
Abstract
Diabetes mellitus is one of the prime risk factors for cardiovascular complications and is linked with high morbidity and mortality. Diabetic cardiomyopathy (DCM) often manifests as reduced cardiac contractility, myocardial fibrosis, diastolic dysfunction, and chronic heart failure. Inflammation, changes in calcium (Ca2+) handling and cardiomyocyte loss are often implicated in the development and progression of DCM. Although the existence of DCM was established nearly four decades ago, the exact mechanisms underlying this disease pathophysiology is constantly evolving. Furthermore, the complex pathophysiology of DCM is linked with exosomes, which has recently shown to facilitate intercellular (cell-to-cell) communication through biomolecules such as micro RNA (miRNA), proteins, enzymes, cell surface receptors, growth factors, cytokines, and lipids. Inflammatory response and Ca2+ signaling are interrelated and DCM has been known to adversely affect many of these signaling molecules either qualitatively and/or quantitatively. In this literature review, we have demonstrated that Ca2+ regulators are tightly controlled at different molecular and cellular levels during various biological processes in the heart. Inflammatory mediators, miRNA and exosomes are shown to interact with these regulators, however how these mediators are linked to Ca2+ handling during DCM pathogenesis remains elusive. Thus, further investigations are needed to understand the mechanisms to restore cardiac Ca2+ homeostasis and function, and to serve as potential therapeutic targets in the treatment of DCM.
Similar content being viewed by others

Introduction
Diabetic cardiomyopathy (DCM) is one of the end-stage consequences of mortality and morbidity in patients with diabetes mellitus. Diabetes stimulates chronic inflammation, alters Ca2+ homeostasis, activates cardiac fibroblast transformation into myofibroblast leading to left ventricular dysfunction and worsening clinical outcomes [1, 2]. Cardiac function is partly dependent on the rhythmic contractions of cardiac muscle, which incessantly goes through contraction and relaxation cycles. As cardiac contractility is regulated by the intracellular calcium concentrations [Ca2+]i, which also changes during contraction/relaxation (systolic and diastolic) cycles, the regulators of [Ca2+]i are the major determinants of cardiac function. In the ventricular myocyte, Ca2+ moves around the sarcoplasmic reticulum, mitochondrial membrane and sarcolemma through different ion channels and ion transporters (Fig. 1). During myocardial excitation–contraction (EC) coupling, extracellular Ca2+ moves into the cardiomyocyte via L-type voltage-dependent Ca2+ channels (LTCC) and reverse sodium-calcium (Na+/Ca2+) exchanger [3, 4]. This influx of Ca2+ serves as a trigger and induces release of Ca2+ from the sarcoplasmic reticulum (intracellular Ca2+ store), through the ryanodine receptors (RyR2), a process known as calcium-induced calcium release (CICR) [5, 6]. This sudden availability of cytosolic free Ca2+ in large amounts results in simultaneous Ca2+ binding to multiple cardiac troponin C molecules, which is part of the troponin complex attached to the thin filament that regulates myosin heavy chain (MHC) filament binding to the actin in the thin filament. When Ca2+ binds to troponin C, it results in activation of myofilaments eventually leading to contraction. Cytosolic Ca2+ concentration must decline before the occurrence of relaxation and diastolic filling. Therefore, as soon as cytosolic Ca2+ dissociates from troponin C, the Ca2+ is cleared from the cytosol leading to the termination of contraction. Four different transporters remove Ca2+ from the cytosol: (i) the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a), (ii) sarcolemmal Na+/Ca2+ exchanger, (iii) sarcolemmal/plasma membrane Ca2+-ATPase, and (iv) the mitochondrial Ca2+ uniporter (MCU).

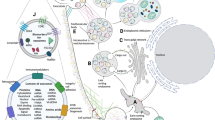
Potential effects of inflammation, exosomes, and microRNA (miRNA) on Ca2+ transport, storage and mitochondrial Ca2+ handling. Excitation–contraction (EC) coupling is initiated by an action potential which depolarizes the sarcolemma by rapid sodium (Na+) influx. Depolarization activates voltage-gated L-type Ca2+ channels (LTCC), and Ca2+ influx triggering calcium-induced calcium release (CICR) from the sarcoplasmic reticulum (SR) via the ryanodine receptor (RyR2). Rapid release of Ca2+ from the SR increases free intracellular Ca2+, enabling muscle contraction. Cardiomyocyte relaxation is regulated by signaling pathways that restore intracellular and SR Ca2+ to resting concentrations. Ca2+-activated kinases phosphorylate phospholamban (PLB), relieving its repression on Sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2a). Consequently, SERCA2a rapidly imports Ca2+ into the SR, decreasing the intracellular Ca2+ concentration. Na+/Ca2+ exchangers (NCX) are allosterically activated by Ca2+ and aid in restoring resting Ca2+ concentrations; decreased cytosolic Ca2+ leads to relaxation of the sarcomere. Genes downregulated in DCM are denoted by a blue downward arrow and genes upregulated during DCM are denoted by a red upward arrow. Mitochondria is an energy mobilization and Ca2+-buffering organelle. The Ca2+ homeostasis is controlled by its uptake through the mitochondrial Ca2+ uniporter (MCU) complex and voltage-dependent channel proteins, Ca2+ efflux is controlled by NCX. Exosomes and miRNAs control the gene expression of certain inflammatory cytokines, Ca2+ handling and signaling proteins. DHPR Dihydropyridine receptor; BIN1 bridging integrator 1; PMCA Sarcolemmal/plasma membrane Ca2+-ATPase; CSQ calsequestrin, mNCX Mitochondrial N+/Ca2+ exchanger; TNF-α Tumor necrosis factor-α; IL1b Interleukin 1β
Pathophysiological alterations during the early stages of DCM involve asymptomatic left ventricular dysfunction with a near normal ejection fraction, which eventually progresses to impaired cardiac contractility and detrimental arrhythmias. Table 1 summarizes cardiac structural and functional changes observed in clinical studies and in different animal models in two forms of diabetes mellitus (insulin-dependent diabetes or Type I and insulin-resistance diabetes or Type II). Given the multitude of Ca2+ handling proteins governing the Ca2+ transients in the cytosol of the cardiomyocyte, DCM has been known to adversely affect many of these Ca2+ handling proteins either qualitatively and/or quantitatively [7]. These changes are either adaptive or maladaptive in nature depending on the stage of the DCM. The etiology and pathophysiology of diabetes is very complex in nature, which is also reflected in contradicting observations made by various researchers, who have studied Ca2+ handling and subcellular organelle remodeling in DCM. Interestingly, disturbances in Ca2+ homeostasis is noticeable during the early stages of DCM, further emphasizing the detrimental role of Ca2+ in the pathophysiology of the DCM [8]. As demonstrated by Pereira et al. in the obese model of diabetes [9], defective Ca2+ influx through reduced LTCC resulted in reduced stimulation of Ca2+ currents, which further led to reduced Ca2+release from the endoplasmic reticulum. In addition to the attenuated Ca2+ current trigger, they showed chronic suppression of sarcoplasmic reticulum Ca2+ load due to the inhibition of SERCA2a leading to sarcoplasmic reticulum Ca2+ uptake and enhanced efflux of Ca2+ through Na+/Ca2+ exchanger, culminating in chronically reduced sarcoplasmic reticulum Ca2+ load leading to Ca2+ moving out of the cardiomyocytes.
Myocardial inflammation is one of the contributing factors for the development of DCM triggering many inflammatory signaling pathways. Moreover, abnormalities in Ca2+ homeostasis is involved in pathogenesis of cardiac inflammation that could be related to the increased Ca2+ signals and inflammatory responses [10]. Several proinflammatory cytokines, such as TNF-α, IL6, IL8, IL1β, and IL1 and other molecules such as IFNγ, chemokines (MCP-1, IL8 and biglycan) actively contribute to the myocardial oxidative stress, fibrosis, and cardiac dysfunction [11]. However, the effect of inflammation on Ca2+ signaling in DCM needs further investigation.
While the interplay of heart disease and diabetes is very complex, recent reports show that extracellular vesicles such as exosomes play a crucial role in the pathophysiology of DCM as well. Exosomes are nanosized vesicles which contain different types of cargo molecules: mRNAs, DNAs, proteins, lipids, miRNAs, released by the fusion of multivesicular body with the cell membrane [12]. Their function solely depends upon the origin of cell/tissue, and they play a critical role in angiogenesis, inflammation, and coagulation. Their beneficial role has been explored in various pathophysiological processes such as improving cardiac function, mitigating inflammatory response, and regulating immune responses [13].
In diabetes, structural composition and exosome cargo are modified as the original cells are altered by the diabetic milieu [14]. Recent studies demonstrate the role of heat shock protein 20 (Hsp20) in increasing the production of cardiomyocyte exosomes by interacting with Tumor Susceptibility 101 (TSG101), suggesting the contribution of pathogenic exosomes in the development of DCM [15]. Although there are few reports that suggest the role of exosome-mediated cellular communication in DCM, their actual role in pathophysiology of DCM remains unknown.
Rationale
In last couple of decades, numerous studies have been conducted in the space of DCM, but very limited information is available about the effect of Ca2+ signaling in DCM. In this review, we discuss the remodeling of subcellular organelles in DCM and how this remodeling potentially contributes to disruption in Ca2+ dynamics/homeostasis. We also review the effect of inflammation and potential role of exosomes in Ca2+ signaling and its impact on pathogenesis of DCM. Furthermore, we also briefly discuss the role of miRNA and its regulation of Ca2+ signaling in DCM.
Literature review methodology
The systematic search for all recent relevant literature was done using Pubmed (https://www.ncbi.nlm.nih.gov/PubMed), Google scholar (https://www.scholar.google.com) with the keywords such as Ca2+ signaling, EC coupling, diabetes-associated cardiomyopathy, myocardial inflammation, exosomes, Ca2+ reflux, sarcolemma, sarcoplasmic reticulum, Ca2+ binding proteins, effect of inflammation on Ca2+, and changes in mitochondria and extracellular matrix (ECM). Thorough screening of titles and abstracts was done to see the potential relevance. After identifying the deemed relevancy, full-fledged papers were reviewed in-detail to be considered for inclusion. English was the only publication language considered by the authors. Articles that were not peer-reviewed, were excluded, and were not considered. Additionally, we focused on articles published in the last 20 years and the selection was made based on their citation frequency. All the articles cited throughout the manuscript have been mentioned in the reference section along with their journal information.
I. DCM prevalence and risk factors
Diabetes mellitus is associated with increased cardiovascular complications, including hypertension, coronary artery disease and heart failure [16, 17]. However, there is increasing evidence of association of diabetes in development of primary myocardial disease known as DCM, characterized by abnormal myocardial structure, dilated, and impaired contraction of ventricles [18, 19]. Large population-based data show that heart failure occurs in approximately 19–26% of patients with diabetes [20, 21]. Moreover, studies have demonstrated an increase in the rate of heart failure is independent of other comorbidities such as obesity, hypertension, and other types of heart diseases [22, 23]. Furthermore, the data from Cardiovascular Health Study suggests detrimental cardiac remodeling in diabetic patients, as evidenced by increase in left ventricular mass and left ventricular wall thickness with diastolic and systolic dysfunction compared to normal individuals [24,25,26].
DCM can occur at any age, it can occur in children, adults and elderly [27]. The patients affected by DCM could present with various symptoms, including asymptomatic cardiomegaly, sudden death, peripheral edema and orthopnea [28].
DCM prevalence is increasing parallelly with the increase in diabetes mellitus. Diabetes is a complex disease characterized by impaired cardiac function because of imbalance in antioxidants and pro-oxidants at the cellular level. Also, high sugar diet induces cardiomyocyte autophagy, oxidative stress and fibrosis [29]. Although large evidence initially reported detrimental structural changes in the heart in diabetic patients with concomitant obesity and hypertension [30], studies have shown Type II diabetes mellitus independently increases left ventricular mass and causes detrimental cardiac remodeling by itself [31].
II. Sarcolemmal changes in DCM
IIa. L-type Ca2+ channels (LTCC)
The LTCC, CaV1.2 plays a key role in the initiation of the Ca2+ currents to kickstart the EC coupling [32, 33]. Therefore, the level of Ca2+ current generated eventually determines the intensity of the CICR and contributes to the extent of contractile force generated. It was shown that DCM due to both Type I and Type II diabetes involves contractile impairment. Consistent with this impairment, it was noted that the Ca2+ currents generated by the LTCC were also reduced. In the case of Type I diabetes, the surface density of this channel was reduced owing to the decreased trafficking to the cell surface [33]. In addition to lowered trafficking to the cell surface, the expression levels of CaV1.2 was also reduced in Type II diabetic mouse models [33]. Moreover, reports suggested that decline in caveolin-3 (Cav3), a known interactor of LTCC and organizer of the macromolecular complexes in the caveolae, may also contribute to the reduced presence of LTCC at the T-tubular membrane [8, 34]. Alternatively, it is possible that Cav3 depletion might have diminished the LTCC interactions with other signal transducers leading to diminished function [35]. Furthermore, CaV1.2, Ras-related G-proteins were also reported to interact with the LTCC and modulate its trafficking or function [36, 37]. However, the functional relevance of such interactions in DCM is not clear. Interestingly, it was demonstrated that LTCC negatively auto-regulates its expression through its truncated c-terminal fragment through a feedback mechanism [38]. It is unclear whether the lower LTCC expression in Type II diabetes is due to the negative after effects of a failed compensatory mechanism in response to the lack of sufficient contractile force (due to mounting demand).
IIb. Na+/Ca2+ exchanger
Although Na+/Ca2+ exchanger can function as a bidirectional Ca2+ pump, under normal physiological conditions, Na+/Ca2+ exchanger serves as the main Ca2+ extruder at the end of the EC coupling cycle [39]. Its dysfunction or depletion leads to Ca2+ overloading and contributes as one of the pathophysiologic mechanisms in DCM [40]. The diabetes-mediated regulation of Na+/Ca2+ exchanger activity is complicated, and accumulated evidence suggests that diabetes may increase or decrease or do not change the Na+/Ca2+ exchanger activity. Schaffer et al. in their experiments using insulin-independent diabetic rat (a condition generated by the injection of streptozotocin) has shown a decrease in cardiomyocyte sarcolemmal Na+/Ca2+ exchanger activity without affecting the mRNA levels [41]. In contrast to this report, Hattori et al. suggested that both the Na+/Ca2+ exchanger activity and mRNA levels were reduced in diabetes, while insulin supplementation reversed these effects [42]. This indicates that the lower Na+/Ca2+ exchanger function observed in diabetic myocytes maybe in part due to the quantitative decrease in Na+/Ca2+ exchanger machinery through decreased expression [42]. A recent study reported that Na+/Ca2+ exchanger levels were in fact elevated in the genetic models of Type I diabetic hearts [43]. However, in the obese diabetic models (db/db mice), the Na+/Ca2+ exchanger activity was not significantly altered [44]. Therefore, to reconcile the differences in observed effects, it might be worth determining if diabetes alters Na+/Ca2+ exchanger activity through epigenetic mechanisms in different diabetic models. In this context, diabetes was shown to enhance miRNA that target Na+/Ca2+ exchanger [45, 46]. Furthermore, changes in acetylation of transcription factor by histone deacetylases (HDACs) also contributes to alterations in the expression of Na+/Ca2+ exchanger at the transcriptional level [47]. Such regulation might play an important role in differential expression of Na+/Ca2+ exchanger in different diabetic models. Additional complexity might also exist, as it was demonstrated that diminished SERCA2a function also leads to elevations in the Na+/Ca2+ exchanger activity through Ca2+/calmodulin-dependent protein kinase (CaMK)/PKB/FoxO3a/miR-1 pathway and may lead to further deterioration of cardiac function in diabetes [48].
Direct post-translational modifications of Na+/Ca2+ exchanger may also regulate its activity. Consistent with this hypothesis, Na+/Ca2+ exchanger has been shown to form macromolecular complexes at its large intracellular loop, consisting of PKA, PKC and phosphatases (PP1 and PP2A). The complex seems to regulate phosphorylation status of Na+/Ca2+ exchanger and presumably its activity [49,50,51]. The possibility of existence of macromolecular complex with kinases and phosphatases suggest highly dynamic regulation of Na+/Ca2+ exchanger activity in relation to the [Ca2+]i levels. Such dynamic regulation is necessary, as Na+/Ca2+ exchanger is one of the major Ca2+ extruder channels, which regulate cardiomyocyte contraction ability and pathologic responses. Though the direct phosphorylation status was not assessed, it was shown that enhanced PKC activity was associated with the decline in Na+/Ca2+ exchanger activity in diabetes [52]. Nonetheless, the role of direct phosphorylation of Na+/Ca2+ exchanger and its effect on its activity during diabetes remains elusive. In summary, the decrease in Na+/Ca2+ exchanger activity observed in diabetic myocytes may be in part due to the quantitative decrease in functional Na+/Ca2+ exchanger machinery and decreased expression. However, other possible mechanisms might include reduced activation of PKCα and/or transfer of PKCβ and compositional changes in cell membrane phospholipids.
IIc. Sarcolemmal/Plasma membrane Ca2+-ATPase
The plasma membrane Ca2+-ATPase pump plays a less critical role in extrusion of the [Ca2+]i when compared to the Na+/Ca2+ exchanger. Takeda et al. have shown that decreased activity of sarcolemmal Ca2+ pump occurs earlier compared to sarcoplasmic reticulum Ca2+ pump activity and myofibrillar Ca2+ stimulated ATPase activity [53]. The investigation by Golfman et al. supports this hypothesis as well [54]. In contrast, Sheikh et al. did not observe any significant changes in the plasma membrane Ca2+-ATPase activity during DCM, specifically in the cardiac endothelial cells [55]. It is possible that different cell types adapt differently during the DCM. These diverging results demonstrate the limiting ability of the cell to release Ca2+ through Ca2+ pump and Na+/Ca2+ exchanger in sarcolemma, therefore initiating the [Ca2+]i overload contributing to detrimental cardiovascular outcomes.
IId. Sarcolemmal membrane changes
As discussed previously, the EC coupling starts through LTCC of the sarcolemma following the entry of Ca2+. The sarcolemma can bind to the Ca2+ and thus regulate Ca2+ exchanges during EC coupling cycles. In the heart, Ca2+ binding pool of sarcolemma is linked with the residues of sialic acid [56]. The lack of sarcolemmal sialic acid content was shown to enhance Ca2+ exchange and may impair precise regulation of EC coupling cycles [57]. Thus, sarcolemmal Ca2+ binding ability might be critically important for the normal functioning of the heart. Accordingly, a pathophysiological modulation in the efficiency of this superficial Ca2+ pool could influence the mechanical performance of the heart. In diabetic cardiomyopathies, similar conclusion has also been reported by Pierce et al. [58, 59]. In hearts of diabetic rats, the level of sialic acid was significantly downregulated in myocardial sarcolemmal membrane, which was reversible by insulin therapy. Suppression in Ca2+ binding may be partly due to the lower content of neuraminidase-sensitive sialic acid residues, since neuraminidase treatment also failed to reduce the Ca2+ binding activity [58]. Therefore, sarcolemmal defect may contribute to the precise regulation of Ca2+ transits through the sarcolemma during diabetes and may lead to improper EC coupling.
IIe. Maturation of cardiomyocytes
The normal functioning of the heart depends on a complex network of cells called cardiomyocytes, which exists in three-dimensional network of multiple cells and drive cardiac contractility. These cells are connected to the ECM produced by the supporting fibroblast cells, which transduces the force and coordinates with the contraction of the heart.
During maturation, the cardiomyocytes undergo several structural, metabolic, and physiological changes from conversion of fetal cardiomyocytes into the adult cardiomyocytes. Existing cardiomyocytes proliferate to regenerate the cardiomyocytes. Multiple factors are involved in progression of cardiomyocyte maturation [60, 61] but forced proliferation or maturation by inhibition/overexpression of cofactors, miRNA, molecules such as activated Yap (Yes-associated protein), cyclin B1-CDC2 complex, certain G1/S-phase molecules including CDK2, E2F1, cyclin D1 [62,63,64] may cause cardiac dysfunction. Therefore, it is very important to understand the tuning between proliferation and maturation to strategically design the parameters for enhancing cardiomyocyte regeneration and minimizing its side effects. The role of active cardiomyocytes is not well studied in the inflammatory responses underlying the DCM development and progression.
Diabetes-related inflammation induces mitochondrial dysfunction, impaired cardiomyocyte Ca2+ handling, oxidative stress, collagen-induced cardiomyocyte, and ECM stiffness. ECM accumulation at the cellular level in the heart leads to a cardiomyopathic phenotype resulting in heart failure with preserved ejection fraction (HFpEF) [65]. Moreover, homeostasis in myocardial tissue requires the balance between inflammatory damage and healing but diabetes mellitus promotes different inflammatory responses which further delays the healing process. Reduction in glycemic condition limits DCM and associated cardiac diseases as well. According to Tate et al., modification of normal glycemia with insulin reduced the collagen content, cardiomyocyte hypertrophy and controlled the progression of DCM in rats [66]. Additionally, abnormal expression of contractile and regulatory proteins contributes to impaired cardiac contraction. For example, phosphorylation of troponin is responsible for defective myocardial contractility since troponin and myosin both regulate the cardiomyocyte contraction [67].
Cardiomyocytes demand high energy due to its continuous contractions, which allows cells to utilize multiple substrates for energy production [68] in the heart. Previous reports suggests the reduction of glucose transporter (GLUT) 4 levels and depletion in glucose intake during hyperglycemia and insulin resistance [69]. The biopsies from the Type II diabetes patients also demonstrate significant reduction of GLUT4 and activation of PI3K/Akt signaling pathways at sarcolemma in diabetic mice and patients with non-insulin-dependent diabetes mellitus. Whereas, patients with left-ventricular dysfunction had limited activation of PI3K but not Akt and increased GLUT4 expression at sarcolemma [70].
Not only cardiomyocytes play a pivotal role in cardiac inflammation in DCM, cardiomyocytes exposed to excessive sugar/lipids generate a meta-inflammation like milieu as well [71]. The inflammasome activation is linked with the production of IL1β and IL18 which in turn induce the cardiomyocyte apoptosis [72].
IIf. Transverse tubule (T-tubule)
T-tubules are highly branched invaginations of the sarcolemma in ventricular myocytes that are rich in ion channels and play critical role in EC coupling, signal transduction, initiation and regulation of action potential, and maintenance of resting membrane potential. T-tubules are critical for normal cardiac physiology and reported to be structurally and functionally compromised during disease. Disorganized/lost T-tubules has been shown in animal models of heart failure [73], this loss results in systolic and diastolic dysfunction, disrupted Ca2+ homeostasis leading to loss of contractility in failing myocardium [74, 75]. The subcellular mechanisms of dysfunction in DCM have been extensively investigated and numerous studies have linked alterations in T-tubule structure and function with cardiac disease etiopathologies [76]. Typically, the disease progression includes the reduced T-tubule density [77], T-tubule dilatation, loss of tubule opening at the cell surface, appearance of broad T-tubule sheets and changed orientation [78]. In a recent report it has been demonstrated that the density of T-tubules remain unchanged specifically in diabetic patients with HFpEF, while increased in non-diabetic controls with HFpEF, and decreased in heart failure with reduced ejection fraction (HFrEF). The T-tubules were found to be dilated in all the heart failure entities [79]. Moreover, the disruption in T-tubules promotes the asynchronous and slower Ca2+ absorption and release in rodents with HFrEF which resulted in diastolic dysfunction in HFpEF and HFrEF with diabetes.
Due to limited available data on T-tubule comparative studies in diabetic heart, a new confocal based laser scanning method has been used to examine the labelling of T-tubules, which showed a modest decrease in T-Power (also known as sarcomere power) in diabetic cardiomyocytes [80]. Another study by Setterberg et al. demonstrated the association of reduced T-tubule density with the asynchronous EC coupling in diabetic cardiomyocytes [81].
III. Changes occurring in sarcoplasmic reticulum
The sarcoplasmic reticulum regulates [Ca2+]i and cardiac contractility, as it participates in Ca2+ release, reuptake, and storage during the Ca2+ transits associated with the cardiac contraction-relaxation cycles. Each of these functions are achieved by three-special class of proteins: (1) cytosolic Ca2+ spikers/releasers: sarcoplasmic reticulum Ca2+ release channels (RyR2); (2) Ca2+storage by the luminal Ca2+-binding proteins (histidine-rich Ca2+-binding protein, calsequestrin (CSQ), sarcalumenin and junctate); and (3) Ca2+ uptakers: SERCA2a pumps for Ca2+ reuptake. As proper Ca2+ storage, release and reuptake are essential for normal cardiac function, any qualitative and quantitative changes in these regulators lead to impaired cardiac function. Several studies have shown that sarcoplasmic reticulum function is adversely affected during diabetes. For instance, sarcoplasmic reticulum-release and uptake activities play a crucial role in regulation of [Ca2+]i, which are decreased in the diabetic heart.
IIIa. Sarcoplasmic reticulum Ca2+-ATPase (SERCA2a)
At the end of the EC coupling cycle, the [Ca2+]i has to be reset to low levels to facilitate cardiac dilatation and ventricular filling. SERCA2a, the major isoform that is present in the cardiac tissue, plays a central role in pumping >70% of the sarcoplasmic Ca2+ back to the sarcoplasmic reticulum lumen. SERCA2a is negatively regulated by a peptide known as phospholamban (PLN). The PLN association with SERCA2a is determined by the phosphorylation status, which causes its dissociation from SERCA2a leading to higher transportation rate of Ca2+ through the pump. Classically, β-adrenergic stimulation is involved in enhancing the heart function through the PKA-mediated PLN phosphorylation [82]. Interestingly, SERCA2a also exists as a multimeric protein complex involving several regulators of SERCA2a function, which interacts either directly or indirectly with the SERCA2a [83].
Multiple defects in the SERCA2a expression and function were observed during diabetes. It has been demonstrated that reduced activity and expression of SERCA2a contributes to the reduced Ca2+ return into the sarcoplasmic reticulum, leading to early stage diastolic dysfunction during diabetes [84]. Such chronic reduction in Ca2+ returns lead to depletion of sarcoplasmic reticulum Ca2+ reserves resulting in systolic dysfunction and heart failure. It was further demonstrated that SERCA2a upregulation can ameliorate the diabetic cardiac dysfunction, which restores the Ca2+ transient to the normal levels [85]. Recent studies have provided several mechanistic insights regarding the diminished SERCA2a function during diabetes. Despite controversial reports on the SERCA2a levels and quantitative changes in the phosphorylation status of the PLN, it was consistently demonstrated that SERCA2a function was diminished in Type I and Type II diabetes mellitus [86]. There were some commonalities that exist in these two scenarios. For instance, AGE (Advanced glycation end products) and their receptor RAGE (Receptor for AGE) are highly upregulated in diabetic hearts and were found to modify SERCA2a [87]. Hence, it is most probable that AGE [88] can also alter SERCA2a interactions in such a way that SERCA2a function is compromised during diabetes. Alternatively, oxidative stress might downregulate SERCA2a expression through inactivation of its transcription factor, Sp1 [86]. In addition, high glucose levels might enhance O-GlcNAcylation of Sp1 transcription factor, which is the crucial regulator of SERCA2a gene, ATP2A2, in the heart. Such alteration in Sp1 was shown to reduce its transcriptional activity. Furthermore, enhanced O-GlcNAcylation of PLN can also enhance its association with SERCA2a leading to inhibition [86]. Recently, a role for histone acetylation was noted in regulation of SERCA2a expression [89]. Given that HDACs contribute to adverse diabetic cardiac remodeling [90], it is plausible that there might be involvement of epigenetic regulation in SERCA2a expression. Additionally, a recent report has shown that cardiac-specific deletion in PKBα/β inhibits the insulin-dependent phosphorylation of striated muscle preferentially expressed protein kinase (SPEG) and SERCA2a inducing Ca2+ re-uptake by sarcoplasmic reticulum leading to cardiac dysfunction [91]. In summary, based on various assessments and studies, diabetes reduces SERCA2a function and activity at both cellular and protein levels and alters its interactions with its regulators leading to DCM.
IIIb. Ryanodine receptor 2 (RYR2)
RYR2 is a Ca2+ releasing protein present in the sarcoplasmic reticulum of the cardiomyocytes and is responsible for the Ca2+ sparks following the Ca2+ entry into the cytosol following membrane depolarization. Upon binding with the Ca2+ and entering the cytosol as part of Ca2+ currents, RYR2 is responsible for the Ca2+ sparks, known as CICR, which determines the extent of cardiac contraction. Thus, the extent of Ca2+ spike is dependent on the RYR2 function as well as the Ca2+ stores in the endoplasmic reticulum. As RYR2 function is critical in determining the free Ca2+ spikes in the cytosol, any disruptions in the RYR2 levels and/or function are associated with the diminished cardiac contractile response. In fact, diabetes has been reported to cause changes in RYR2 function leading to contractile dysfunction. During the early stages of dysfunction, there were arrhythmias owing to the Ca2+ leak from the sarcoplasmic reticulum and during the later stages, contractile dysfunction owing to the depleted RYR2 levels. Studies in the mouse models simulating cardiac lipid overload (a frequently noted abnormal shift to enhanced fatty acid oxidation, observed in the Type II diabetes mellitus patient’s heart) have noted that enhanced mitochondrial oxidative stress and lipid overload leads to RYR2 oxidation leading to Ca2+ leak and arrhythmias [92]. Interestingly, other studies also emphasized the significance of oxidative stress in cardiac arrhythmias and demonstrated that glucose intolerance leads to inappropriately enhanced Ca2+/calmodulin-dependent protein kinase II (CaMKII) mediated phosphorylation-dependent activation of RYR2, which is also dependent on the oxidative stress leading to Ca2+ leakage [93, 94]. During the later stages, depletion of the RYR2 levels and its stabilizer FKBP12.6 were noted, which potentially could lead to Ca2+ release during diastole, leading to abnormal contractility and Ca2+ loss [95,96,97]. Although much is known about the RYR2 interactors, less is known about the mechanisms of its downregulation and altered interactions during diabetes [98].
IIIc. Ca2+ binding proteins
The main Ca2+ binding/storage protein in the cardiac sarcoplasmic reticulum is the CSQ [99]. Interestingly, it was noted that diabetes-induced cardiac dysfunction also involves reduced levels of CSQ, and rescue of the CSQ levels were associated with the amelioration of the dysfunction [100]. However, others have not observed such changes in the cardiac tissue [101]. Nonetheless, CSQ polymorphisms were suggested to influence the risk of Type II diabetes mellitus in certain human populations [102]. Further studies are needed to demonstrate the relationship between CSQ and its role in DCM.
IV. Epicardial adipose tissue (EAT)
Epicardial adipose tissue (EAT) is a multifaceted fat depot that confers the mechanical protection to the coronary arteries from distortion and compression during the excitation and contraction of the myocardium [103,104,105]. EAT displays a higher rate of lipogenesis and fatty acid metabolism which are critical for the proper functioning of heart. EAT related functional and morphological changes are age and disease specific [106]. Recent evidence has shown that increased levels of EAT can induce various pathologies and can alter the Ca2+ handling that eventually lead to contractile dysfunction of cardiomyocytes [107]. Greulich et al. have reported the reduced contractility and Ca2+ activity in the cardiomyocytes isolated from the animals eating a high fat diet, as compared to the animals on normal diet [108]. Although the metabolic crosstalk between EAT and the Ca2+signaling in context of DCM is poorly understood, the existence of compelling evidence suggests the importance of correct functioning of EAT is required for proper Ca2+ signaling and cardiac activity. Thus, these interactions between EAT and Ca2+ signaling should be investigated on a larger scale to identify clinically relevant molecules that might uncover the novel pharmacologic interventions for the treatment of DCM.
V. Changes occurring in mitochondria
Although mitochondrial dysfunction has been implicated in the DCM nearly three decades ago, underlying mechanisms of functional and structural changes associated with DCM are not fully understood [109, 110]. The main features of such dysfunction include reduced energy production and excessive generation of reactive oxygen species (ROS). Interestingly, mitochondrial Ca2+ load influences these two interlinked processes to a certain extent. Optimal levels of mitochondrial Ca2+ leads to enhanced metabolism and ATP production leading to reduced ROS production [111]. However, Ca2+ overload leads to abnormal mitochondrial permeability transition pore (MPTP) opening, thereby reducing ATP production culminating in enhanced ROS production. Interestingly, excessive ROS production also leads to enhanced mitochondrial permeability leading to Ca2+ overload [112]. Excessive ROS also leads to [Ca2+]i overload through Ca2+ leak from RYR2 and reduced cellular efflux [113]. As ROS and Ca2+ overload can regulate each other in a positive reciprocal fashion and both can adversely open MPTP independently, it has been postulated that during diabetes and ischemic conditions, excessive ROS and mitochondrial Ca2+ overload jointly trigger mitochondrial death [111]. Consistent with this hypothesis, reduced ATP production in diabetic hearts was observed, presumably due to dysfunctional mitochondria. These findings suggest that mitochondrial Ca2+ load acts as a sensor of Ca2+ homeostasis in the cell, thereby triggering the pathological outcomes associated with diabetes. Diabetic cardiac mitochondrial dysfunction and reduced productivity also involves extensive remodeling of mitochondrial structure, lipid and protein composition. This has been evident in two different animal models of diabetes [114,115,116]. Although the abnormal remodeling in the mitochondria is well appreciated, the underlying cause for such changes and the possibility of reversal of these changes need to be established. Nonetheless, most of these changes might have occurred due to the sustained damage inflicted by the enhanced Ca2+ overload and ROS. It is interesting to note that certain degree of uncoupling of mitochondrial oxidative phosphorylation exists in different types of diabetes and ROS has been implicated in such uncoupling adaptations [117, 118]. Furthermore, in the animal models of diabetes, inducing exercise was shown to reverse cardiac mitochondrial dynamics to certain extent [119]. Also, others have reported that exercise enhances antioxidant capacity and potentially prevent mitochondrial Ca2+ overload, enhanced endoplasmic reticulum Ca2+ uptake and reduced endoplasmic reticulum Ca2+ leak [120]. In this context, it is of great significance to develop exercise mimetics to better translate exercise-induced diabetic cardiac benefits, especially for advanced DCM patients.
In addition to the above findings, the mitochondrial Ca2+depletion was also proposed to cause mitochondrial defects leading to DCM. The mitochondrial Ca2+ uptake is mainly regulated by the MCU. The MCU activity is highly tissue-specific, which is tightly controlled by the active cells with intensive cytosolic Ca2+ signaling required for the integrity of the mitochondria [121]. Among others, one of the physiological roles of MCU complex is controlling the ATP production through activation of Ca2+-dependent dehydrogenase in the mitochondrial matrix, the manipulation in any of the MCU components could alter the pyruvate dehydrogenase activity and intracellular ATP levels in various human cells such as HeLa cells [122], and pancreatic β cells [123]. Alteration of MCU complex could also modulate the cellular metabolism [124], its presence and/or absence also controls the cell death [125, 126]. The inhibition and/or overexpression of MCU leads to abnormal pathophysiological disease states such as [127] reduced cardiac performance and enhanced energy demand [128, 129], alter the beat-to-beat amplitude of cytoplasmic Ca2+ oscillations [130]. It has been noted that higher glucose levels was associated with reduced MCU levels, which might be the reason behind the reduced mitochondrial Ca2+ levels [131]. Furthermore, it was shown that restoration of the MCU levels in the cardiomyocytes resulted in amelioration of mitochondrial metabolic deficiencies and heightened oxidative stress induced by high glucose levels [131]. Alternatively, enhanced mitochondrial Ca2+efflux might also lead to mitochondrial Ca2+depletion most likely due to increased Na+ levels (as discussed below). Although further understanding is necessary, it is possible that both mitochondrial Ca2+ depletion and overload might be occurring at different stages of diabetes, the former in the early stages and the later in the late stages of diabetes.
VI. Changes occurring in the myofibrils
Myofibrils are the structural units responsible for contraction and relaxation cycles and occupy more than half of the total volume of myocardial cells. The myofibrils participate at the end stage of the EC coupling process and are made of thin (actin) and thick (myosin) filaments apart from other regulatory and supportive proteins such as tropomyosin (TM), troponins, and titin. The major function of cardiomyocyte involves cyclic contraction and relaxation that is tightly regulated by complex interaction of contractile proteins and different membrane proteins in the heart, a process known as EC coupling [132]. During EC coupling following stimulation, the release of Ca2+ from sarcoplasmic reticulum to the myofilament and binds to troponin C, for a conformational change in the location of TM on actin, thereby exposing the myosin-binding site. During muscle relaxation, TM blocks the myosin-binding site on actin when the cytoplasmic levels of Ca2+ is low. It has been demonstrated that the expression of myofibrillar proteins and their isoforms is highly regulated and dynamically changed depending on the age, species, physiological and pathological conditions, including diabetes mellitus [133,134,135]. Previous reports confirm the relationship between myosin ATPase activity, the maximum velocity of shortening, myosin isoenzyme composition and speed of cardiac muscle shortening in rat hearts [136, 137].
Two main features of diabetic hearts with regard to the functional decline include- reduced Ca2+ sensing ability of the regulatory proteins of the actomyosin system and myosin isoform shift [67]. The decline or lack of MHC was associated with decline in functional efficiency [67, 138]. Some of these changes (enhanced isoform switching from the normally expressed α-MHC to the fetal isoform β-MHC) can be noticed even before the overt diabetic cardiac phenotype [139]. Perhaps, in rodents, this isotype switching could be an adaptive mechanism in response to the mitochondrial dysfunction and lower ATP production, as these isoforms, α-MHC and β-MHC generate same force, but the former consumes more ATP [133, 140]. The regulatory mechanism of MHC isoform switch in diabetes might be similar to that of thyroid dysfunction [141]; in diabetes and hypothyroid state, the antisense RNA and α-MHC transcription are turned-off. Insulin treatment in different animal models was shown to reverse the decreased myofibrillar ATPase activity and MHC isoform changes [142].
Apart from changes in the isotype switching, alterations in posttranslational modifications of the regulatory proteins of actomyosin formation have been involved in cardiac dysfunction during diabetes. The activities of depressed myofibrillar ATPase may be related to changes in the cardiac troponin I subunits of diabetic hearts, as cardiac troponin I phosphorylation is reported to modify the ATPase activity. Previous investigations suggests the increase (by 40%) in cardiac troponin I phosphorylation in the diabetic hearts, which can be reversed by administration of insulin [143]. Furthermore, PKC mediated cardiac troponin I phosphorylation was shown to reduce Ca2+ sensitivity and force generation, which is higher during diabetes due to [Ca2+]i depletion and enhanced PKC phosphorylation [144,145,146]. Like cardiac troponin I, phosphorylation changes in myosin light chain (MLC) protein may also have a modulatory role in altering the myofibrillar ATPase activity [147]. In this regard, Liu et al. [147] reported that in the diabetic rat heart, the protein contents of MLC, MLC-kinase and MLC phosphorylation were significantly decreased (40% to 45% and 30% to 45%, respectively), and insulin administration can reverse these changes. Phosphorylation of MLC at a regulatory site near to the binding domain of calmodulin increased the concentration of Ca2+/calmodulin required for MLC kinase activity, whereas non-phosphorylated MLC have opposite effect in diabetic heart, which may partially explain the reason of decreased myofibrillar ATPase activity and impaired contractile function [148]. All these observations highlight the central role of myofibrillar protein changes and their post-translational modifications in diabetes and thus revealing their contribution in decreased cardiac contractility.
VII. Role of Na+ in Ca2+ signaling
Intracellular Na+ levels not only regulate osmotic strength, but also contribute to the net positivity of the cell. Because of these reasons, cells use Na+ flux to regulate Ca2+ transits, often in an opposite direction. Recent report suggests that increased dependency of the diabetic hearts on the Na+-glucose cotransporter (SGLT) for glucose uptake resulted in Na+ overload in the hearts, which has been postulated to contribute to the arrhythmia and enhanced oxidative stress [149]. In another study, it was found that Na+/H+ exchanger could potentially contribute to the Na+ overload in the presence of high glucose levels [150]. While elevated levels/function of the SGLT and Na+/H+ exchanger are the main cause for Na+ overload during Type II diabetes, the decreased activity of Na+/K+ pump and Na+/Ca2+ exchanger contributes to the Na+ overload during Type I diabetes [150, 151]. Nonetheless, the downstream adverse effects of Na+ overload seem to be common in these models of diabetes.
The enhanced intracellular Na+ levels/Na+ overload might cause efflux of Ca2+ from the mitochondria through the mitochondrial Na+/Ca2+ exchanger and thus may deprive the Ca2+ mediated enhancement in oxidative phosphorylation. It has been known that moderate increments in the mitochondrial Ca2+ can enhance activities of dehydrogenases and ATP synthase culminating in enhanced ATP production [152]. Hence, Na+ overload might cause cardiac contractile dysfunction through reduced mitochondrial Ca2+ levels and ATP production.
VIII. The effect of inflammation on Ca2+ signaling
The landmark studies correlating inflammation with diabetes were conducted in early 90’s by Hotamisligil group, who reported the critical role of TNF-α in obesity and Type II diabetes [153]. Following this, several investigators have studied inflammation in relation to Type II diabetes by measuring the circulating concentrations of inflammatory markers/mediators [154]. Studies conducted in human and animal models in the past decade have supported this correlation by providing further evidences for the role of inflammation in initiation, development and progression of diabetes [155]. A recent study has shown association of activated pro-inflammatory pathways in response to insulin action with obesity and other metabolic disorders including Type II diabetes [156]. Inflamed β cell pancreatic islets also known as insulitis is a characteristic feature of Type I diabetes [157]. A study by Anderson et al., suggests the failure of central and peripheral immune tolerance results in the activation of autoreactive T cells in diabetic mice [157]. Involvement of potential inflammatory pathways in pathophysiology of diabetes has advanced the interest of targeting inflammation/inflammatory biomarkers for the prevention and control of diabetes .
Cardiac inflammation contributes to cardiomyocyte loss, fibrosis, and dysfunction leading to DCM [24, 158]. Various molecular mechanisms associated with diabetic myocardial inflammation is related to activation of NF-κB signaling pathway and the renin–angiotensin–aldosterone system [159]. Several neurohormones and pro-inflammatory molecules, such as IL6 and IL8, TNF-α, monocyte chemotactic protein 1 (MCP1), adhesion molecule intercellular adhesion molecule 1 (ICAM1), and vascular cell adhesion molecule 1 (VCAM1), actively contribute to the myocardial oxidative stress, fibrosis, and cardiac dysfunction [11, 160, 161]. The increased levels of these inflammatory responses in the heart have been shown to directly influence cardiac function through multiple mechanisms. The resulting inflammatory mediators alter intracellular signaling mechanisms in cardiomyocytes for the development of DCM. Myocardial overexpression of TNF-α transgenic mice resulted in heart failure due to Ca2+ handling defects and cardiac dilation [160].
Inflammatory cytokines such as TNF-α and IL1β has shown to decrease the expression of Ca2+-regulating genes (SERCA2a and Ca2+ release channel) thus leading to a negative ionotropic effect due to modification in [Ca2+]i homeostasis in adult cardiomyocytes [162]. Abnormalities in sarcoplasmic reticulum Ca2+ release promote myocardial remodeling (hypertrophy, substantial fibrosis, ventricular dilation, pump failure) resulting in heart failure due to pressure overload. These findings demonstrate that inflammation-triggered Ca2+ imbalance can contribute to cardiac remodeling [163]. Acute exposure to IL1β caused NLRP3-signaling activation and CaMKII-dependent RyR2/PLN hyperphosphorylation and enhanced spontaneous sarcoplasmic reticulum Ca2+-release events in both postoperative atrial fibrillation cardiomyocytes and HL-1-cardiomyocytes [164]. Previous studies have also shown that murine Ca2+-sensing receptor (CASR) activates the NLRP3 inflammasome which is mediated by the increase in [Ca2+]i and decrease in cellular cyclic AMP [165]. The pressure overload activates the CaMKIIδ which in turn triggers the inflammatory gene response and activates the NLRP3 inflammasome in cardiomyocytes [166]. Recently, TNF-α signaling pathway in cardiomyocytes were associated with Ca2+ signaling in Type II diabetes obese (db/db) mice. TNF-α upregulated transient [Ca2+]i amplitude and expedited its decay without changing the sarcoplasmic reticulum Ca2+ load or its spark frequency in mice [167]. Also, alteration of Ca2+ signaling and TNF-α, is gender specific, displaying the increase in TNF-α cardio-protective effect in male mice.
On the other hand, it is not clear whether Ca2+ signaling affects inflammatory response. Previous report has shown that the regulation of cell proliferation, energy, cell death of T-cells [168] and different steps of the inflammatory responses are associated with active Ca2+ [169,170,171,172]. CAMKII is known as the key regulator of the generation of inflammatory response, as it can act as a [Ca2+]i sensor [173,174,175]. Cardiac stress activates the NF-κB-dependent inflammatory transcription pathway and oxidative injury, whereas the CaMKII is triggered by oxidation [176]. Singh et al. has also shown the association of increased CAMKII activity and inflammation in heart failure [177]. The increased levels of TNF-α during ischemia/reperfusion injury is related to Ca2+ overload resulting in cardiac dysfunction [178, 179]. The different forms of CAMKII have been shown to display distinct functions such as Ca2+-independent form can enhance the formation of T-cells and help in modulation of cell death [180]. CaMKII regulates the production of IL2, IL4, and IL10 by T-lymphocytes, which is also involved in the Ca2+-dependent IL2 transcriptional arrest [174, 181]. Studies on macrophages suggested role of CAMKII as a booster of pro-inflammatory cytokines and production of interferons on stimulation with toll-like receptors (TLRs) [182]. Moreover, the previous studies suggests that G protein-coupled CaSR-dependent inflammation activates the NLRP3 inflammasome that induces the maturation and secretion of IL1β but the inhibition of ERK pathway reduced the activation of CaSR-dependent NLRP3 inflammasome [183]. Increased levels of Ca2+ through CaSR can stimulate multiprotein inflammasomes which helps in the maturation of proinflammatory cytokine IL1β through Caspase-1, making CaSR a promoter and responder of inflammation [184].
In vascular smooth muscle cells, angiotensin II induces the activation of NLRP3 inflammasome associated with CaSR and collagen synthesis, the inhibition of both CaSR and NLRP3 inflammasome promotes the secretion of proinflammatory cytokines [185]. Additionally, in monocytes and macrophages, CaSR can activate the NLRP3 inflammasome mediated by the increase in [Ca2+]i [186].
IX. Epigenetic regulation of Ca2+ mediated processes
Epigenetic mechanisms such as histone modification and DNA methylation regulate gene expression and play an important role in different cellular processes.
Histone modification has a major impact on chromatin structure and gene expression. Of the several types, histone acetylation is the most widely studied and robustly associated modification. It is regulated by histone acetyl transferases (HATs) and HDACs. HDACs are known as the key modulators that controls the proteostasis by changing the acetylation status; altered proteostasis has been studied in cardiovascular diseases including hypertrophy, heart failure in the past [187, 188]. According to a study by Chen et al. in diabetic mice, reduction in HDACs attenuated the cardiac hypertrophy and fibrosis in diabetic heart disease and inhibited the apoptosis by increasing the GLUT1 acetylation and decreased Caspase-3 activity [189]. Whereas in another study, increased HDAC levels led to myocardial ischemia and reduced mitochondrial dysfunction in diabetic heart [190]. Recent study showed the role of HDAC in regulation of Na+/Ca2+ exchanger, which is responsible for Ca2+ flux and efflux in cardiomyocytes [191, 192]. In porcine model of heart failure, the downregulation of HDAC affected the potassium channels [193].
DNA methylation regulates gene expression by altering the DNA stability, chromosomal structure, and DNA conformation. It works in proximity of histone modifications and miRNA to regulate the transcription. DNA methylation is catalyzed by a set of DNA methyltransferases (DNMTs) and previous studies suggest an important role of DNMTs in maintaining the homeostasis of cardiomyocytes in normal and stressed conditions [194]. In a study by Kumar et al., decreased Sirtuin 1 (SIRT1) and DNMT3b activity could increase the levels of histone H3 acetylation and CpG demethylation in diabetes-induced oxidative stress [195] and ROS-mediated stress, which ultimately lead to myocardial inflammation [196]. Reduced global methylation and increased hypomethylation have been previously associated with the development of atherosclerosis and other cardiac complications [197,198,199].
Growing evidence supports the hypothesis that during diabetes, the heart goes through epigenetic reprogramming. Although much has not been studied about these processes in pathogenesis of DCM, emerging evidence suggests that they might play a crucial role. Once identified, these epigenetic regulatory mechanisms could act as a potential target for drug discovery but detailed elucidation of these mechanisms in pathogenesis/manifestation of DCM needs further elucidation.
X. Exosome and micro-RNA regulation of Ca2+ dynamics
Exosomes are nanosized extracellular vesicles (40–140 nm in diameter) that originate from multivesicular bodies, secreted by different cells, and found in body fluids such as plasma, saliva, urine, and serum. Exosomes play crucial role in intercellular communication by promoting the transport of macromolecules such as miRNA, noncoding RNA, DNA, lipids and proteins between the cells [200, 201].
Tissue microenvironment, including diabetes-induced effects modulate exosome cargo and has been shown to regulate communication between various cardiac cells (cardiomyocytes, fibroblasts, and endothelial cells) and among the heart and peripheral tissues/organs such as bone marrow, lungs, vasculature, kidney, and immune cells [202]. Our recent report demonstrates that under diabetic conditions, macrophage-secreted exosomes are enriched in HuR (mRNA-stabilizing protein) which activates profibrogenetic response in the heart [203]. miRNA has emerged as a key regulator of gene expression at the post-transcriptional level and has been shown to regulate several cardiac pathologic changes [204,205,206]. Different reports suggest the role of miRNAs in controlling the gene expression of certain inflammatory cytokines, Ca2+ handling and signaling proteins [46, 207]. Yildirim et al. found the upregulated expression of muscle-specific miRNA-1 in diabetic heart due to its binding to [208] molecular target Junctin (a key component of RyR2 Ca2+ release channel complex). Previous studies have already shown the role of miRNA-1 in impairing the cardiac relaxation and induction of cardiac hypertrophy and arrhythmia [209, 210]. Wahlquist et al. has reported that use of miRNA-25 impairs the Ca2+ uptake and aggravates cardiac dysfunction by interacting with SERCA2a [211]. The overexpression and inhibition of miRNA-1 influences Ca2+ flux in cardiomyocytes [212]. The increase in [Ca2+]i concentration directly upregulates the expression of certain apoptotic genes. miRNA-145 could inhibit the Ca2+ overload and the overexpression of miRNA-145 has a protective effect against the ROS-induced cardiomyocyte apoptosis [213, 214]. miRNA-25 [38], miRNA-1, miRNA-138, miRNA-133a and miRNA-214 have been shown to influence mitochondrial Ca2+ homeostasis [213]. Macrophage-derived miRNA-155 promotes cardiac inflammation by inducing the secretion of inflammatory cytokines such as IL1β, IL6 and TNF-α [215]. It is plausible that exosomes carrying this miRNA might alter Ca2+ signaling in target cells like cardiomyocytes in DCM. However, very little is known about such a possibility. Mayourian et al. [216], reported that MSC-derived exosomal miRNA-21-5p could improve the contractile force of ischemic cardiomyocytes by regulating the Ca2+ homeostasis [216]. Also, it is not known so far if Ca2+ signaling related proteins or transcripts are enriched in exosomes under pathologic conditions. Interestingly, Ca2+ regulates multivesicular body formation and exosome release. For example, increased Munc13-4 (Ca2+-dependent Rab binding protein) is associated with increased Ca2+ uptake, multivesicular body formation and exosome release [217]. Whether DCM-induced alterations in Ca2+ signaling could impact exosome biogenesis, release and cellular uptake needs further investigation.
XI. Therapeutic agents targeting Ca2 + dynamics in DCM
As this review clearly focusses on the importance of Ca2+ handling proteins in the development of hypertrophy and heart failure in DCM, we summarize here some of the novel therapeutic approaches targeting Ca2+ handling proteins in modulating this aberrant expression and activity to improve symptoms in patients with DCM.
By modulating expression of Ca2+ handling proteins, gene therapy targeting RyR2, SERCA2a and PLN is gaining fresh impetus recently, as one of the modalities to treat contractile dysfunction in DCM. S100A1 is a Ca2+ binding protein, which modulates RyR2 and SERCA2a activity [218]. In animal models of acute and chronic ischemic heart failure, adenoviral-based delivery of S100A1 has shown to improve cardiac function by restoring Ca2+ homeostasis [219,220,221,222,223]. Like this approach, several studies have explored the use of adenoviral delivery of AC6 in the setting of congestive heart failure as well. AC6 has shown to downregulate the expression of PLN [224]. In a pig model of heart failure, adenoviral delivery of AC6 mitigated adverse ventricular remodeling and improved cardiac function [225]. Following these promising results, Hammond et al. and Penny et al. conducted a randomized controlled clinical trial in 56 HFrEF patients and demonstrated improvement in ejection fraction 4 weeks after the delivery of AC6 adenoviral vector [ClinicalTrials.gov identifier NCT00787059] [226, 227]. Although both S100A1 and AC6 delivered viral vectors have shown beneficial effects in animal models and early clinical trials, gene therapy using these vectors requires further elucidation in large multi-center clinical trials. Additionally, direct gene transfer using adenoviral-associated SERCA2a has also shown to improve Ca2+ transients and cardiac function in animal models of heart failure [228,229,230,231]. Given that SERCA2a gene transfer improved outcomes in animal models of heart failure, AAV1.SERCA2a was administered in Phase I and Phase II clinical trials [ClincalTrials.gov identifiers NCT00534703 and NCT00454818] [232,233,234]. The results of the initial Phase I and IIa trials suggested that AAV1.SERCA2a treatment reduced the number of cardiac events in heart failure patients [232, 235]. With these initial promising results, clinical trials are using AAV1.SERCA2a vector, istaroxime, a dual functional lusoinotropic agent which stimulates SERCA2a and inhibits Na+/K+ ATPase [236]. Also, by inhibiting PLN-mediated SERCA2a activity through cAMP/PKA-dependent mechanism, istaroxime has shown to increase Ca2+ uptake into the SR [237]. Animal studies in heart failure have shown improvement in cardiac function using I.V. administration of istaroxime [238, 239]. Additionally, early clinical studies and HORIZON-HF concurred beneficial effects on cardiac contractility and showed reduction in diastolic stiffness in heart failure patients using istaroxime [240, 241]. Large scale clinical trials for istaroxime are currently on-going [ClincalTrials.gov identifiers NCT02617446 and NCT02477449].
Another interesting approach is by modifying SERCA2a and PLN using nitroxyl (HNO/NO−). BMS-986231 has undergone extensive clinical testing using HNO derivative. Early clinical trials have demonstrated BMS-986231 to be safe and efficacious in HFrEF patients [242] [ClincalTrials.gov identifier NCT02157506]. Based on these promising results, currently, 3 clinical trials are on-going to address the efficacy and safety of BMS-986231 in patients with various forms of heart failure [ClincalTrials.gov identifiers NCT03016325, NCT03357731, and NCT03730961] [243].
Conclusions
DCM involves failure of Ca2+ handling at multiple transports and ion channels located at sarcolemma, sarcoplasmic reticulum, and mitochondria (Fig. 2). Though there are discrepancies regarding the level of involvement in each of the above discussed Ca2+ transit regulators, it is evident that Ca2+ mishandling remains the major cause in the development of DCM. Inflammatory mediators and miRNAs are shown to interact with regulators of Ca2+ signaling as well. Some of these miRNAs and inflammatory mediators are shown to be enriched in exosomes, however, it’s not clear if transcripts/proteins directly related to Ca2+ signaling are modulated in exosomes during DCM pathogenesis. Reversal of certain changes (for example SERCA2a over-expression) has been proven to be beneficial in ameliorating DCM. However, to find wide applicability, it is necessary to develop highly selective modulators to correct aberrant cardiomyocyte Ca2+ transits and Ca2+ handling proteins.

Salient molecular changes in the regulators of the Ca2+ transits during excitation-contraction (EC) coupling associated with DCM. Arbitrary separations of the early and late-stage changes are also indicated. These changes eventually lead to the diabetic cardiomyopathic phenotypes, mitochondrial changes, inflammation, and exosome mediated effects on Ca2+ transits leading to heart failure. NCX N+/Ca2+ exchanger; PMCA Sarcolemmal/plasma membrane Ca2+-ATPase; SERCA2a Sarco(endo)plasmic reticulum Ca2+-ATPase; RYR2 Ryanodine receptor 2; LTCC L-type calcium channels; SR Sarcoplasmic reticulum; MCU Mitochondrial Ca2+uniporter; mCa2+ mitochondrial Ca2+; ROS Reactive oxygen species; mPTP Mitochondrial permeability transition pore; iCa2+ Intracellular Ca2+; MHC Myosin heavy chain
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Abbreviations
- Ca2+ :
-
Calcium
- [Ca2+]i:
-
Intracellular calcium concentrations
- CICR:
-
Calcium-induced calcium release
- MCU:
-
Mitochondrial Ca2+ uniporter
- DCM:
-
Diabetic cardiomyopathy
- EC coupling:
-
Excitation–contraction coupling
- ECM:
-
Extracellular matrix
- RyR2:
-
Ryanodine receptor 2
- SERCA2a:
-
Sarco(endo)plasmic reticulum Ca2+-ATPase
- SPEG:
-
Striated muscle preferentially expressed protein kinase
- EAT:
-
Epicardial adipose tissue
- PLN:
-
Phospholamban
- TM:
-
Tropomyosin
- HATs:
-
Histone acetyl transferases
- HDACs:
-
Histone deacetylases
- DNMTs:
-
DNA methyltransferases
- MHC:
-
Myosin heavy chain
- Na+ :
-
Sodium
- SGLT:
-
Na+-glucose cotransporter
- POAF:
-
Postoperative atrial fibrillation
- CASR:
-
Ca2+-sensing receptor
- CaMK:
-
Ca2+/calmodulin-dependent protein kinase
- VSMC:
-
Vascular smooth muscle cell
- Ang II:
-
Angiotensin II
- HFpEF:
-
Heart failure with preserved ejection fraction
- HFrEF:
-
Heart failure with reduced ejection fraction
- Yap:
-
Yes-associated protein
- CSQ:
-
Calsequestrin
- MPTP:
-
Mitochondrial permeability transition pore
- Cav3:
-
Caveolin-3
- LTCC:
-
L-type calcium channels
- CSQ:
-
Calsequestrin
- TNF-α:
-
Tumor necrosis factor-α
- MCP1:
-
Monocyte chemotactic protein-1
- ICAM1:
-
Adhesion molecule intercellular adhesion molecule-1
- VCAM1:
-
Vascular cell adhesion molecule-1
- CASR:
-
Ca2+-sensing receptor
- miRNA:
-
microRNA
- AGE:
-
Advanced glycation end products
- RAGE:
-
Receptor for advanced glycation end products
- Hsp20:
-
Heat shock protein 20
- GLUT:
-
Glucose transporter
- SIRT1:
-
Sirtuin 1
- ROS:
-
Reactive oxygen species
- MLC:
-
Myosin light chain
- TLR:
-
Toll-like receptor
- TSG101:
-
Tumor Susceptibility 101
References
Frangogiannis NG. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. 2019;125(1):117–46.
Nagaraju CK, Robinson EL, Abdesselem M, Trenson S, Dries E, Gilbert G, et al. Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure. J Am Coll Cardiol. 2019;73(18):2267–82.
Sheu SS, Sharma VK, Korth M. Voltage-dependent effects of isoproterenol on cytosolic Ca concentration in rat heart. Am J Physiol. 1987;252(4 Pt 2):H697-703.
Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245(1):C1-14.
Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation–contraction coupling in the heart. Circ Res. 2017;121(2):181–95.
Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys J. 2007;92(10):3541–55.
Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca 2+ handling in heart failure. J Clin Investig. 2005;115(3):556–64.
Murfitt L, Whiteley G, Iqbal MM, Kitmitto A. Targeting caveolin-3 for the treatment of diabetic cardiomyopathy. Pharmacol Therapeut. 2015;151:50–71.
Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, et al. Mechanisms of [Ca2+](i) transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55(3):608–15.
Bergantin LB. Diabetes and inflammatory diseases: An overview from the perspective of Ca2+/3′-5′-cyclic adenosine monophosphate signaling. World J Diabetes. 2021;12(6):767.
Lind M, Bounias I, Olsson M, Gudbjörnsdottir S, Svensson A-M, Rosengren A. Glycaemic control and incidence of heart failure in 20 985 patients with type 1 diabetes: an observational study. Lancet. 2011;378(9786):140–6.
Zhang Q, Higginbotham JN, Jeppesen DK, Yang Y-P, Li W, McKinley ET, et al. Transfer of functional cargo in exomeres. Cell Rep. 2019;27(3):940-54. e6.
Lakkaraju A, Rodriguez-Boulan E. Itinerant exosomes: emerging roles in cell and tissue polarity. Trends Cell Biol. 2008;18(5):199–209.
Bellin G, Gardin C, Ferroni L, Chachques JC, Rogante M, Mitrečić D, et al. Exosome in cardiovascular diseases: a complex world full of hope. Cells. 2019;8(2):166.
Wang X, Gu H, Huang W, Peng J, Li Y, Yang L, et al. Hsp20-mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes. 2016;65(10):3111–28.
Kannel WB, McGee DL. Diabetes and cardiovascular risk factors: the Framingham study. Circulation. 1979;59(1):8–13.
Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999;100(10):1134–46.
Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. 2003;107(3):448–54.
Liu JE, Palmieri V, Roman MJ, Bella JN, Fabsitz R, Howard BV, et al. The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J Am Coll Cardiol. 2001;37(7):1943–9.
Rydén L, Armstrong P, Cleland J, Horowitz J, Massie B, Packer M, et al. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J. 2000;21(23):1967–78.
Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, et al. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. 2005;28(3):612–6.
Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34(1):29–34.
Aronow WS, Ahn C. Incidence of heart failure in 2,737 older persons with and without diabetes mellitus. Chest. 1999;115(3):867–8.
Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12(3):144–53.
Zadok OIB, Kornowski R, Goldenberg I, Klempfner R, Toledano Y, Biton Y, et al. Admission blood glucose and 10-year mortality among patients with or without pre-existing diabetes mellitus hospitalized with heart failure. Cardiovasc Diabetol. 2017;16(1):1–9.
Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50(6):1035–43.
Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331(23):1564–75.
Abelmann WH, Lorell BH. The challenge of cardiomyopathy. J Am Coll Cardiol. 1989;13(6):1219–39.
Mellor KM, Bell JR, Ritchie RH, Delbridge LM. Myocardial insulin resistance, metabolic stress and autophagy in diabetes. Clin Exp Pharmacol Physiol. 2013;40(1):56–61.
Bluemke DA, Kronmal RA, Lima JA, Liu K, Olson J, Burke GL, et al. The relationship of left ventricular mass and geometry to incident cardiovascular events: the MESA (multi-ethnic study of atherosclerosis) study. J Am Coll Cardiol. 2008;52(25):2148–55.
Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, et al. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol. 2008;101(12):1787–91.
Pereira L, Ruiz-Hurtado G, Rueda A, Mercadier JJ, Benitah JP, Gomez AM. Calcium signaling in diabetic cardiomyocytes. Cell Calcium. 2014;56(5):372–80.
Lu ZJ, Ballou LM, Jiang YP, Cohen IS, Lin RZ. Restoration of defective L-type Ca2+ current in cardiac myocytes of type 2 diabetic db/db mice by Akt and PKC-iota. J Cardiovasc Pharm. 2011;58(4):439–45.
Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103(19):7500–5.
Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal. 2014;3(2):15–38.
Buda P, Reinbothe T, Nagaraj V, Mahdi T, Luan C, Tang Y, et al. Eukaryotic translation initiation factor 3 subunit e controls intracellular calcium homeostasis by regulation of cav1.2 surface expression. PLoS ONE. 2013;8(5):e64462.
Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol. 2010;588(10):1665–81.
Schroder E, Byse M, Satin J. L-type calcium channel C terminus autoregulates transcription. Circ Res. 2009;104(12):1373–81.
Pott C, Eckardt L, Goldhaber JI. Triple threat: the Na+/Ca2+ exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr Drug Targets. 2011;12(5):737–47.
Dhalla NS, Rangi S, Zieroth S, Xu YJ. Alterations in sarcoplasmic reticulum and mitochondrial functions in diabetic cardiomyopathy. Exp Clin Cardiol. 2012;17(3):115–20.
Schaffer SW, Ballard-Croft C, Boerth S, Allo SN. Mechanisms underlying depressed Na+/Ca2+ exchanger activity in the diabetic heart. Cardiovasc Res. 1997;34(1):129–36.
Hattori Y, Matsuda N, Kimura J, Ishitani T, Tamada A, Gando S, et al. Diminished function and expression of the cardiac Na + -Ca 2+ exchanger in diabetic rats: implication in Ca 2+ overload. J Physiol. 2000;527(1):85–94.
LaRocca TJ, Fabris F, Chen J, Benhayon D, Zhang S, McCollum L, et al. Na+/Ca2+ exchanger-1 protects against systolic failure in the Akitains2 model of diabetic cardiomyopathy via a CXCR4/NF-kappaB pathway. Am J Physiol Heart Circ Physiol. 2012;303(3):H353–67.
Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. 2004;53(12):3201–8.
Wang X, Shen E, Wang Y, Li J, Cheng D, Chen Y, et al. Cross talk between miR-214 and PTEN attenuates glomerular hypertrophy under diabetic conditions. Sci Rep. 2016;6:31506.
Harada M, Luo X, Murohara T, Yang B, Dobrev D, Nattel S. MicroRNA regulation and cardiac calcium signaling: role in cardiac disease and therapeutic potential. Circ Res. 2014;114(4):689–705.
Chandrasekaran S, Peterson RE, Mani SK, Addy B, Buchholz AL, Xu L, et al. Histone deacetylases facilitate sodium/calcium exchanger up-regulation in adult cardiomyocytes. FASEB J. 2009;23(11):3851–64.
Battiprolu PK, Lopez-Crisosto C, Wang ZV, Nemchenko A, Lavandero S, Hill JA. Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci. 2013;92(11):609–15.
Schulze DH, Muqhal M, Lederer WJ, Ruknudin AM. Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem. 2003;278(31):28849–55.
Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka HI, Shigekawa M. Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J Biol Chem. 1996;271(23):13609–15.
Zhang YH, Hancox JC. Regulation of cardiac Na+–Ca2+ exchanger activity by protein kinase phosphorylation—still a paradox? Cell Calcium. 2009;45(1):1–10.
Schaffer S. Mechanisms underlying depressed Na+/Ca2+ exchanger activity in the diabetic heart. Cardiovasc Res. 1997;34(1):129–36.
Takeda N, Dixon IM, Hata T, Elimban V, Shah KR, Dhalla NS. Sequence of alterations in subcellular organelles during the development of heart dysfunction in diabetes. Diabetes Res Clin Pract. 1996;30(Suppl):113–22.
Golfman L, Dixon IM, Takeda N, Lukas A, Dakshinamurti K, Dhalla NS. Cardiac sarcolemmal Na(+)-Ca2+ exchange and Na(+)-K+ ATPase activities and gene expression in alloxan-induced diabetes in rats. Mol Cell Biochem. 1998;188(1–2):91–101.
Sheikh AQ, Hurley JR, Huang W, Taghian T, Kogan A, Cho H, et al. Diabetes alters intracellular calcium transients in cardiac endothelial cells. PLoS ONE. 2012;7(5): e36840.
Matsukubo MP, Singal PK, Dhalla NS. Negatively charged sites and calcium binding in the isolated rat heart sarcolemma. Basic Res Cardiol. 1981;76(1):16–28.
Frank JS, Langer GA, Nudd LM, Seraydarian K. The myocardial cell surface, its histochemistry, and the effect of sialic acid and calcium removal on its stucture and cellular ionic exchange. Circ Res. 1977;41(5):702–14.
Pierce GN, Kutryk MJ, Dhalla NS. Alterations in Ca2+ binding by and composition of the cardiac sarcolemmal membrane in chronic diabetes. Proc Natl Acad Sci USA. 1983;80(17):5412–6.
Pierce GN, Ganguly PK, Dzurba A, Dhalla NS. Modification of the function of cardiac subcellular organelles by insulin. Adv Myocardiol. 1985;6:113–25.
Chattergoon NN, Giraud GD, Louey S, Stork P, Fowden AL, Thornburg KL. Thyroid hormone drives fetal cardiomyocyte maturation. FASEB J. 2012;26(1):397–408.
Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–79.
Bicknell KA, Coxon CH, Brooks G. Forced expression of the cyclin B1–CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem J. 2004;382(Pt 2):411.
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019;569(7756):418–22.
Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, et al. YAP partially reprograms chromatin accessibility to directly induce adult cardiogenesis in vivo. Dev Cell. 2019;48(6):765-79. e7.
Castillo EC, Vázquez-Garza E, Yee-Trejo D, García-Rivas G, Torre-Amione G. What is the role of the inflammation in the pathogenesis of heart failure? Curr Cardiol Rep. 2020;22(11):1–15.
Tate M, Deo M, Cao AH, Hood SG, Huynh K, Kiriazis H, et al. Insulin replacement limits progression of diabetic cardiomyopathy in the low-dose streptozotocin-induced diabetic rat. Diab Vasc Dis Res. 2017;14(5):423–33.
Malhotra A, Sanghi V. Regulation of contractile proteins in diabetic heart. Cardiovasc Res. 1997;34(1):34–40.
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–58.
Russell RR III, Yin R, Caplan MJ, Hu X, Ren J, Shulman GI, et al. Additive effects of hyperinsulinemia and ischemia on myocardial GLUT1 and GLUT4 translocation in vivo. Circulation. 1998;98(20):2180–6.
Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J. 2010;31(1):100–11.
Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–68.
Somanna NK, Yariswamy M, Garagliano JM, Siebenlist U, Mummidi S, Valente AJ, et al. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell Signal. 2015;27(10):1928–38.
Brette F, Despa S, Bers DM, Orchard CH. Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol. 2005;39(5):804–12.
Louch WE, Mørk HK, Sexton J, Strømme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574(2):519–33.
Heinzel FR, MacQuaide N, Biesmans L, Sipido K. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol. 2011;50(3):390–400.
Stølen TO, Høydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105(6):527–36.
Frisk M, Le C, Shen X, Røe ÅT, Hou Y, Manfra O, et al. Etiology-dependent impairment of diastolic cardiomyocyte calcium homeostasis in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2021;77(4):405–19.
Frisk M, Ruud M, Espe EK, Aronsen JM, Røe ÅT, Zhang L, et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc Res. 2016;112(1):443–51.
Jones PP, MacQuaide N, Louch WE. Dyadic plasticity in cardiomyocytes. Front Physiol. 2018;9:1773.
Ward M-L, Crossman DJ. Mechanisms underlying the impaired contractility of diabetic cardiomyopathy. World J Cardiol. 2014;6(7):577.
Setterberg IE, Le C, Frisk M, Perdreau-Dahl H, Li J, Louch WE. The physiology and pathophysiology of T-tubules in the heart. Front Physiol. 2021;12:718404.
MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Bio. 2003;4(7):566–77.
Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circ Res. 2012;110(12):1646–60.
Kim HW, Cho YS, Lee HR, Park SY, Kim YH. Diabetic alterations in cardiac sarcoplasmic reticulum Ca2+-ATPase and phospholamban protein expression. Life Sci. 2001;70(4):367–79.
Trost SU, Belke DD, Bluhm WF, Meyer M, Swanson E, Dillmann WH. Overexpression of the sarcoplasmic reticulum Ca2+-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes. 2002;51(4):1166–71.
Zarain-Herzberg A, Garcia-Rivas G, Estrada-Aviles R. Regulation of SERCA pumps expression in diabetes. Cell Calcium. 2014;56(5):302–10.
Bidasee KR, Zhang YN, Shao CH, Wang M, Patel KP, Dincer UD, et al. Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53(2):463–73.
Liebisch M, Bondeva T, Franke S, Daniel C, Amann K, Wolf G. Activation of the receptor for advanced glycation end products induces nuclear inhibitor of protein phosphatase-1 suppression. Kidney Int. 2014;86(1):103–17.
Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, et al. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. 2015;10(5):418–30.
Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. 2015;14:99.
Quan C, Du Q, Li M, Wang R, Ouyang Q, Su S, et al. A PKB-SPEG signaling nexus links insulin resistance with diabetic cardiomyopathy by regulating calcium homeostasis. Nat Commun. 2020;11(1):1–14.
Joseph LC, Subramanyam P, Radlicz C, Trent CM, Iyer V, Colecraft HM, et al. Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm. 2016;13(8):1699–706.
Sommese L, Valverde CA, Blanco P, Castro MC, Rueda OV, Kaetzel M, et al. Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca2+ release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int J Cardiol. 2016;202:394–406.
Tuncay E, Okatan EN, Toy A, Turan B. Enhancement of cellular antioxidant-defence preserves diastolic dysfunction via regulation of both diastolic Zn2+ and Ca2+ and prevention of RyR2-leak in hyperglycemic cardiomyocytes. Oxid Med Cell Longev. 2014;2014:290381.
Zhao SM, Wang YL, Guo CY, Chen JL, Wu YQ. Progressive decay of Ca2+ homeostasis in the development of diabetic cardiomyopathy. Cardiovasc Diabetol. 2014;13:75.
Yaras N, Ugur M, Ozdemir S, Gurdal H, Purali N, Lacampagne A, et al. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54(11):3082–8.
Waddingham MT, Edgley AJ, Tsuchimochi H, Kelly DJ, Shirai M, Pearson JT. Contractile apparatus dysfunction early in the pathophysiology of diabetic cardiomyopathy. World J Diabetes. 2015;6(7):943–60.
Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2(11): a003996.
Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol. 2011;3(6): a004317.
Cheng YS, Dai DZ, Ji H, Zhang Q, Dai Y. Sildenafil and FDP-Sr attenuate diabetic cardiomyopathy by suppressing abnormal expression of myocardial CASQ2, FKBP12.6, and SERCA2a in rats. Acta Pharmacol Sin. 2011;32(4):441–8.
Howarth FC, Glover L, Culligan K, Qureshi MA, Ohlendieck K. Calsequestrin expression and calcium binding is increased in streptozotocin-induced diabetic rat skeletal muscle though not in cardiac muscle. Pflugers Arch. 2002;444(1–2):52–8.
Fu M, Damcott CM, Sabra M, Pollin TI, Ott SH, Wang J, et al. Polymorphism in the calsequestrin 1 (CASQ1) gene on chromosome 1q21 is associated with type 2 diabetes in the old order Amish. Diabetes. 2004;53(12):3292–9.
Hong YS, Lee H, Oh J-Y, Sung Y-A, Kim Y. Increased epicardial adipose tissue thickness in type 2 diabetes mellitus and obesity. Diabetes Metab J. 2015;39(5):405–13.
Dubois SG, Heilbronn LK, Smith SR, Albu JB, Kelley DE, Ravussin E, et al. Decreased expression of adipogenic genes in obese subjects with type 2 diabetes. Obesity. 2006;14(9):1543–52.
Nasarre L, Juan-Babot O, Gastelurrutia P, Llucia-Valldeperas A, Badimon L, Bayes-Genis A, et al. Low density lipoprotein receptor–related protein 1 is upregulated in epicardial fat from type 2 diabetes mellitus patients and correlates with glucose and triglyceride plasma levels. Acta Diabetol. 2014;51(1):23–30.
Chechi K, Richard D. Thermogenic potential and physiological relevance of human epicardial adipose tissue. Int J Obes Suppl. 2015;5(1):S28–34.
Hatem SN, Redheuil A, Gandjbakhch E. Cardiac adipose tissue and atrial fibrillation: the perils of adiposity. Cardiovasc Res. 2016;109(4):502–9.
Greulich S, de Wiza DH, Preilowski S, Ding Z, Mueller H, Langin D, et al. Secretory products of guinea pig epicardial fat induce insulin resistance and impair primary adult rat cardiomyocyte function. J Cell Mol Med. 2011;15(11):2399–410.
Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion. 2012;12(1):77–85.
Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–23.
Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–71.
Shahbaz AU, Zhao T, Zhao W, Johnson PL, Ahokas RA, Bhattacharya SK, et al. Calcium and zinc dyshomeostasis during isoproterenol-induced acute stressor state. Am J Physiol Heart Circ Physiol. 2011;300(2):H636–44.
Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure—a rational approach for disease treatment. Cell Metab. 2015;21(2):183–94.
Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, et al. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58(9):1986–97.
Turko IV. Quantitative protein profiling in heart mitochondria from diabetic rats. J Biol Chem. 2003;278(37):35844–9.
Shi Y. Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J Biomed Res. 2010;24(1):6–15.
Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, et al. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57(11):2924–32.
Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56(10):2457–66.
Veeranki S, Givvimani S, Kundu S, Metreveli N, Pushpakumar S, Tyagi SC. Moderate intensity exercise prevents diabetic cardiomyopathy associated contractile dysfunction through restoration of mitochondrial function and connexin 43 levels in db/db mice. J Mol Cell Cardiol. 2016;92:163–73.
Hafstad AD, Boardman N, Aasum E. How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22(17):1587–605.
Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3(1):1–12.
Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell. 2012;151(3):630–44.
Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, et al. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem. 2012;287(41):34445–54.
Samanta K, Douglas S, Parekh AB. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS ONE. 2014;9(7): e101188.
De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40.
Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–72.
Rimessi A, Pozzato C, Carparelli L, Rossi A, Ranucci S, De Fino I, et al. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci Adv. 2020;6(19):e9093.
Wu Y, Rasmussen TP, Koval OM, Mei-ling AJ, Hall DD, Chen B, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6(1):1–13.
Rasmussen TP, Wu Y, Mei-ling AJ, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci. 2015;112(29):9129–34.
Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci. 2012;109(32):12986–91.
Diaz-Juarez J, Suarez J, Cividini F, Scott BT, Diemer T, Dai A, Dillmann WH. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am J Physiol Cell Physiol. 2016;311(6):C1005–C1013.
Gregorio CC, Antin PB. To the heart of myofibril assembly. Trends Cell Biol. 2000;10(9):355–62.
Gupta MP. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol. 2007;43(4):388–403.
Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101(19):2271–6.
Jorgensen PG, Jensen MT, Mogelvang R, Fritz-Hansen T, Galatius S, Biering-Sorensen T, et al. Impact of type 2 diabetes and duration of type 2 diabetes on cardiac structure and function. Int J Cardiol. 2016;221:114–21.
Schwartz K, Lompre AM, Bouveret P, Wisnewsky C, Whalen RG. Comparisons of rat cardiac myosins at fetal stages in young animals and in hypothyroid adults. J Biol Chem. 1982;257(23):14412–8.
Walklate J, Ujfalusi Z, Geeves MA. Myosin isoforms and the mechanochemical cross-bridge cycle. J Exp Biol. 2016;219(Pt 2):168–74.
Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86(4):386–90.
Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, et al. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol. 2000;32(6):985–96.
Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15(1):11–9.
Haddad F, Bodell PW, Qin AX, Giger JM, Baldwin KM. Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J Biol Chem. 2003;278(39):37132–8.
MacLean IM, Rajotte RV, Belcastro AN. Insulin and islet cell transplants: effects on diabetic rat cardiac myofibril ATPase. Am J Physiol. 1987;252(2 Pt 1):E244–7.
Liu X, Takeda N, Dhalla NS. Troponin I phosphorylation in heart homogenate from diabetic rat. Biochim Biophys Acta. 1996;1316(2):78–84.
Wattanapermpool J, Guo X, Solaro RJ. The unique amino-terminal peptide of cardiac troponin I regulates myofibrillar activity only when it is phosphorylated. J Mol Cell Cardiol. 1995;27(7):1383–91.
Wijnker PJ, Sequeira V, Witjas-Paalberends ER, Foster DB, dos Remedios CG, Murphy AM, et al. Phosphorylation of protein kinase C sites Ser42/44 decreases Ca(2+)-sensitivity and blunts enhanced length-dependent activation in response to protein kinase A in human cardiomyocytes. Arch Biochem Biophys. 2014;554:11–21.
Noland TA Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, et al. Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry. 1996;35(47):14923–31.
Liu X, Takeda N, Dhalla NS. Myosin light-chain phosphorylation in diabetic cardiomyopathy in rats. Metabolism. 1997;46(1):71–5.
Morano I, Piazzesi G, Ruegg JC. Myofibrillar calcium sensitivity modulation: influence of light chain phosphorylation and positive inotropic drugs on skinned frog skeletal muscle. Adv Exp Med Biol. 1988;226:129–37.
Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc. 2015;4(9): e002183.
Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia. 2017;60(3):568–573. https://doi.org/10.1007/s00125-016-4134-x.
Kjeldsen K, Braendgaard H, Sidenius P, Larsen JS, Norgaard A. Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes. 1987;36(7):842–8.
Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium. 2012;52(1):28–35.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.
Kengne AP, Batty GD, Hamer M, Stamatakis E, Czernichow S. Association of C-reactive protein with cardiovascular disease mortality according to diabetes status: pooled analyses of 25,979 participants from four UK prospective cohort studies. Diabetes Care. 2012;35(2):396–403.
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Investig. 2006;116(7):1793–801.
Marques-Vidal P, Schmid R, Bochud M, Bastardot F, Von Känel R, Paccaud F, et al. Adipocytokines, hepatic and inflammatory biomarkers and incidence of type 2 diabetes. The CoLaus study. Plos ONE. 2012;7(12):e51768.
Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85.
Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40(3):315–27.
Frati G, Schirone L, Chimenti I, Yee D, Biondi-Zoccai G, Volpe M, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. 2017;113(4):378–88.
Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the α calcium-calmodulin kinase II in LTP and learning. Science. 1998;279(5352):870–3.
Stratton IM, Adler AI, Neil HAW, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321(7258):405–12.
Stangl V, Baumann G, Stangl K, Felix SB. Negative inotropic mediators released from the heart after myocardial ischaemia–reperfusion. Cardiovasc Res. 2002;53(1):12–30.
Van Linthout S, Tschöpe C. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep. 2017;14(4):251–65.
Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, et al. Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res. 2020;127(8):1036–55.
Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca 2+ and cAMP. Nature. 2012;492(7427):123–7.
Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 Inflammasome activation initiated in response to pressure overload by CaMKIIδ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138(22):2530.
Delgado C, Gomez A-M, El Hayek MS, Ruiz-Hurtado G, Pereira L. Gender-dependent alteration of Ca2+ and TNFα signaling in db/db mice, an obesity-linked type 2 diabetic model. Front Physiol. 2019;10:40.
McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. Biochem Biophys Res Commun. 1997;239(2):357–66.
Sloan-Lancaster J, Allen PM. Altered peptide ligand–induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14(1):1–27.
Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184(1):1–8.
Bullens DM, Rafiq K, Charitidou L, Peng X, Kasran A, Warmerdam PA, et al. Effects of co-stimulation by CD58 on human T cell cytokine production: a selective cytokine pattern with induction of high IL-10 production. Int Immunol. 2001;13(2):181–91.
Rafiq K, Charitidou L, Bullens D, Kasran A, Lorre K, Ceuppens J, et al. Regulation of the IL-10 production by human T cells. Scand J Immunol. 2001;53(2):139–47.
Hama N, Paliogianni F, Fessler BJ, Boumpas DT. Calcium/calmodulin-dependent protein kinase II downregulates both calcineurin and protein kinase C-mediated pathways for cytokine gene transcription in human T cells. J Exp Med. 1995;181(3):1217–22.
Nghiem P, Ollick T, Gardner P, Schulman H. lnterleukin-2 transcriptional block by multifunctional Ca 2+/calmodulin kinase. Nature. 1994;371(6495):347–50.
Lin MY, Zal T, Chen IL, Gascoigne NR, Hedrick SM. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: from activation to unresponsiveness. J Immunol. 2005;174(9):5583–92.
Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. MyD88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52(5):1135–44.
Singh MV, Anderson ME. Is CaMKII a link between inflammation and hypertrophy in heart? J Mol Med. 2011;89(6):537–43.
Rathi SS, Xu Y-J, Dhalla NS. Mechanism of cardioprotective action of TNF-α in the isolated rat heart. Exp Clin Cardiol. 2002;7(2–3):146.
Zhang M, Xu Y-J, Saini HK, Turan B, Liu PP, Dhalla NS. TNF-α as a potential mediator of cardiac dysfunction due to intracellular Ca2+-overload. Biochem Biophys Res Commun. 2005;327(1):57–63.
Bui JD, Calbo S, Hayden-Martinez K, Kane LP, Gardner P, Hedrick SM. A role for CaMKII in T cell memory. Cell. 2000;100(4):457–67.
Boubali S, Liopeta K, Virgilio L, Thyphronitis G, Mavrothalassitis G, Dimitracopoulos G, et al. Calcium/calmodulin-dependent protein kinase II regulates IL-10 production by human T lymphocytes: a distinct target in the calcium dependent pathway. Mol Immunol. 2012;52(2):51–60.
Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X. CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood. 2008;112(13):4961–70.
D’Espessailles A, Mora YA, Fuentes C, Cifuentes M. Calcium-sensing receptor activates the NLRP3 inflammasome in LS14 preadipocytes mediated by ERK1/2 signaling. J Cell Physiol. 2018;233(8):6232–40.
Hendy GN, Canaff L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin Cell Dev Biol. 2016;49:37–43.
Zhang X, Hong S, Qi S, Liu W, Zhang X, Shi Z, Chen W, Zhao M, Yin X. NLRP3 inflammasome is involved in calcium-sensing receptor-induced aortic remodeling in SHRs. Mediat Inflamm. 2019;2019:6847087.
Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca 2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3(1):1–9.
Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, et al. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci. 2013;110(24):9806–11.
Bush EW, McKinsey TA. Targeting histone deacetylases for heart failure. Expert Opin Ther Targets. 2009;13(7):767–84.
Chen Y, Du J, Zhao YT, Zhang L, Lv G, Zhuang S, et al. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol. 2015;14(1):1–13.
Wu Y, Leng Y, Meng Q, Xue R, Zhao B, Zhan L, Xia Z. Suppression of excessive histone deacetylases activity in diabetic hearts attenuates myocardial ischemia/reperfusion injury via mitochondria apoptosis pathway. J Diabetes Res. 2017;2017:8208065.
Harris LG, Wang SH, Mani SK, Kasiganesan H, Chou CJ, Menick DR. Evidence for a non-canonical role of HDAC5 in regulation of the cardiac Ncx1 and Bnp genes. Nucleic Acids Res. 2016;44(8):3610–7.
Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018;24(1):62–72.
Syren P, Rahm A-K, Schweizer PA, Bruehl C, Katus HA, Frey N, et al. Histone deacetylase 2-dependent ventricular electrical remodeling in a porcine model of early heart failure. Life Sci. 2021;281: 119769.
Madsen A, Höppner G, Krause J, Hirt MN, Laufer SD, Schweizer M, et al. An important role for DNMT3A-mediated DNA methylation in cardiomyocyte metabolism and contractility. Circulation. 2020;142(16):1562–78.
Kumar S, Kim Y-R, Vikram A, Naqvi A, Li Q, Kassan M, et al. Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proc Natl Acad Sci. 2017;114(7):1714–9.
Wei D, Loeken MR. Increased DNA methyltransferase 3b (Dnmt3b)-mediated CpG island methylation stimulated by oxidative stress inhibits expression of a gene required for neural tube and neural crest development in diabetic pregnancy. Diabetes. 2014;63(10):3512–22.
Turunen MP, Aavik E, Ylä-Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta. 2009;1790(9):886–91.
Yideng J, Jianzhong Z, Ying H, Juan S, Jinge Z, Shenglan W, et al. Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol. 2007;26(8):603–11.
Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49(8):1292–6.
Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9(1):1–18.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83.
Patil M, Henderson J, Luong H, Annamalai D, Sreejit G, Krishnamurthy P. The art of intercellular wireless communications: exosomes in heart disease and therapy. Front Cell Dev Biol. 2019;7:315.
Govindappa PK, Patil M, Garikipati VNS, Verma SK, Saheera S, Narasimhan G, et al. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 2020;34(2):2238–51.
Babu SS, Thandavarayan RA, Joladarashi D, Jeyabal P, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci Rep. 2016;6(1):1–12.
Joladarashi D, Garikipati VNS, Thandavarayan RA, Verma SK, Mackie AR, Khan M, et al. Enhanced cardiac regenerative ability of stem cells after ischemia-reperfusion injury: role of human CD34+ cells deficient in microRNA-377. J Am Coll Cardiol. 2015;66(20):2214–26.
Jeyabal P, Thandavarayan RA, Joladarashi D, Babu SS, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016;471(4):423–9.
Hathaway QA, Pinti MV, Durr AJ, Waris S, Shepherd DL, Hollander JM. Regulating microRNA expression: at the heart of diabetes mellitus and the mitochondrion. Am J Physiol Heart Circ Physiol. 2018;314(2):H293–310.
Tijskens P, Jones LR, Franzini-Armstrong C. Junctin and calsequestrin overexpression in cardiac muscle: the role of junctin and the synthetic and delivery pathways for the two proteins. J Mol Cell Cardiol. 2003;35(8):961–74.
Yildirim SS, Akman D, Catalucci D, Turan B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: junctin as a target protein of miR-1. Cell Biochem Biophys. 2013;67(3):1397–408.
Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS, et al. Cardiac remodeling and atrial fibrillation in transgenic miceoverexpressing junctin. FASEB J. 2002;16(10):1310–2.
Wahlquist C, Jeong D, Rojas-Muñoz A, Kho C, Lee A, Mitsuyama S, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508(7497):531–5.
Su X, Liang H, Wang H, Chen G, Jiang H, Wu Q, et al. Over-expression of microRNA-1 causes arrhythmia by disturbing intracellular trafficking system. Sci Rep. 2017;7(1):1–12.
De Giusti CJ, Roman B, Das S. The influence of MicroRNAs on mitochondrial calcium. Front Physiol. 2018;9:1291.
Carbonell T, Gomes AV. MicroRNAs in the regulation of cellular redox status and its implications in myocardial ischemia-reperfusion injury. Redox Biol. 2020;36:101607.
Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. 2017;25(1):192–204.
Mayourian J, Ceholski DK, Gorski PA, Mathiyalagan P, Murphy JF, Salazar SI, et al. Exosomal microRNA-21-5p mediates mesenchymal stem cell paracrine effects on human cardiac tissue contractility. Circ Res. 2018;122(7):933–44.
Messenger SW, Woo SS, Sun Z, Martin TF. A Ca2+-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J Cell Biol. 2018;217(8):2877–90.
Ritterhoff J, Most P. Targeting S100A1 in heart failure. Gene Ther. 2012;19(6):613–21.
Most P, Seifert H, Gao E, Funakoshi H, Völkers M, Heierhorst J, et al. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation. 2006;114(12):1258–68.
Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115(19):2506–15.
Pleger ST, Remppis A, Heidt B, Völkers M, Chuprun JK, Kuhn M, et al. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005;12(6):1120–9.
Pleger ST, Shan C, Ksienzyk J, Bekeredjian R, Boekstegers P, Hinkel R, et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011;3(92):92ra64.
Kraus C, Rohde D, Weidenhammer C, Qiu G, Pleger ST, Voelkers M, et al. S100A1 in cardiovascular health and disease: closing the gap between basic science and clinical therapy. J Mol Cell Cardiol. 2009;47(4):445–55.
Gao MH, Tang T, Guo T, Sun SQ, Feramisco JR, Hammond HK. Adenylyl cyclase type VI gene transfer reduces phospholamban expression in cardiac myocytes via activating transcription factor 3. J Biol Chem. 2004;279(37):38797–802.
Lai NC, Roth DM, Gao MH, Tang T, Dalton N, Lai YY, et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation. 2004;110(3):330–6.
Hammond HK, Penny WF, Traverse JH, Henry TD, Watkins MW, Yancy CW, et al. Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: a randomized clinical trial. JAMA Cardiol. 2016;1(2):163–71.
Penny WF, Henry TD, Watkins MW, Patel AN, Hammond HK. Design of a Phase 3 trial of intracoronary administration of human adenovirus 5 encoding human adenylyl cyclase type 6 (RT-100) gene transfer in patients with heart failure with reduced left ventricular ejection fraction: the FLOURISH clinical trial. Am Heart J. 2018;201:111–6.
Del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100(23):2308–11.
del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci. 2004;101(15):5622–7.
Del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation. 2001;104(12):1424–9.
Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51(11):1112–9.
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID) a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–13.
Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, et al. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res. 2014;114(1):101–8.
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387(10024):1178–86.
Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Cardiac Fail. 2009;15(3):171–81.
Micheletti R, Mattera G, Rocchetti M, Schiavone A, Loi M, Zaza A, et al. Pharmacological profile of the novel inotropic agent (E, Z)-3-((2-aminoethoxy) imino) androstane-6, 17-dione hydrochloride (PST2744). J Pharmacol Exp Ther. 2002;303(2):592–600.
Ferrandi M, Barassi P, Tadini-Buoninsegni F, Bartolommei G, Molinari I, Tripodi MG, et al. Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br J Pharmacol. 2013;169(8):1849–61.
Micheletti R, Palazzo F, Barassi P, Giacalone G, Ferrandi M, Schiavone A, et al. Istaroxime, a stimulator of sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am J Cardiol. 2007;99(2):S24–32.
Sabbah HN, Imai M, Cowart D, Amato A, Carminati P, Gheorghiade M. Hemodynamic properties of a new-generation positive luso-inotropic agent for the acute treatment of advanced heart failure. Am J Cardiol. 2007;99(2):S41–6.
Ghali JK, Smith WB, Torre-Amione G, Haynos W, Rayburn BK, Amato A, et al. A phase 1–2 dose-escalating study evaluating the safety and tolerability of istaroxime and specific effects on electrocardiographic and hemodynamic parameters in patients with chronic heart failure with reduced systolic function. Am J Cardiol. 2007;99(2):S47–56.
Shah SJ, Blair JE, Filippatos GS, Macarie C, Ruzyllo W, Korewicki J, et al. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the hemodynamic, echocardiographic, and neurohormonal effects of istaroxime, a novel intravenous inotropic and lusitropic agent: a randomized controlled trial in patients hospitalized with heart failure (HORIZON-HF) trial. Am Heart J. 2009;157(6):1035–41.
Tita C, Gilbert EM, Van Bakel AB, Grzybowski J, Haas GJ, Jarrah M, et al. A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur J Heart Fail. 2017;19(10):1321–32.
Felker GM, Borentain M, Cleland JG, DeSouza MM, Kessler PD, O’Connor CM, et al. Rationale and design for the development of a novel nitroxyl donor in patients with acute heart failure. Eur J Heart Fail. 2019;21(8):1022–31.
Galderisi M, Anderson KM, Wilson PW, Levy D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am J Cardiol. 1991;68(1):85–9.
Hadour G, Ferrera R, Sebbag L, Forrat R, Delaye J and de Lorgeril M. Improved myocardial tolerance to ischaemia in the diabetic rabbit. J Mol Cell Cardiol. 1998;30(9):1869–75.
Joffe, II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, et al. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. J Am Coll Cardiol. 1999;34(7):2111–9.
Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279(5):E1104–13.
Ilercil A, Devereux RB, Roman MJ, Paranicas M, O'Grady M J, Welty TK, et al. Relationship of impaired glucose tolerance to left ventricular structure and function: The Strong Heart Study. Am Heart J. 2001;141(6):992–8.
Carugo S, Giannattasio C, Calchera I, Paleari F, Gorgoglione MG, Grappiolo A, et al. Progression of functional and structural cardiac alterations in young normotensive uncomplicated patients with type 1 diabetes mellitus. J Hypertens. 2001;19(9):1675–80.
Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50(6):1414–24.
Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology. 2002;98(1-2):33–9.
Nielsen LB, Bartels ED, Bollano E. Overexpression of apolipoprotein B in the heart impedes cardiac triglyceride accumulation and development of cardiac dysfunction in diabetic mice. J Biol Chem. 2002;277(30):27014–20.
Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADPribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–21.
Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283(3):H976–82.
Young ME, Wilson CR, Razeghi P, Guthrie PH, Taegtmeyer H. Alterations of the circadian clock in the heart by streptozotocin-induced diabetes. J Mol Cell Cardiol. 2002;34(2):223–31.
Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52(2):434–41.
Barouch LA, Berkowitz DE, Harrison RW, O'Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation. 2003;108(6):754–9.
Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, et al. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144(8):3483–90.
Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes care. 2003;26(8):2433–41.
Conti M, Renaud IM, Poirier B, Michel O, Belair MF, Mandet C, et al. High levels of myocardial antioxidant defense in aging nondiabetic normotensive Zucker obese rats. Am J Physiol Regul Integr Comp Physiol. 2004;286(4):R793–800.
Desrois M, Sidell RJ, Gauguier D, Davey CL, Radda GK, Clarke K. Gender differences in hypertrophy, insulin resistance and ischemic injury in the aging type 2 diabetic rat heart. J Mol Cell Cardiol. 2004;37(2):547–55.
Kristiansen SB, Lofgren B, Stottrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, et al. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004;47(10):1716–21.
Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53(9):2366–74.
Broderick TL, Hutchison AK. Cardiac dysfunction in the euglycemic diabetic prone BB Wor rat. Metabolism. 2004;53(11):1391–4.
Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–95.
Broderick TL, Poirier P. Cardiac function and ischaemic tolerance during acute loss of metabolic control in the diabetic BB Wor rat. Acta diabetologica. 2005;42(4):171–8.
Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146(12):5341–9.
Kralik PM, Ye G, Metreveli NS, Shem X, Epstein PN. Cardiomyocyte dysfunction in models of type 1 and type 2 diabetes. Cardiovasc Toxicol. 2005;5(3):285–92.
Vasanji Z, Cantor EJ, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am J Physiol Cell Physiol. 2006;291(4):C772–80.
Suarez J, Scott B, Dillmann WH. Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1439–45.
Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, et al. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol. 2009;297(6):H2096–108.
Daniels A, van Bilsen M, Janssen BJ, Brouns AE, Cleutjens JP, Roemen TH, et al. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol (Oxf). 2010;200(1):11–22.
Bupha-Intr T, Oo YW, Wattanapermpool J. Increased myocardial stiffness with maintenance of length-dependent calcium activation by female sex hormones in diabetic rats. Am J Physiol Heart Circ Physiol. 2011;300(5):H1661–8.
Howarth FC, Qureshi MA, Hassan Z, Al Kury LT, Isaev D, Parekh K, et al. Changing pattern of gene expression is associated with ventricular myocyte dysfunction and altered mechanisms of Ca2+ signalling in young type 2 Zucker diabetic fatty rat heart. Exp Physiol. 2011;96(3):325–37.
Patel VB, Bodiga S, Basu R, Das SK, Wang W, Wang Z, et al. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res. 2012;110(10):1322–35.
Salem KA, Adrian TE, Qureshi MA, Parekh K, Oz M, Howarth FC. Shortening and intracellular Ca2+ in ventricular myocytes and expression of genes encoding cardiac muscle proteins in early onset type 2 diabetic Goto-Kakizaki rats. Exp Physiol. 2012;97(12):1281–91.
Takada A, Miki T, Kuno A, Kouzu H, Sunaga D, Itoh T, et al. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. Plos One. 2012;7(6):e39893.
Namekata I, Hamaguchi S, Wakasugi Y, Ohhara M, Hirota Y, Tanaka H. Ellagic acid and gingerol, activators of the sarco-endoplasmic reticulum Ca(2)(+) ATPase, ameliorate diabetes mellitus-induced diastolic dysfunction in isolated murine ventricular myocardia. Eur J Pharmacol. 2013;706(1–3):48–55.
Lamberts RR, Lingam SJ, Wang HY, Bollen IA, Hughes G, Galvin IF, et al. Impaired relaxation despite upregulated calcium-handling protein atrial myocardium from type 2 diabetic patients with preserved ejection fraction. Cardiovasc Diabetol. 2014;13:72.
Cioffi G, Rossi A, Targher G, Zoppini G, de Simone G, Devereux RB, et al. Usefulness of subclinical left ventricular midwall dysfunction to predict cardiovascular mortality in patients with type 2 diabetes mellitus. Am J Cardiol. 2014;113(8):1409–14.
Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130(7):554–64.
Thomas CM, Yong QC, Rosa RM, Seqqat R, Gopal S, Casarini DE, et al. Cardiacspecific suppression of NF-kappaB signaling prevents diabetic cardiomyopathy via inhibition of the renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2014;307(7):H1036–45.
Tocchetti CG, Stanley BA, Sivakumaran V, Bedja D, O'Rourke B, Paolocci N, et al. Impaired mitochondrial energy supply coupled to increased H2O2 emission under energy/redox stress leads to myocardial dysfunction during Type I diabetes. Clin Sci (Lond). 2015;129(7):561–74.
Ruiz M, Coderre L, Lachance D, Houde V, Martel C, Thompson Legault J, et al. MK2 Deletion in Mice Prevents Diabetes-Induced Perturbations in Lipid Metabolism and Cardiac Dysfunction. Diabetes. 2016;65(2):381–92.
Funding
None.
Author information
Authors and Affiliations
Contributions
SKS planned and designed the overall framework of the review article. SD, SV and KN gathered and analyzed available literature, original studies, extracted the required information, and wrote the first draft of the manuscript. SD and SV prepared the figures and table for the manuscipt. SKS and PK corrected and validated the content of the manuscript in its entirety. The authors declare that there is no duality of interest associated with this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors listed on this manuscript have declared that there is no conflict of interest associated with this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sanganalmath, S.K., Dubey, S., Veeranki, S. et al. The interplay of inflammation, exosomes and Ca2+ dynamics in diabetic cardiomyopathy. Cardiovasc Diabetol 22, 37 (2023). https://doi.org/10.1186/s12933-023-01755-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-023-01755-1