfy the production demand of increased number of target-genes. Previously, promoter PsrfA had been displayed broad host range and used to construct auto-inducible expression system in B. subtilis and E. coli. In this work, the feasibility of PsrfA in LAB was estimated.
Results
Plasmid with the green fluorescent protein (GFP) inserting downstream of PsrfA was transformed into L. casei 5257, L. plantarum 97, L. fermentum 087 and Weissella confusa 10, respectively. The recombinant strains grew well and displayed different fluorescence which could be detected by spectrophotometer and laser scanning confocal microscope. Moreover, the promoter activity was strain- specifically influenced by particular carbon and nitrogen sources. Heterologous laccase CotA could be expressed by PsrfA in L. casei 5257-05 and L. plantarum 97-06. By adjusting the pH value from 4.5 to 6.5 during incubation, the CotA activity detected from L. plantarum 97-05 and L. casei 5257-05 was increased by 137.7% and 61.5%, respectively. Finally, the fermentation pH was variably up-regulated along with the production of NADH oxidase which was controlled by the PsrfA and its derivative mutated with core regions.
Conclusions
These data suggested that PsrfA was valid for gene expression in different species of LAB. Moreover, PsrfA could be used as an attractive candidate for fine-tuning gene expression in a broad range of prokaryotic expression plants.
Similar content being viewed by others

Introduction
Expression host systems are developed for the expression of heterologous proteins and are becoming more and more attractive along with the development of molecular biology and the emergence of the synthetic biology [1]. So far, the extensively used expression systems were developed in E. coli, Bacillus subtilis and Saccharomyces cerevisiae due to their specific genetic background, mature genetic manipulation and capacity for continuous fermentation [2, 3]. The developed expression systems generally have inherent advantages and drawbacks, such as intracellular accumulation of heterologous proteins, lack of post-transcriptional modification, production of endotoxin, instability of plasmids and so on. It is necessary to exploit novel expression host platforms to satisfy the demand of producing the increased number of target-genes for various industrial productions.
Lactic acid bacteria (LAB) are a heterogeneous group of bacteria characterized by their ability to produce lactic acid as a fermentation product, including genera such as Lactococcus, Lactobacillus and Streptococcus. LAB are exploited for development of gene expression system owing to its health promoting properties and a high degree of safety status [4, 5]. Recently, remarkable achievements have been made towards the development of genetic engineering tools and therapeutic delivery systems to produce many different proteins of health interest, such as antigens, cytokines, vitamin, etc. [6,7,8]. The most successfully and widely used expression system is NICE (nisin controlled gene expression) system in L. lactis using promoter PnisA which is proved to be a valuable tool for production of heterologous proteins including antimicrobial peptides, membrane proteins and vaccines [9]. Besides, some other inducible expression systems were constructed in LAB with promoters activated by lactose (PlacA), zinc (Pzn), xylose (PxylT), fructo-oligosaccharide (Pfos), etc. [10]. However, limited constitutive promoters were isolated which were able to be used for the production of food additives or application in the intestinal environment as LAB comprise a probiotic background. For example, promoter P11 (a synthetic sequence based on an rRNA promoter from L. plantarum WCSF1), promoter Ptuf33 and Ptuf34 (the promoter regions upstream of the gene encoding the putative translation elongation factor TU from L. plantarum CD033 and L. buchneri CD034) were demonstrated to be feasible for strong constitutive gene expression in L. plantarum [11].
The preferred expression system is selected depending upon the required quality and quantity of the target protein. Furthermore, the cost, availability and convenience of an optimal expression system should also be taken into consideration before making an informed choice of a system with which to express a foreign protein [12]. Therefore, it is necessary to evaluate the preliminary expression of the interest gene in the possible host cells. As the developed host cells have inherent genetic diversity, construction of cross-species expression system using the same genetic tools is a time- and cost-saving strategy to select the preferred expression cell for the target gene. The promoter, playing the role of expression switch, is commonly strain and context specific and can vary significantly within species. Hence, promoters available for a broad range of host strains should be developed to simultaneously examine gene expression in different genetic and physiological backgrounds and thus identify the most suitable expression strain. Only a limited number of promoters have been used in both Gram-positive and Gram-negative species, such as PT7, Pxyl, Plac and PBAD, etc. [13,14,15]. To date, several promoters with cross species activity were found in LAB. Promoter P11 was demonstrated to be component in L. plantarum and L. sakei and promoter Ptuf was highly active in some tested Lactobacillus strains as well as in E. coli [16].
Previously, the promoter of the srf operon (PsrfA) originated from B. subtilis, had been used to construct expression systems in B. subtilis and E. coli in which heterologous proteins were highly produced without inducer [17, 18]. In this work, promoter PsrfA was used for construction of expression systems in LAB. The feasibility of promoter PsrfA was studied including the range of host strains, the factors affecting the promoter strength and the applicability of producing heterologous proteins. This work will enrich the efficient genetic toolbox for gene expression in LAB. Meanwhile, it will be helpful for cross-species gene expression or module construction in E. coli, B. subtilis and LAB.
Results and discussion
Feasibility of promoter PsrfA in different species of LAB
Hitherto, genetic engineering tools used for cloning heterologous genes in LAB have mainly exploited with L. lactis which has been used as the model microorganism in LAB research. Recently, some other LAB are identified to be indispensable in dairy and health applications which are potential to be used as production organisms for antimicrobials, exopolysaccharides, flavor-compound and texturing compound. With the increased interest in the probiotic function of the LAB, it is necessary to develop novel expression systems which could work in a broad range of LAB especially in the strains with proven probiotic [19]. Promoter PsrfA, originated from B. subtilis, had already been used to develop expression system for production of heterologous proteins in B. subtilis and E. coli in our previous work [17, 18]. In this study, using the GFP as the report protein, the feasibility of PsrfA was evaluated in four representative genera of LAB which were wildly used in food production.
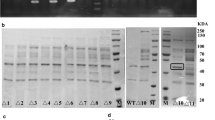
The recombinant plasmid pMY01 containing the fragment PsrfA-gfp was separately transformed into L. casei 5257, L. plantarum 97, L. fermentum 087 and W. confusa 10, yielding the recombinant strain L. casei 5257-01, L. plantarum 97-01, L. fermentum 087-01 and W. confusa 10-01, respectively. After 24 h cultivation, the cell growth, the fluorescence and the fluorescence intensity (defined as fluorescence per biomass of OD600) were measured (Fig. 1). Although the wild strains were grew the same as the corresponding recombinant strains, there were no fluorescence to be detected (data not show). However, the recombinant strains displayed obviously different fluorescence (Fig. 1a). The highest fluorescence was obtained from L. plantarum 97-01 which was 1.76-, 3.08- and 3.23-times higher than that of L. casei 5257-01, L. fermentum 087-01 and W. confusa 10-01, respectively. The fluorescence intensity of L. plantarum 97-01 was 1.06- to 1.49-times higher than that of the other recombinant strains. Compared with the other three host strains, L. plantarum 97 was demonstrated to be most feasible to express GFP under the control of PsrfA. It was equally shown in different studies that promoter strength was strain and context specific varied significantly within LAB [20]. Moreover, the recombinant strains with fluorescence could be obviously detected by laser scanning confocal microscope (Fig. 1b). These data suggested that promoter PsrfA had a broad transcriptional compatibility in different species of LAB.

Expression of GFP controlled by PsrfA in recombinant strains (a) and fluorescence analyzed by laser scanning confocal microscope (b). Plasmid pMY01was transformed into L. casei 5257, L. plantarum 97, L. fermentum 087 and W. confusa 10, respectively. The cell growth, fluorescence and fluorescence intensity of the recombinant strains were detected
The effect of carbon and nitrogen sources on the activity of promoter PsrfA
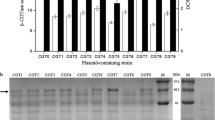
The usual used carbon and nitrogen components for the cultivation of LAB were employed to estimated their effect on the expression of GFP controlled by PsrfA. Firstly, the recombinant strains L. casei 5257-01, L. plantarum 97-01, L. fermentum 087-01 and W. confusa 10-01were cultivated in the medium containing glucose, maltose, fructose and lactose as the solo carbon source, respectively. The strain biomass was ranged from 0.36 (W. confusa 10-01 with lactose) to 2.09 (L. plantarum 97-01 with maltose) (Fig. 2 a). L. plantarum 97-01 showed better growth with these carbon sources than the other recombinant strains, especially than that of W. confusa 10-01. In addition, L. casei 5257-01 preferred glucose and fructose while L. plantarum 97-01, L. fermentum 087-01 and W. confusa 10-01 preferred maltose. Meanwhile, the fluorescence and the fluorescence intensity of recombinant strains were varied from 1032.95 Au (W. confusa 10-01 with fructose) to 9102.83 Au (L. plantarum 97-01 with maltose) and 843.96 Au/OD600 (L. casei 5257-01 with fructose) to 10,085.96 Au/OD600 (W. confusa 10-01 with lactose), respectively. Similarly, the recombinant strains were separately cultivated in the medium containing tryptone, soy peptone and polypeptone as the solo nitrogen sources. The particular recombinant strain showed consistent growth with the nitrogen sources. The strain biomass, fluorescence and the fluorescence intensity were ranged from 0.84 (W. confusa 10-01 with soy peptone) to 1.89 (L. plantarum 97-01 with polypeptone), 7996.13 Au (L. plantarum 97-01 with polypeptone) to 696.52 Au (W. confusa 10-01 with polypeptone) and 704.98 Au/OD600 (W. confusa 10-01 with polypeptone) to 4270.99 Au/OD600 (L. casei 5257-01 with soy peptone), respectively (Fig. 2b).

The effects of carbon (a) and nitrogen (b) sources on the biomass of recombinant strains and the activity of PsrfA. The recombinant strains were individually cultivated in the medium containing various carbon and nitrogen sources. The strain biomass and expression of GFP were detected. 1: L. casei 5257-01, 2: L. plantarum 97-01, 3: L. fermentum 087-01, 4: W. confusa 10-01
The varied fluorescence intensity was not directly proportional to the changed biomass among the recombinant strains with different carbon and nitrogen sources (Fig. 2). For instance, the biomass, fluorescence and fluorescence intensity of L. plantarum 97-01 cultivated with maltose were 0.63-, 6.33- and 3.50-times higher than that of L. casei 5257-01, respectively. In addition, the biomass, fluorescence and fluorescence intensity of L. plantarum 97-01 with maltose were 0.10-, 0.45- and 0.31-times higher than that of glucose. Moreover, to one specific stain, the difference of biomass and promoter activity with nitrogen sources were less than that of with carbon sources. Carbon and nitrogen components played different roles in the growth and survival of the bacteria, synthesis of enzymes and metabolites and regulation of the intracellular and extracellular environment, etc. All these factors directly or indirectly played different role in the activity of the promoter [21,22,23]. For instance, in B. subtilis, it was demonstrated that the activity of promoter PsrfA was negatively influenced by hydrogen peroxide (H2O2) because the H2O2 stress inactivated the DNA-binding activity of PerR which was required for activation of promoter PsrfA [24]. While in LAB, H2O2 is a highly relevant metabolite varied along with the carbon and nitrogen sources [25]. Therefore, carbon and nitrogen sources might affect the activity of promoter PsrfA by means of influencing H2O2 metabolism. These data suggested that the carbon and nitrogen source not only played an important role in the cell growth but also in the strength of the promoter PsrfA.
Application of promoter PsrfA in LAB
Expression of heterologous enzyme CotA originated from B. subtilis
In spite of feasibility in various host strains, the other crucially important criteria for a mature expression system are the capacity to produce various target proteins [18]. Laccase represent an interesting group of oxidative enzymes among various enzymes owing to their great potential for biotechnological and environmental applications in biodegrading fields and food industry, such as wine stabilization, baking, etc. [26]. So far, laccase was mainly produced by the wild and recombinant fungus and there were scarce reports about laccase expression in LAB. Here, laccase (CotA), originated from B. subtilis, was used to estimate the suitability of PsrfA for the production of heterologous proteins in LAB. To express CotA, the gene fragment cotA was inserted downstream of PsrfA to replace gfp on plasmid pMY01. The yielded plasmid pMY05 was separately transformed into L. plantarum 97 and L. casei 5257 obtaining the recombinant strains L. plantarum 97-05 and L. casei 5257-05, respectively. After cultivation, the enzyme activity of CotA was detected. Compared with L. plantarum 97 and L. casei 5257, enzyme activity of CotA was obviously detected from L. plantarum 97-05 and L. casei 5257-05 (Fig. 3). The CotA activity of strain L. plantarum 97-05 was 0.40 times higher than that of L. casei 5257-05.

Expression of heterologous enzyme CotA controlled by PsrfA. The recombinant strains containing CotA were incubated and the enzyme activity was detected. Further, the expression of CotA was tested by up-regulating the fermentation pH to 6.5 when the pH reduced below 4.5 during incubation
Moreover, by adjusting the pH of the medium to 6.5 when the pH reduced below 4.5 during incubation, the enzyme activity of L. plantarum 97-05 and L. casei 5257-05 was increased by 137.7% and 61.5%, respectively. Though there was enzyme activity, the corresponding protein band could not be detected on the SDS-PAGE. Considering on CotA and promoter PsrfA originated from B. subtilis, one explanation for the low expression might due to the different genetic background between Lactobacilus and Bacillus. Similarly, when nattokinase originated from B. subtilis was expressed in L. lactis, even though active nattokinase was markedly detected by western blotting and fibrinolytic activity analyses, it could not be detected with coomassie brilliant blue [27]. In addition, it was reported that various carbon and nitrogen sources were used to increase the production of laccase [28]. Therefore, to enhance the expression of laccase in LAB, the culture components and the strength of promoter PsrfA would be optimized in our subsequent work.
Expression of NADH oxidase to regulate the fermentation pH
Usually, LAB produce lactic acid during fermentation and the accumulated acid is a stress for the cell growth of LAB [29]. Normally, lactic acid is produced through glycolysis by converting pyruvate to lactate. Nevertheless, the pyruvate pool could be rerouted consequently through NADH-independent pathways when the intracellular pool of NADH was reduced by NADH oxidase (NOX) in the presence of oxygen. It was reported that when NOX was overexpressed, H+ was consumed as well as the reduced pyruvate to lactate flux was almost abolished [30]. Therefore, in this work, the pH of the supernatant was attempted to be regulated by the expression of NOX controlled by the promoter PsrfA.
To express NOX, gene fragment gfp on the plasmid pMY01 was replaced by gene nox, obtaining plasmid pMY06. After transforming the pMY06 into L. plantarum 97 and L. casei 5257, the corresponding recombinant strains L. plantarum 97-06 and L. casei 5257-06 were cultured for 15 h as usual procedure. During fermentation, the recombinant strains grew similar to the wild-type strains and the NOX activity was detected from both the wild-type and recombinant strains (Fig. 4a, b). However, the NOX activity and fermentation pH measured from L. plantarum 97-06 were higher than that of L. plantarum 97 during fermentation (Fig. 4a). The NOX activity of L. plantarum 97-06 detected at 3 h, 9 h and 15 h was 1.03-, 0.18- and 0.24-times higher than that of L. plantarum 97, respectively. Meanwhile, the fermentation pH of L. plantarum 97-06 measured at 3 h, 9 h and 15 h was separately 0.13-, 0.19- and 0.15-∆pH higher than that of L. plantarum 97. These data suggested that gene nox was successfully expressed in L. plantarum 97 under the control of promoter PsrfA and the pH of the supernatant could be upregulated along with the expression of NOX. However, there was scarce difference of NOX activity and fermentation pH gap between L. casei 5257 and L. casei 5257-06 during incubation (Fig. 4b). It was speculated that gene nox expressed by promoter PsrfA in L. casei 5257 was too low to obviously affect the fermentation pH. The data indicated that the genetic background of L. plantarum 97 was more suitable than L. casei 5257 for promoter PsrfA to trigger the expression of NOX.

Regulation of the fermentation pH by production of NOX. The recombinant plasmids containing promoter PsrfA and its derivatives were transformed into L. plantarum 97 (a) and L. casei 5257 (b), respectively. The sequence of promoter PsrfA and its derivatives P07, P08, P09, P10, P11 and P12 which were mutated with -35 region, -10 region, -35 and -10 region, RBS, deletion of 20 bp of the CodY binding sites and deletion the entire region of Cody binding sites, respectively (c). The biomass, fermentation pH and NOX activity of the recombinant strains were detected at 3 h, 9 h and 15 h during fermentation. 1: wild strain, 2-8: recombinant strains containing promoter PsrfA, P07, P08, P09, P10, P11 and P12, respectively
Fine-tunning the fermentation pH by promoter mutation
Metabolic engineering and synthetic biology approaches are becoming more and more attractive owing to its capacity to improve industrial application of microbes [1]. Generally, comparing with strong over-expression, strategies for the fine-tuning of gene expression are required to control the aimed metabolic fluxes in metabolic engineering. The most used method for accurate regulation of gene expression is promoter engineering by modifying the specific sequences of the promoter which is essential for the efficiency of transcription initiation and then for the expression of a targeted gene [30, 31]. To evaluate the validity of PsrfA for accurate gene expression, the sequence of PsrfA was analyzed. Besides the usual core regions of -10 box, -35 box and RBS (ribosome recognition site), there was a small sequence downstream of the promoter PsrfA which was probably the CodY binding site. CodY, a protein has proved to be ubiquitous in the low GC gram-positive bacteria and has been shown to regulate lots of genes expression by combining to target promoters to repress transcription of many genes while activate others [32]. Here, the conserved regions of the promoter PsrfA mentioned above were mutated using plasmid pMY06 as the template (Fig. 4c). The corresponding plasmids were individually transformed into L. plantarum 97 and L. casei 5257. The cell growth of the recombinant strains was similar to the wild strains (Fig. 4a, b). The NOX activity of the recombinant L. plantarum 97 containing the derivatives of PsrfA was varied from 9.02 U/mg to 62.55 U/mg which was accordingly 1.04- and 4.99-times higher than that of L. plantarum 97 during fermentation. Correspondingly, the pH of the supernatant detected from the recombinant strains was approximately 0.02- to 0.56-∆pH higher than that of the wide-type strain. Furthermore, the NOX activity and the fermentation pH of the recombinant strains containing the mutant promoters were diversely enhanced compared with that of PsrfA. The mutant promoters P09 (mutations in -10 box and -35 box) and P10 (mutations in RBS region) could obviously enhance the enzyme expression and the fermentation pH of the corresponding recombinant strains was markedly up-regulated. The promoter P08 ((mutations in -35 box) showed higher enzyme expression than the promoter P07 ((mutations in -10 box). One of the explanation might be the identity of the original sequence of -10 box (TAAACT) with the prokaryotic conserved -10 region (TATAAT) was higher than the original -35 box (GGTGAT) with the prokaryotic conserved -35 box (TTGACA). When combining the mutations in -10 and -35 regions together (promoter P09), the NOX activity controlled by promoter P09 was 3.19- and 2.05-times higher than that of promoters P07 and P08, respectively. Besides, the promoter P10 with mutation in the RBS region showed brilliant performance of NOX expression and pH up-regulation. It was suggested that the optimized RBS sequence provided optimal complementation to the 3′-end of the 16S rRNA, thereby increasing the ribosome’s affinity to the mRNA and enhancing the translation initiation efficiency. In addition, the promoters P11 and P12 mutated with the predicted CodY binding site showed slight higher NOX activity than that of promoter PsrfA, suggesting that the CodY protein in LAB was likely to repress the activity of promoter PsrfA. Currently, despite the detailed studies on CodY-mediated regulation, which revealed direct interaction between CodY and the regulatory regions of its targets, no CodY recognition sequence could be deduced [33]. Hence, the enhanced activity of the mutated promoter P11 and P12 might have no relation with CodY. It was supposed that the truncation of the predicted CodY binding sites was helpful for the RNA polymerase and the mutated promoters to generate a more stable topological structure and then strengthen the transcription. These predictions need to be verified in our followed work. Likewise, Guo constructed a constitutive promoter library by randomizing the -35 and -10 region of the promoter sequence of the noxE gene (H2O-forming NADH oxidase) in L. lactis and the fermentation pH was fine-tunned ranging from 5.93 to 6.32 [30]. Furthermore, the similar varied trend of enzyme activity and the supernatant pH were also found in the recombinant host strains L. casei 5257. These data demonstrated that the core regions of PsrfA were valid in LAB exhibiting different promoter strength and were able to regulate the fermentation pH by governing the expression of NOX.
The main focus of metabolic engineering has been on E. coli and S. cerevisiae as microbial workhorse. Recently, LAB has gained increasing attention because of its indispensable in dairy and health applications. Firstly, many engineering efforts were made concentrating on L. lactis in which the glycolytic highway and its uncoupling of other catabolic pathways were efficiently rerouted resulting in the high level production of both natural and novel end products, such as vitamin, polyol, extracellular polymeric substances, etc. [34]. But there were only a few genetic tools developed in other LAB except L. lactis which were used in probiotic products. Secondly, most expression system developed in LAB were using inducible promoters, such as PnisA, Pzn, Plac, P170 which were induced by nisin, heavy metals, sugar and pH, respectively [35,36,37,38]. Inducible expression can be preferable in cases where the aim is to overproduce a desired protein at high levels, e.g. at a specific moment during fermentation. However, as LAB comprise a food grade background, inducible systems are less suitable in the intestinal environment or when steady-state gene expression is required, instead, constitutive promoters providing expression of a target gene at a suitable level are desirable [39]. In addition, constitutive promoters are preferred to construct synthetic promoter library for the fine-tuning of gene expression which is important for applications as metabolic optimization and metabolic control analysis [40].
In general, promoter PsrfA used in this work was able to constitutively trigger gene expression at distinct times during growth in different species of LAB. Moreover, PsrfA had been proved to be valid to express various heterologous proteins in B. subtilis and E. coli. Thus, promoter PsrfA was feasible for the construction of circuits functional in multiple species as well as convenient preliminary screening of before deployment in E. coli, Lactobacillus spp. or Bacillus spp.
Conclusion
Developing of genetic engineering tools for gene expression have been mainly made in L. lactis and the limited Lactobacillus spp. In this work, promoter PsrfA originated from B. subtilis, was verified to be valid to trigger the expression of GFP and heterologous protein CotA in different species of LAB. Moreover, PsrfA and its derivatives were able to accurately tune the fermentation pH by managing the production of NOX. Besides, in our previous work, promoter PsrfA was available to produce heterologous proteins in different genus of B. subtilis and E coli. Hence, promoter PsrfA could be used as an attractive candidate in metabolic engineering for tuning gene expression in broad range of prokaryotic expression plants.
Materials and methods
Strains, plasmids and incubated conditions
E. coli JM109 was used as a host for gene cloning. L. casei 5257, L. plantarum 97, L. fermentum 087 and Weissella confusa 10 were used for the gene expression. The recombinant strains and plasmids used in this study are listed in Table 1. The vector pColdII-Z containing gene cotA for enzyme Laccase was kindly given by Zhang YZ. (Yancheng Institute of Technology, Jiangsu, China). The media used for E. coli and LAB were LB (Luria–Bertani) medium and MRS (de Man, Rogosa and Sharp) medium. When appropriate, growth media for recombinant strains were supplied with ampicillin (10 μg/mL) and chloramphenicol (10 μg/mL).
Recombinant DNA techniques
Plasmid construction was performed in E. coli and DNA extraction was performed by following a standard procedure as previously described [17]. Recombinant plasmids were transformed into LAB as previously described [16]. The primers used in this study are listed in Table 2. PCR was performed using primerSTAR max DNA polymerase (Takara, Japan). All of the recombinant plasmids constructed in this work were confirmed by DNA sequencing (Shanghai Sangon Biotech Co., Ltd., China).
Plasmid pMY01 was linearized using inverse PCR with primers F1 and R1. Gene cotA flanked by a 30-bp homology sequence upstream and downstream of the inserted position in pMY01 was amplified from plasmid pColdII using primers F5 and R5. The purified linearized pMY01 and cotA fragment were connected according to the manufacturers’ protocols of the Clone ExpressTM II One Step Cloning Kit (Vazyme Biotech Co., Ltd, Nanjing, China). The product was transformed into E. coli and the sequenced clone was named as pMY05. Plasmid pMY06 was constructed according to the steps described above. Gene nox was cloned from L. plantarum genome using primers F6 and R6 which was then connected with the purified linear pMY01, yielding plasmid pMY06.
To construct the plasmids containing PsrfA with mutation in the core region, plasmid pMY06 was used as the template by means of a megaprimer PCR as previously described [18]. The derivatives of pMY06 (from pMY07 to pMY12 containing the related promoters P07–P12) with mutation in the core sequences of the promoter were obtained with the corresponding primers.
Cultivation of LAB
A single colony of the appropriate recombinant LAB strain was picked from an MRS agar plates, inoculated in 10 mL of MRS liquid medium and cultured at 37 °C for 24 h. Then, 0.3 mL of the culture were inoculated in 10 mL of MRS liquid medium and cultured at 37 °C for 12 h to serve as a preculture. Then, 6 mL of the preculture was transferred to 250-mL flasks that contained 200 mL of MRS liquid medium and cultivated at 37 °C for 24 h during which the culture medium was sampled with time intervals. After cultivation, the cells and the supernatant were separated by centrifugation. The supernatant was used for pH detection. The harvested cells were washed by PSB buffer (50 mM Tris–HCl, 100 mM NaCl, pH 7.5) for three times, and suspended in an appropriate diluted ratio to be used for detection of fluorescence and enzyme activity.
Detection of cell growth and pH
The cell density was determined by measuring the OD600 with a UV-1800/PC spectrophotometer (MAPADA Instrument Co., Shanghai, Ltd., China). The pH was measured using a laboratory pH meter (Mettler Toledo FE20, Shanghai, China). Each trial was performed in triplicate.
Effect of carbon source, nitrogen source and pH
To analyze the effect of carbon and nitrogen sources, MRS medium with glucose was separately replaced by the equal weight of maltose, fructose and lactose while tryptone replaced by equal weight of soy peptone and polypeptone, respectively. To assay the effect of the pH, sodium hydroxide solution (3 mol/L) was used to control the pH. The strains were cultivated in the MRS medium with the initial pH of 6.5, 7.5, 8.0 and 9.0 to analyze the effect of the initial pH. Besides, to evaluate the effect of the pH during fermentation, the strains were cultivated in the MRS medium and then the pH were separately regulated to 5.5, 6.5 and 7.5 when the pH declined to 4.5 during cultivation. For the above cultivation, assays were done with the usual procedure.
Assay of GFP fluorescence
The fluorescence and the image of the harvested cells were separately detected by spectrophotometer and laser scanning confocal microscope as previously described [18]. All the data were averaged from three independent samples of the same time points.
Assay for NADH oxidase
Three milliliters of the preculture of the suitable strains were incubated in a 250-mL flask loading 100 mL MRS medium and cultivated in a shaker incubator at rotate speed of 120 r/min at 37 °C for 15 h. The sample was obtained at 3 h, 9 h and 15 h. After centrifugation, the supernatant and the cells were used for detection of pH and the activity of NADH oxidase, respectively. The harvested cells were disrupted on ice by a cell disruptor (Constant System Ltd., England) and then centrifugated for 10 min (4 °C, 10,000 r/min). The corresponding supernatant was used for the detection of the protein concentration and the enzyme activity according to the instruction of the NADH oxidase test kit and total protein quantitative assay kit (Jiancheng Bioengineering Institute, Nanjing, China). One unit of activity was defined as the absorbance increase equal to 0.01 in 1 min at 600 nm per milligram protein. The results are the averages of triplicate assays.
Availability of data and materials
All data used this study are included in this published article.
References
van Tilburg AY, Cao H, van der Meulen SB, Solopova A, Kuipers OP. Metabolic engineering and synthetic biology employing Lactococcus lactis and Bacillus subtilis cell factories. Curr Opin Biotechnol. 2019;59:1–7.
Navale GR, Dharne MS, Shinde SS. Metabolic engineering and synthetic biology for isoprenoid production in Escherichia coli and Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2021;105:457–75.
Liu Y, Liu L, Li J, Du G, Chen J. Synthetic biology toolbox and chassis development in Bacillus subtilis. Trends Biotechnol. 2019;37:548–62.
Douglas GL, Goh YJ, Klaenhammer TR. Integrative food grade expression system for lactic acid bacteria. Methods Mol Biol. 2011;765:373–87.
Kleerebezem M, Hols P, Hugenholtz J. Lactic acid bacteria as a cell factory: rerouting of carbon metabolism in Lactococcus lactis by metabolic engineering. Enzyme Microb Technol. 2000;26:840–8.
Reuter MA, Hanniffy S, Wells JM. Expression and delivery of heterologous antigens using lactic acid bacteria. Methods Mol Med. 2003;87:101–14.
Gackowska L, Michalkiewicz J, Krotkiewski M, Helmin-Basa A, Kubiszewska I, Dzierzanowska D. Combined effect of different lactic acid bacteria strains on the mode of cytokines pattern expression in human peripheral blood mononuclear cells. J Physiol Pharmacol. 2006;57(Suppl 9):13–21.
Sybesma W, Starrenburg M, Kleerebezem M, Mierau I, de Vos WM, Hugenholtz J. Increased production of folate by metabolic engineering of Lactococcus lactis. Appl Environ Microbiol. 2003;69:3069–76.
Mierau I, Kleerebezem M. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol. 2005;68:705–17.
Landete JM. A review of food-grade vectors in lactic acid bacteria: from the laboratory to their application. Crit Rev Biotechnol. 2017;37:296–308.
Tauer C, Heinl S, Egger E, Heiss S, Grabherr R. Tuning constitutive recombinant gene expression in Lactobacillus plantarum. Microb Cell Fact. 2014;13.
Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–22.
Karsi A, Lawrence ML. Broad host range fluorescence and bioluminescence expression vectors for Gram-negative bacteria. Plasmid. 2007;57:286–95.
Wu W, Liu F, Singh S. Toward engineering E. coli with an autoregulatory system for lignin valorization. Proc Nat Acad Sci. 2018;115:2970–5.
Browning DF, Godfrey RE, Richards KL, Robinson C, Busby SJW. Exploitation of the Escherichia coli lac operon promoter for controlled recombinant protein production. Biochem Soc Trans. 2019;47:755–63.
Spangler JR, Caruana JC, Phillips DA, Walper SA. Broad range shuttle vector construction and promoter evaluation for the use of Lactobacillus plantarum WCFS1 as a microbial engineering platform. Synth Biol. 2019;4.
Guan C, Cui W, Cheng J, Zhou L, Guo J, Hu X, Xiao G, Zhou Z. Construction and development of an auto-regulatory gene expression system in Bacillus subtilis. Microb Cell Fact. 2015;14:150.
Guan C, Ma Y, Chen X, Zhao R, Huang X, Su J, Chen D, Lu Z, Li Q, Gu R. Broad-host-range application of the srfA promoter from Bacillus subtilis in Escherichia coli. J Microbiol Methods. 2020;168:105798.
Mays ZJS, Nair NU. Synthetic biology in probiotic lactic acid bacteria: at the frontier of living therapeutics. Curr Opin Biotechnol. 2018;53:224–31.
McCracken A, Turner MS, Giffard P, Hafner LM, Timms P. Analysis of promoter sequences from Lactobacillus and Lactococcus and their activity in several Lactobacillus species. Arch Microbiol. 2000;173:383–9.
Dzikowska A, Kacprzak M, Tomecki R, Koper M, Scazzocchio C, Weglenski P. Specific induction and carbon/nitrogen repression of arginine catabolism gene of Aspergillus nidulans-functional in vivo analysis of the otaA promoter. Fungal Genet Biol. 2003;38:175–86.
Bren A, Park JO, Towbin BD, Dekel E, Rabinowitz JD, Alon U. Glucose becomes one of the worst carbon sources for E-coli on poor nitrogen sources due to suboptimal levels of cAMP. Sci Rep. 2016;6.
Schulz AA, Collett HJ, Reid SJ. Nitrogen and carbon regulation of glutamine synthetase and glutamate synthase in Corynebacterium glutamicum ATCC 13032. FEMS Microbiol Lett. 2001;205:361–7.
Mongkolsuk S, Helmann JD. Regulation of inducible peroxide stress responses. Mol Microbiol. 2002;45:9–15.
Hayashi K, Ohsawa T, Kobayashi K, Ogasawara N, Ogura M. The H2O2 stress-responsive regulator PerR positively regulates srfA expression in Bacillus subtilis. J Bacteriol. 2005;187:6659–67.
Patel N, Shahane S, Shivam MR, Mishra U. Mode of action, properties, production, and application of laccase: a review. Recent Pat Biotechnol. 2019;13:19–32.
Liang X, Zhang L, Zhong J, Huan L. Secretory expression of a heterologous nattokinase in Lactococcus lactis. Appl Microbiol Biotechnol. 2007;75:95–101.
Kumar R, Kaur J, Jain S, Kumar A. Optimization of laccase production from Aspergillus flavus by design of experiment technique: partial purification and characterization. J Genet Eng Biotechnol. 2016;14:125–31.
Wu C, Huang J, Zhou R. Progress in engineering acid stress resistance of lactic acid bacteria. Appl Microbiol Biotechnol. 2014;98:1055–63.
Guo T, Kong J, Zhang L, Zhang C, Hu S. Fine tuning of the lactate and diacetyl production through promoter engineering in Lactococcus lactis. PLoS ONE. 2012;7:e36296.
Nevoigt E, Kohnke J, Fischer CR, Alper H, Stahl U, Stephanopoulos G. Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl Environ Microbiol. 2006;72:5266–73.
Levdikov VM, Blagova E, Young VL, Belitsky BR, Lebedev A, Sonenshein AL, Wilkinson AJ. Structure of the branched-chain amino acid and GTP-sensing global regulator, CodY, from Bacillus subtilis. J Biol Chem. 2017;292:2714–28.
den Hengst CD, van Hijum SA, Geurts JM, Nauta A, Kok J, Kuipers OP. The Lactococcus lactis CodY regulon: identification of a conserved cis-regulatory element. J Biol Chem. 2005;280:34332–42.
Papagianni M. Metabolic engineering of lactic acid bacteria for the production of industrially important compounds. Comput Struct Biotechnol J. 2012;3:e201210003.
Mierau I, Olieman K, Mond J, Smid EJ. Optimization of the Lactococcus lactis nisin-controlled gene expression system NICE for industrial applications. Microb Cell Fact. 2005;4.
Llull D, Poquet I. New expression system tightly controlled by zinc availability in Lactococcus lactis. Appl Environ Microbiol. 2004;70:5398–406.
Ma SJ, Li K, Li XS, Guo XQ, Fu PF, Yang MF, Chen HY. Expression of bioactive porcine interferon-alpha in Lactobacillus casei. World J Microbiol Biotechnol. 2014;30:2379–86.
Jorgensen CM, Vrang A, Madsen SM. Recombinant protein expression in Lactococcus lactis using the P170 expression system. Fems Microbiol Lett. 2014;351:170–8.
Rud I, Jensen PR, Naterstad K, Axelsson L. A synthetic promoter library for constitutive gene expression in Lactobacillus plantarum. Microbiology. 2006;152:1011–9.
Xu N, Wei L, Liu J. Recent advances in the applications of promoter engineering for the optimization of metabolite biosynthesis. World J Microbiol Biotechnol. 2019;35:33.
Acknowledgements
The authors want to thank Zhang Y.Z. (Yancheng Institute of Technology, Jiangsu, China) for giving us the vector pColdII-Z.
Funding
The investigation was supported by a Project Funded by the Natural Science Foundation of Jiangsu Province (BK20170496), the National Natural Science Foundation of China (31700079) and the fund of China Scholarship Council.
Author information
Authors and Affiliations
Contributions
CG supervised the experiment and wrote the draft manuscript, YY accomplished the determination of the recombinant expression system and analyzed the data; YM constructed the recombinant vectors; RG and CZ participated in the detection of gene expression; XW polished the draft manuscript; ML and DC conceived of the study, participated in its design, and coordination. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guan, C., Yuan, Y., Ma, Y. et al. Development of a novel expression system in lactic acid bacteria controlled by a broad-host-range promoter PsrfA. Microb Cell Fact 21, 23 (2022). https://doi.org/10.1186/s12934-022-01754-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-022-01754-z